一组稳定高效表达活性融合蛋白的Gateway真核表达系统
文献发布时间:2023-06-19 12:27:31
技术领域
本发明涉及一组稳定高效表达活性融合蛋白的Gateway真核表达系统。
背景技术
Gateway系统是一套高效精确的分子克隆技术。该套系统只需要两个简单的LR或BP反应,就能使目标序列在表达载体(Destination vector)和入门载体(ENTR vector)之间随意高效准确转换,且能保证每个目标序列的旁侧序列都高度一致。蛋白质的活性功能是生命力的体现。研究蛋白的结构,功能和活性一直是生物学研究的基本目标之一。将某一功能蛋白在非变性条件下纯化,测试其各种生物属性,是实现这一基本目标的主要途径。但现有的蛋白原核表达载体系统,往往采用低效过时的酶切连接系统,耗时费力,而且不能保证每种表达出来的重组蛋白都有完全一致的旁侧序列,对照性不强。因此,我们希望开发一种能利用Gateway分子克隆技术的新的载体系统,帮助我们更加高效精确的表达蛋白,为纯化蛋白做好前期工作。之前我们已开发出一套Gateway载体系统用于原核表达各种蛋白(专利申请号202110472170.X),但鉴于少数真核生物的蛋白并不能在原核系统中正确表达和组装,我们因而紧接着开发出以下两个载体,12xHis-GFP-GW和12xHis-GW-GFP,用于对这些少数特殊蛋白的真核系统表达。
发明内容
本发明的目的在于解决上述背景技术中提出的问题。
为解决上述技术问题,本发明提供的技术方案为:一组稳定高效表达活性融合蛋白的Gateway真核表达系统,具体方法包括制作以下A,B两款Gateway通用载体:
A:12xHis-GFP-GW
1)以pK7WGF2载体为底物,利用限制性内切酶SpeI对其进行酶切,得到酶切产物,并对酶切后片段进行纯化;
2)设计合成带有重组位点和组蛋白标签部分编码序列的的正向引物321-F1(5’-TCGACCTGCAGGCGGCCGCACTAGTATGCACCATCATCATCATCATCACCACCACCACCA-3’)以及反向引物321-R2(5’-CTCCTCGCCCTTGCTCACCATACTAGTGTGGTGGTGGTGGTGGTGATGATGATGATGATG-3’),由于正反引物彼此部分重叠,因此可以彼此相互作为模板,扩增延伸成完整的平展末端PCR产物;
3)检测PCR产物,并将正确片段长度的PCR产物纯化;
4)将纯化后的pK7WGF2酶切片段和纯化后的PCR产物按一定比例混合,加入重组酶
5)将反应好的重组产物导入大肠杆菌DB3.1感受态细胞,加入适量SOC液态培养基,37度摇床200rpm/min孵育1小时后,涂抹适量该生长液于含有奇霉素的固体LB培养基平板上,并放入37度生长箱过夜培养;
6)设计一条位于pK7WGF2骨架上35S启动子内的正向引物P35SF3(5’-CGCACAATCCCACTATCCTT-3’),以及一条位于GFP的反向引物GFP-R3(5’-GTAGGTCAGGGTGGTCACGA-3’),用于对单克隆的检测;
7)培养皿平板上长出适当大小的克隆后,挑取部分单克隆溶于20ul水中,取其中1.5ul作为样品模板,利用设计好的一对检测引物,配制成20ul PCR反应体系,对这些克隆进行检测,若PCR能扩增出预期大小的正确片段,则说明重组反应可能成功;
8)挑取能扩增出预期正确片段的单克隆接种于5ml含奇霉素的LB液体培养基中,37度摇床200rpm/min过夜培养;
9)利用试剂盒提取单克隆质粒;
10)将提取好的单克隆质粒进行测序检验,若测序结果和理论设计完全一致,则证明产品开发成功,可以大量留存该质粒,等待分子克隆制作某一特定蛋白表达载体时使用。
B:12xHis-GW-GFP
1)以pK7FWG2载体为底物,利用限制性内切酶SpeI对其进行酶切,得到酶切产物,并对酶切后片段进行纯化;
2)设计合成带有重组位点和组蛋白标签部分编码序列的的正向引物321-F1(5’-TCGACCTGCAGGCGGCCGCACTAGTATGCACCATCATCATCATCATCACCACCACCACCA-3’)以及反向引物321-R3(5’-TTTGTACAAACTTGTGATATCACTAGTGTGGTGGTGGTGGTGGTGATGATGATGATGATG-3’),由于正反引物彼此部分重叠,因此可以彼此相互作为模板,扩增延伸成完整的平展末端PCR产物;
3)检测PCR产物,并将正确片段长度的PCR产物纯化;
4)将纯化后的pK7FWG2酶切片段和纯化后的PCR产物按一定比例混合,加入重组酶
5)将反应好的重组产物导入大肠杆菌DB3.1感受态细胞,加入适量SOC液态培养基,37度摇床200rpm/min孵育1小时后,涂抹适量该生长液于含有奇霉素的固体LB培养基平板上,并放入37度生长箱过夜培养;
6)设计一条位于pK7FWG2骨架上35S启动子内的正向引物P35SF3(5’-CGCACAATCCCACTATCCTT-3’),以及一条位于attR1位点的反向引物attR1R(5’-CATTTTACGTTTCTCGTTCAGC-3’),用于对单克隆的检测;
7)培养皿平板上长出适当大小的克隆后,挑取部分单克隆溶于20ul水中,取其中1.5ul作为样品模板,利用设计好的一对检测引物,配制成20ul PCR反应体系,对这些克隆进行检测,若PCR能扩增出预期大小的正确片段,则说明重组反应可能成功;
8)挑取能扩增出预期正确片段的单克隆接种于5ml含奇霉素的LB液体培养基中,37度摇床200rpm/min过夜培养;
9)利用试剂盒提取单克隆质粒;
10)将提取好的单克隆质粒进行测序检验,若测序结果和理论设计完全一致,则证明产品开发成功,可以大量留存该质粒,等待分子克隆制作某一特定蛋白表达载体时使用。
本发明优点在于:本发明作为对前面申请专利(专利申请号202110472170.X)的必要补充,提供了一种方便快捷的分子克隆系统,用于少数不能在原核系统中表达的真核蛋白的高效准确表达,使蛋白表达纯化系统更全面和完善,功能更强大,可有力促进活性蛋白的纯化工作,为蛋白的功能学研究建立基础。
附图说明
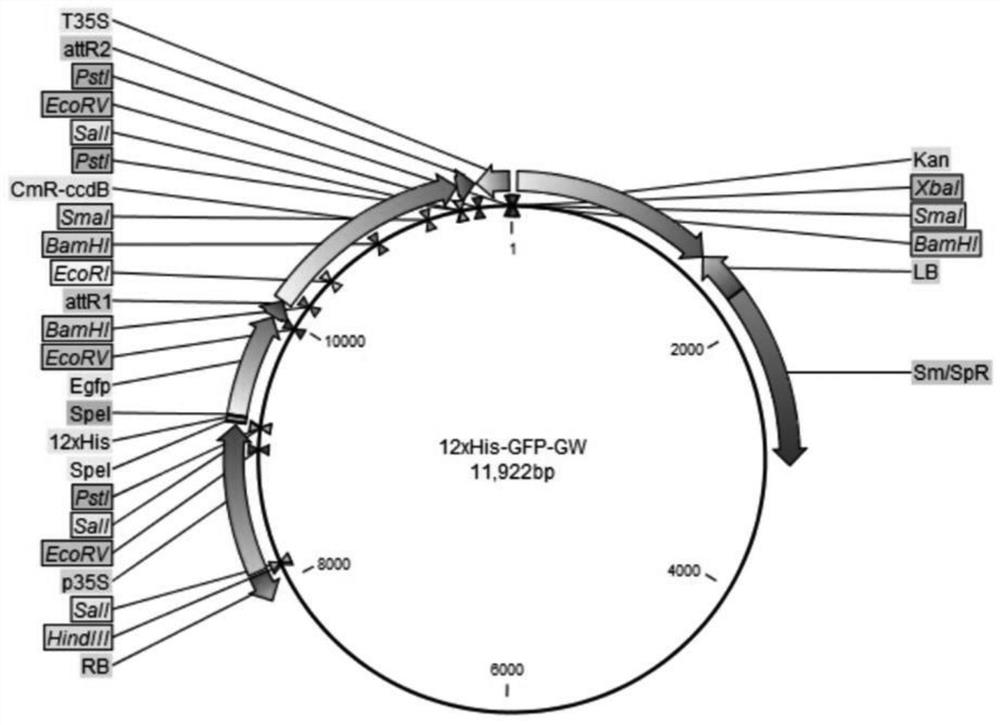
图1是本发明A载体(12xHis-GFP-GW)的结构图。
图2是本发明B载体(12xHis-GW-GFP)的结构图。
图3是本发明实施例1(12xHis-GFP-GW)的流程示意图。
图4是本发明实施例2(12xHis-GW-GFP)的流程示意图。
具体实施方式
下面用具体实施例说明本发明,并不是对本发明的限制。
实施例1(12xHis-GFP-GW)载体
如图3所示,一组稳定高效表达活性融合蛋白的Gateway真核表达系统中的A通用载体,具体制作方法包括以下内容:
1)以pK7WGF2载体为底物,利用限制性内切酶SpeI对其进行酶切,得到酶切产物,并对酶切后片段进行纯化;
2)设计合成带有重组位点和组蛋白标签部分编码序列的的正向引物321-F1(5’-TCGACCTGCAGGCGGCCGCACTAGTATGCACCATCATCATCATCATCACCACCACCACCA-3’)以及反向引物321-R2(5’-CTCCTCGCCCTTGCTCACCATACTAGTGTGGTGGTGGTGGTGGTGATGATGATGATGATG-3’),由于正反引物彼此部分重叠,因此可以彼此相互作为模板,扩增延伸成完整的平展末端PCR产物;
3)检测PCR产物,并将正确片段长度的PCR产物纯化;
4)将纯化后的pK7WGF2酶切片段和纯化后的PCR产物按一定比例混合,加入重组酶
5)将反应好的重组产物导入大肠杆菌DB3.1感受态细胞,加入适量SOC液态培养基,37度摇床200rpm/min孵育1小时后,涂抹适量该生长液于含有奇霉素的固体LB培养基平板上,并放入37度生长箱过夜培养;
6)设计一条位于pK7WGF2骨架上35S启动子内的正向引物P35SF3(5’-CGCACAATCCCACTATCCTT-3’),以及一条位于GFP的反向引物GFP-R3(5’-GTAGGTCAGGGTGGTCACGA-3’),用于对单克隆的检测;
7)培养皿平板上长出适当大小的克隆后,挑取部分单克隆溶于20ul水中,取其中1.5ul作为样品模板,利用设计好的一对检测引物,配制成20ul PCR反应体系,对这些克隆进行检测,若PCR能扩增出预期大小的正确片段,则说明重组反应可能成功;
8)挑取能扩增出预期正确片段的单克隆接种于5ml含奇霉素的LB液体培养基中,37度摇床200rpm/min过夜培养;
9)利用试剂盒提取单克隆质粒;
10)将提取好的单克隆质粒进行测序检验,若测序结果和理论设计完全一致,则证明产品开发成功,可以大量留存该质粒,等待分子克隆制作某一特定蛋白表达载体时使用。
实施例2(12xHis-GW-GFP)载体
如图4所示,一组稳定高效表达活性融合蛋白的Gateway真核表达系统,具体制作方法包括以下内容:
1)以pK7FWG2载体为底物,利用限制性内切酶SpeI对其进行酶切,得到酶切产物,并对酶切后片段进行纯化;
2)设计合成带有重组位点和组蛋白标签部分编码序列的的正向引物321-F1(5’-TCGACCTGCAGGCGGCCGCACTAGTATGCACCATCATCATCATCATCACCACCACCACCA-3’)以及反向引物321-R3(5’-TTTGTACAAACTTGTGATATCACTAGTGTGGTGGTGGTGGTGGTGATGATGATGATGATG-3’),由于正反引物彼此部分重叠,因此可以彼此相互作为模板,扩增延伸成完整的平展末端PCR产物;
3)检测PCR产物,并将正确片段长度的PCR产物纯化;
4)将纯化后的pK7FWG2酶切片段和纯化后的PCR产物按一定比例混合,加入重组酶
5)将反应好的重组产物导入大肠杆菌DB3.1感受态细胞,加入适量SOC液态培养基,37度摇床200rpm/min孵育1小时后,涂抹适量该生长液于含有奇霉素的固体LB培养基平板上,并放入37度生长箱过夜培养;
6)设计一条位于pK7FWG2骨架上35S启动子内的正向引物P35SF3(5’-CGCACAATCCCACTATCCTT-3’),以及一条位于attR1位点的反向引物attR1R(5’-CATTTTACGTTTCTCGTTCAGC-3’),用于对单克隆的检测;
7)培养皿平板上长出适当大小的克隆后,挑取部分单克隆溶于20ul水中,取其中1.5ul作为样品模板,利用设计好的一对检测引物,配制成20ul PCR反应体系,对这些克隆进行检测,若PCR能扩增出预期大小的正确片段,则说明重组反应可能成功;
8)挑取能扩增出预期正确片段的单克隆接种于5ml含奇霉素的LB液体培养基中,37度摇床200rpm/min过夜培养;
9)利用试剂盒提取单克隆质粒;
10)将提取好的单克隆质粒进行测序检验,若测序结果和理论设计完全一致,则证明产品开发成功,可以大量留存该质粒,等待分子克隆制作某一特定蛋白表达载体时使用。
本发明的原理:实施例1表达载体12xHis-GFP-GW和实施例2表达载体12xHis-GW-GFP通过与ENTR入门载体的LR反应,目标序列能高效精确插入这其中的任一表达载体。将构建好的表达载体导入农杆菌并注射浸染烟草后,通过观察烟草叶片的绿色荧光蛋白(GFP)信号,可以方便快速判断目标融合蛋白的表达情况。若融合蛋白的表达符合要求,便可大量收集具有GFP信号的烟草叶片并制成组织匀浆,并利用组蛋白标签与镍柱的特异亲和特性,将目标蛋白纯化出来。综上,本发明开发了一套Gateway分子克隆体系,用于对一些难以原核表达的单一真核生物蛋白成分,尤其是植物源蛋白成分的高效特异表达纯化。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 一组稳定高效表达活性融合蛋白的Gateway真核表达系统
- 新的基因工程抗体真核高效表达系统、其制备方法及其应用