通过具有不定误差的读段的追踪重构
文献发布时间:2023-06-19 10:36:57
背景技术
当今世界上许多数据被存储在磁性和光学介质上。磁带技术最近发现存储185TB的单个磁带盒的显著密度提高,并且是当今商业上可用的最密集存储形式,大约为10GB/mm
对数据存储的需求呈指数增长,但是现有存储介质的容量没有跟上。脱氧核糖核酸(DNA)的聚合物能够以高密度存储信息。理论密度极限为1艾字节/mm
尽管DNA存储系统具有优点,但它必须克服若干挑战。例如,DNA合成、存储期间的降解以及测序都是潜在的误差来源。因此,由测序仪输出的DNA序列可能与最初被提供给寡核苷酸合成仪的DNA序列不同。
发明内容
该发明内容被提供来以简化的形式介绍对于下面在详细描述中进一步被描述的概念的选择。该发明内容不旨在标识所要求保护的主题的关键特征或者必要特征,也不旨在被用于限制所要求保护的主题的范围。
当前由计算机用于存储文本文件、音频文件、视频文件、软件等种类的二进制数据可以被表示为多核苷酸中的一系列核酸(即,DNA或核糖核酸(RNA))。有多种技术用于将二进制数据的0和1表示为一系列核苷酸。多种技术是本领域的普通技术人员已知的。多核苷酸序列被设计为保存二进制数据,然后用寡核苷酸合成仪来合成。合成的多核苷酸被放入存储装置中,最终由多核苷酸测序仪读取。由多核苷酸测序仪生成的数据被译码以恢复所存储的二进制数据。写入和读取多核苷酸序列的机器并非100%准确,并且会引入误差。一些类型的误差(诸如核苷酸的插入、缺失或取代)可以被标识和校正。其他类型的误差(尤其是“突发性”误差,其中在彼此相邻或接近的局部“突发”中存在多个误差)可能很难或无法校正。因此,利用一些译码技术,包括突发性误差的序列读段(read)可能无法使用。本公开提供了从多核苷酸序列的读段提取有用信息的技术,该多核苷酸序列包括突发性误差或其他类型的不定误差(indeterminant error)。
一批多核苷酸的多核苷酸测序可以生成的读段的总长度比测序批次中所有多核苷酸的总长度长很多倍。这被称为“覆盖深度”。覆盖深度大于1(例如10×、20×、30×等)指示平均而言,每个多核苷酸被提供给测序仪的次数由覆盖深度指示。因为这是一个平均值,所以一些多核苷酸可能根本无法被测序,而其他多核苷酸被测序的次数却比覆盖级别多出许多倍。针对单次测序运行,覆盖深度可以通过所测序的碱基对总数除以提供给测序机器的多核苷酸的总长度来计算。相同多核苷酸的多个测序读段中的每个读段可能具有略微不同的序列。彼此类似且因此很可能都是通过对同一多核苷酸进行测序而生成的读段可以一起被分组在集群中以进行进一步分析。平均而言,集群中的读段数目与覆盖深度相同。
对集群中的读段的分析被执行,以从集群中的多个读段中标识单个共有输出序列。这种类型的分析可以被称为“追踪重构(trace reconstruction)”。与任何单个读段相比,共有输出序列更有可能表示源多核苷酸分子中的实际核苷酸序列。用于从多个读段创建共有序列的技术是本领域技术人员已知的。一些技术在读段的单个位置中标识误差,并基于同一位置处的其他读段的序列来校正误差。可以被标识和校正的误差包括插入、缺失和取代。然而,当多个误差靠近在一起时,诸如插入后是取代,然后是两个缺失,用于创建共有输出序列的当前技术无法正确标识误差,并且在确定共有输出序列时整个读段可能会被忽略。本公开中讨论的技术包括标识不定误差的位置,跳过读段的包含不定误差的部分,以及在读段中使用后续碱基调用确定共有输出序列,而不是忽略或丢弃来自具有不定误差的读段的数据。
不定误差可以在读段中与从相同集群中的其他读段得出的共有序列不匹配并且误差的类型无法被确定的位置处被标识。例如,如果读段中的给定碱基调用不同于同一位置处所有其他读段的碱基调用,但该差异无法被标识为插入、缺失或取代误差,那么该碱基调用可能被表征为不定误差。设定数目的位置可以被跳过,并且读段往下的搜索窗口被评估,以确定具有不定误差的读段与集群中的其他读段之间是否存在匹配的子序列。搜索窗口内的给定长度的每个子序列可以被评估,以确定它是否与来自其他读段的序列相匹配。匹配可以部分地通过具有不定误差的读段中的碱基调用与共有序列的碱基调用之间的匹配来确定。附加地,匹配可以通过将具有不定误差的读段的碱基调用与其他读段的简单多数投票进行比较来确定。简单多数投票实际上会考虑给定位置处最常见的碱基调用,而不考虑诸如插入、缺失和取代等误差。
如果匹配被找到,那么匹配子序列内的单个位置被作为候选位置,并且与候选位置相邻的后续位置被用作比较的位置,以使用读段来继续以用于确定共有输出序列。因此,读段中不定误差之前的位置可以与集群中的其他读段中的一些或所有一起被使用,以确定共有输出序列的一部分。读段具有不定误差的部分以及不定误差之后的一些数目的位置被忽略,因此,这些位置的共有输出序列由集群中的其他读段确定。在候选位置被定位之后,下一位置将再次与集群中的其他读段一起被使用,以确定共有输出序列。因此,不是忽略由读段提供的包括不定误差的所有数据,而是仅该读段的不定误差附近的部分被忽略,并且读段的误差前后的部分可以被用于对共有输出序列的确定。
集群中的多个读段的比对(alignment)可以基于读段中的第一位置或读段中的最后一个位置。随着共有输出序列的生成逐个位置进行(例如“前”到“后”或“后”到“前”),由于插入和缺失会使读段相对于彼此异相,误差和不确定性可能会累积。在合成时有意插入所有读段中的已知序列用作比对锚,并提供除末端以外的参考定位(location)以重新比对多个读段。因此,集群中的多个读段可以基于比对锚以及基于读段的第一位置和最后一个位置来相对于彼此比对。读段可以被设计为包括一个或多个比对锚。因为比对锚除了读段的开始和末端之外还包括替代比对点,所以读段可能在比对锚处被拆分为较短的分段,并且较短分段中的每个分段可以分别被评估以确定共有输出序列。较短分段中的每个分段的共有输出序列可以被重新联合,以为原始读段的全长创建单个共有输出序列。
本公开中的特定实现和示例可以仅指DNA;然而,要被理解的是,本公开的内容同样适用于包括DNA、RNA、DNA-RNA杂合体的任何多核苷酸,以及包括合成或非天然碱基的多核苷酸。
附图说明
详细描述是参照附图来陈述的。在附图中,附图标记的最左边的(多个)数字标识附图标记首次出现的附图。在不同附图中使用相同的附图标记指示类似或相同的项目。
图1示出了用于追踪重构系统的操作的说明性架构。
图2是示出了追踪重构系统的使用的说明性原理图。
图3示出了根据本公开的技术标识的取代误差的说明性表示。
图4示出了根据本公开的技术标识的缺失误差的说明性表示。
图5示出了根据本公开的技术标识的插入误差的说明性表示。
图6示出了根据本公开的技术的不定误差和非活性追踪的说明性表示。
图7示出了将延迟引入非活性追踪中并在非活性追踪内定位候选位置的说明性表示。
图8示出了读段内的比对锚的放置位置。
图9示出了通过由比对锚划分的读段的分段生成部分共有序列。
图10示出了说明性追踪重构系统的框图。
图11A和图11B示出了用于从多个读段确定共有输出序列的说明性过程。
图12A和图12B示出了用于根据从多核苷酸测序仪接收的读段生成二进制数据的说明性过程。
图13示出了用于通过省略读段的包含突发性误差的一部分来生成共有输出序列的说明性过程。
图14示出了用于在确定是否存在超过与其他读段相匹配的不定误差的定位的子序列时分析包含不定误差的读段的说明性过程。
具体实施方式
如上面所提及的,DNA作为数字信息的存储介质具有巨大的潜力。然而,应对可能损坏数据的误差是使用DNA存储数字数据的挑战中的一个挑战。存在在将数字数据转换为合成DNA分子,然后从合成DNA分子恢复数字数据时涉及的许多步骤。本公开中描述的技术提供了用于当该读段包括不定误差时使用DNA读段中的一些信息的技术。
术语“DNA链(strand)”或简称为“链”是指DNA分子。如本文所使用的,“DNA读段”、“序列读段”或简称为“读段”是指当多核苷酸测序仪读取DNA链的序列时由多核苷酸测序仪生成的数据字符串。因为读段是数据字符串,所以它们也可以被称为“字符串”。然而,由多核苷酸测序仪产生的读段经常包含误差,因此不能以100%的准确度表示DNA链的结构。幸运的是,大多数DNA测序技术都能够产生DNA链的多个读段。读段被称为“噪声读段”,因为每个读段可能包含一个或多个误差,这些误差的分布大致是随机的。给定的读段也可能没有误差,但是除非彼此参照,否则可能不知道哪些读段没有误差以及哪些包含误差。本公开的技术对单个DNA链使用多个不同的噪声读段来创建可能表示DNA链的真实序列的共有输出序列。共有输出序列是类似于任何读段的数据字符串,但是共有输出序列是通过对读段的分析来生成的,而不是直接从多核苷酸测序仪输出的。从许多噪声读段变为一个大概准确的共有输出序列的过程被称为“追踪重构”。
天然存在的DNA链由四种类型的核苷酸组成:腺嘌呤(A)、胞嘧啶(C)、鸟嘌呤(G)和胸腺嘧啶(T)。DNA链或多核苷酸是这些核苷酸的线性序列。DNA链的两个末端(被称为5'和3'末端)在化学上是不同的。DNA序列照惯例是从5'核苷酸末端开始来表示的。不同链之间的相互作用基于序列是可预测的:两个单链可以彼此结合并形成双螺旋(如果它们是互补的):一个链中的A与另一链中的T比对,并且C和G同样地比对。双螺旋中的两个链具有相反的方向性(5'末端附接至另一链的3'末端),因此,这两个序列是彼此的“反向互补”。两个链不需要完全互补就能够相互结合。核糖核酸(RNA)具有与DNA类似的结构,并且天然存在的RNA由四个核苷酸A、C、G和尿嘧啶(U)组成,而不是T。为了简洁和易读起见,本公开中的讨论仅提及DNA,但是RNA可以代替DNA或与DNA结合被使用。
追踪重构问题可以使用如下数学记号来陈述。令∑表示有限字母,例如∑={A,C,G,T}。令X∈∑
诸如通过合成测序(在下面有更多讨论)等一些测序技术具有误差分布,其中噪声至少在一定程度上被独立地分布。因此,噪声可以被建模为在整个链中独立且相同地分布(i.i.d.)。这种类型的噪声可以利用合成的测试数据来创建。这样做,令Y
(无误差),概率为1-p,将X[j]复制到Y的末端,并将j增加1;
(缺失),概率为p
(插入),概率为p
(取代),概率为p
诸如纳米孔(nanopore)测序(下面更多讨论的)等其他测序技术具有不同的误差分布,其中误差不是独立分布的而是倾向于被聚类,使得如果存在一个误差,则其他误差很可能在附近。纳米孔测序比合成测序有更多的误差。与大约为1-2%相比,大约为12%。通过创建局部化的高误差区域,可以在字符串中创建误差的“突发”。除了单个位置误差外,纳米孔测序结果还可能具有较大的误差,诸如邻近碱基块的调换。
从多个噪声读段中标识单个共有序列-追踪重构-输出估计读段
图1示出了用于实现追踪重构系统102的说明性架构100。简而言之,旨在用于DNA分子存储的数字信息被转换为表示核苷酸字符串的信息。表示核苷酸字符串的信息(即,表示核苷酸碱基顺序的字母字符串)被用作DNA合成模板,其指示寡核苷酸合成仪104通过核苷酸化学合成DNA分子核苷酸。人工合成DNA允许创建具有任意系列碱基的合成DNA分子,其中碱基的各个单体一起被组装为核苷酸的聚合物。寡核苷酸合成仪104可以是使用任何公认的DNA合成技术的任何寡核苷酸合成仪。本文使用的术语“寡核苷酸”被定义为包括两个或更多个核苷酸的分子。
合成过程的偶联(coupling)效率是核苷酸在该过程的每个步骤与现有的部分链结合的概率。尽管每个步骤的偶联效率都可以高于99%,但是这个小误差仍然会导致产物产量随长度的增加而呈指数下降,并将目前可以被有效合成的寡核苷酸的大小限制在200个核苷酸左右。因此,所存储的DNA链的长度约为100至200个碱基对(bp)。该长度将随寡核苷酸合成技术的发展而增加。
由寡核苷酸合成仪104产生的合成DNA可以被转移到DNA存储库106。有许多可能的方式来构造DNA存储库106。除了在分子级别上通过将标识序列附加到DNA链的结构之外,DNA存储库106可以通过将DNA链物理分离为一个或多个DNA池108来构造。此处,DNA池108被示出为翻盖管,其表示用于多个DNA链的物理容器。当DNA被存储在液体溶液中时,DNA链通常最易于通过生物技术进行操作。因此,DNA池108可以被实现为填充有液体的腔室,在许多实现中是水,并且在DNA池108中可以存在数千个、数百万个或更多的单独DNA分子。
除了处于液体悬浮液中之外,DNA存储库106中的DNA链还可以呈玻璃状(或玻璃体)状态,作为冻干产物,作为盐的一部分,被吸附在纳米颗粒的表面上或另一格式存在。DNA池108的结构可以被实现为将包括DNA的一定体积的液体保持在物理定位的任何类型的机械、生物或化学布置。存储装置也可以是非液体形式,诸如固体珠或通过封装。例如,在其上存在液滴的单个平坦表面是DNA池108的一个实现,即使没有被完全封闭在容器内,液滴也部分地通过液体的表面张力而保持。DNA池108可以包括单链DNA(ssDNA)、双链DNA(dsDNA)、单链RNA(ssRNA)、双链RNA(dsRNA)、DNA-RNA杂合链或包括使用非天然碱基的任何组合。
从DNA存储库106中删除的DNA链可以用多核苷酸测序仪110来测序。在一些实现中,DNA链可以被制备以通过使用聚合酶链反应(PCR)的扩增来测序以创建大量的DNA链,该DNA链是彼此相同的副本。测序之前的PCR扩增的需求可能取决于所使用的特定测序技术。尽管PCR的级别远低于当前的测序技术,但PCR本身可能是误差源。目前,PCR技术通常每10,000个碱基引入大约一个误差。因此,平均而言,针对100个碱基的每100个读段,都会有作为PCR结果的一个误差。由PCR引入的误差通常是随机分布的,因此追踪重构系统能够校正一些PCR引起的误差。
如上面所提及的,多核苷酸测序仪110读取DNA链中的核苷酸碱基的顺序,并从该链生成一个或多个读段。多核苷酸测序仪110使用多种技术来解译分子信息,并且可以以系统和随机的方式将误差引入数据中。误差通常可以被归类为取代误差,其中真实核苷酸被不正确的碱基调用(例如A与G交换)、插入或缺失取代,其中随机碱基调用被插入(例如AGT变为AGCT)或缺失(例如AGTA变为ATA)。交换更长的DNA区域的换位是另一类型的误差。读段中的每个位置是由多核苷酸测序仪110基于由多核苷酸测序仪110的组件感测的性质确定的单个碱基调用。由多核苷酸测序仪110感测的各种性质取决于所使用的特定测序技术而变化。碱基调用表示确定DNA(或RNA)链中的四个核苷酸碱基(A、G、C和T(或U))中的哪一个存在于链中的给定位置处。有时,碱基调用是错误的,并且这是由测序引入的误差源。多核苷酸测序包括被用于从DNA或RNA链生成碱基调用的任何方法或技术。
可以被使用的测序技术是合成测序(
可以被使用的测序技术的另一示例是纳米孔测序。纳米孔是直径约1纳米的小孔。将纳米孔浸入导电流体中并在纳米孔上施加电势导致由于离子通过纳米孔的传导而产生的微小电流。流过纳米孔的电流量对纳米孔的大小敏感。当DNA分子穿过纳米孔时,DNA分子上的每个核苷酸都会不同程度地阻塞纳米孔。因此,当DNA分子穿过纳米孔时,穿过纳米孔的电流的变化表示DNA序列的读数。
当前已知的其他测序技术包括来自Pacific Biosciences的单分子实时(SMRT
用于测序DNA的所有技术都与某种程度的误差相关联,并且误差的类型和频率因测序技术而异。例如,合成测序会在大约2%的碱基调用中创建误差,并且误差倾向于被独立且均匀地分布(i.i.d.)。这些误差大多数是取代误差。纳米孔测序的误差率更高,大约为15%至40%,由这种测序技术引起的大多数误差都是缺失。由纳米孔测序生成的序列中的误差倾向于会出现在集群中,因此,如果一个位置有误差,则相邻位置也很有可能也会出现误差。因此,误差可能被认为是“突发”。特定测序技术的误差分布可以描述误差的总体频率、误差的分布特性以及各种类型误差的相对频率。
在一些实现中,多核苷酸测序仪110提供质量信息,其指示给定碱基调用的准确度的置信度级别。质量信息可以指示在特定碱基调用中存在高置信度级别或低置信度级别。例如,质量信息可以被表示为碱基调用的准确性的百分比,诸如80%置信度。附加地,质量信息可以被表示为四个碱基中的每个碱基对于DNA链中的给定位置是正确碱基调用的置信度级别。例如,质量信息可以指示碱基调用为T的80%置信度,碱基调用为A的18%置信度,碱基调用为G的1%置信度,并且碱基调用为C的1%置信度。因此,该碱基调用的结果将为T,因为与任何其他核苷酸相比,该核苷酸为正确碱基调用的置信度更高。质量信息不能标识误差源,而只是建议哪些碱基调用或多或少可能是准确的。
多核苷酸测序仪110以电子格式向追踪重构系统102提供输出,多个噪声读段(通常是多个DNA链)。输出可以包括质量信息作为元数据,否则与由多核苷酸测序仪110产生的读段相关联。
追踪重构系统102可以被实现为多核苷酸测序仪110的组成部分。多核苷酸测序仪110可以包括实现追踪重构系统102的板载计算机。备选地,追踪重构系统102可以被实现为分离的计算设备112的一部分,该计算设备112是通过不跨网络的有线或无线连接被直接连接至多核苷酸测序仪110。例如,计算设备112可以是台式计算机或笔记本计算机,其被用于从多核苷酸测序仪110接收数据和/或控制多核苷酸测序仪110。有线连接可以包括将计算设备112物理连接至多核苷酸测序仪110的一个或多个电线或电缆。有线连接可以通过耳机电缆、电话电缆、SCSI电缆、USB电缆、以太网电缆、FireWire等来创建。无线连接可以通过无线电波(例如蓝牙、ANT、Wi-Fi IEEE 802.11等的任何版本)、红外光等来创建。追踪重构系统102还可以使用经由网络116与多核苷酸测序仪110进行通信的一个或多个服务器114被实现为基于云或网络系统的一部分。网络116可以被实现为任何类型的通信网络,诸如局域网、广域网、网状网络、自组织网络、对等网络、互联网、电缆网络、电话网络等。附加地,追踪重构系统102可以部分地由多核苷酸测序仪110、计算设备112和服务器114的任何组合来实现。
图2示出了使用追踪重构系统102作为对存储在合成DNA链200中的信息进行译码的过程的一部分。合成DNA链200是具有特定核苷酸碱基序列的分子。如图1所示,合成DNA链200可以被存储在DNA池108中。合成DNA链200可以作为单链分子存在于DNA池108中,或者可以与互补的ssDNA分子杂合以形成dsDNA。多核苷酸测序仪110从单个合成DNA链200产生多个噪声读段202的输出。读段的数目与测序的覆盖深度相关。测序的深度越大,导致从DNA链生成的平均序列就越多。由于测序误差,同一DNA链的多个读段中的许多读段可能具有不同的序列。读段中的每个读段具有长度(n),在该示例中,其为九(对应于合成DNA链200中的九个碱基)。在实际的测序仪数据中,噪声读段可以具有不是全部彼此相等的任意长度。缺失和插入是读段长度变化的一个原因。针对给定的读段,该读段的长度可以被表示为n,但针对所有读段,n不一定相同。在实际实现中,由于当前对可以被人工合成的DNA链的最大长度的限制,读段的长度可能在100到200之间。读段上的定位可以被称为“位置”,诸如在该示例中从位置一到位置九。如本文所使用的,“碱基”是指给定单体在DNA分子中的定位,而“位置”是指沿着诸如读段等数据字符串的定位。因此,假设没有误差,合成DNA链200中的第三碱基对应于由多核苷酸测序仪110生成的读段中的第三位置。
在该示例中,被提供给追踪重构系统102的噪声读段202的数目(m)为5。然而,任何数字可以被使用。在一些实现中,被提供给追踪重构系统102的噪声读段202的数目可以是10、20或100。被提供给追踪重构系统102的噪声读段202的数目可以小于由多核苷酸测序仪110产生的读段的总数。由多核苷酸测序仪110产生的读段总数的子集可以被随机选择或使用启发式以进行追踪重构系统102的分析。除了随机选择,其他技术也可以被用于选择读段的哪个子集被传递给追踪重构系统102。例如,质量信息可以被用于从由多核苷酸测序仪110生成的所有读段中标识对在碱基调用具有最高置信度的m个读段。在一些实现中,仅具有某些长度的读段被选择。
追踪重构系统102根据本公开的技术分析噪声读段202,并生成共有输出序列204。共有输出序列204表示合成DNA链200中的核苷酸序列,其误差小于单个噪声读段202中的任何一个,并且理想情况下没有误差。
转换器206将共有输出序列204转换为二进制数据208,从而取回存储在DNA存储库106中的数字信息。转换器206可以使用附加纠错技术来校正可能保留在共有输出序列204中的任何误差。因此,追踪重构系统102不必校正所有类型的误差,因为存在可以被用于恢复二进制数据208的其他纠错技术。
尽管本文讨论的实现涉及从合成DNA链200的读段获得二进制数据208,但是追踪重构系统102在天然DNA链的读段上同样操作良好。来自多核苷酸测序仪110的输出是针对合成DNA和天然DNA两者的多个噪声读段202。因此,在不涉及使用合成DNA来存储二进制数据208的实现中,追踪重构系统102可以被用于从由多核苷酸测序仪110生成的读段中删除误差。
图3示出了用于标识取代误差的技术。读段可以在起始位置或任何其他位置(诸如下面更详细地讨论的比对锚)处被比对。起始位置可能对应于生成读段的DNA链的5'末端。在本公开的附图中,5′末端被定向到左侧。跨越读段的比较的位置300由实心矩形框表示。当读段中的每个位置被依次分析时,比较的位置300可以沿着读段从左到右移动。紧随比较的位置300的是由两个虚线矩形框表示的前瞻窗口302。前瞻窗口302在比较的位置300的右侧或朝向3'末端“前向看”。即,如果读段被表示为Y
多数共有碱基304是比较的位置300处最频繁的碱基调用。此处,五个读段中的四个读段在该位置处具有G,并且一个读段具有T。因为G是数目最多的碱基调用,所以多数共有碱基304是G。在一些实现中,多数共有碱基304可以通过考虑比较的位置300处的相应碱基调用的质量信息来确定。比较的位置300处的每个碱基调用可以基于关联的质量信息来加权。例如,如果给定碱基调用为G有80%置信度,那么在确定多数共有碱基304时可以算作0.8G,而在确定多数共有碱基304时给定碱基调用是C的30%置信度将算作0.3C。因此,在针对给定比较的位置300标识多数共有碱基304时,单个碱基调用的置信度可以被考虑。附加地或备选地,具有指示碱基调用的置信度小于阈值级别(例如15%)的质量信息的所有碱基调用可以从多数碱基调用304的确定中省略。如果以相等的频率存在两个或更多个不同的碱基调用,则多数共有碱基304可以是从中随机选择的。
在比较的位置300处具有不同于多数共有碱基304的碱基调用的读段被称为变体读段。因此,针对变体读段Y
前瞻窗口共有306是以与多数共有碱基304类似的方式从前瞻窗口302确定的。前瞻窗口共有306的确定也可能被质量信息影响。前瞻窗口共有306可以基于通过其相应置信度级别加权的碱基调用和/或通过省略具有低于阈值的置信度级别的碱基调用。前瞻窗口共有306是通过考虑比较的位置300的不是变体读段的读段来确定的。因此,此处,前瞻窗口302被示出为覆盖非变体读段但不覆盖变体读段308。在该示例中,前瞻窗口302的第一位置中最常见的碱基调用是C,并且前瞻窗口302的第二位置中最常见的碱基调用是T。因此,前瞻窗口共有306是碱基调用的两个位置的字符串:CT。
接下来,变体读段(CT)的前瞻窗口310中的碱基调用与前瞻窗口共有306(CT)进行比较。因为它们相匹配,所以第三读段308中的第二位置处的不匹配被分类为取代。在数学记号中,如果Y
在变体读段的误差类型被分类之后,比较的位置300被移动以继续分析读段。针对不是变体读段的每个读段,比较的位置300被向右移动一个位置。在该示例中,这些是第一读段、第二读段、第四读段和第五读段。针对误差类型被分类为取代的变体读段(此处为第三读段308),比较的位置300也被向右移动了一个位置。因此,如图3的下部所示,针对所有读段,比较的位置300被向右移动一个位置。分析在该新比较的位置312处重复,并且在该迭代中,第二读段314被标识为变体读段。
图4示出了用于标识缺失误差的技术。比较的位置400被再次分析以确定该位置处最频繁的碱基调用。在该示例中,五个链中的三个链具有碱基调用T,一个链具有碱基调用G,并且一个链具有碱基调用C。因此,最常见的碱基调用是T,并且针对读段中的该位置的多数共有碱基402为T。第一链410和第四链被标识为变体读段。
非变体读段的链的前瞻窗口404中的碱基调用被比较,以确定前瞻窗口共有406。在该示例中,前瞻窗口404中的两个碱基调用的值针对三个非变体读段是GA、GA和TG。因此,最常见的碱基调用系列是GA,并且这成为了前瞻窗口共有406。
在第一链410的前瞻窗口408中的碱基调用(AG)的值与前瞻窗口共有406(GA)不同。因此,负责第一链410中的不匹配的误差类型不被分类为取代。然而,比较的位置400中的碱基调用以及除前瞻窗口404(GA)的最终位置之外的所有位置与前瞻窗口共有406(GA)相匹配。因此,第一链410的该位置的误差类型被分类为缺失。在该示例中,前瞻窗口404的长度(w)为2,因此,除前瞻窗口404的最终位置的所有位置均为w-1或前瞻窗口404的第一碱基。如果前瞻窗口404的长度(w)为3,那么当确定变体读段中的误差类型是否为缺失时,前瞻窗口404的前两个碱基(3-1)将被考虑。在数学记号中,如果Y
在变体读段的误差类型被分类之后,针对不是变体读段的读段中的每个读段以及误差被分类为取代的读段中的每个读段,比较的位置400被向右移动一个位置。针对被分类为缺失的第一链410,比较的位置400不被移动。它保留在位于第一链410的第五位置中的相同G处。在针对第一链410(即,零)和其他链(即,1)将差分移动之后的链重新比对到新比较的位置412之后,如图4的下部所示,该缺失变得明显。由于基于误差类型的分类将新比较的位置412移动不同量而导致的这种重新比对使链保持同相,从而进一步改进沿着链的分析。在新比较的位置412被移动之后(或不取决于误差类型),分析可以重复,此处为标识第五链414中的不匹配。
图5示出了用于标识插入误差的技术。如上面所讨论的,三种可能的误差类型是取代、缺失和插入。如同标识取代和缺失误差一样,插入误差的标识开始于在比较的位置500中分析碱基调用以确定多数共有碱基502,并在前瞻窗口504中分析碱基调用以标识前瞻窗口共有506。在该示例中,多数共有碱基502为T。第五读段510为变体读段,因为它在比较的位置500处具有A而不是T。前瞻窗口共有506的碱基调用是GA。第五读段510的前瞻窗口508中的碱基调用与前瞻窗口共有506不匹配,因此误差类型未被分类为取代。比较的位置500中的碱基调用和前瞻窗口508中针对变体读段(AT)的第一碱基调用与前瞻窗口共有506(GA)不匹配,因此误差类型未被分类为缺失。
然而,第五读段510的前瞻窗口508中的碱基调用与多数共有碱基502的碱基调用和除前瞻窗口共有506的最终碱基调用(即,w-1位置)之外的所有碱基调用相匹配(即,两者都是TG)。因此,该误差被分类为在第五读段510的第5位置处插入A。在数学记号中,如果Y
图3至图5所示的示例分别图示了仅一种误差类型的分析。然而,本公开的技术同样适用于在一个比较的位置中具有多个误差的读段组。跨单个比较的位置也可能存在多种误差类型,例如在总共20个读段(m=20)中,三个读段可能具有取代,一个读段可能具有缺失,并且一个读段可能具有插入。
图6图示了无法标识误差类型的情况。读段可能在比较的位置中具有碱基调用,该碱基调用与多数共有碱基调用不匹配,因此这是变体读段,但是在比较的位置和该前瞻窗口中的碱基调用可能不会表现出被分类为取代、缺失或插入的任何关系。这是无法根据上述技术被分类的误差,因此被称为“不定误差”604。即使除本公开中描述的那些技术以外的其他技术被用于标识误差类型,无法被解析为特定类型的误差的未能与多数共有碱基调用相匹配的误差可以被分类为不定误差。
使用图3至图5所示的技术无法将第五读段602中的不定误差604的定位处的碱基调用分类为取代、插入或缺失。紧邻不定误差604的定位之前的比较的位置600是不定误差604之前的最后一个位置。
处置具有不定误差的读段的一种方式是从进一步处理中丢弃读段。因此,如果读段具有误差,并且它无法被解析为单种误差类型,则该读段将从进一步的分析中被省略。如果读段602被丢弃,则它可以被指示为“非活性追踪”,因为读段602对追踪重构没有贡献任何信息。一旦读段602被标识为非活性追踪,共有输出序列就从剩余的活性追踪606中生成。活性追踪606可以是来自同一集群的所有其他读段,或者如果由于还包括不定误差,低置信度级别或其他原因而有一些被丢弃,则可以小于所有其他读段。
然而,完全丢弃读段还丢弃了可能被包含在读段的没有不定误差的部分中的有用信息。针对一些测序技术,诸如纳米孔测序,丢弃具有不定误差的每个读段可以显著减少恢复的集群数目,因为这甚至可能会留下很少的读段,其无法执行追踪重构。
处置歧义误差的另一方式是使用偏差或决胜局以便推动分类。该偏差可以基于用于生成读段的多核苷酸测序仪的误差分布。例如,如果多核苷酸测序仪已知生成取代误差的频率要比缺失或插入误差的频率高得多,则所有歧义误差可以被分类为取代。如果误差可以被标识为两种可能的误差类型中的一种类型,则多核苷酸测序仪技术的那些误差类型的相对频率可以被用于在它们之间进行选择。例如,如果误差已经被标识为缺失或插入(但不是取代),并且多核苷酸测序仪发生80%取代误差、15%缺失误差和5%插入误差,则该误差可以被分类为缺失误差,因为在该示例中,缺失误差比插入误差更有可能发生。然而,这可能会降低共有输出序列的可靠性,因为可能不正确的碱基调用可能会被包括在共有碱基调用的计算中。
附加地或备选地,各个碱基调用的质量信息可以被用于对歧义误差进行分类。在一个实现中,当误差无法被解析为单种误差类型时,比较的位置和具有质量信息的前瞻窗口中的所有碱基调用可以从多数共有碱基和前瞻窗口共有中被省略,该质量信息指示碱基调用置信度小于阈值级别。因此,相关位置的共有碱基调用是在多个读段的最可靠的碱基调用上确定的。忽略低置信度的碱基调用可能会导致上述技术能够将误差解析为单种误差类型。然而,如果大多数碱基调用的置信度级别很低,甚或即使置信度级别是一致的,那么基于置信度级别选择特定的碱基调用也可能不会产生准确的结果。
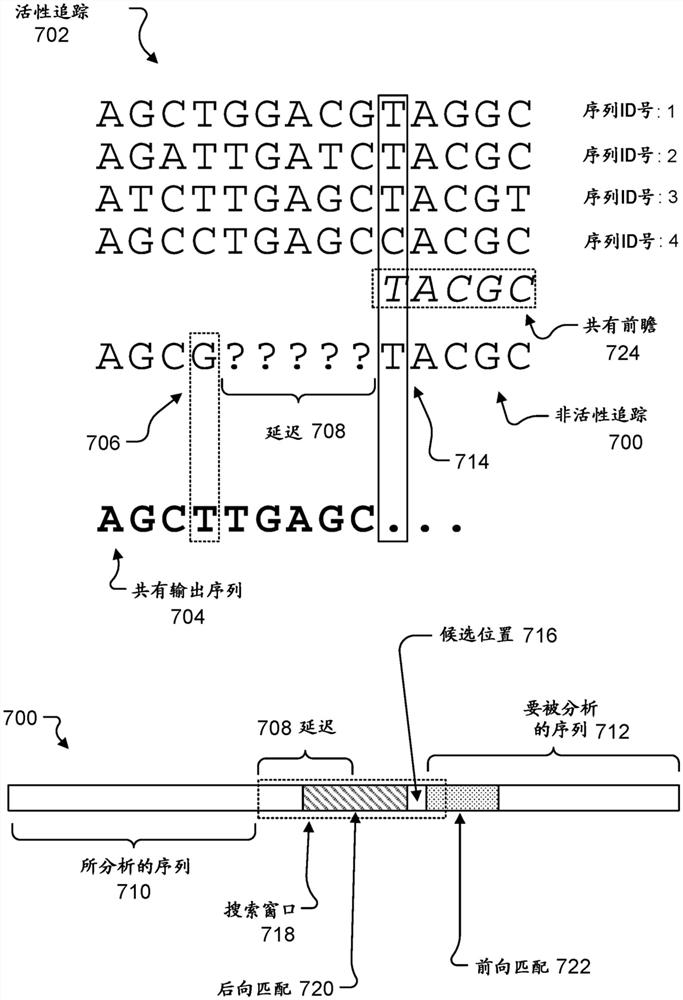
图7示出了用于跳过包含不定误差的非活性追踪700的一部分以在沿着非活性追踪700更远的较晚位置处搜索与活性追踪702的匹配的技术。非活性追踪700可以被标记为由于确定不定误差而导致的非活性追踪,如图6所示。集群中的读段中的每个读段所表示的碱基调用的字符串可以被表示为Y
共有输出序列704可以从活性追踪702和非活性追踪700的部分而被生成。共有输出序列704可以被表示为对提供给寡核苷酸测序仪的多核苷酸的序列X的最佳估计
位置706是非活性追踪700中的位置,该位置是在最后一个读段变为非活性时估计的。这可以是图6所示的比较的位置600。该位置可以被表示为i
图7的底部的非活性追踪700的示意性表示示出了非活性追踪700可以如何被评估以标识匹配序列的一种说明性布置。当不定误差被标识时,非活性追踪700的一些部分可能已经被过多分析,并被用于开发共有输出序列704。非活性追踪700的这一部分被指示为分析序列710。这接着是延迟708,然后经过是非活性追踪700的一部分的延迟708,该部分具有要被分析的序列712。一旦标识了经过延迟708多远,则非活性追踪700可以被重新活化,追踪重构可以被继续。
在延迟708之后存在比较的位置714,其可以被表示为i。为了确定该比较的位置714是否在延迟708区域内,i与i
针对共有输出序列704尚未被计算的读段的比对,共有前瞻序列724可以从活性追踪702创建。通过在比较的位置处对活性追踪702进行多数共有投票,共有前瞻序列724可以通过与早前描述的前瞻窗口相同的方式来创建。该技术仅在每个位置处使用多数共有碱基调用作为共有序列的碱基调用。如果有相等数目的碱基调用(例如5个T和5个C),那么连结会被任意地打破。然而,与前瞻窗口不同,共有前瞻序列724不被限于两个或三个位置的窗口。
一旦经过延迟708区域,候选位置716就被寻找。候选位置716是非活性追踪700中的单个位置,其可以与活性追踪702比对,并被用于继续确定共有输出序列704。候选位置716可以被表示为k。候选位置716可以在比较的位置处714紧随延迟708,或者可以位于延迟708的末端之后更远的地方。
如果从位置706到候选位置716以相等的频率发生插入和缺失,那么非活性追踪700可以再次与活性追踪702比对的位置将位于比较的位置714处或附近。该比较的位置714可以被表示为k
搜索窗口718可以包括在k
搜索窗口718内的非活性追踪700的子序列与比较序列Z进行比较,以确定是否存在匹配。比较序列Z包括
后向匹配720区域位于位置i之前,并且与共有输出序列704的部分比对。后向匹配720序列的长度可以被实验式地确定,并且范围可以是例如0至10个位置。因此,后向匹配720序列可以被省略。后向匹配720序列也可以与延迟708重叠。在由延迟708表示的位置处,共有输出序列704由活性追踪702确定,而在延迟708的定位处不使用来自非活性追踪700的碱基调用。
Z的前mb+1个位置是
前向匹配722序列延伸经过位置i,因此在对应位置处没有共有输出序列704。因此,出于与非活性追踪700中的序列比较的目的,来自活性追踪702的共有前瞻序列724序列被使用。前向匹配722序列的长度可以是例如0至10个位置。因此,如果mf=0,那么前向匹配722序列不被使用。尽管后向匹配720序列和前向匹配722序列与从活性追踪702生成的共有序列进行比较,但是每个与不同的共有序列进行比较。后向匹配720序列与
与Z比较的非活性追踪700的子序列的长度为mb+mf+1,该长度与Z相同。因此,匹配是在相等长度的两个字符串之间执行的。回顾一下,非活性追踪700可以被表示为Y
利用Z和
如果搜索窗口718内的候选位置716中没有一个关联于与对应的Z子序列相匹配的
如果在搜索窗口内存在关联于与对应的Z子序列相匹配的
可能存在多个候选位置716(k的多个定位),在该位置处匹配被发现。如果是这样,则候选位置716中的一个候选位置被选择以用作重启追踪重构的定位。一种方式是选择最接近k
给定的读段可以具有多于一个不定误差,并且标识不定误差然后发现候选位置716以重启追踪重构的过程可以在单个读段中被多次重复。此外,可能包括数十个序列的读段集群可以具有包括不定误差的多个读段。因此,该技术可以被用于集群中的多于一个读段(包括所有读段)。因为在多个位置处,针对多个读段中的i个不同读段可以是活性的或非活性的,所以构成活性追踪702的读段集合在不同的比对定位处可以是不同的。
图8是指示比对锚的定位的数个DNA链800、802、804的示意图。比对锚除了读段的起始和末端之外还提供参考定位,其可以比对多个噪声读段,从而辅助构建共有输出序列。
通常,共有输出序列的准确度朝着读段的开始或末端更加准确,其中在链中彼此正确比对存在较高置信度。不被理论束缚,任何误差可能可以累积并在沿着读段分析后续位置时引起其他误差。例如,如果缺失误差被错误地标识为取代误差,则该读段中的剩余碱基调用可能异相,并对后续共有碱基调用的准确度产生负面影响。最小化该影响的一种方式是使用“正向”分析的前半部分和“反向”分析的前半部分。例如,假定要被分析的读段的长度为200个位置。从左到右的分析可以被执行,其提供位置1至100的共有输出序列。从右到左的分析也可以被执行,其提供位置101至200的共有输出。两种分析可以被并行执行。所得的共有输出序列通过组合从左到右的分析和从右到左的分析为所有位置1至200提供碱基调用。这被称为组合共有输出序列。
因为利用组合共有输出序列离读段的末端最不可靠,所以一种布置是在DNA链800的中心处的比对锚806处添加。这提供了在彼此相对比对最不可靠的定位处重新比对读段的位置。标记为共有比对提供了“锚”,以重置并重新开始,而不会受到先前累积的误差的影响。
比对锚用作定位的标记,并且即使在存在由测序创建的噪声和误差的情况下,在每个读段中也应该可识别。比对锚可能是例如4至8个位置的相对较短的序列。比对锚中的碱基序列和比对锚的长度的选择可以是任意的。在聚类在一起的多个DNA链上一致的任何序列都可以用作比对锚。因为比对锚使用一些数目的碱基,所以这是以减少可用于存储数字信息的DNA链中的碱基数目为代价的。然而,在DNA链中包括比对锚可能使其可以用于追踪重构以生成具有较低覆盖级别的准确共有输出序列,从而减少恢复存储在DNA中的信息所需的测序总量。
在实现中,比对锚的序列被创建以避免重复碱基并避免内部比对。因为比对锚被用于比对多个读段,所以减少两个不同读段中的比对锚序列之间未比对的可能性可能是有益的。例如,ACGT是没有任何重复碱基并且没有任何内部比对的序列。但是,如果一个读段中的第一ACG与不同读段中的最后一个ACG比对,则ACGACG可以与自身形成部分比对。例如,如果在比对锚内的位置的碱基调用中有误差,或者与比对锚相邻的读段序列与比对锚的一部分相同,则可能会发生未比对。
一个或多个比对锚可以被包括在DNA链的多核苷酸序列中。DNA链内的比对锚有多种可能的布置。一种布置是将比对锚806放置为在DNA链800的中间居中。将比对锚806放置在DNA链800的中心当然会导致比对锚序列位于对应读段的中间或附近。例如,比对锚806可以位于从DNA链800的开始到DNA链800的末端的距离的40%至60%、45%至55%、49%至51%或50%之间。
DNA链802和804示出了多个比对锚的示例。如果多个比对锚被使用,则它们可以以各种方式被布置。DNA链802图示了比对锚808、810和812等距的示例。因此,在DNA链802的起始与比对锚808之间,在比对锚808与比对锚810之间,在比对锚810与比对锚812之间以及在比对锚812与DNA链末端之间的位置数目全部基本相同。为了比对多个读段,比对锚可以充当DNA链802的附加“末端”。因此,如果DNA链802长200bp,而不是创建100个位置的一个从右到左的共有输出序列和100个位置的一个从左到右的共有输出序列,则比对锚806、810和812会将DNA链802划分为四个区域,每个区域约50bp。因此,在比对点被达到之前的测序长度从200bp被缩短到大约50bp,并且将读段中的一个读段放置为与其他读段异相的错误碱基调用的机会也被减少了。
在由DNA链804所图示的不同布置中,比对锚的间隔可以偏向DNA链的中心。因此,与远离中心的比对锚820和822相比,靠近DNA链804的中心的比对锚814、816和818被定位得更靠近在一起。而且,比对锚820和822可以距DNA链804的相应末端更远。一起使用的每个比对标记应该是唯一的,使得它可以与其他标记区分开。因此,锚814至822将分别具有不同的序列。各个锚的长度也可以不同。
图9使用读段的示意图来图示从由比对锚划分的读段部分生成的部分共有序列可以如何被联合在一起以创建完整的共有输出序列。图9所图示的读段900可以例如从图8所示的DNA链800来生成。比对锚902可以位于读段900的中间,大约在读段900的左端和右端之间的路的50%。即使比对锚902沿着DNA链的长度正好位于50%,在测序期间引入的插入和/或缺失也可能会使比对锚902在读段中的位置偏移,从而不再精确地位于中间。因此,比对锚902将读段900划分为大致相等长度的两个部分。
因为比对锚902的序列是已知的(假设在读段900的该部分中没有误差),所以对该已知序列(例如AGCT)的搜索可以标识整个读段900中的一个或多个匹配。比对锚902的放置也是已知的,因此不在距读段900的中间的阈值距离之内的任何匹配都可以被忽略。因此,比对锚902的定位在同一集群中的读段900和其他读段中可以被标识。
如果读段900长180个位置并且比对锚902长四个位置且正好位于中间,那么位于比对锚902的左侧的读段900的左侧子序列将长88个位置,并且位于比对锚902的右侧的右侧子序列也将长88个位置。左侧子序列可以通过正向和反向分析来分析,以生成从左到右的共有输出序列904和从右到左的共有输出序列906。类似地,右侧子序列可以被分析,以生成共有输出序列908和910。
如上面所讨论的,这四个共有输出序列904至910中的每个序列的仅前半部分可以被用于最小化由测序导致的插入和缺失引起的累积相移引入的误差。在该实现中,在切换到以相反方向运行的共有输出序列之前,仅44个位置将被分析。备选地,相应的共有输出序列中的每个序列的一半以上可以被使用,使得在子序列的中间附近存在一定程度的重叠。在重叠区域中,来自从左到右和从右到左的共有输出序列的碱基调用可以被用于确定将被分配给某个位置的共有输出碱基调用。
从左到右的共有输出序列904和从右到左的共有输出序列906的组合可以为读段900的左半部分生成部分共有序列912。类似地,共有输出序列908和910可以以任何合适的方式被组合以为读段900的右半部分生成部分共有序列914。部分共有序列912可以通过参照比对锚902与部分共有序列914比对。
一旦比对,部分共有序列912和部分共有序列914就可以使用比对锚902来联合,以创建单个联合的共有序列916。联合的共有序列916表示读段900的共有输出序列,其是从正向和反向共有输出序列904至910开发的。这与不使用比对锚的实现形成对比,其中读段900的共有输出序列是仅从一个正向和一个反向共有输出序列创建的。
因为比对锚902不像读段的900的其他部分那样编码数字信息,所以与比对锚902相对应的碱基调用可以被删除以创建没有比对锚918的共有输出序列。没有比对锚918的共有输出序列表示碱基调用字符串,其可以被译码以恢复读段900中所包含的数字信息。
图10示出了图1所示的追踪重构系统102的说明性框图1000。回顾一下,追踪重构系统102可以全部或部分地在计算设备112、多核苷酸测序仪110和服务器114中的任何一个中被实现。因此,追踪重构系统102可以在包含(多个)一个或多个处理单元1002和存储器1004的系统中被实现,两者均可以跨一个或多个物理或逻辑定位被分布。(多个)处理单元1002可以包括中央处理单元(CPU)、图形处理单元(GPU)、单核处理器、多核处理器、专用集成电路(ASIC)、诸如现场可编程门阵列(FPGA)等可编程电路等的任何组合。除了硬件实现之外,(多个)处理单元1002中的一个或多个可以以软件和/或固件被实现。(多个)处理单元1002的软件或固件实现可以包括以任何合适的编程语言编写的计算机或机器可执行指令,以执行所描述的各种功能。(多个)处理单元1002的软件实现可以全部或部分地被存储在存储器1004中。
备选地或附加地,追踪重构系统102的功能性可以至少部分地由一个或多个硬件逻辑组件执行。例如而非限制,可以被使用的说明性类型的硬件逻辑组件包括现场可编程门阵列(FPGA)、专用集成电路(ASIC)、专用标准产品(ASSP)、片上系统(SOC)、复杂可编程逻辑设备(CPLD)等。作为硬件逻辑组件的实现可能被特别适用于追踪重构系统102的部分,这些部分被包括为多核苷酸测序仪110的板载部分。
追踪重构系统102可以包括一个或多个输入/输出设备1006,诸如键盘、指向设备、触摸屏、麦克风、相机、显示器、扬声器、打印机等。
追踪重构系统102的存储器1004可以包括可移除存储装置、不可移除存储装置、本地存储装置和/或远程存储装置,以提供对计算机可读指令、数据结构、程序模块和其他数据的存储。存储器1004可以被实现为计算机可读介质。计算机可读介质包括至少两种类型的介质:计算机可读存储介质和通信介质。计算机可读存储介质包括以用于存储信息(诸如计算机可读指令、数据结构、程序模块或其他数据)的任何方法或技术实现的易失性和非易失性介质、可移除和不可移除介质。计算机可读存储介质包括但不被限于RAM、ROM、EEPROM、闪速存储器或者其他存储器技术、CD-ROM、数字通用光盘(DVD)或者其他光学存储装置、磁盒、磁带、磁盘存储装置或者其他磁性存储设备或者可以被用于存储信息以由计算设备访问的任何其他非传输介质。
相比之下,通信介质可以将计算机可读指令、数据结构、程序模块或其他数据实施在调制后的数据信号中,诸如载波或其他传输机构。如本文所定义的,计算机可读存储介质和通信介质是互斥的。
追踪重构系统102可以通过序列数据接口1008通过直接连接和/或网络连接而被连接至一个或多个多核苷酸测序仪110。直接连接可以被实现为有线连接、无线连接或两者。网络连接可以遍历网络116。序列数据接口1008从多核苷酸测序仪110接收一个或多个读段。
追踪重构系统102包括多个模块,这些模块可以被实现为存储在存储器1004中以由(多个)处理单元1002执行和/或由一个或多个硬件逻辑组件或固件全部或部分地实现的指令。
随机化模块1010在用寡核苷酸合成仪104将输入数字数据编码到DNA中之前将输入数字数据随机化。随机化模块1010可以通过进行输入字符串和随机字符串的异或(XOR)从输入数字数据创建随机的更准确的伪随机字符串。随机字符串可以使用基于函数和种子的种子化伪随机生成器来生成。输入数字数据的这种随机化增加了合成DNA链200的随机性,导致来自多核苷酸测序仪110本身的噪声读段202具有A、G、C和T伪随机序列。随机性支持译码(即,聚类和追踪重构)。
聚类模块1012基于多个读段的子集是从同一DNA链得出的可能性对多个读段的子集进行聚类。在序列数据接口1008处从多核苷酸测序仪110接收的数据可以包含从提供给多核苷酸测序仪110的每个DNA链生成的读段集合。尽管在许多读段中可能存在误差,但是来自同一DNA链的读段与来自另一DNA链的读段相比通常彼此更类似。如果要被分析的读段集合包括不同DNA链的读段,则进一步分析将被阻碍。因此,聚类可以被执行以便将用于进一步分析的数据限制在被认为表示相同DNA链的读段子集。不良形成的集群可能由于包括过多或不足而被“不良地”形成。包括过多的不良形成的集群是将单个集群中的多于一个链的读段分组在一起的一个集群。包括不足的不良形成的集群是应该被分组为单个大型集群但又被划分为多个较小集群的多个集群中的一个集群。聚类模块1012可以使用任何合适的聚类技术,诸如基于连接性的聚类(例如层次聚类)、基于质心的聚类(例如k均值聚类)、基于分布的聚类(例如高斯混合模型)、基于密度的聚类(例如具有噪声的应用的基于密度的空间聚类(DBSCAN))等。追踪重构系统102可以分析一个或多个,包括从由多核苷酸测序仪110输出的数据得出的所有集群。如上面所提及的,集群中的读段数目可能取决于测序的深度。利用Nanopore测序,每个集群可能有大约10至100个读段。
读段比对模块1014在跨越多个读段的比较的位置处比对多个读段。最初,读段的左端(对应于DNA链的5'末端)可以被比对。读段中的该第一位置可以被用作初始比较的位置。随着分析的进行,读段比对模块1014基于所标识的误差类型沿着每个读段移动比较的位置多个位置。
读段比对模块1014针对在比较的位置处具有多数共有碱基调用的读段将比较的位置提前一个位置,如果误差类型被分类为取代,则将变体读段提前一个位置,如果误差类型被分类为缺失,则将变体读段提前零个位置,如果误差类型被分类为插入,则将变体读段提前两个位置。
读段比对模块1014还可以生成“反向”比对,该“反向”比对开始于在右端(对应于DNA链的3'末端)比对的读段。然后,分析以相同的方式进行,除了向“右”的移动变为向左的移动之外。当从左到右与从右到左相比被分析时,针对同一读段集合,共有输出序列204可能不同。
通常,共有输出序列204的准确度朝着读段的开始更加准确,其中在比对中较高的置信度是正确的。不被理论束缚,任何误差可能可以累积并在沿着读段分析后续位置时引起其他误差。例如,如果缺失误差被错误地标识为取代误差,则该读段中的剩余碱基调用可能异相,并对后续误差标识的准确度产生负面影响。最小化该影响的一种方式是使用“正向”分析的前半部分和“反向”分析的前半部分。例如,假定要被分析的读段的长度为100个位置。从左到右的分析可以被执行,该分析提供针对前50个位置的共有输出序列204。从右到左的分析可以被执行,该分析提供了针对最后50个位置的共有输出序列204。两种分析可以被并行执行。所得的共有输出序列204是通过从左到右分析标识的50个碱基对和通过从右到左分析标识的50个碱基对的组合。这被称为组合共有输出序列。
比对锚模块1016通过在存在比对锚序列时标识除读段末端以外的定位处的比对定位来起作用。当存在比对锚时,比对锚模块1016可以与读段比对模块1014一起起作用,以在读段的末端中的一个末端与比对锚之间以及两个比对锚(如果存在)之间创建读段的分段的比对。比对锚模块1016可以标识读段中存在的比对锚序列。比对锚序列是预定序列,诸如图8和图9所图示的。比对锚序列可以是3至8bp的序列。在一些实现中,比对锚序列可以长4至6bp。比对锚序列可以是由寡核苷酸合成仪104合成的DNA链中的设计所包括的任意序列。因此,DNA链中的一些核苷酸碱基不被用于编码数字信息,而是被用于插入已知序列-比对锚序列。为了减少两个比对锚序列之间未比对的概率,用作比对锚序列的序列可以被设计而无需重复核苷酸,使得它不与自身比对。例如,序列“AGCT”可以是比对锚的合适序列,而序列“ACACAC”和“TTGG”将更有可能未比对。
一旦比对锚被标识并且读段在比对锚处被比对,比对锚模块1016还可以将读段划分为多个子读段,诸如第一子读段和第二子读段。如果在读段中仅存在一个比对锚序列,那么第一子读段可以从读段的开始(例如5'末端)延伸到比对锚序列,并且第二子读段可以从比对锚序列延伸到读段的末端(例如3'末端)。比对锚模块1016可以针对分析中的读段中的每个读段进行该操作,从而创建第三子读段、第四子读段等。
读段比对模块1014然后可以以与比对整个读段类似的方式来比对子读段,除了比对不仅基于读段的末端而且还基于划分读段的比对锚之外。因此,读段比对模块1014可以将来自第一读段的前半部分的第一子序列与来自第二读段的前半部分的第三子序列比对。由第二子序列表示的第一读段的后半部分也可以与表示第二读段的后半部分的第四子序列比对。因此,代替比对第一读段和第二读段的全部,读段的子序列被比对。当然,在许多实现中,多于两个读段将被比对。
因为子序列比整个读段短,所以在共有输出序列的生成中累积错误的影响被减小。存在较少数目的位置,在这些位置上,误差可能会使读段中的一个读段与其他读段异相。如果误差的确累积了,或者读段与同一集群中的其他读段异相,则比对将在比对锚序列处“重置”。子读段可以在正向和反向定向上被比对,如上面针对整个读段所描述的并且如图9所示。因此,子序列的每个比对(例如来自以上示例的第一子序列、第二子序列、第三子序列和第四子序列)可以以与整个序列的比对相同的方式来分析。
变体读段标识模块1018在比较的位置处确定多数共有碱基调用,并将在比较的位置处具有不同碱基调用的读段标记为变体读段。在一些实现中,变体读段标识模块1018可以使用与上面讨论的多核苷酸测序仪110相关联的误差分布来确定多数共有碱基调用。变体读段标识模块1018可以标示或以其他方式标识对于给定比较的位置而言为变体读段的每个读段。然后,该标示将该读段标识为应该为确定误差类型而被标识的读段。随着比较的位置的每次移动,哪些读段是变体读段而哪些不是变体读段的标识改变。
误差分类模块1020将用于变体读段的误差类型分类为取代、缺失或插入。如果误差无法被唯一地分类,则误差分类模块1020可以指示误差的类型不确定,或者误差类型被限于两种可能性中的一种可能性。误差分类模块1020对误差的分类可以部分地基于在比较的位置处具有多数共有碱基调用的多个读段的子集的前瞻窗口中的碱基调用的共有字符串的比较(即,非变体读段)和变体读段中的碱基调用。在确定误差类型时,不同于在给定迭代中被分析的变体读段未被使用。如果误差分类模块1020将读段中的误差分类为不定误差,则变体读段标识模块1018可以将读段标记为非活性追踪。
如上面在图3中所描述的,基于与比较的位置之后的变体读段中的碱基调用字符串相匹配的前瞻窗口中的碱基调用的共有字符串,误差类型被分类为取代。如上面在图4中所描述的,基于与变体读段中的比较的位置和一个或多个以下位置处的碱基调用相匹配的前瞻窗口共有,误差类型被分类为缺失。
如上面在图5中所描述的,误差类型被分类为插入。它被分类为插入是因为(1)在比较的位置之后的变体读段中的碱基调用(即,在变体读段的前瞻窗口中的第一碱基调用)与多数共有碱基调用相匹配,并且(2)与变体读段中的碱基调用字符串相匹配的前瞻窗口中的碱基调用的共有字符串在长度上与前瞻窗口相等,并在比较的位置之后开始两个位置。如上面在图6中所描述的,如果误差类型无法被分类为取代、缺失或插入,则它被分类为不定误差。
共有输出序列生成器1022至少部分地基于多个共有碱基和沿着比对的读段标识的误差类型来确定共有输出序列204。鉴于由于误差类型分类而导致的读段的调整比对,共有输出序列204中的每个位置是该位置处的多个共有碱基。多核苷酸测序仪110的误差分布和/或各个碱基调用的质量信息也可以被用于通过影响多个共有碱基和误差类型的标识来确定共有输出序列204。
共有输出序列生成器1022还可以生成和组装共有输出序列,以用于由比对锚序列定义的子序列的比对。为读段集群的两个或更多个子序列组装共有输出序列可以包括生成图9所示的每个子序列的正向共有序列和反向共有序列。正向共有序列和反向共有输出序列可以如上所述被组合,以为每个读段集群创建多个组合共有序列。(多个)比对锚的序列被用于将组合共有序列附加在一起。例如,表示读段的前半部分的第一组合共有序列和表示读段的后半部分的第二组合共有序列可以通过在比对锚序列处的比对被附加到彼此。如果存在多个比对锚,则多个组合共有序列可以通过相应比对锚序列处的比对以正确的顺序被附加。在将两个或更多个组合共有序列附加在一起之后,(多个)比对锚序列可以被删除以留下没有比对锚序列的作为读段的组合共有序列的字符串。由多个子序列组装的组合共有序列可能比从没有比对锚的整个读段长度制造的组合共有序列更准确,因为通过任何正向共有序列或反向共有序列遍历的位置数目(以及彼此异相的读段机会的数目)较少。减少累积误差的数目还可以减少形成可以产生准确共有序列的集群所需的覆盖范围或测序深度。
纠错模块1026可以应用附加的纠错技术来译码共有输出序列204。在一些实现中,纠错模块1026基于被编码为链的冗余数据使用非二进制纠错码来译码共有输出序列204。这类纠错的一个示例是Reed-Solomon纠错。在示例实现中,Reed-Solomon外码可以被添加到起始二进制数据,并在被存储时最终被分布在许多DNA链(例如10,000–100,000个链)上。如果超过阈值数目的误差,则可能的是Reed-Solomon纠错可能无法译码共有输出序列204。如果发生这种情况,则追踪重构可以利用改变参数中的一个参数来重复。改变参数可能会导致Reed-Solomon纠错能够译码的不同共有输出序列204。前瞻窗口的长度(w)是可以被改变的一个参数。长度为3的前瞻窗口可以被使用来代替长度为2的前瞻窗口(反之亦然)。通过使阈值更宽容或更严格,用于将读段标记为非活性追踪的切断阈值、延迟的长度、基于质量信息接受碱基调用以及偏置歧义误差的误差类型分类可以被改变。在改变一个或多个参数之后,可以确定共有输出序列204是否不同于先前的共有输出序列204,并且如果是,则Reed-Solomon纠错可以被应用于新的共有输出序列204以查看它是否能够译码序列。
转换模块1028将共有输出序列转换为表示数字文件的至少一部分的二进制数据208。通过反转最初被用于将二进制数据208编码为一系列碱基调用的操作,从一系列碱基调用到二进制数据字符串208的转换被执行。这些操作对于操作DNA存储库106的实体是已知的。在一些实现中,图2中引入的转换器206可以包括与转换模块1028以及纠错模块1026以及可能的其他模块相同的功能性。二进制数据208可以以与任何其他类型的二进制数据相同的方式被使用。如果各种纠错技术足够,则二进制数据208将表示原始二进制数据的完美再现。
说明性过程
为了便于理解,本公开所讨论的过程被划定为表示为独立块的单独操作。然而,这些单独划定的操作不应被解释为在其性能上必定与顺序相关。过程被描述的顺序不旨在被解释为限制,并且任何数目的所描述的过程框可以以任何顺序被组合以实现该过程或替代过程。而且,所提供的一个或多个操作还可以被修改或省略。
图11A和图11B示出了用于校正由多核苷酸测序仪生成的序列数据中的插入、缺失和取代误差的过程1100。过程1100可以由图1、图2和图10所示的追踪重构系统102来实现。
在1102中,要被编码为一个或多个DNA链的二进制数据被可逆地随机化。这种随机化发生在DNA链合成之前,并且在一些实现中,存在于所有读段中。读段可以经由图10所示的序列数据接口1008来接收。读段可以由图10所示的随机化模块1010来随机化。
在1104中,由多核苷酸测序仪生成的序列数据是使用聚类技术来聚类的。任何合适的聚类技术可以被使用,并且本领域的普通技术人员将能够标识合适的聚类技术。聚类创建从相同源DNA链得出的读段组。聚类可以由图10所示的聚类模块1012执行。在一些实现中,对随机化数据执行聚类提高了聚类技术将多个读段准确地分离为不同组的能力。不良形成的集群是包含从不同DNA链得出的读段的一个集群。诸如丢弃与共有序列偏离超过阈值量的读段等技术可以防止不良形成的集群影响最终的共有输出序列。然而,丢弃整个读段会丢失可能从该读段来获得的任何有用信息。
在1106中,被分类为表示DNA链的多个读段被接收以用于进一步分析。基于在1104中执行的聚类,读段可以被分类为表示相同的DNA链。由于使用了多核苷酸测序仪的输入仅是单个DNA链(或通过PCR产生的基本上相同的副本)的测序技术,多个读段也可以被分类为表示相同的DNA链。在一些实现中,多个读段可以经由图10所示的序列数据接口1008来接收。在其他实现中,多个读段可以在由聚类模块1012执行的聚类之后被接收。
在1108中,跨越多个读段的比较的位置被标识。比较的位置可以类似于图3所示的比较的位置300、图4所示的比较的位置400、图5所示的比较的位置500或图6所示的比较的位置600。在实现中,比较的位置可以由图10所示的读段比对模块1014标识。
在1110中,比较的位置处的多数共有碱基调用是通过标识该位置处的最常见的碱基调用来确定的。连结可以被任意地破坏。如上所述,最常见的碱基调用可以通过考虑存在于比较的位置处的碱基调用的质量信息来部分地标识。在实现中,多数共有碱基调用可以由图10所示的变体读段标识模块1018确定。
在1112中,确定比较的位置处的碱基调用是否与多数共有碱基调用相同。如果相同,那么被分析的读段在该位置处具有预期的碱基调用,不是相对于该位置的变体读段,并且过程1100遵循到1114的“是”路径。
在1114中,过程1100沿着读段提前到下一位置。然而,如果在比较的位置处的碱基调用与多数共有碱基调用不匹配,则过程1100遵循从1112到1116的“否”路径。
在1116中,来自比较的位置中的碱基调用不同于多数共有碱基调用的多个读段的读段被标识为变体读段。在实现中,该标识可以由图10所示的变体读段标识模块1018执行。
移动到图11B,在1118中,与比较的位置相邻的前瞻窗口中的碱基调用的共有字符串与变体读段中的碱基调用被比较。前瞻窗口中的碱基调用的共有字符串可以被限于来自在比较的位置处具有多数共有碱基调用的读段子集的碱基调用。例如,在10或20个读段的集合中,由于在比较的位置处的碱基调用与多数共有碱基调用不匹配,因此可能有多于一个变体读段。当比较是针对变体读段中的一个变体读段进行时,其他变体读段的前瞻窗口中的碱基调用不被考虑。这是因为其他变体读段可能具有缺失或插入误差,这将导致前瞻窗口中的碱基调用异相并且可能不正确。至少部分地基于该比较,误差的类型可以针对变体读段来确定。在实现中,该比较可以由图10所示的误差分类模块1020执行。在一个实现中,前瞻窗口的长度可能是2到4个位置。
在1120中,基于前瞻窗口中的碱基调用的共有字符串与变体读段的比较的位置之后的前瞻窗口中的碱基调用的字符串相同,变体读段的误差类型被确定为取代。因此,变体读段的前瞻窗口与非变体读段的前瞻窗口相匹配。该关系例如在图3中被示出。
在1122中,变体读段的比较的位置被提前一个位置。
在替代方案中,在1124中,基于前瞻窗口中的碱基调用的共有字符串与包括比较的位置中的碱基调用和相邻碱基调用的变体读段中的碱基调用字符串相同,变体读段的误差类型被确定为缺失。变体读段中的该碱基调用字符串的长度在长度上与前瞻窗口的长度相等。因此,例如如果前瞻窗口的长度为三个位置,则变体读段中的碱基调用字符串包括比较的位置中的碱基调用和接下来的两个碱基调用。该关系例如在图4中被示出。
在1126中,变体读段的比较的位置被提前零个位置。因为存在缺失,所以不将变体读段的比较的位置提前会重新比对链,使得链将同相以用于后续分析。
作为又一替代方案,在1128中,基于与两种特定方式相匹配的碱基调用,变体读段的误差类型被确定为插入。首先,在比较的位置之后的变体读段中的碱基调用与多数共有碱基调用相同,其次,前瞻窗口中的碱基调用的共有字符串与在变体读段序列中的碱基调用字符串相同。变体读段序列中的碱基调用字符串在长度上与前瞻窗口相等,并且该碱基调用字符串的起始位置是比较的位置之后的两个位置。该关系例如在图5中被示出。
在1130中,变体读段的比较的位置被提前两个位置。比较的位置被提前一个位置以说明插入,并且被提前第二位置,因为比较的位置针对所有非变体链都提前一个位置。这维护了链之间的比对以进行后续分析。
在1120、1124和1128的每一个中,至少部分地基于与多核苷酸测序仪相关联的误差分布,感兴趣位置处的变体读段的误差类型可以被确定。考虑多核苷酸测序仪的误差分布可以改变在前瞻窗口中的多数共有碱基调用和碱基调用的共有字符串的确定中的一个或两个。在实现中,误差分布的考虑可以由共有输出序列生成器1024执行。
在1132中,确定变体读段是否小于阈值可靠性级别。阈值级别可能是变体读段中的误差数目;无法唯一地被分类的变体中的误差数目;变体读段中的碱基调用的置信度级别的最小值、中值或模式;或(多个)其他因素。阈值数目可以是变体读段中从一个位置到位置总数的多个位置。阈值数目也可以是从1%到100%的百分比。如果变体读段小于阈值可靠性级别,则过程1100沿着“是”路径进行到1134。
在1134中,变体读段被省略,并且来自多个读段的单个共有输出序列被使用而不使用变体读段。在省略变体读段之后,过程1100进行到1136。备选地,如果变体读段不小于阈值可靠性级别(即,被认为是可靠的),则变体读段被用于进一步分析,并且过程1100沿着“否”路径进行到1136。如果由于存在不定误差而无法被分类为插入、缺失或取代,则变体读段被分类为这种,而不是丢弃读段,它可以被进一步分析,如下面的图13和/或图14所示。
在1136中,确定读段中是否还有附加未分析的位置。因此,确定是否读段的“末端”已经被达到,并且多数共有碱基调用已经针对读段的位置被标识。如果被用于将读段分解为多个较小的分段,则读段的末端也可以通过比对锚来标识。如果分析尚未到达末端,那么过程1100沿着“是”路径进行到1138。
在1138中,在比较的位置处具有多数共有碱基调用的多个读段的子集的位置(即,非变体读段)被提前一个。变体读段的新比较的位置在1122、1126或1130中基于所标识的误差类型被提前一定量。新比较的位置可能类似于新比较的位置312、412、512和608,如图3至图6所示。然后,过程1100返回到1108,并且分析继续。
在1136中,如果读段中没有未分析的位置,那么过程1100沿着“否”路径进行到1140。
在1140中,单个共有输出序列是部分地基于多数共有碱基调用和误差类型来确定的。单个共有输出序列可以由图10所示的共有输出序列生成器1024来确定。
图12A和图12B示出了用于通过考虑插入、缺失和/或取代误差来恢复合成DNA链中编码的二进制数据的过程1200。过程1200可以由图1、图2和图10所示的追踪重构系统102来实现。
在1202中,通过取二进制数据的异或(XOR)以及由种子和函数生成的随机序列,要被编码为DNA的二进制数据被可逆地随机化。该操作会影响DNA链,这些DNA链在读取时会产生还具有被随机化的特性的读段。在实现中,随机化可以由图10所示的随机化模块1010执行。
在1204中,多个读段是从多核苷酸测序仪接收的。在实现中,多个读段可以由图10所示的序列数据接口1008接收。
在1206中,多个读段通过序列相似性被聚类为多个集群。类似的序列很可能源自相同DNA链的测序(由于由多核苷酸测序仪引入的误差,导致序列不相同)。因此,一个集群应该表示来自同一DNA链的所有读段。回顾一下,多核苷酸测序仪可以同时对多个不同的DNA链进行测序,因此来自多核苷酸测序仪的序列数据的原始输出可能包括与多个不同的DNA链相对应的读段。在实现中,聚类可以由图10所示的聚类模块1012执行。
在1208中,集群是从多个集群中选择的。集群包含读段的聚类集合。如果聚类是准确的,则读段的聚类集合中的所有读段均来自同一DNA链的测序。此时,在进行附加分析之前,集群是仅通过具有聚类在一起的读段的特性来标识的。因此,在一些实现中,每个集群被依次分析,并且选择单个集群的顺序可以是任意的。集群中的多个集群也可以被并行分析。在实现中,集群的选择可以由图10所示的聚类模块1012来执行。分析可以继续直到追踪重构是对来自多个集群的所有集群执行的为止。
在1210中,读段的聚类集合在跨越读段的聚类集合的比较的位置处被比对。在实现中,比较的位置可以是在读段的聚类集合之间共享的第一位置。因此,该原始比对可以定义第一比较的位置。第一位置可以是最左侧的位置(对应于5'末端),或者备选地可以是最右侧的位置(对应于3'末端)。在实现中,比对可以由图10所示的读段比对模块1014执行。
在1212中,第一比较的位置处的多数共有碱基调用被确定。多数共有碱基调用至少部分基于跨读段的聚类集合的最常见的碱基调用。多数共有碱基调用还可以部分地基于与多核苷酸测序仪相关联的误差分布。即,碱基调用可以基于关联的误差分布来加权(例如更多的某些碱基调用更有价值,并且更少的某些碱基调用对确定多数共有碱基调用的影响较小)。
在1214中,来自读段的聚类集合的变体读段被标识。变体读段在比较的位置处具有与多数共有碱基调用不同的碱基调用。在实现中,变体读段可以由图10所示的变体读段标识模块1018来标识。
现在移动到图12B,在1216中,前瞻窗口中的碱基调用的共有字符串被标识。共有字符串基于读段的聚类集合的子集的碱基调用,其在比较的位置处具有多数共有碱基调用(即,非变体读段)。前瞻窗口与比较的位置相邻。在一些实现中,前瞻窗口可以是两个或三个位置长。
在1218中,至少部分地基于与碱基调用的共有字符串相匹配的变体读段的前瞻窗口中的基础调用,确定比较的位置处的变体读段的误差类型是取代。该关系的示例在图3中被示出。在实现中,误差类型可以由误差分类模块1020确定。
在1220中,变体链的比较的位置被前向移动一个位置。
在1222中,至少部分地基于变体读段中的一系列碱基调用(包括比较的位置处的碱基调用)以及与碱基调用的共有字符串相匹配的比较的位置之后的一个或多个碱基调用,确定比较的位置处的变体读段的误差类型是缺失。该关系的示例在图4中被示出。在实现中,误差类型可以由误差分类模块1020确定。
在1224中,变体链的比较的位置被前向移动零个位置。
在1226中,确定比较的位置处的变体读段的误差类型是插入。插入误差是至少部分地基于在与多数共有碱基调用相匹配的比较的位置之后的变体读段中的碱基调用以及在从与碱基调用的共有字符串相匹配的比较的位置之后的两个位置开始的变体读段中的一系列碱基调用来标识的。该关系的示例在图5中被示出。在实现中,误差类型可以由误差分类模块1020确定。
在1228中,变体链的比较的位置被前向移动两个位置。
在1230中,在读段的聚类集合的子集中的读段(即,非变体读段)的比较的位置被前向提前一个位置。
在1232中,单个共有输出序列是通过读段的聚类集合确定的。在实现中,单个共有输出序列由图10所示的共有输出序列生成器1024生成。
在1234中,单个共有输出序列被转换为二进制数据。在再次被用作数字计算机文件之前,这可能是对从DNA链得出的信息的最终操纵。在实现中,从序列数据到二进制数据的改变可以由图10所示的转换模块1028执行。
图13示出了用于标识读段的包含突发性误差的部分并利用该读段的除包含突发性误差的部分以外的部分生成共有输出序列的过程1300。作为丢弃整个读段的替代方案,过程1300可以与过程1100或1200一起被执行。如上所述,读段可以从存储数字数据的DNA链被生成。
在1302中,读段的包含突发性误差的一部分的起始被标识。读段的包含突发性误差的部分的起始可以通过检测误差本身来标识。例如,在与共有输出序列不匹配的读段(变体读段)中,未被分类为插入、缺失或取代的位置可能被解译为读段的包含突发性误差的部分的起始。标识不定误差的示例在图6中被示出。
在1304中,包含突发性误差的定位的末端被标识。标识包含突发性误差的定位的末尾会发现读段中超过突发性误差的定位的位置,使得读段往下的进一步分析该会发现可以与集群中的其他读段比对的序列。突发性误差的末端由读段中的候选位置标识。候选位置与后向匹配区域(mb)或前向匹配区域(mf)中的一个或两个侧面相接。如上所述,后向匹配区域与从同一集群中的多个读段中的其他读段生成的共有序列相匹配,并且前向匹配序列与通过多数投票从集群中的其他读段的序列生成的序列相匹配。
在1306中,读段的包含突发性误差的部分从共有输出序列的生成中被省略。因此,在被标识为突发性误差的起始的位置与被标识为突发性误差的末端的该位置之间的读段的对应位置无助于确定共有输出序列。针对共有输出序列中的这些位置,碱基调用是基于使用诸如图11和图12所图示的技术的集群中的其他读段中的一些或所有来确定的。
在1308中,共有输出序列是利用读段的包含突发性误差的部分的任一侧起的部分来生成的。因此,即使读段的一部分不被用于生成共有输出序列,读段的其他部分也被使用。通过在比较的位置处将多个读段的值进行比较,同时基于插入、缺失和取代相对于彼此比对多个读段,读段的具有突发性误差的部分的任一侧的部分被用于生成共有输出序列。共有输出序列可以由共有输出序列生成器1024生成。
图14示出了用于确定具有不定误差的读段是否可以通过发现读段往下的匹配序列来“带回”的过程1400。作为如果读段包括不定误差则丢弃整个读段的替代方案,过程1400可以与过程1100或1200一起被执行。不定误差可以是突发性误差,其不被分类为插入、缺失或取代中的任何一个。
在1402中,在作为多核苷酸测序仪的多个读段中的一个读段的读段中,不定误差被标识。多个读段可以是与例如由聚类模块1012形成的相同集群中的一组读段。不定误差是相对于多个读段的共有输出序列的误差。共有输出序列可以由共有输出序列生成器1024生成。
不定误差可以如图6所示被标识。附加地,将误差标识为不定误差可以由误差分类模块1020执行。不定误差位于读段中的第一位置处。例如,不定误差可能位于具有200个碱基对的总长度的读段中的位置100处。因此,在确定位置100处存在不定误差之前,共有输出序列可以针对位置1至99来标识。
在1404中,搜索窗口被定义,该搜索窗口包括读段中的第二位置,该第二位置位于至少延迟超过第一位置的一个距离处。在实现中,第二位置是由k
将搜索限于搜索窗口而不是该读段的全部其余部分将对匹配序列的搜索集中在匹配序列(如果存在)应该被发现的附近的区域。较短的搜索窗口使匹配序列将被标识的可能性降低,但是较长的搜索窗口增加了误报匹配的概率。在实现中,搜索窗口的长度可以在9到20个位置之间。如上所述,搜索窗口可以包括两个部分:用于k
在1406中,编辑距离是针对从比较序列确定的搜索窗口中的子序列来计算的。比较序列可以包括后向匹配序列(mb),其是多个序列读段中的其他读段中的一些或所有中的对应位置的共有输出序列。后向匹配序列的长度可能约为1至11个位置。
多个序列读段的共有输出序列可以是通过标识考虑位置的多数投票来确定的,同时考虑插入误差、缺失误差和取代误差,并且从当前考虑的碱基调用位置顺序地进行到相邻的碱基调用位置。因此,来自相同集群的所有其他读段(或在该位置处也没有不定误差的所有其他读段)可以被用于生成如上所述的共有输出序列。
比较序列还可以包括前向匹配序列(mf)。如上所述,前向匹配序列与后向匹配序列相邻,并被定位为超过后向匹配序列的定位(即,远离不定误差的定位)。前向匹配序列对应于比对读段的一部分,其共有输出序列尚未被确定。因为共有输出序列尚未针对前向匹配序列中所包括的位置被确定,所以来自集群中的其他读段的碱基调用的多数共有投票被用于确定是否存在匹配。多数共有投票简单地标识每个位置处最频繁的碱基调用,而不用尝试考虑诸如插入、缺失和取代等误差。如果前向匹配序列被包括,则前向匹配序列的长度为1至10个位置。
在实现中,滑动窗口可以在搜索窗口上移动,并且滑动窗口内的序列可以被用作与共有输出序列来比较的子序列。编辑距离可以针对滑动窗口的位置中的每个位置计算。取决于其位置,滑动窗口可以包括相对更多或相对更少的后向匹配序列和前向匹配序列。
在1408中,确定匹配是否是在对应位置处的子序列中的任何一个与共有输出序列之间发现的。匹配是通过编辑距离小于阈值来标识的。例如,如果阈值为0,那么仅完全匹配被视为匹配。如果编辑距离的阈值为1,那么具有单个碱基调用的两个序列可以被分类为匹配项。
如果匹配未被发现,那么过程1400沿着“否”路径进行到1410,并且读段从考虑中被删除。缺少匹配指示搜索窗口内没有与共有输出序列/多数共有投票相匹配的子序列。因此,该读段可能具有大量误差,并且最准确的共有输出序列可以通过忽略该读段来获得。
然而,如果匹配被发现,那么过程1400沿着“是”路径进行到1412。在1412中,确定多个匹配是否被发现。在搜索窗口内,可能有多于一个子序列与共有输出序列的对应碱基调用相匹配。如果是这种情况,那么过程1400沿着“是”路径进行到1414。
在1414中,匹配子序列中的一个匹配子序列被选择。在两个或更多个匹配子序列中,一个子序列可以通过多种技术中的任何一种来选择,诸如随机选择,选择子序列中最接近读段的开始的一个子序列(例如5'-末端),选择子序列中距离读段的制造最远的一个子序列或者通过另一标准。
在1416中,搜索窗口中的编辑距离小于阈值的单个子序列被标识。这是匹配子序列,指示在不定误差的定位之后,读段中存在可以再次与集群中的其他读段比对的部分。
在1418中,匹配子序列内的候选位置被选择。匹配子序列的长度可能约为5至15个位置,并且这些位置中的一个位置被选择以用作候选位置。候选位置可以与图7所示的候选位置(k)相同。在实现中,候选位置可以是在后向匹配序列中距不定误差开始的位置最远的位置。因此,如果不存在前向匹配序列,那么候选位置可以是在匹配子序列的末端的匹配子序列内的位置。如果存在前向匹配序列,那么候选位置可以是后向匹配序列中与前向匹配序列直接相邻的位置。
在1420中,比较的位置被设置为超过候选位置一个位置。比较的位置可以与图6所示的比较的位置600相同。因此,匹配子序列的定位和候选位置被用于确定如何在不定误差之后将该读段与其他读段重新比对。比较的位置位于读段中的第三位置处,使用上面引入的记号,该位置可以被指示为位于k+1处。
在1422中,来自多个读段的共有输出序列是在第三位置处确定的。该比较的位置被用于比较集群中的读段以及其他读段,以确定如上所述的共有输出序列。读段的分析可以继续,直到读段的末端被到达为止,并且来自输出序列的读段中的任何变型可以如本公开中早前描述的那样处置。
被用于标识匹配子序列并最终确定比较的位置的许多参数(诸如延迟、bsw、fsw、mb和mf)具有可以变化的长度。被用于分析序列数据的给定集合的长度可以通过测试延迟、搜索窗口、mb和mf中的每一个的不同值来实验式地确定。每个可以在值的范围内变化,并且不同的组合可以被测试。
针对长度的每个组合,可以从由多核苷酸测序仪产生的读段生成的集群数目被确定。生成大量集群指示即使有不定误差,更多的读段也能够被使用,并且由多核苷酸测序仪输出的更多数据有助于生成共有输出序列。因此,导致形成来自多个序列读段的最大数目的读段集群的值的组合可以被用作参数的长度。
示例
本公开中描述的技术是在由纳米孔测序生成的三个不同文件上测试的,其大小为32kB、115kB和1500kB。这些文件中的每个文件都包含以DNA链的序列被编码的数字信息。这不是合成数据,而是以DNA被编码的实际计算机文件。因此,该技术的定性测试部分地是对从DNA链中成功恢复文件的能力的测试。定量度量是从多核苷酸合成仪的输出正确恢复的集群数目以及测序的覆盖深度。
忽略了读段的包含不定误差的部分而使用不包含不定误差的部分(“新技术”)的该新颖技术与尝试将误差分类为插入、缺失或取代并且如果误差无法被分类为这三种类型中的一种类型(“旧技术”)则丢弃来自进一步考虑的整个读段的早前技术进行了比较。针对两个较小文件中的每个文件,新技术产生了由集群恢复和覆盖测量的更好结果。
正确恢复的集群数目指示了唯一的DNA链的数目,分析可以针对其生成共有序列。所形成的集群总数包括不产生共有序列的集群。因此,恢复更大数目的集群指示从更大数目的DNA链恢复序列。比较在下面的表1中被呈现。
表1.用于从多个噪声读段的追踪重构的两种不同技术之间的集群恢复比较。
因此,新技术正确地将更多集群恢复了2%至4%。1500kB的最大文件无法使用旧技术来译码(因此,没有比较的集群恢复值),但已使用新技术成功译码了。通过尝试延迟、搜索窗口前向(swf)、搜索窗口后向(swb)、后向匹配(mb)和前向匹配(mf)的各种值,大约5000个不同的参数组合被测试,以查看哪个组合会生成最大数目的正确集群。针对表1所示的所有测试结果,距离阈值(dt)被设置为零,因此需要精确匹配。swf和swb被设置为与sw(f/b)列中指示的值相同。产生32kB文件和115kB文件的最大数目的正确集群的参数在下面的表2中被示出。成功译码1500kB文件的四个不同参数组合也在表2中被示出。
表2.用于新技术的参数值的比较以成功译码文件并最大化正确恢复的集群数目。
针对所测试的文件,延迟长度为5至8,并且搜索窗口(前向和后向)也是5至8。如长度为0的示例所指示的,前向匹配区域或后向匹配区域可以被省略。然而,针对表2所示的所有示例,后向匹配区域和前向匹配区域的组合长度为7至10。
此外,新技术能够恢复覆盖率级别较低的文件。针对32kB文件,它是使用新技术以22×覆盖范围被成功译码,但旧技术未能以38×覆盖范围对其进行译码。115kB文件以27×覆盖范围被成功译码,但使用旧技术32×覆盖范围是不足的。针对1500kB的大文件,它是以27.2×覆盖范围成功译码的。利用1500kB文件的随机子样本进行的测试确定,它可以以低至23.1×的覆盖范围来恢复。使用该新技术,所有测试样本可以以27×或更小的覆盖范围来恢复,而旧技术在可以成功恢复文件时需要覆盖深度大大超过30×。与更高的覆盖级别相比,减少的覆盖范围允许以更少的测序和更少的费用对存储在DNA中的数字信息进行译码。
说明性实施例
以下条款描述了用于实现本公开所描述的特征的多个可能的实施例。本文描述的各种实施例不是限制性的,也不是来自要求存在于另一实施例中的任何给定实施例的每个特征。除非上下文另外清晰指示,否则任何两个或更多个实施例可以被组合在一起。如本文所使用的,在本文档中,“或”是指和/或。例如,“A或B”是指没有B的A,没有A的B,或者A和B。如本文所使用的,“包括”是指包括所有列出的特征,并可能包括未被列出的其他特征的添加。“基本上由...组成”是指包括所列出的特征以及不会实质性影响所列出特征的基础特性和新颖特性的那些附加特征。“由…组成”仅表示所列出的特征,而排除未列出的任何特征。
条款1.一种从存储数字数据的脱氧核糖核酸(DNA)链的多个读段生成共有序列的方法,该多个读段是通过如下测序技术以小于30×的覆盖范围而被生成的,该测序技术将突发性误差引入多个读段中的读段,该方法包括:从共有输出序列的生成中省略读段中包含突发性误差的一部分;以及利用从读段中包含突发性误差的部分的任一侧起的读段的部分来生成共有输出序列。
条款2.条款1的方法,其中测序技术是纳米孔测序。
条款3.条款1至2中任一项的方法,其中多个读段是以小于25×的覆盖范围而被生成的。
条款4.条款1至3中任一项的方法,还包括:通过标识读段中不与共有序列相匹配并且未被分类为插入、缺失或取代的位置来标识读段中包含突发性误差的部分的起始。
条款5.条款1至4中任一项的方法,还包括:通过标识读段中与以下至少一项侧面相接的位置来标识读段中包含突发性误差的部分的末端:与共有序列相匹配的后向匹配区域、或者与通过从多个读段中的至少两个其他读段的序列进行多数投票而生成的序列相匹配的前向匹配区域。
条款6.条款1至5中任一项的方法,其中共有序列是通过以下而被生成的:将比较的位置处的针对多个读段的值进行比较,同时基于插入、缺失和取代而将多个读段彼此进行比对。
条款7.一种对指令进行编码的计算机可读介质,该指令在由处理单元执行时使计算设备执行条款1至6中任一项的方法。
条款8.一种系统,包括被配置为实现条款1至6中任一项的方法的处理单元使用和存储器。
条款9.一种方法,包括:在来自多核苷酸测序仪的多个序列读段中的读段中,标识相对于多个序列读段的共有输出序列的不定误差,该不定误差位于第一位置处;定义包括读段中的第二位置的搜索窗口,该第二位置位于至少延迟超过第一位置的一个距离处;计算针对搜索窗口中的子序列与比较序列的编辑距离,该比较序列包括后向匹配序列,该后向匹配序列是多个序列读段中的至少两个序列读段中的对应位置的共有输出序列;标识搜索窗口中具有小于阈值的编辑距离的子序列;基于后向匹配序列的长度,选择子序列内的候选位置;将与来自多个序列读段的至少两个其他读段的比较的位置设置为读段中的第三位置,该第三位置是超过候选位置的一个位置;以及在第三位置处从多个序列读段确定共有输出序列。
条款10.条款9中任一项的方法,其中不定误差是未被分类为插入、缺失或取代中的任何一种的突发性误差。
条款11.条款9至10中任一项的方法,其中多个序列读段的共有输出序列是通过以下而被确定的:标识针对考虑位置的多数投票,同时考虑插入误差、缺失误差和取代误差,并且多个序列读段的共有输出序列从当前考虑的碱基调用位置顺序地进行到相邻的碱基调用位置。
条款12.条款9至11中任一项的方法,还包括:标识搜索窗口中具有小于阈值的编辑距离的第二子序列,并且选择子序列或第二子序列中最接近候选位置的一个子序列。
条款13.条款9至12中任一项的方法,其中编辑距离的阈值是0。
条款14.条款9至13中任一项的方法,其中比较序列还包括前向匹配序列,该前向匹配序列是从来自多个序列读段的至少两个其他读段的针对碱基调用的多数共有投票。
条款15.条款14的方法,其中延迟的长度在4到10个位置之间,搜索窗口的长度在9到20个位置之间,后向匹配序列的长度在1到11个位置之间,并且前向匹配序列的长度在1到10个位置之间。
条款16.条款15的方法,其中后向匹配序列的长度和前向匹配序列的长度的总和在5到15个位置之间。
条款17.条款15或16的方法,其中延迟的长度、搜索窗口的长度、后向匹配序列的长度和前向匹配序列的长度分别通过以下来实验确定:测试针对每个长度的不同值,并且选择导致从多个序列读段恢复出最大数目的读段集群的值的组合。
条款18.一种对指令进行编码的计算机可读介质,该指令在由处理单元执行时使计算设备执行条款9至17中任一项的方法。
条款19.一种系统,包括被配置为实现条款9至17中任一项的方法的处理单元使用和存储器。
条款20.一种用于序列读段的比对的系统,包括:一个或多个处理单元;耦合至一个或多个处理单元的存储器;比对锚模块,被存储在存储器中并且由一个或多个处理单元实现以:标识具有预定序列的比对锚序列,该预定序列存在于第一序列读段和第二序列读段中,将第一序列读段划分成第一子读段和第二子读段,第一子读段从第一序列读段的开始延伸到比对锚序列,第二子读段从比对锚序列延伸到第一序列读段的末端,将第二序列读段划分成第三子读段和第四子读段,第三子读段从第二序列读段的开始延伸到比对锚序列,第四子读段从比对锚序列延伸到第二序列读段的末端;以及读段比对模块,被存储在存储器中并且在一个或多个处理单元上被实现以:基于第一序列读段的开始、第二序列读段的开始、和比对锚序列,将第一子读段与第三子读段进行比对,以及基于比对锚序列、第一序列读段的末端、和第二序列读段的末端,将第二子读段与第四子读段进行比对。
条款21.条款20的系统,其中比对锚序列具有不与自身比对的序列。
条款22.条款20至21中任一项的系统,其中比对锚序列位于从第一序列读段的开始到第一序列读段的末端的距离的45%到55%之间,并且位于从第二序列读段的开始到第二序列读段的末端的距离的45%到55%之间。
条款23.条款20至22中任一项的系统,还包括寡核苷酸合成仪,该寡核苷酸合成仪被配置为用包括比对锚序列的仅一个实例的第一序列读段第一多核苷酸分子,并且合成第二多核苷酸序列,其中第二多核苷酸序列包括比对锚序列的仅一个实例。
条款24.条款20至23中任一项的系统,还包括共有输出序列生成器模块,该共有输出序列生成器模块被存储在存储器中并且在一个或多个处理单元上被实现以:从第一子读段和第三子读段生成第一正向共有序列;从第一子读段和第三子读段生成第一反向共有序列;将第一正向共有序列和第一反向共有序列组合成第一组合共有序列;从第二子读段和第四子读段生成第二正向共有序列;从第二子读段和第四子读段生成第二反向共有序列;将第二正向共有序列和第二反向共有序列组合成第二组合共有序列;通过在比对锚序列处的比对,将第二组合共有序列附加到第一组合共有序列的末端;以及删除比对锚序列。
条款25.一种用于序列读段的比对的系统,包括:用于处理的一个或多个部件;耦合至用于处理的一个或多个部件的存储器;用于标识具有预定序列的比对锚序列的部件,该预定序列存在于第一序列读段和第二序列读段中,用于将第一序列读段划分为从第一序列读段的开始延伸到比对锚序列的第一子读段、以及从比对锚序列延伸到第一多核苷酸序列的末端的第二子读段的部件,用于将第二序列读段划分为从第二序列读段的开始延伸到比对锚序列的第三子读段、以及从比对锚序列延伸到第二多核苷酸序列的末端的第四子读段的部件;用于基于第一序列读段的开始、第二序列读段的开始和比对锚序列将第一子读段与第三子读段进行比对的部件,以及用于基于比对锚序列、第一序列读段的末端和第二序列读段的末端将第二子读段与第四子读段进行比对的部件。
结论
尽管主题已经用特定于结构特征和/或方法动作的语言进行描述,但是应当理解,在所附权利要求中限定的主题并不一定被限于上述特定特征或动作。相反,特定特征和动作被公开为实现权利要求的示例形式。
- 通过具有不定误差的读段的追踪重构
- 使用具有独特分子索引(UMI)的冗余读段在测序DNA片段中抑制误差