一种手性1,4-二苯基-2-羟基-1,4-二丁酮类化合物的制备方法
文献发布时间:2023-06-19 11:30:53
技术领域
本发明属于有机合成技术领域,涉及一种手性1,4-二苯基-2-羟基-1,4-二丁酮类化合物的制备方法。
背景技术
不对称催化反应是制备手性物质的关键手段,不对称Mukaiyama aldol反应更是构建C-C键的重要方法,在生物医药、天然产物的合成制备上都有关键应用。
Aldol反应自发现以来,在构建各种重要的碳骨架方面都有重要的应用,aldol反应的研究仍在深入。2016年,Ikemoto等人选用乙醛酸酯作为亲电试剂参与不对称aldol反应,产物结构是抗AIDS药物的关键骨架(Org.Process Res.Dev.,2016,20,1615.)。2020年,Da课题组利用酶催化乙醛酸酯和醛类化合物的aldol反应,可以高效地制备高对映选择性的2-羟基双羰基化合物(Org.Lett.2020,22,4444.)。1973年,Mukaiyama小组使用烯醇硅醚与羰基化合物的aldol反应制备β-羟基酮化合物,进一步深化了aldol反应的研究,并开拓了Mukaiyama aldol反应的新领域。但是二羰基化合物参与的Mukaiyama aldol反应由于副反应的发生,其应用受到了很大的限制。2010年,Feng课题组使用手性镍络合物催化Mukaiyama aldol反应制备得到了本发明中的1,4-二苯基-2-羟基-1,4-二丁酮类化合物(Synlett,2011,7,903)。该化合物经过衍生后,在生物医药、天然产物骨架的构建上都有重要的应用。该化合物首先是由该课题组使用酰胺或烯氨基甲酸酯与苯酮醛反应制备得到(Chem.Commun.,2010,46,3771)。该反应为制备这类化合物提供了氮烯型反应的方法,反应对映选择性高,便捷高效。但是,使用Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物却存在着对映选择性不突出、反应条件苛刻的问题,反应需要使用严格的惰性气体保护,尤其在反应的中间体向最终产物转化的过程需要使用盐酸这样强酸,这使得该反应在环保和应用等方面都有很大的限制。而本发明提供的制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物的方法有高对映选择性的特点,产物ee值也比较突出;且反应可以一步转化为最终产物,无需中间体转化,反应条件温和,绿色环保。
尽管aldol反应有了以上的突破性的进展,但是双羰基化合物的不对称Mukaiyamaaldol反应仍有待研究发展,且底物的适用性、反应的对映选择性也有待进一步提高。
发明内容
本发明的目的在于提供一种手性1,4-二苯基-2-羟基-1,4-二丁酮类化合物的制备方法。本发明同时提供一种脯氨酸衍生的手性配体及其制备方法;及利用该配体与金属络合形成的手性金属复合物;以及利用该手性金属复合物作为催化剂催化苯酮醛一水合物或取代的苯酮醛一水合物的不对称Mukaiyama aldol反应,制备了一系列高对映选择性的1,4-二苯基-2-羟基-1,4-二丁酮类化合物的方法。
为此,本发明提供了一种手性1,4-二苯基-2-羟基-1,4-二丁酮类化合物的制备方法,所述方法包括:在手性金属复合物的存在下,烯醇硅醚与苯乙二醛一水合物或取代的苯乙二醛一水合物在溶剂中混合反应,得到手性1,4-二苯基-2-羟基-1,4-二丁酮类化合物;其中烯醇硅醚与手性金属复合物的摩尔比例为(10-20):1,反应如下式1所示:
所述烯醇硅醚的R
进一步地,所述的烯醇硅醚与手性金属复合物的摩尔比例为(10-18):1。
进一步地,所述的手性金属复合物具有如下A或A’表示的通式结构,
其中X
进一步地,所述的苯乙二醛一水合物或取代的苯乙二醛一水合物的浓度为0.1-10mmol/mL;优选地,所述的苯乙二醛一水合物或取代的苯乙二醛一水合物的浓度为0.1-5mmol/mL。
进一步地,所述混合反应的温度为-20-30℃;优选地,所述混合反应的温度为-20-20℃;更优选地,所述混合反应的温度为-10-20℃。
进一步地,所述混合反应的时间为1-2.5h。
进一步地,所述的手性金属复合物参与反应的浓度为0.5-5mmol/mL。
进一步地,所述的溶剂为酮、腈或含卤化合物;优选地,所述溶剂为丙酮、乙腈或氯仿。
同时本发明还提供了如下技术方案:
<1>.一种脯氨酸衍生的手性配体,其特征在于,所述手性配体由L或L’表示:
R
符合上述条件的配体可有如下所示结构:
<2>.一种用于制备上述<1>所述的手性配体的方法,所述方法包括如下步骤:
将带有保护基的脯氨酸甲酯A与溴化芳烃通过格氏反应,得到氨基醇类化合物B;
将氨基醇类化合物B使用还原剂还原,脱去保护得到氨基醇C;
氨基醇C与水杨醛化合物D反应得到手性配体L;
或者将带有保护基的脯氨酸甲酯A’与溴化芳烃通过格氏反应,得到氨基醇类化合物B’;
将氨基醇类化合物B’使用还原剂还原,脱去保护得到氨基醇C’;
氨基醇C’与水杨醛化合物D反应得到手性配体L’;
在上述式中,溴化芳烃中的取代基R选自氢原子、C1-C5的烷基、C1-C5的烷氧基和C1-C5的全氟烷基中的一种或多种,m为1~5的整数,R
<3>.一种手性金属复合物,其具有如下A或A’表示的通式结构,并且由根据上述<1>所述的手性配体与金属盐配位络合反应形成,
其中X
<4>.一种手性金属复合物的制备方法,其特征在于,所述方法包括:在反应溶剂中,加入金属盐、碱和根据上述<1>所述的手性配体,混合反应,所述金属盐中的金属离子选自铜、铁和锌中的至少一种;所述碱选自三乙胺、DABCO、碳酸钠和碳酸锂中的一种或多种。三乙胺、DABCO为有机碱,碳酸钠、碳酸锂为无机碱中。
<5>.根据上述<4>的方法,所述手性配体与金属盐及碱的摩尔比为(0.2-2.0):(0.2-2.0):(0.2-2.0)。
<6>.根据上述<4>的方法,所述的反应溶剂选自酮、腈或含卤化合物中的一种或多种;优选地,所述反应溶剂选自丙酮、乙腈、二氯甲烷或氯仿中的一种或多种。
本发明提供的手性金属复合物能高效地催化苯乙二醛一水合物及取代的苯乙二醛一水合物的不对称Mukaiyama aldol反应。反应得到了高对映选择性的1,4-二苯基-2-羟基-1,4-二丁酮类化合物。本发明提供的金属配体复合物解决了现有1,4-二苯基-2-羟基-1,4-二丁酮类化合物的Mukaiyama aldol反应中条件严苛的问题,为制备重要的有机合成中间体开拓了新的催化剂,对有机反应的发展和应用有重要意义。
附图说明
图1为实施例7中的得到的产物s-3aa的核磁共振氢谱;
图2为实施例7中的得到的产物s-3aa的核磁共振碳谱;
图3为实施例8中的得到的产物s-3ba的核磁共振氢谱;
图4为实施例8中的得到的产物s-3ba的核磁共振碳谱;
图5为实施例9中的得到的产物s-3ca的核磁共振氢谱;
图6为实施例9中的得到的产物s-3ca的核磁共振碳谱;
图7为实施例10中的得到的产物s-3da的核磁共振氢谱;
图8为实施例10中的得到的产物s-3da的核磁共振碳谱;
图9为实施例10中的得到的产物s-3da的核磁共振氟谱;
图10为实施例11中的得到的产物s-3ea的核磁共振氢谱;
图11为实施例11中的得到的产物s-3ea的核磁共振碳谱;
图12为实施例12中的得到的产物s-3fa的核磁共振氢谱;
图13为实施例12中的得到的产物s-3fa的核磁共振碳谱;
图14为实施例13中的得到的产物s-3ga的核磁共振氢谱;
图15为实施例13中的得到的产物s-3ga的核磁共振碳谱;
图16为实施例13中的得到的产物s-3ga的核磁共振氟谱;
图17为实施例14中的得到的产物s-3ha的核磁共振氢谱;
图18为实施例14中的得到的产物s-3ha的核磁共振碳谱;
图19为实施例15中的得到的产物s-3ia的核磁共振氢谱;
图20为实施例15中的得到的产物s-3ia的核磁共振碳谱;
图21为实施例16中的得到的产物s-3ja的核磁共振氢谱;
图22为实施例16中的得到的产物s-3ja的核磁共振碳谱;
图23为实施例17中的得到的产物s-3ka的核磁共振氢谱;
图24为实施例17中的得到的产物s-3ka的核磁共振碳谱;
图25为实施例18中的得到的产物s-3la的核磁共振氢谱;
图26为实施例18中的得到的产物s-3la的核磁共振碳谱;
图27为实施例19中的得到的产物s-3ma的核磁共振氢谱;
图28为实施例19中的得到的产物s-3ma的核磁共振碳谱;
图29为实施例20中的得到的产物s-3na的核磁共振氢谱;
图30为实施例20中的得到的产物s-3na的核磁共振碳谱;
图31为实施例20中的得到的产物s-3na的核磁共振氟谱;
图32为实施例21中的得到的产物s-3oa的核磁共振氢谱;
图33为实施例21中的得到的产物s-3oa的核磁共振碳谱;
图34为实施例22中的得到的产物s-3ab的核磁共振氢谱;
图35为实施例22中的得到的产物s-3ab的核磁共振碳谱。
具体实施方式
以下参照附图及具体实施方式对本发明进行进一步说明。
第一方面,本发明提供的一种手性金属复合物。所述的手性金属复合物具有如下A或A’表示的通式结构:
X
其中,配体是脯氨酸衍生的化合物,具有如下L或L’所示结构通式:
符合上述条件的配体可有如下所示结构:
本发明由脯氨酸酯出发,经过简单的三步法可以制备配体L或L’:
带有保护基的脯氨酸甲酯A(A’)与溴化芳烃通过格氏反应,得到氨基醇类化合物B(B’);
氨基醇类化合物B(B’)经氢气还原,脱保护得到氨基醇C(C’);
氨基醇C(C’)与水杨醛化合物D(D)反应可得到手性配体L(L’)。
R
第二方面,本发明提供了通式(1)所示的手性金属复合物的制备方法:使用金属盐与配体可在碱的作用下原位生成。
制备上述所示的手性金属复合物使用的配体与金属盐、碱的摩尔比例为(0.2-2.0):(0.2-2.0):(0.2-2.0)。优选为(0.2-1.5):(0.2-1.5):(0.2-1.5)。
本发明所述的金属盐可为从事本技术领域人员所熟知的铜、铁或锌。多选自于三氟甲磺酸铜、卤化铜、碘化亚铜、三氟甲磺酸铁、卤化铁、三氟甲磺酸锌或卤化锌等。通过实验结果,为实现金属的多配位、优异催化效果,以三氟甲磺酸盐为优选。
本发明提供的手性金属复合物,制备该手性技术复合物所用的碱,是从事本技术领域人员所熟知的各类有机碱、无机碱中的一种或多种,对于该手性金属复合物的制备并无限制。其中,以三乙胺、DABCO等有机碱,碳酸钠、碳酸锂等无机碱最为常用。考虑碱在溶剂中的溶解情况,一般优选使用有机碱。
制备上述所示的手性金属复合物所使用的溶剂可为酮类、腈类、含卤化合物中的一种或多种。其中,各类溶剂以丙酮、乙腈、二氯甲烷、氯仿为优选。
第三方面,本发明的手性金属复合物对于不对称Mukaiyama aldol反应展现出高效优良的催化效果。由下面各实例可以看出,选用配体L2与金属铜原位生成的手性金属复合物对于苯乙二醛一水合物及取代的苯乙二醛一水合物的Mukaiyama aldol反应有高效的催化效果,反应展现出优异的对映选择性。使用本发明中所述的手性金属配体复合物解决了这类反应的条件苛刻、中间体转换多步反应的问题,更加高效、环保、便捷地制备了β-羟基二羰基化合物。
因此,本发明还提供了一种1,4-二苯基-2-羟基-1,4-二丁酮类化合物的简便环保的制备方法,所述方法包括:将本发明所述的手性金属复合物或根据本发明所述制备的手性金属复合物与烯醇硅醚及苯乙二醛一水合物或取代的苯乙二醛一水合物混合反应,可以顺利得到1,4-二苯基-2-羟基-1,4-二丁酮类化合物,所述烯醇硅醚的R
本发明中所述的手性金属复合物,其参与反应的浓度可为0.5-5mmol/L;所述Mukaiyama aldol反应的温度为-10-25℃;所述Mukaiyama aldol反应的时间可为1-2.5h。
在本发明中,上述的烯醇硅醚与手性金属复合物的摩尔比例可为(10-20):1,以(10-18):1为优选。
本发明所述的手性金属复合物制备的具体实施方案如下,以手性配体L2-铜复合物为例:在反应容器中加入选定溶剂,按比例加入铜盐,搅拌一小时,让铜盐充分溶解;随后加入手性配体L2和碱,反应容器转移到0℃下搅拌一小时,降低温度减缓反应放热,有助于配体L2和铜均匀反应,保留铜的配位点。溶剂为本技术领域人员所熟知的溶剂,可为酮类、腈类、含卤化合物,以丙酮、乙腈、氯仿为优选。铜盐可以选用三氟苯磺酸铜、卤化铜、硝酸铜中的一种或多种。铜盐在溶剂中的浓度可为0.5–5mmol/L。
本发明提供的上述手性铜-配体L2复合物作为苯乙二醛一水合物或取代的苯乙二醛一水合物的不对称Mukaiyama aldol反应的催化剂的应用,以及一种制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物的方法,所述苯乙二醛一水合物或取代的苯乙二醛一水合物结构如下所示:
具体地,将手性铜复合物,烯醇硅醚与式(2)所示的一种苯乙二醛一水合物混合反应:烯醇硅醚与手性铜复合物的摩尔比例可为(10-20):1,以(10-18):1为优选;所述溶剂为本技术领域人员所熟知的溶剂,可优选为酮类、腈类或含卤化合物,其中最为常用的为丙酮、乙腈或氯仿;所述结构如上述式(2)所示的苯乙二醛一水合物的浓度可为0.1-10mmol/mL,优化为0.1-5mmol/mL;所述混合反应的温度可为-20-30℃,可优化为-20-20℃,反应时间可为1-2.5h。
反应方程式如下所示(以苯基烯醇硅醚作为亲核试剂参与反应为例):
优选条件的混合反应分离提纯后,得到手性1,4-二苯基-2-羟基-1,4-二丁酮类化合物;所述提纯方法均为本技术领域人员所熟知的分离提纯方法,包括萃取分液、液相色谱、气相色谱、重结晶、蒸馏、柱层析分离的方法,可优化为蒸馏和柱层析分离;所述柱层析分离方法的淋洗剂为本技术领域人员所熟知的溶剂,可选自石油醚、乙酸乙酯、甲醇、二氯甲烷、苯等溶剂中的一种或多种,可优化为石油醚与乙酸乙酯的混合溶剂;所述石油醚与乙酸乙酯的优化体积比可为(50-5):1。
从上述内容可知,本发明提供了使用一种手性铜配体L复合物的Lewis酸催化苯乙二醛一水合物的不对称Mukaiyama aldol反应、制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物的方法。并且这种方法具有高对映选择性、操作简便、原料和催化剂较为环保易得等优点。
本发明的所有原料可以来自市售,也可以根据文献报道自行制备,没明确限制。
以下实施例中所用的设备:
核磁共振:布鲁克400MHzfor
质谱仪:沃特斯Waters
为进一步说明本发明,下面结合具体实例对手性金属复合物在苯乙二醛一水合物的不对称Mukaiyama aldol反应中的高效催化作用进行详细备述。
下面实施例中所用的式3和式4所示的烯醇硅醚结构如下所示:
实施例1
脯氨酸衍生的手性配体的制备,以带有保护基的L-脯氨酸制备手性配体L2为例;
合成路线如下所示:
在充满氮气氛围的三口烧瓶中加入溶剂四氢呋喃(20mL),加入镁条(0.36g,15mmol)和对甲基溴苯(1.3g,7.5mmol),吹风机加热至反应引发。20min内逐滴加入剩余对甲基溴苯(1.3g,7.5mmol)。滴加结束后,继续加热至70℃回流2h。向反应体系中滴加带苄基保护的L-脯氨酸A(3.3g,15mmol)的四氢呋喃溶液(20mL),继续在70℃回流,反应7h。利用薄层色谱法监测反应结束,用饱和氯化铵水溶液淬灭反应。用乙酸乙酯萃取,饱和食盐水反萃。无水硫酸钠干燥后浓缩得到苄基保护的氨基醇化合物B。
将上述制备的化合物B(1.11g,3mmol)溶解在乙醇(30mL)中,加入浓盐酸(0.3mL,12mmol),反应体系中通入氢气。并在氢气氛围下,室温反应8h。薄层色谱法监测反应结束后,终止反应,脱离氢气氛围,反应混合物过滤后,用乙醇(30mL)洗涤滤渣,收集滤液,减压浓缩。随后向体系中加入乙酸50mL,用适量氢氧化钠溶液调节PH>9。混合物用乙酸乙酯萃取,饱和食盐水反萃,加入无水硫酸钠干燥,减压浓缩得到氨基醇C备用。
将上一步骤制备的氨基醇C(0.56g,2mmol)溶解在叔丁醇(30ml)中,加入2-羟基-3-三氟甲基苯甲醛(2mmol),加热至30℃,搅拌反应6小时。薄层析色谱监测反应结束,减压浓缩,剩余物利用硅胶柱柱层析分离提纯,淋洗剂为石油醚/乙酸乙酯,二者的体积比为5:1,得到手性配体L2。
实施例2
脯氨酸衍生的手性配体的制备,以带有保护基的L-脯氨酸制备手性配体L3为例;
合成路线如下所示:
在充满氮气氛围的三口烧瓶中加入溶剂四氢呋喃(20mL),加入镁条(0.36g,15mmol)和对甲氧基溴苯(1.4g,7.5mmol),吹风机加热至反应引发。20min内逐滴加入剩余对甲氧基溴苯(1.4g,7.5mmol)。滴加结束后,继续加热至70℃回流2h。向反应体系中滴加带苄基保护的L-脯氨酸A(3.3g,15mmol)的四氢呋喃溶液(20mL),继续在70℃回流,反应7h。利用薄层色谱法监测反应结束,用饱和氯化铵水溶液淬灭反应。用乙酸乙酯萃取,饱和食盐水反萃。无水硫酸钠干燥后浓缩得到苄基保护的氨基醇化合物B。
将上述制备的化合物B(1.21g,3mmol)溶解在乙醇(30mL)中,加入浓盐酸(0.3mL,12mmol),反应体系中通入氢气。并在氢气氛围下,室温反应8h。薄层色谱法监测反应结束后,终止反应,脱离氢气氛围,反应混合物过滤后,用乙醇(30mL)洗涤滤渣,收集滤液,减压浓缩。随后向体系中加入乙酸50mL,用适量氢氧化钠溶液调节PH>9。混合物用乙酸乙酯萃取,饱和食盐水反萃,加入无水硫酸钠干燥,减压浓缩得到氨基醇C备用。
将上一步骤制备的氨基醇C(0.63g,2mmol)溶解在叔丁醇(30ml)中,加入2-羟基-3-三氟甲基苯甲醛(2mmol),加热至30℃搅拌反应6小时。薄层析色谱监测反应结束,减压浓缩,剩余物利用硅胶柱进行柱层析分离提纯,淋洗剂为石油醚/乙酸乙酯,二者的体积比为5:1,得到手性配体L3。
实施例3
脯氨酸衍生的手性配体的制备,以带有保护基的L-脯氨酸制备手性配体L4为例;
合成路线如下所示:
在充满氮气氛围的三口烧瓶中加入溶剂四氢呋喃(20mL),加入镁条(0.36g,15mmol)和对三氟甲基溴苯(1.7g,7.5mmol),吹风机加热至反应引发。20min内逐滴加入剩余对甲基溴苯(1.7g,7.5mmol)。滴加结束后,继续加热在70℃回流2h。向反应体系中滴加带苄基保护的L-脯氨酸A(3.3g,15mmol)的四氢呋喃溶液(20mL),继续在70℃回流,反应7h。利用薄层色谱法监测反应结束,用饱和氯化铵水溶液淬灭反应。用乙酸乙酯萃取,饱和食盐水反萃。无水硫酸钠干燥后浓缩得到苄基保护的氨基醇化合物B。
将上述制备的化合物B(1.44g,3mmol)溶解在乙醇(30mL)中,加入浓盐酸(0.3mL,12mmol),反应体系中通入氢气。并在氢气氛围下,室温反应8h。薄层色谱法监测反应结束后,终止反应,脱离氢气氛围,反应混合物过滤后,用乙醇(30mL)洗涤滤渣,收集滤液,减压浓缩。随后向体系中加入乙酸50mL,用适量氢氧化钠溶液调节PH>9。混合物用乙酸乙酯萃取,饱和食盐水反萃,加入无水硫酸钠干燥,减压浓缩得到氨基醇C备用。
将上一步骤制备的氨基醇C(0.78g,2mmol)溶解在叔丁醇(30ml)中,加入2-羟基-3-三氟甲基苯甲醛(2mmol),加热至70℃,搅拌反应6小时。薄层析色谱监测反应结束,减压浓缩,剩余物利用硅胶柱进行柱层析分离提纯,淋洗剂为石油醚/乙酸乙酯,二者的体积比为5:1,得到手性配体L4。
实施例4
手性铜配体络合物制备
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时,通过降低温度减缓反应放热,促进配体L和铜均匀反应,保留铜的配位点。
实施例5
手性金属铁配体络合物制备
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铁(10.0mg,0.02mmol),搅拌一小时,让铁盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时,通过降低温度减缓反应放热,促进配体L和铜均匀反应,保留铁的配位点。
实施例6
手性金属锌配体络合物的制备
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸锌(7.27mg,0.02mmol),搅拌一小时,让锌盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时,通过降低温度减缓反应放热,促进配体L和铜均匀反应,保留铜的配位点。
实施例7
铜-手性配体L2络合物催化苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3aa
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体L2(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1a(苯乙二醛一水合物)(15.2mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3aa(89%收率,94%ee)。
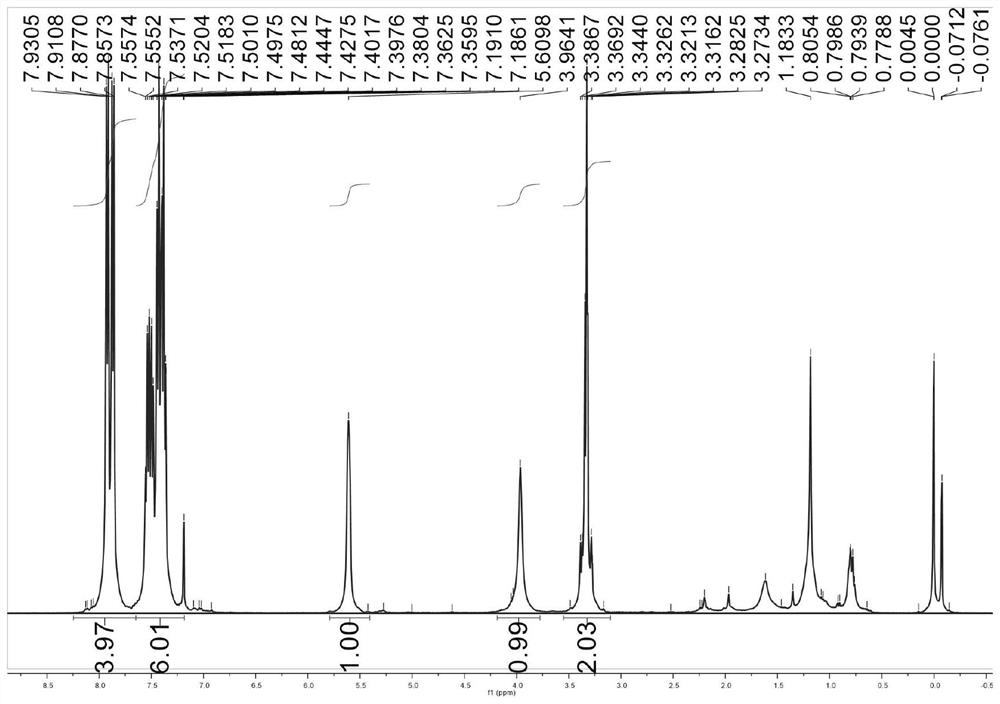
利用核磁共振对实施例7所得到的目标产物s-3aa进行分析,得到核磁共振氢谱,如图1所示。1H NMR(400MHz,CDCl3):δ7.93-7.86(dd,4H),7.56-7.36(m,6H),5.52(s,1H),4.02(s,1H),3.33(m,2H)。
利用核磁共振对实施例7所得到的目标产物s-3aa进行分析,得到核磁共振碳谱,如图2所示。13C NMR(100MHz,CDCl3):δ200.8,197.2,136.6,134.0,133.6,133.5,129.0,128.8,128,7,128.3,100.0,70.1,43.6。
利用质谱仪对实施例7所得到的目标产物s-3aa进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为277.0841,测定值为277.0843。
实施例8
铜-手性配体L2络合物催化4-甲基苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ba
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1b(4-甲基苯乙二醛一水合物)(16.6mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ba(88%收率,95%ee)。
利用核磁共振对实施例8所得到的目标产物s-3ba进行分析,得到核磁共振氢谱,如图3所示。1H NMR(400MHz,CDCl3):δ7.85,(m,4H),7.50(t,1H),7.38(t,2H),7.22(d,2H),5.60(d,1H),3.30(qd,2H),2.36(d,3H)。
利用核磁共振对实施例8所得到的目标产物s-3ba进行分析,得到核磁共振碳谱,如图4所示。13C NMR(100MHz,CDCl3):δ200.3,197.1,145.1,136.6,133.5,130.8,129.6,128.8,128.6,128.3,69.8,43.8,21.7。
利用质谱仪对实施例8所得到的目标产物s-3ba进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为291.0997,测定值为291.0999。
实施例9
铜-手性配体L2络合物催化4-甲氧基苯乙二醛一水合物Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ca
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1c(4-甲氧基苯乙二醛一水合物)(18.2mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ca(87%收率,91%ee)。
利用核磁共振对实施例9所得到的目标产物s-3ca进行分析,得到核磁共振氢谱,如图5所示。1H NMR(400MHz,CDCl3):δ7.98(ddd,4H),7.57(t,1H),7.46(t,2H),6.97(t,2H),5.67(dd,1H),4.09(dd,1H),3.87(s,3H)。
利用核磁共振对实施例9所得到的目标产物s-3ca进行分析,得到核磁共振碳谱,如图6所示。13C NMR(100MHz,CDCl3):δ199.1,197.3,164.3,136.7,133.6,131.2,128.7,128.4,126.1,114.2,69.6,55.6,44.1。
利用质谱仪对实施例9所得到的目标产物s-3ca进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为307.0946,,测定值为307.0949。
实施例10
铜-手性配体L2络合物催化4-氟苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3da
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1d(4-氟苯乙二醛一水合物)(17.0mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3da(83%收率,87%ee)。
利用核磁共振对实施例10所得到的目标产物s-3da进行分析,得到核磁共振氢谱,如图7所示。1H NMR(400MHz,CDCl3):δ8.06(m,2H),7.95(m,2H),7.59(m,1H),7.47(t,2H),7.17(m,2H),5.61(td,1H),4.03(d,1H),3.41(qd,2H)
利用核磁共振对实施例10所得到的目标产物s-3da进行分析,得到核磁共振碳谱,如图8所示。13C NMR(100MHz,CDCl3):δ199.1,197.5,167.4,164.9,136.5,133.7,131.7,131.6,131.5,128.7,128.3,116.3,116.1,70.2,43.3
利用核磁共振对实施例10所得到的目标产物s-3da进行分析,得到核磁共振氟谱,如图9所示。核磁共振氟谱显示产物中有氟原子。
利用质谱仪对实施例10所得到的目标产物s-3da进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为295.0746,,测定值为295.0749。
实施例11
铜-手性配体L2络合物催化4-氯苯乙二醛一水合物Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ea
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1e(4-氯苯乙二醛一水合物)(18.6mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ea(75%收率,95%ee)。
利用核磁共振对实施例11所得到的目标产物s-3ea进行分析,得到核磁共振氢谱,如图10所示。1H NMR(400MHz,CDCl3):δ7.97-7,。94(dd,4H),7.61-7.57(t,1H),7.52-7.44(t,4H),5.58(dd,1H),4.01(d,1H),4.11-3.24(qd,2H)
利用核磁共振对实施例11所得到的目标产物s-3ea进行分析,得到核磁共振碳谱,如图11所示。13C NMR(100MHz,CDCl3):δ199.5,197.5,140.4,136.5,133.8,132.1,130.3,129.3,128.8,128.3,70.3,43.2
利用质谱仪对实施例11所得到的目标产物s-3ea进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为311.0451,测定值为311.0451。
实施例12
铜-手性配体L2络合物催化4-溴苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3fa
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1f(4-溴苯乙二醛一水合物)(23.1mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3fa(80%收率,98%ee)。
利用核磁共振对实施例12所得到的目标产物s-3fa进行分析,得到核磁共振氢谱,如图12所示。1H NMR(400MHz,CDCl3):δ7.87(d,2H),7.81(d,2H),7.57(d,2H),7.52(t,1H),7.40(t,2H),5.50(dd,1H),3.93(d,1H),3.35(qd,2H)
利用核磁共振对实施例12所得到的目标产物s-3fa进行分析,得到核磁共振碳谱,如图13所示。13C NMR(100MHz,CDCl3):δ199.6,197.5,133.8,132.3,130.3,128.8,128.3,70.4,43.1
利用质谱仪对实施例12所得到的目标产物s-3fa进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为354.9946,测定值为354.9941。
实施例13
铜-手性配体L2络合物催化4-三氟甲基苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ga
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1g(4-三氟甲基苯乙二醛一水合物)(22.0mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ga(70%收率,89%ee)。
利用核磁共振对实施例11所得到的目标产物s-3ga进行分析,得到核磁共振氢谱,如图13所示。1H NMR(400MHz,CDCl3):δ8.0(t,2H),7.87(m,2H),7.70(d.2H),7.53(t,1H),7.41(m,2H),5.50(d,1H),3.96(t,1H),3.41(m,2H)
利用核磁共振对实施例13所得到的目标产物s-3ga进行分析,得到核磁共振碳谱,如图15所示。13C NMR(100MHz,CD3OD):δ199.1,198.0,138.5,136.7,134.0,133.2,129.2,128.4,127.9,125.3,125.2,69.4,42.0
利用核磁共振对实施例13所得到的目标产物s-3ga进行分析,得到核磁共振氟谱,如图16所示。核磁共振氟谱显示产物中含有氟原子。
利用质谱仪对实施例13所得到的目标产物s-3ga进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为345.0714,测定值为345.0710。
实施例14
铜-手性配体L2络合物催化3-甲基苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ha
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1h(3-甲基苯乙二醛一水合物)(16.6mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ha(90%收率,93%ee)。
利用核磁共振对实施例14所得到的目标产物s-3ha进行分析,得到核磁共振氢谱,如图17所示。1H NMR(400MHz,CDCl3):δ7.91-7.88(m,2H),7.62-7.54(m,2H),7.46-7.39(m,3H),7.31-7.29(m,2H),5.47(dd,1H),3.46-3.30(ddd,2H),2.55(d,3H)
利用核磁共振对实施例14所得到的目标产物s-3ha进行分析,得到核磁共振碳谱,如图18所示。13C NMR(100MHz,CD3OD):δ201.1,197.1,139.0,136.7,134.8,133.6,133.5,129.2,128.8,128.7,128.4,125.9,70.0,43.7,21.4
利用质谱仪对实施例14所得到的目标产物s-3ha进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为291.0997,测定值为291.1003。
实施例15
铜-手性配体L2络合物催化3-甲氧基苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ia
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1i(3-甲氧基苯乙二醛一水合物)(18.2mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ia(87%收率,93%ee)。
利用核磁共振对实施例15所得到的目标产物s-3ia进行分析,得到核磁共振氢谱,如图19所示。1H NMR(400MHz,CDCl3):δ7.88-7.85(dd,2H),7.49-7.40(m,3H),7.39-7.30(m,3H),7.09-7.06(m,1H),5.60-5.56(dd,1H),3.79(s,3H),3.39-3.28(m,2H)
利用核磁共振对实施例15所得到的目标产物s-3ia进行分析,得到核磁共振碳谱,如图20所示。13C NMR(100MHz,CD3OD):δ200.7,197.1,160.2,136.6,134.9,133.6,130.0,128.7,128.3,121..2,120.5,113.0,70.1,55.5,43.7
利用质谱仪对实施例15所得到的目标产物s-3ia进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为307.0941,测定值为307.0943。
实施例16
铜-手性配体L2络合物催化3-氯苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ja
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1j(3-氯苯乙二醛一水合物)(18.7mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ja(73%收率,89%ee)。
利用核磁共振对实施例16所得到的目标产物s-3ja进行分析,得到核磁共振氢谱,如图21所示。1H NMR(400MHz,CDCl3):δ7.99(s,1H),7.96-7.93(d,2H),7.89-7.86(d,2H),7.61-7.56(t,2H),7.49-7.42(dd,3H),5.56(s,1H),3.99(s,1H),3.60-3.27(m,2H)
利用核磁共振对实施例16所得到的目标产物s-3ja进行分析,得到核磁共振碳谱,如图22所示。13C NMR(100MHz,CD3OD):δ199.6,197.5,136.4,135.5,135.3,133.8,130.2,128.9,128.8,128,4,126.9,70.5,43.0,29.7
利用质谱仪对实施例16所得到的目标产物s-3ja进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为311.0451,测定值为311.0453。
实施例17
铜-手性配体L2络合物催化2-甲基苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ka
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1k(2-甲基苯乙二醛一水合物)(16.6mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ka(89%收率,95%ee)。
利用核磁共振对实施例17所得到的目标产物s-3ka进行分析,得到核磁共振氢谱,如图23所示。1H NMR(400MHz,CDCl3):δ7.90(d,2H),7.62-7.54(m,2H),7.46-7.39(m,2H),7.32-7.26(m,3H),5.46(m,1H),4.05(d,1H),3.46-3.30(ddd,2H),2.58(s,3H)
利用核磁共振对实施例17所得到的目标产物s-3ka进行分析,得到核磁共振碳谱,如图24所示。13C NMR(100MHz,CD3OD):δ201.1,197.1,139.0,136.7,134.8,133.6,133.5,129.2,128.8,128.7,128.4,125.9,70.0,43.7,21.4
利用质谱仪对实施例17所得到的目标产物s-3ka进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为291.0997,测定值为291.0999。
实施例18
铜-手性配体L2络合物催化2-硝基苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3la
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1l(2-硝基苯乙二醛一水合物)(19.7mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3la(79%收率,91%ee)。
利用核磁共振对实施例18所得到的目标产物s-3la进行分析,得到核磁共振氢谱,如图25所示。1H NMR(400MHz,CDCl3):δ8.08(d,1H),7.90(m,2H),7.72(m,1H),7.60-7.50(m,2H),7.48-7.40(m,3H),4.96(d,1H),3.66(d,2H),3.42(d,1H)
利用核磁共振对实施例18所得到的目标产物s-3la进行分析,得到核磁共振碳谱,如图26所示。13C NMR(100MHz,CD3OD):δ205.4,199.7,134.4,134.1,130.9,129.1,128.8,128.3,123.6,73.4,42.8,
利用质谱仪对实施例18所得到的目标产物s-3la进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为322.0691,测定值为322.0693。
实施例19
铜-手性配体L2络合物催化2-氯苯乙二醛一水合物Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ma
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入2-氯苯乙二醛一水合物1m(18.7mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ma(72%收率,94%ee)。
利用核磁共振对实施例19所得到的目标产物s-3ma进行分析,得到核磁共振氢谱,如图27所示。1H NMR(400MHz,CDCl3):δ7.99(s,1H),7.94(d,2H),7.88(d,1H),7.61-7.56(t,2H),7.49-7.44(t,3H),5.56(s,1H),3.99(s,1H),3.44(s,2H)
利用核磁共振对实施例19所得到的目标产物s-3ma进行分析,得到核磁共振碳谱,如图28所示13C NMR(100MHz,CD3OD):δ199.6,197.5,136.4,135.5,135.3,133.8,130.2,128.9,128.8,128,4,126.9,70.5,43.0
利用质谱仪对实施例19所得到的目标产物s-3ma进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为311.0451,测定值为311.0457。
实施例20
铜-手性配体L2络合物催化2-氟苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3na
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1n(2-氟苯乙二醛一水合物)(17.0mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3na(80%收率,94%ee)。
利用核磁共振对实施例20所得到的目标产物s-3na进行分析,得到核磁共振氢谱,如图29所示。1H NMR(400MHz,CDCl3):δ7.91-7.83(m,3H),7.51-7.45(m,2H),7.39-7.34(t,2H),7.24-7.18(dd,1H),7.10-7.04(dd,1H),5.32-5.28(dt,1H),3.99(s,1H),3.57-3.51(ddd,1H),3.31-3.25(dd,1H)
利用核磁共振对实施例20所得到的目标产物s-3na进行分析,得到核磁共振碳谱,如图30所示。13C NMR(100MHz,CD3OD):δ199.6,199.5,197.1,162.3,159.8,136.5,135.3,135.2,133.6,131.5,131.4,128.7,128.3,125.1,125.0,116.7,116.5,73.4,73.3,42.3,29.7
利用核磁共振对实施例20所得到的目标产物s-3na进行分析,得到核磁共振碳谱,如图31所示。核磁共振氟谱显示产物中含有氟原子。
利用质谱仪对实施例20所得到的目标产物s-3na进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为295.0746,测定值为295.0743。
实施例21
铜-手性配体L2络合物催化2-甲氧基苯乙二醛一水合物的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3oa
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1o(2-甲氧基苯乙二醛一水合物)(18.2mg,0.1mmol)以及上述式3所示的烯醇硅醚2a(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3oa(83%收率,99%ee)。
利用核磁共振对实施例21所得到的目标产物s-3oa进行分析,得到核磁共振氢谱,如图32所示。1H NMR(400MHz,CDCl3):δ7.87-7.83(m,3H),7.50-7.43(m,2H),7.39-7.34(t,2H),7.03-6.98(t,1H),6.91-6.88(d,1H),5.54(d,1H),4.04(s,1H),3.81(s,3H),3.41-3.32(dd,1H),3.19-3.12(dd,1H)
利用核磁共振对实施例21所得到的目标产物s-3oa进行分析,得到核磁共振碳谱,如图33所示。13C NMR(100MHz,CD3OD):δ202.2,197.1,158.5,136.9,134.8,133.3,131.7,128.6,128.3,124.0,121.3,111.7,73.5,55.7,42.9
利用质谱仪对实施例21所得到的目标产物s-3oa进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为307.0946,测定值为307.0953。
实施例22
铜-手性配体L2络合物催化呋喃衍生的烯醇硅醚的Mukaiyama aldol反应制备1,4-二苯基-2-羟基-1,4-二丁酮类化合物S-3ab
向10mL反应试管中加入1mL丙酮,加入三氟甲磺酸铜(7.22mg,0.02mmol),搅拌一小时,让铜盐充分溶解;加入上述实施例1中所制备的手性配体(4.2mg,0.1mmol)和三乙胺(2.8μL,0.02mmol),反应转移到0℃下搅拌一小时得到手性铜复合物。向体系中加入苯乙二醛一水合物1a(2-(呋喃-2-基)-2-氧代乙醛)(15.2mg,0.1mmol)以及上述式4所示的呋喃衍生的烯醇硅醚2b(42μL,0.2mmol),反应体系转移至5℃下反应2.5h。薄层色谱法监测反应结束后,用乙酸乙酯萃取,饱和食盐水反萃,无水硫酸钠干燥,经减压浓缩,剩余物利用柱层析分离,使用石油醚/乙酸乙酯以20/1到5/1梯度淋洗,得到白色固体产物S-3ab(89%收率,94%ee)。
利用核磁共振对实施例22所得到的目标产物s-3ab进行分析,得到核磁共振氢谱,如图34所示。1H NMR(400MHz,CDCl3):δ7.94-7.91(m,2H),7.57-7.51(m,2H),7.46-7.41(dd,2H),7.17-7.15(dd,1H),6.48-6.46(dd,1H),5.57(t,1H),3.93(s,1H),3.21-3.11(m,2H)
利用核磁共振对实施例22所得到的目标产物s-3ab进行分析,得到核磁共振碳谱,如图35所示。13C NMR(100MHz,CD3OD):δ200.5,185.8,152.5,147.0,134.1,133.4,129.0,128.8,118.2,112.6,69.9,43.7
利用质谱仪对实施例22所得到的目标产物s-3ab进行分析,得到结果HRMS(ESI)m/z,对于C16H14O3[M+Na]+的计算值为267.0633,测定值为267.0636。
需要说明的是,按照本发明上述各实施例,本领域技术人员是完全可以实现本发明独立权利要求及从属权利的全部范围的,实现过程及方法同上述各实施例;且本发明未详细阐述部分属于本领域公知技术。以上所述,仅为本发明部分具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本领域的人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。
- 一种手性1,4-二苯基-2-羟基-1,4-二丁酮类化合物的制备方法
- 一种手性噁二嗪杂环稠合1,4-苯并二氮䓬-2-酮类化合物及制备方法