一种猪流行性腹泻病毒S1D片段蛋白的编码基因及其应用
文献发布时间:2023-06-19 12:27:31
技术领域
本发明涉及猪流行性腹泻技术领域,具体地,涉及一种猪流行性腹泻病毒S1D片段蛋白的编码基因及其应用。
背景技术
流行性腹泻(Porcine epidemic diarrhea)由猪流行性腹泻病毒(PEDV)感染导致仔猪呕吐、腹泻、脱水及死亡的高度传染性疾病,不同年龄的猪均可感染,但以10日龄的仔猪死亡率最高。在我国,该病最早报道于2010年秋,之后蔓延全国。目前,该病已导致大量哺乳仔猪死亡,给养殖户造成了严重的经济损失,已成为影响我国养猪业健康发展的重要疫病之一。
PEDV具有典型的冠状病毒结构,含有4种主要结构蛋白,分别为核衣壳蛋白(N)、膜蛋白(M)、纤突蛋白(S)和小囊膜蛋白(E)。其中S蛋白携带主要的B淋巴细胞抗原决定簇,能够诱导机体产生中和抗体。因此,S蛋白在病毒感染、致病性和宿主细胞嗜性方面起关键作用。PEDV进入细胞前,在细胞蛋白酶的作用下,S蛋白裂解为S1(1~789aa)和S2 (790~1383aa),S1折叠成球状结构域。S1蛋白的COE(胶原酶等效域)区域(499~638 aa)是诱导机体产生中和抗体的主要抗原,而S1蛋白的S1D(636~789aa)区包含1个线性抗原表位(697~742aa)和2个B细胞表位(744~759aa和756~771aa),两者都被视为是优秀的疫苗候选抗原。
为进一步研发PEDV疫苗,需要获得大量的S1D片段蛋白作为抗原。而作为最早被人们采用、目前掌握最为成熟的技术,就是原核表达系统。它主要是将已带有克隆目的基因片段的载体转化细菌(通常是大肠杆菌)通过诱导表达获得目的蛋白。由于其遗传背景清楚,成本低、周期短,因此最常用的就是大肠杆菌的表达系统。然而,在大肠杆菌里表达外源蛋白,经常会遇到表达量低的情况,主要原因是由于外源基因的密码子是大肠杆菌非偏爱的密码子,细菌里面缺乏识别这些密码子的tRNA,而蛋白的翻译速度往往取决于tRNA,其减少会导致蛋白质合成速率降低,因此表达量不高,甚至有可能提前终止翻译。
同时,在在大量表达蛋白后,纯化方法的选择显得尤为重要,常用的方法是镍柱亲和纯化或切胶纯化,两种方法各有利弊。前一种方法可以获得较纯且具有良好活性的目的蛋白,但是在过柱过程中会损耗较多,导致纯化后的蛋白浓度不高,不利于后续小鼠的免疫;后一种方法也可以获得较纯的目的蛋白且损失不会太大,但是纯化后的蛋白不具有生物活性。
发明内容
本发明的目的是为了克服现有技术的上述不足,提供一种猪流行性腹泻病毒S1D片段蛋白的编码基因及其应用,本发明运用大肠杆菌常用密码子(偏爱密码子)替代稀有密码子,可在原核表达系统中显著提高重组蛋白的表达水平。解决了重组蛋白在原核表达系统(大肠杆菌) 表达量低的技术问题,以达到重组蛋白在细胞内大量表达的目的,并利用其制备单克隆抗体制备及建立乳清IgA ELISA检测方法。
本发明的第一个目的是提供一种猪流行性腹泻病毒S1D片段蛋白的编码基因。
本发明的第二个目的是提供一种重组载体。
本发明的第三个目的是提供一种工程菌。
本发明的第四个目的是提供一种大量表达猪流行性腹泻病毒S1D片段蛋白的制备方法。
本发明的第五个目的是提供任一所述的制备方法制备得到的猪流行性腹泻病毒S1D片段蛋白。
本发明的第六个目的是提供所述的猪流行性腹泻病毒S1D片段蛋白在制备猪流行性腹泻病毒检测试剂盒和/或抗猪流行性腹泻病毒抗体中的应用。
为了实现上述目的,本发明是通过以下方案予以实现的:
本发明成功构建了含有S1D片段蛋白密码子优化后序列ΔS1D基因的重组质粒pET-△S1D,利用大肠杆菌外源基因表达系统成功表达出重组蛋白,经SDS-PAGE、Westernblotting鉴定,重组蛋白为猪流行性腹泻病毒S1D片段蛋白。经过密码子优化后,与未优化的S1D片段蛋白相比,密码子优化后的△S1D片段蛋白表达量显著提高。采用的方法是经优化后的包涵体纯化方法,配制包涵体的洗涤缓冲液、溶解缓冲液及复性缓冲液,并严格调节其pH值;室温用洗涤缓冲液对粗制包涵体进行洗涤3次,此过程洗掉了较多的杂蛋白,制成精制包涵体;用溶解缓冲液于4℃对精制包涵体进行溶解2h~12h,具体时间视包涵体溶解程度决定,待大部分溶于溶解缓冲液时离心取上清进行透析复性,按照梯度降低复性的方法用复性缓冲液于4℃使目的蛋白恢复活性,最终成功制备具有活性且纯度良好的△S1D片段蛋白,经BCA方法测定,纯化后△S1D片段蛋白浓度能达到1mg/mL左右,可以作为良好的抗原免疫小鼠以制备单克隆抗体,或包被酶标板进行ELISA检测。
进一步,制备△S1D片段蛋白的单克隆抗体,将纯化的△S1D片段蛋白免疫BALB/c小鼠,采用细胞融合、筛选及亚克隆方法,成功筛选出一株单克隆细胞株,利用体内诱生法制备小鼠腹水。ELISA结果显示,腹水的抗体效价达到了1:1000000,腹水与PEDV病毒粒子和纯化后的△S1D片段蛋白反应均呈阳性,与PEDV N蛋白、pET-32a空载蛋白和PRRSV、TGEV、CSFV、 PDCoV、SADs 5种病毒粒子反应均呈阴性。Western-blotting表明腹水与△S1D片段蛋白、PEDV S蛋白反应均有特异性条带,与pET-32a空载蛋白、正常Vero细胞蛋白均无特异性条带出现。 IFA结果显示,腹水及阳性对照组均能使细胞出现特异性绿色荧光信号,而空白及阴性组均未见绿色荧光信号。
以及,建立乳清IgA ELISA检测方法,基于纯化后的△S1D片段蛋白作为包被抗原,经过不同条件的优化,确定了该方法的最佳反应条件。最佳抗原包被浓度及条件为4μg/mL、4℃过夜包被,最佳乳清稀释倍数为1∶160,最佳封闭液及封闭时间分别为5%(w/v)BSA、90min,乳清最佳作用时间为120min,HRP-山羊抗猪IgA最佳稀释浓度及作用时间分别为1:10000、 60min,底物显色液(TMB)最佳显色时间为25min。运用平均值与标准差的组合方法,确定了阳性乳清临界值为0.144,重复性实验表明,批内及批间重复性实验两者变异系数均不超过10%,敏感性实验表明,4份阳性乳清样品均按一定条件稀释至1:160时,结果仍呈阳性,对比实验表明,本研究建立的方法敏感性为85.4%,特异性为80.0%,两种方法符合率为85.0%,说明本研究建立的检测方法敏感性和特异性良好,其阳性检出率略低于国产试剂盒,为临床上监测母猪初乳乳清抗PEDV IgA的水平提供了技术支撑。
因此本发明要求保护一种猪流行性腹泻病毒S1D片段蛋白的编码基因,其核苷酸序列如 SEQ ID NO:1所示。
经过密码子优化的基因序列△S1D(438bp),SEQ ID NO:1:
ATG ACC CTG GAT GTG TGC ACC AAA TAT ACC ATT TAT GGC TTT AAA GGC GAAGGC ATT ATT ACC CTG ACC AAT AGC AGC TTT CTG GCG GGC GTG TAT TAT ACC AGC GATAGC GGC CAG CTG CTG GCG TTT AAA AAT GTG ACC AGC GGC GCG GTG TAT AGC GTG ACCCCG TGC AGC TTT AGC GAA CAG GCG GCG TAT GTG GAT GAT GAT ATT GTG GGC GTG ATTAGC AGC CTG AGC AGC AGC ACC TTT AAT AGC ACC CGC GAA CTG CCG GGC TTT TTT TATCAT AGC AAT GAT GGC AGC AAT TGC ACC GAA CCG GTG CTG GTG TAT AGC AAT ATT GGCGTG TGC AAA AGC GGC AGC ATT GGC TAT GTG CCG AGC CAG AGC GGC CAG GTG AAA ATTGCG CCG ATG GTG ACC GGC AAT ATT AGC ATT CCG ACC AAT TTT AGC
本发明还要求保护一种重组载体,所述重组载体为连接有所述的编码基因的载体。
优选地,所述载体为表达载体。
更优选地,所述载体为pET-32a。
本发明还要求保护一种工程菌,含有所述的重组载体的菌株。
优选地,所述菌株为大肠杆菌。
本发明还要求保护一种大量表达猪流行性腹泻病毒S1D片段蛋白的方法,利用含有携带所述的编码基因的质粒的菌株进行表达。
优选地,所述载体为表达载体。
更优选地,所述载体为pET-32a。
优选地,所述菌株为大肠杆菌。
优选地,当菌液OD
更优选地,当菌液OD
优选地,诱导表达后菌液以6000~8000r/min离心10~20min,弃上清取菌体,用0.01mol/L, pH为7.4,含1~2%(v/v)PMSF(苯甲基磺酰氟)的PBS重悬菌体,在冰浴中超声破碎细菌, 11000~12000r/min离心15~20min后分别收获沉淀,得到包涵体粗品。
更优选地,诱导表达后菌液以6000r/min离心15min,弃上清取菌体,用0.01mol/L,pH为 7.4,含1%(v/v)PMSF(苯甲基磺酰氟)的PBS重悬菌体,在冰浴中超声破碎细菌,12000r/min 离心20min后分别收获沉淀,得到包涵体粗品。
优选地,依次使用包涵体洗涤液和Tris-HCl洗涤包涵体粗品,其中包涵体洗涤液含有Tris -HCl、NaCl,尿素、和Triton X-100;使用包涵体变性溶解液溶解,其中包涵体变性溶解液含有Tris-HCl、NaCl、尿素、Triton X-100、和DTT。。
更优选地,包涵体洗涤液含有15~30mmol/L pH 7.5~8.0Tris-HCl、0.4~0.6mol/L NaCl、 1~2mol/L尿素、和1~2%(v/v)Triton X-100。
进一步优选地,包涵体洗涤液含有20mmol/L pH 8.0Tris-HCl、0.5mol/L NaCl、2mol/L 尿素、和2%Triton X-100。
更优选地,依次使用包涵体洗涤液进洗涤包涵体粗品两次,洗涤2~4h,再使用Tris-HCl 洗涤包涵体粗品。
更优选地,包涵体变性溶解液含有15~30mmol/L pH 7.5~8.0Tris-HCl、、0.4~0.6mol/L NaCl、6~8mol/L尿素、1~2%Triton X-100,和0.1~0.3mmol/L DTT。
进一步优选地,包涵体变性溶解液含有20mmol/L pH 8.0Tris-HCl、0.5mol/LNaCl、6mol/L 尿素、2%Triton X-100,和0.2mmol/L DTT。
优选地,依次使用含有浓度递减的尿素TGE缓冲液透析。
更优选的,依次使用含有6~8M、4~6M、2~4M、和0~2M的尿素TGE缓冲液透析。
进一步优选地、依次使用含有6M、4M、2M、和0M的尿素TGE缓冲液透析。
所述的编码基因、所述的重组载体和/或所述的工程菌在制备猪流行性腹泻病毒S1D片段蛋白的应用,也属于本发明的保护范围。
本发明还要求保护任一所述的制备方法制备得到的猪流行性腹泻病毒S1D片段蛋白。
所述的猪流行性腹泻病毒S1D片段蛋白在制备猪流行性腹泻病毒检测试剂盒和/或抗猪流行性腹泻病毒抗体中的应用,也属于本发明的保护范围。
优选地,所述抗体为单克隆抗体。
本发明还提供所述猪流行性腹泻病毒S1D片段蛋白制备得到的单克隆抗体建立的猪流行性腹泻病毒的检测试剂盒,含有所述单克隆抗体,作为一抗。
还含有PBS和FITC标记的羊抗鼠IgG二抗。
本发明还提供所述猪流行性腹泻病毒S1D片段蛋白制备得到乳清IgA ELISA检测试剂盒。
其还含有稀释液(磷酸盐缓冲液)、封闭液(含有5%(w/v)的BSA的PBST)、PBST 洗涤液(0.05%(v/v)Tween-20的0.01mol/L的PBS,pH 7.4)、TMB底物液、2mol/L 硫酸。
其使用方法为:以4μg/mL包被浓度,纯化的△S1D蛋白包被酶标版,4℃包被过夜,用 5%(w/v)的BSA的PBST作为封闭液封闭酶标板,封闭时间为90min;封闭后,乳清按照稀释1∶160加入,37℃作用120min,将HRP-山羊抗猪IgA分别进行1:10000稀释,37℃作用60min,TMB室温避光作用25min。
更优选地,以4μg/mL包被浓度,纯化的△S1D蛋白包被酶标版,每孔包被100μL,37℃孵育1h,4℃包被过夜;PBST洗涤液洗涤4次,每次1min,洗涤后拍干,用5%(w/v)的 BSA的PBST作为封闭液封闭酶标板,200μL/孔,封闭时间为90min;封闭后,PBST洗涤液洗涤4次,每次1min,洗涤后拍干,乳清按照稀释1∶160加入,37℃作用120min;PBST 洗涤液洗涤4次,每次1min,洗涤后拍干,将HRP-山羊抗猪IgA分别进行1:10000稀释, 100μL/孔,37℃作用60min;PBST洗涤液洗涤4次,每次1min,洗涤后拍干,100μL/ 孔,TMB室温37℃避光反应避光作用25min;加入2mol/L硫酸50μL/孔,终止反应,于酶标仪630nm波长读数。
当
与现有技术相比,本发明具有以下有益效果:
本发明利用密码子优化后的S1D片段蛋白的编码基因,建立了一种大量表达猪流行性腹泻病毒S1D片段蛋白的制备方法,利用该制备方法可以高效表达S1D片段蛋白,纯度高,生物学活性好,利用其制备得到单克隆抗体效价高、特异性强;作为抗原建立的乳清IgAELISA方法重复性、敏感性和特异性均良好。
附图说明
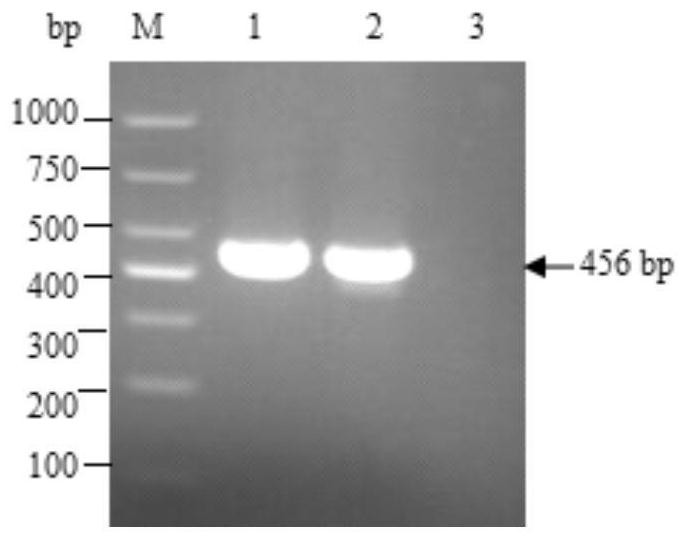
图1为S1D及△S1D基因PCR扩增;M:DNA分子质量标准;1:S1D基因;2:ΔS1D 基因;3:阴性对照。
图2为重组质粒双酶切鉴定;M:DNA分子质量标准;1:pET-S1D;2:pET-ΔS1D;3:阴性对照。
图3为S1D及△S1D重组蛋白的SDS-PAGE分析;M:蛋白质分子质量标准;1:BL21(DE3) 宿主菌;2:pET-32a空载;3:未诱导pET-S1D;4:未诱导pET-ΔS1D;5:诱导pET-S1D;6诱导pET-ΔS1D。
图4为S1D及△S1D重组蛋白的Western-blotting分析;M:蛋白质分子质量标准;1:BL21(DE3) 宿主菌;2:pET-32a空载;3:未诱导pET-S1D;4:未诱导pET-ΔS1D;5:诱导pET-S1D;6:诱导pET-ΔS1D。
图5为S1D及△S1D重组蛋白的Western-blotting分析(PEDV S蛋白单抗);M:蛋白质分子质量标准;1:BL21(DE3)宿主菌;2:pET-32a空载;3:未诱导pET-S1D;4:未诱导pET-△S1D; 5:诱导pET-S1D;6:诱导pET-△S1
图6为诱导温度的优化;M:蛋白质分子质量标准;1~5:含PET-S1D菌液诱导温度分别为16℃、22℃、28℃、37℃、42℃;6~10:含PET-△S1D菌液诱导温度分别为16℃、22℃、 28℃、37℃、42℃。
图7为诱导时间的优化;M:蛋白质分子质量标准;1~5:含PET-S1D菌液诱导时间分别为2h、3h、4h、5h、6h;6~10含PET-△S1D菌液诱导时间分别为2h、3h、4h、5h、6h。
图8为IPTG诱导浓度的优化;M:蛋白质分子质量标准;1~6:含PET-S1D菌液诱导IPTG 浓度分别为0.1mmol/L、0.5mmol/L、0.8mmol/L、1.0mmol/L、1.2mmol/L、1.5mmol/L;7~12 含PET-△S1D菌液诱导IPTG浓度分别为0.1mmol/L、0.5mmol/L、0.8mmol/L、1.0mmol/L、 1.2mmol/L、1.5mmol/L。
图9为S1D与△S1D片段蛋白的可溶性分析;M:蛋白质分子质量标准;1、7:pET-32a上清;2、8:pET-32a沉淀;3、9:S1D上清;4、10:S1D沉淀;5、11:ΔS1D上清:6、12:ΔS1D沉淀。
图10为S1D与△S1D片段蛋白表达量的差异;**:S1D与ΔS1D片段蛋白表达量差异显著 (t检验法,p<0.01)。
图11为包涵体的洗涤;M:蛋白质分子质量标准;1:第一次洗涤上清;2:第一次洗涤沉淀;3:第二次洗涤上清;4:第二次洗涤沉淀;5:第三次洗涤上清;6:第三次洗涤沉淀。
图12为包涵体溶解;M:蛋白质分子质量标准;1:△S1D片段蛋白溶解上清。
图13为包涵体的透析复性;M:蛋白质分子质量标准;1:△S1D片段蛋白溶解上清。
图14为△S1D的质谱分析。
图15为三免后小鼠抗体水平。
图16为不同抗原包被量结果比较。
图17为阴性小鼠血清测定。
图18为融合后不同时期的杂交瘤细胞生长状态(×100,500um);a:第4d;b:第7d;c:第10d。
图19为腹水的抗体效价。
图20为腹水特异性检测。
图21为Western-blotting检测腹水与△S1D片段蛋白反应的特异性;M:蛋白质分子质量标准;1:pET-32a;2:纯化的△S1D片段蛋白。
图22为Western-blotting检测腹水与PEDV反应的特异性;M:蛋白质分子质量标准;1: Vero细胞;2:PEDV。
图23为FA检测腹水的特异性;a腹水:1:500稀释;b腹水:1:1000稀释;c:腹水1:2000稀释;d:阳性对照;e:阴性对照;f:空白对照。
图24为最佳抗原包被条件。
图25为最佳封闭液的选择。
图26为最佳封闭时间。
图27为最佳乳清作用时间。
图28为最佳酶标二抗作用时间。
图29为最佳酶标二抗作用稀释度。
图30为底物最佳显色时间。
图31为临界值的确定。
图32为敏感性实验。
具体实施方式
下面结合说明书附图及具体实施例对本发明作出进一步地详细阐述,所述实施例只用于解释本发明,并非用于限定本发明的范围。下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
实施例1 S1D基因序列的优化
一、实验方法
1、S1D基因序列的优化
根据大肠杆菌密码子偏嗜性,对PEDV流行野毒株ZH02的S1D基因进行全序列核苷酸同义替换,优化的DNA由生工生物工程(上海)股份有限公司广州分公司合成,命名为PUC-△S1D。
2、引物的合成
根据PEDV S1D基因和优化后的△S1D基因序列,利用Oligo软件,分别设计特异性引物用于S1D及△S1D基因的扩增。在两对引物的5′端引入酶切位点BamHI与XhoI。引物序列如表1(下划线为酶切位点)。
表1引物序列:
3、S1D及△S1D的PCR扩增
以PEDV ZH02株的cDNA为模板扩增S1D基因,PCR反应体系(25μL)为:2×Taq PlusMaster Mix酶13μL,上、下游引物各0.5μL,DNA模板1μL,去离子水10μL。PCR反应条件:95℃预变性5min;95℃变性15s;63.5℃退火15s;72℃延伸1min,共进行35个循环;最后72℃再延伸5min。
以质粒PUC-△S1D为模板扩增△S1D基因,PCR反应体系(25μL)为:2×Taq PlusMaster Mix 酶13μL,上、下游引物各0.5μL,DNA模板1μL,去离子水10μL。PCR反应条件:95℃预变性5min;95℃变性15s;67.5℃退火15s;72℃延伸1min,共进行35个循环;最后72℃再延伸5min。
上述PCR产物分别经1.5%琼脂糖凝胶电泳鉴定,按胶回收试剂盒步骤回收目的基因。
二、实验结果
分别以PEDV ZH02株的cDNA、质粒PUC-△S1D为模板,采用表1中的引物进行PCR,扩增出S1D和△S1D基因,在约456bp(图1)处出现条带,与预期大小相符。
实施例2原核表达载体pET-S1D及pET-△S1D的构建
一、实验方法
S1D及△S1D DNA片段经BamH I与Xho I双酶切和切胶回收后,分别与用相同酶切的 pET-32a载体连接,构建重组质粒pET-S1D及pET-△S1D,并将其转化至BL21(DE3)感受态细胞。经PCR和双酶切鉴定均为阳性的质粒送生工生物工程(上海)股份有限公司测序。
二、实验结果
原核表达载体pET-S1D及pET-△S1D经双酶切后均在5.7kb和456bp出现条带(图2),测序结果显示正确无误,得到重组菌株pET-S1D及pET-△S1D。
实施例3 S1D及△S1D片段蛋白的原核表达与鉴定
一、实验方法
将实施例2测序正确的阳性重组菌株pET-S1D及pET-△S1D分别接种至含氨苄青霉素的液体LB培养基中,当菌液OD
其中,SDS-PAGE的具体方法为:
按照雅酶PAGE凝胶制备试剂盒方法配制SDS-聚丙烯酰胺凝胶,随后进行电泳,上层胶 80v,20~30min,下层胶120V,60min。电泳结束后,将凝胶取下,用考马斯亮蓝染色液对其染色30~40min,结束后反复进行脱色,待凝胶颜色背景变得较浅时观察结果。
Western-blotting的具体方法为:
按照雅酶PAGE凝胶试剂盒方法配制SDS-聚丙烯酰胺凝胶,随后进行电泳,上层胶80v, 20~30min,下层胶120V,60min。电泳结束后,用常规方法对其进行电转,设置恒流为200mA,时间为40min,结束后将PVDF膜取下,PBST洗涤四次后用5%(w/v)脱脂奶粉进行封闭1~ 2h,PBST洗涤四次后分别加入1:1000稀释的His-tag标签鼠单抗与1:1000稀释的PEDVS蛋白鼠单抗4℃过夜孵育,PBST洗涤四次后加入1:5000稀释的HRP-羊抗鼠IgG二抗,37℃孵育1h,PBST洗涤四次后避光加入曝光液作用2~5min后于FCL化学曝光仪内曝光,观察结果。
二、实验结果
经IPTG诱导的菌液进行SDS-PAGE检测,结果(图3)显示,在34kDa处有1条明显的蛋白条带与预期大小相符,未诱导组、宿主原菌BL21(DE3)与pET-32a空载均无该蛋白条带,表明S1D片段蛋白表达成功。
使用鼠抗His-tag单抗及PEDV S蛋白单抗再次对其进行Western-blotting检测,结果显示,诱导后的重组蛋白在34kDa处均有明显印迹条带,未诱导组均有微量表达,而BL21(DE3)与 pET-32a空载没有印迹条带(图4和图5),说明获得了与预期相符的目的蛋白。
实施例4△S1D片段蛋白的表达量
一、诱导温度对△S1D片段蛋白表达的影响
1、实验方法
将实施例2测序正确的阳性重组菌株pET-S1D及pET-△S1D分别接种至含氨苄青霉素的液体LB培养基中,菌液浓度达到OD600=0.5后加入终浓度为1.0mmol/L的IPTG,分别于42℃、 37℃、28℃、22℃、16℃五个诱导温度条件下诱导表达4h,对表达产物按实施例3的方法处理并进行SDS-PAGE凝胶电泳检测。
2、实验结果
结果如图6所示,含pET-S1D、PET-△S1D的菌液均在28℃时表达的蛋白量明显高于16℃、 22℃、37℃和42℃下表达的蛋白量,表明含pET-S1D、PET-△S1D的菌种其最佳诱导表达温度均为28℃。
二、诱导时间浓度对△S1D片段蛋白表达的影响
1、实验方法
将实施例2测序正确的阳性重组菌株pET-S1D及pET-△S1D分别接种至含氨苄青霉素的液体LB培养基中,菌液浓度达到OD600=0.5后加入终浓度为1.0mmol/L的IPTG,于37℃分别诱导培养2h、3h、4h、5h、6h,对表达产物按实施例3的方法处理并进行SDS-PAGE凝胶电泳检测。
2、实验结果
结果如图7所示,含pET-S1D、PET-△S1D的菌液均在诱导6h时表达的蛋白量明显高于诱导2h、3h、4h、5h表达的蛋白量,表明含pET-S1D、PET-△S1D的菌种其最佳诱导表达时间均为6h。
三、IPTG诱导浓度对△S1D片段蛋白表达的影响
1、实验方法
将实施例2测序正确的阳性重组菌株pET-S1D及pET-△S1D分别接种至含氨苄青霉素的液体LB培养基中,菌液浓度达到OD600=0.5后分别加入终浓度为0.1mmol/L、0.5mmol/L、0.8 mmol/L、1.0mmol/L、1.2mmol/L、1.5mmol/L的IPTG,于37℃诱导表达4h,对表达产物按实施例3的方法处理并进行SDS-PAGE凝胶电泳检测。
2、实验结果
结果如图8所示,含pET-S1D、PET-△S1D的菌液均在IPTG诱导浓度为0.1mmol/L时表达的蛋白量明显高于诱导浓度为0.5mmol/L、0.8mmol/L、1.0mmol/L、1.2mmol/L、1.5mmol/L表达的蛋白量,表明含pET-S1D、PET-△S1D的菌种其最佳IPTG诱导浓度均为0.1mmol/L。
实施例5△S1D片段蛋白的可溶性分析与序列优化前后表达量的比较
一、实验方法
将含pET-32a、pET-S1D及pET-△S1D的重组子分别在含氨苄青霉素的液体LB培养基中大量培养、诱导表达后以6000r/min离心15min,弃上清,用0.01mol/L,pH为7.4,含1%(v/v) PMSF(苯甲基磺酰氟)的PBS重悬菌泥。在冰浴中超声破碎细菌,12000r/min离心20min后分别收获上清和沉淀(沉淀即为包涵体粗品)。SDS-PAGE分析S1D及△S1D片段蛋白,以确定表达蛋白以可溶形式还是以包涵体形式存在。同时,利用Image J软件对密码子优化前S1D 片段蛋白与密码子优化后的△S1D片段蛋白表达量进行灰度扫描后分析两者表达量的差异。
二、实验结果
SDS-PAGE检测结果显示(图9),pET-32a空载表达的蛋白主要在上清,位置在20kDa左右,沉淀中少量表达;S1D与△S1D片段蛋白在细胞上清中均少量表达,主要以包涵体的形式存在。利用Image J软件对S1D与△S1D片段蛋白表达量进行灰度扫描后分析两者差异性(图 10),发现优化后的重组子与未优化的重组子相比,△S1D的表达量明显比S1D片段蛋白高,两者差异较显著(p<0.01)。
实施例6△S1D片段蛋白的纯化
一、实验方法
1、包涵体的洗涤、溶解
将包涵体粗品加入到10ml冷的洗涤液(20mmol/L Tris-HCl,pH 8.0,0.5mol/LNaCl, 2mol/L尿素,2%Triton X-100)中,搅拌20min。4℃,12000rpm离心25min,弃上清,取沉淀。重复一次(洗涤2~4h去除膜碎片和膜蛋白)。所得沉淀再用50mmol/L Tris-HCl洗1次(以去除残留的EDTA),相同离心条件,离心弃去上清,所得的沉淀即为洗涤后的包涵体。每次洗涤后的上清与沉淀均留一小部分样品进行SDS-PAGE凝胶电泳分析。
往上述沉淀加入10ml包涵体变性溶解液溶解(20mmol/L Tris-HCl pH 8.0,0.5mol/L NaCl, 6mol/L尿素,0.2mmol/L DTT,2%Triton X-100),于室温搅拌30-60min以充分溶解,4℃ 12000rpm离心15min,弃沉淀取上清。将上清用0.45um滤膜过滤,于-20℃保存,同时取一小部分样品进行SDS-PAGE凝胶电泳分析。
2、包涵体的透析复性
按梯度降低复性的方法用复性缓冲液于4℃使目的蛋白恢复活性,具体方法如下:
(1)裁剪合适长度(根据透析蛋白的体积决定)的透析膜
(2)将透析膜放进烧杯,加适量的双蒸水,煮沸10min
(3)使用室温的双蒸水洗涤透析膜2~3次
(4)用夹子夹住透析膜一端,将蛋白加进去,赶走气泡后,夹紧另一端,放进装有适量体积含6M尿素的TGE缓冲液烧杯中,4℃透析4~6h或过夜
(5)将6M尿素的TGE缓冲液换成4M的尿素TGE缓冲液,4℃透析4~6h或过夜
(6)将4M尿素的TGE缓冲液换成2M的尿素TGE缓冲液,4℃透析4~6h或过夜
(7)将2M尿素的TGE缓冲液换成0M的尿素TGE缓冲液,4℃透析4~6h或过夜
(8)收集透析后复性的蛋白,分装后于-80℃冰箱保存,取一小部分进行SDS-PAGE凝胶电泳分析,观察蛋白的纯化效果。
3、纯化蛋白浓度测定(BCA)
按照BCA蛋白定量试剂盒的方法对获得的纯化蛋白进行浓度测定
4、表达后纯化产物的质谱分析
将纯化后的表达产物△S1D片段蛋白送生工生物工程(上海)股份有限公司进行Maldi-TOF-TOF质谱鉴定。
二、实验结果
对获得的粗制包涵体△S1D片段蛋白洗涤三次,每次均收集洗涤上清与沉淀,通过SDS-PAGE考马染色分析纯化效果,结果见图11,每次洗涤的上清均有一些杂蛋白,且第一次洗涤的杂蛋白较多,而经过3次洗涤后,获得的精制包涵体纯度良好,杂蛋白较少,表明包涵体的洗涤方法能有效去除杂蛋白。
将上述制得的精制包涵体用溶解缓冲液在4℃条件下进行溶解,收集上清,经SDS-PAGE 考马染色后,分析其纯化效果,结果见图12,所获得的△S1D片段蛋白纯度较高,基本上无其他杂蛋白出现,说明所制备的△S1D片段蛋白可进行下一步的透析复性。
对获得的溶解上清△S1D片段蛋白进行透析复性,收集上清,经SDS-PAGE考马染色后,分析蛋白复性后的纯化效果,结果见图13,收获的△S1D片段蛋白透析上清纯度良好,无明显的杂蛋白出现,经BCA方法测定后,蛋白浓度达到1.0mg/mL,证明复性后的蛋白其纯度与浓度均适用于下一步小鼠的免疫。
生工生物工程(上海)股份有限公司对△S1D片段蛋白进行Maldi-TOF-TOF质谱鉴定的结果如图14所示,肽段覆盖率为31%,红色字体即为鉴定出来的肽段,合计6段,进一步证明△S1D 重组蛋白为PEDV S1蛋白,与预期相符。
实施例7△S1D片段蛋白的抗体的制备
一、BALB/c小鼠的免疫
1、实验方法
免疫程序如表2所示。选取6周龄雌性SPF BALB/c小鼠25只,阳性对照组10只,阴性对照组15只。用弗氏完全佐剂(CFA)与实施例6纯化得到的△S1D片段蛋白等体积的进行乳化一免,对小鼠颈背部皮下多点注射,阴性组小鼠不作处理;分别在第14d和28d,用弗氏不完全佐剂乳化(IFA)与等体积的抗原乳化进行二免及三免;在三免后一星期检测小鼠抗体效价,当阳性抗体效价达到1∶10
表2 BABL/c小鼠免疫程序
2、实验结果
结果见图15,10只阳性小鼠的抗体效价均达到了1:150000,证明抗原△S1D免疫小鼠的效果良好,可准备进行下一步的细胞融合,即融合前3d加强免疫1次阳性小鼠。
二、间接ELISA方法条件摸索
1、实验方法
取三免后7d的小鼠血清进行间接ELISA,进行包被抗原浓度的优化。
取15只阴性对照小鼠血清,以△S1D蛋白为包被抗原进行间接ELISA,测定其OD
包被:用包被液将抗原按1μg/mL、2μg/mL、4μg/mL、8μg/mL、12μg/mL、16μg/mL、 24μg/mL、32μg/mL浓度稀释,加入酶标板中,100μL/孔,湿盒37℃吸附1h后4℃过夜;洗涤:甩干包被液,每孔加入200μL洗涤液振荡洗涤3次,30s/次;封闭:加入封闭液,100 μL/孔,37℃湿盒封闭3h,甩干封闭液,同上洗涤;加样:用抗体稀释液适当稀释血清,将所稀释血清100μL/孔加入上述酶标板内,湿盒37℃反应60min,甩干板内液体,同上洗涤;加二抗:用PBS按1∶10000倍浓度稀释酶标羊抗鼠IgG,100μL/孔加入上述酶标版,湿盒37℃作用30min,甩干板内液体,同上洗涤;加底物:每孔加入底物溶液100μL,室温反应10min;加终止液:每孔加50μL H
读数及结果判定:样品读取结果记为S,若
2、实验结果
结果见图16。在抗原包被浓度一定时,随着阳性血清稀释倍数增加,其OD450值逐渐减小;在其他条件不变的情况下,抗原浓度低于12ug/mL时,随着浓度不断升高,其OD450值也在提高,但当抗原浓度大于12ug/mL时,OD450值略微下降后维持恒定,同时考虑到节省抗原,故在后期间接ELISA检测中采用8ug/mL浓度的抗原进行包被。
如图17所示,根据统计学方法计算出,在血清稀释度为1:100时,其15份血清OD450平均值
实施例8分泌△S1D片段蛋白的单克隆抗体杂交瘤的制备
一、细胞融合
1、实验方法
(1)SP2/0骨髓瘤细胞的准备
融合前4d用1640完全培养基复苏SP2/0骨髓瘤细胞,在37℃、5%CO2条件下培养 1~2代以使其恢复生长性,选择生长状态好的细胞种子进行扩大培养;保证融合当天的SP2/0 细胞至少长满3个直径10cm平皿底面。
(2)制备脾细胞
于细胞融合的前一天,取实施例7的8周龄以上与BALB/c同系的小鼠颈脱位致死,于 75%酒精中浸泡5min,用针头固定于超净台内用新洁尔灭浸泡过的解剖板,腹部向上;分别从两只大腿处剪开,向前、向背部方向剪开一定长度,按住尾部,用镊子夹住皮毛向前撕翻,暴露右侧脾脏,换把镊子夹住脾脏处腹膜,剪一小口(能见脾脏即可),取一半脾脏,用镊子夹住脾脏从小口内牵出,并同时用剪刀去除附着脾脏的脂肪结缔组织等;将脾脏放于含1640平皿中(带盖,剖小鼠前倒好培养基),轻轻洗涤,并细心剥除结缔组织;转移至另一含1640平皿中,用西林瓶底部或注射器内芯轻柔挤压脾脏,充分释放脾细胞;加入5mL HAT培养基,将平皿中的脾细胞全部轻轻吹洗下来,转移至50mL离心管中,补加HAT培养基至30mL,分装于6块96孔板,每孔50μL,37℃、5%CO
(3)融合
取加强免疫3d后的BABL/c小鼠,摘除眼球采血;制备脾细胞悬液;将骨髓瘤细胞与脾细胞按1:10的比例混合在一起,转入50mL离心管内,1000r/m离心10min,弃上清,用滴管吸净残留液体;用滴管轻轻搅动沉淀,使其松散均匀成糊状;在37℃水浴中融合:一手均匀地转动离心管,另一手用吸管吸取50%(w/v)PEG1450溶液1mL,并沿转动的管壁(尽量接近细胞处)加入,从加入到加完的时间控制在1min左右,边加边搅动,用吸管在1min内加入1mL预热至37℃不完全DMEM培养液,重复3次,往后1min加入3mL,直至加满到25 mL为止(注意此时操作应轻柔,边转动边加,尽量不搅散细胞);37℃培养箱静置10min, 800r/m离心10min,弃上清;加入10mL HAT培养液,轻轻吹吸混匀;根据所用96孔培养板的数量,按一块96孔培养板用液15mL计算补加HAT培养液至所需的量;将融合好的细胞悬液加入含有饲养细胞的96孔板,每孔150μL,37℃、5%CO
2、实验结果
融合前3d加强免疫一次阳性小鼠,融合当天取出阳性小鼠脾细胞,与骨髓瘤细胞SP2/0 按照10:1的比例混匀后,加入50%(w/v)PEG 1450使二者完成融合。细胞融合的镜下结果见图18,第4d(a)时成功融合的杂交瘤细胞开始分裂并增殖,形成由20~30个细胞组成的细胞团;第7d(b)时杂交瘤细胞活力较好,增殖较快,已经形成由100~200个细胞组成的细胞簇;第10d(c)时杂交瘤细胞已经长至孔底的1/5~1/2,此时可吸取细胞培养上清用建立好的间接ELISA方法检测抗体。
二、阳性杂交瘤细胞的筛选
1、实验方法
(1)初次检测
用建立的间接ELISA法检测融合细胞上清液中的抗体,初次检测时阴性孔予以淘汰,阳性孔及可疑阳性孔进行二次检测。
(2)假阳性的筛除
以纯化的pET-32a空载体表达产物为抗原对检出的阳性进行假阳性的筛除,去除针对载体上非目的蛋白抗原位点的单克隆抗体。
(3)阳性杂交瘤细胞的克隆化
对筛选所获得的阳性杂交瘤细胞孔以有限稀释法进行克隆化,以获取能稳定分泌抗体的细胞株。
克隆化:取130个细胞放入6.5ml含饲养细胞的HT培养液,即20个细胞/mL,100μL/孔加A、B、C三排,为每孔2个细胞。余下2.9mL细胞悬液补加2.9mL含饲养细胞的HT培养液,细胞数为10个/mL,100μL/孔加D、E、F三排,为每孔1个细胞。余下2mL细胞悬液补加2.2mL含饲养细胞的HT培养液,细胞数5个/mL,100μL/孔,加G、H两排,为每孔0.5 个细胞。培养4~5d后,在倒置显微镜上可见到小的细胞克隆,补加完全培养液200μL/孔。亚克隆9d后,观察培养板中细胞的生长情况,并标记细胞集落数目,当细胞孔上清变淡黄或黄色时即可用间接ELISA法进行检测,取读数较高、孔内细胞克隆数在1~2个的细胞再次进行亚克隆,直至得到稳定分泌单抗的单细胞克隆。
(4)扩大培养
进行亚克隆的同时,多余的杂交瘤细胞转至24孔细胞培养板中进行扩大培养,即细胞原始孔。
(5)阳性杂交瘤细胞冻存
亚克隆之前检测阳性孔的细胞除亚克隆之外的全部剩余细胞进行扩大培养后,进行冻存,保存原始孔。最后获得呈100%阳性的阳性杂交瘤细胞株也要扩大培养并冻存。冻存时将杂交瘤细胞吹下并离心弃去上清,沉淀用含10%(v/v)DMSO、50%(v/v)小牛血清的DMEM培养液重新悬浮,分装于冻存管中,1mL/管,细胞密度不低于2×10
(6)细胞复苏
细胞冻存一周后复苏检测细胞存活率及分泌抗体特性是否丢失,复苏时从液氮罐中取出冻存管后迅速放入37℃水浴锅中,轻轻摇动使其在1min内融化,融化后从水浴中取出放置冰盒上,1000r/min离心5min,移至超净工作台,无菌打开冻存管,将细胞悬液转移至细胞瓶,悬浮于预热的HT完全培养液中,置于37℃,5%CO
2、实验结果
以8ug/mL的△S1D片段蛋白为抗原包被酶标板,采用建立的间接ELISA方法,根据判断标准得到的阴性、空白、阳性对照的结果以及融合的杂交瘤细胞生长状态,将待检孔OD450≥0.1 判断为初步阳性,OD450≥0.07为初步弱阳性,OD450<0.07为阴性。通过后续的假阳性筛除、阳性杂交瘤细胞的3~5次克隆化实验,最终得到稳定分泌△S1D抗体的单克隆细胞株。
三、小鼠腹水制备及效价的检测
1、实验方法
筛选出的细胞株经培养稳定后,取生长状态良好、抗体分泌能力强者采用体内诱生法制备腹水。将弗氏不完全佐剂按500μL/只腹腔BALB/c小鼠,7d后备用。将细胞由培养瓶中吹下后计数,调整至1×10
2、实验结果
以8ug/mL△S1D片段蛋白为抗原包被酶标板,不同稀释度的腹水作一抗,HRP-羊抗鼠IgG 作为二抗,同时设置阴性及对照,用间接ELISA方法进行检测,结果见图19。制备的单克隆抗体在经过1:1000000稀释后仍为弱阳性,证明制备的腹水抗体效价较高。
四、特异性检测
1、实验方法
以PEDV、PRRSV、TGEV、CSFV、PDCoV、SADS、PEDV△S1D片段蛋白、PEDV N蛋白、pET-32a空载蛋白为抗原包被酶标板,同时设置阳、阴性血清对照,用间接ELISA方法检测所得腹水的特异性。
2、实验结果
结果见图20。腹水能与PEDV病毒和纯化后的△S1D片段蛋白反应呈阳性,与PEDV N蛋白、32a空载蛋白和PRRSV、TGEV、CSFV、PDCoV、SADs 5种病毒反应呈阴性,与预期相符,表明所制备的腹水具有良好的特异性。
五、Western blot分析
1、实验方法
分别以纯化后的△S1D、pET-32a、PEDV及Vero细胞蛋白为抗原,制备的腹水1:1000稀释作为一抗,HRP标记的山羊抗鼠IgG作为二抗,进行Western-blotting试验。将PEDV以MOI =1.0的剂量感染Vero细胞,感染48h后,收取细胞并制备蛋白样品。同时,采用纯化后的△S1D及pET-32a空载体蛋白为抗原。以制备的腹水作为一抗,HRP标记的山羊抗鼠IgG作为二抗,进行Western-blotting试验,检测腹水与△S1D、PEDV S蛋白的反应性。
2、实验结果
结果显示,腹水与pET-32a空载体蛋白无反应,与△S1D片段蛋白发生强裂的反应,在34KDa 处出现一特异性条带(图21)。腹水与Vero细胞无反应条带,与PEDV感染的Vero细胞也产生强一条180KDa特异性条带(图22)。结果与预期相符,表明腹水中的单克隆抗体特异性良好。
六、间接免疫荧光(IFA)
1、实验方法
待96孔板的Vero细胞长成单层,接种PEDV。20h后,弃掉培养液,加入预冷的80%(v/v) 丙酮150μL/孔,-20℃固定30min。灭菌的PBS洗涤3次后加入1∶500、1:1000、1:2000PBS 稀释的△S1D单抗50μL/孔,37℃孵育2h,设1:1000PBS稀释的PEDV N单抗为阳性对照,1: 1000稀释的阴性小鼠血清为阴性对照;不加抗体为空白对照。灭菌的PBS洗涤3次,加入1∶ 500PBS 50μL/孔稀释的FITC标记的羊抗鼠IgG二抗,37℃孵育1h。灭菌的PBS洗涤3次,孔内留100μLPBS,于荧光倒置显微镜观察结果。
2、实验结果
结果如图23所示,3个稀释度的腹水及阳性对照组均能使细胞出现特异性绿色荧光信号,而空白及阴性组未见绿色荧光信号,进一步表明制备的单克隆抗体能特异性识别PEDV的S1D 片段蛋白。
实施例9乳清IgA ELISA检测方法的建立
一、抗原最佳包被浓度和乳清稀释度的选择
1、实验方法
采用方阵滴定法,以磷酸盐缓冲液为稀释液将实施例6纯化的△S1D蛋白稀释至终浓度为 16μg/mL,8μg/mL,4μg/mL,2μg/mL,1μg/mL,每个浓度包被两列,每孔包被100μL, 37℃1h后置4℃包被过夜。PBST洗涤液洗涤4次,每次1min,洗涤后拍干,PBST洗涤液为含0.05%(v/v)Tween-20的0.01mol/L的PBS,pH 7.4。加入含0.5%(v/v)牛乳清白蛋白(BSA)的PBST,200μL/孔,37℃封闭2h,洗涤同上。洗涤后拍干。将阴、阳性乳清分别按1∶20,1∶40,1∶80,1∶1 60;1:320五个梯度稀释后加入ELISA反应板,100 μL/孔,37℃作用70min,取出后洗涤同上。拍干后加入1∶10 000倍稀释的HRP标记的羊抗猪IgA酶标二抗,100μL/孔,37℃反应40min,洗涤同上。洗涤后加入TMB底物液, 100μL/孔,37℃避光反应10min。加入2mol/L硫酸50μL/孔,终止反应,于酶标仪630nm波长读数。以阳性乳清OD值接近1,P/N值最大孔的抗原浓度和乳清稀释度作为最佳抗原包被浓度和乳清稀释度。
2、实验结果
方阵滴定结果如表3所示,当抗原包被浓度为4μg/mL,乳清稀释倍数为1∶160时,阳性乳汁OD450nm值接近1.0,且此稀释度的P/N值最大。因此选择最佳抗原包被浓度为4μg /mL,最佳乳清稀释倍数为1∶160。
表3抗原最佳包被浓度和乳清稀释度的确定
二、抗原最佳包被时间的选择
1、实验方法
以最适抗原包被浓度(4μg/mL)分别以4种不同条件将实施例6纯化的△S1D蛋白包被酶标板;4℃过夜;4℃过夜,37℃孵育1h;37℃孵育1h;37℃孵育1h,4℃过夜;包被完成后,其他步骤同上。比较各组阴、阳性乳清P/N值,以选择最佳包被时间。
2、实验结果
用最佳包被浓度进行抗原包被,通过不同的时间包被抗原,结果如图24所示,4℃过夜时P/N最大,确定4℃过夜为抗原的最佳包被条件。
三、最佳封闭液的选择
1、实验方法
以最适抗原包被浓度(4μg/mL)和最佳包被时间(4℃过夜)将实施例6纯化的△S1D蛋白包被酶标板,封闭液分5组,分别为5%(w/v)BSA的PBST,10%(w/v)BSA的PBST, 5%(w/v)脱脂奶粉(SMP)的PBST,10%(w/v)脱脂奶粉(SMP)的PBST,1×无蛋白封闭液。每孔加入100μL封闭液,37℃封闭2h。封闭完成后,乳清按照最佳稀释度1∶160加入,其他步骤同上。比较各组阴、阳性乳清的P/N值以选择最佳封闭液。
2、实验结果
结果如图25所示,5%(w/v)的BSA的PBST作为封闭液时P/N值最大。确定5%(w/v)BSA的PBST为最佳封闭液。
四、最佳封闭时间的选择
1、实验方法
以最适抗原包被浓度(4μg/mL)和最佳包被时间(4℃过夜)将实施例6纯化的△S1D蛋白包被酶标版,用5%(w/v)的BSA的PBST作为封闭液封闭酶标板,37℃分别封闭30min、60min、90min、和120min,封闭完成后,乳清按照确定的最佳稀释度1∶160加入,其他步骤同上。比较各组阴、阳性乳清P/N值,以选择合适的封闭时间。
2、实验结果
结果如图26所示,37℃封闭90min时P/N值最大。确定最佳封闭时间为90min。
五、最佳一抗作用时间的选择
1、实验方法
以最适抗原包被浓度(4μg/mL)和最佳包被时间(4℃过夜)将实施例6纯化的△S1D蛋白包被酶标版,用5%的BSA的PBST作为封闭液封闭酶标板,封闭时间为90min。封闭后,乳清按照确定的最佳稀释度1∶160加入,37℃分别作用30min、60min、90min、和120min,其他步骤同上。比较各组阴、阳性乳清P/N值,以选择合适的乳清作用时间。
2、实验结果
结果如图27所示,乳清作用120min时P/N值最高。确定乳清最佳反应时间为120min。
六、最佳酶标抗体作用时间的选择
1、实验方法
以最适抗原包被浓度(4μg/mL)和最佳包被时间(4℃过夜)将实施例6纯化的△S1D蛋白包被酶标版,用5%(w/v)的BSA的PBST作为封闭液封闭酶标板,封闭时间为90min。封闭后,乳清按照确定的最佳稀释度1∶160加入,37℃作用120min,将酶标抗体按照1:10000 加入酶标板后,37℃分别作用30min,45min,60min,90min,其他步骤同上,比较各组阴、阳性乳清的P/N值,以选择合适的酶标抗体作用时间。
2、实验结果
结果如图28所示,酶标抗体作用60min时P/N值最高。确定酶标抗体最佳反应时间为 60min。
七、酶标二抗最佳作用浓度的确定
1、实验方法
以最适抗原包被浓度(4μg/mL)和最佳包被时间(4℃过夜)将实施例6纯化的△S1D蛋白包被酶标版,用5%(w/v)的BSA的PBST作为封闭液封闭酶标板,封闭时间为90min。封闭后,乳清按照确定的最佳稀释度1∶160加入,37℃作用120min,将HRP-山羊抗猪IgA 分别进行1:5000、1:10000、1:15000、和1:20000梯度稀释,37℃作用40min,其他步骤同上,比较各组阴、阳性乳清的P/N值,以选择合适的酶标抗体作用时间。
2、实验结果
结果如图29所示,酶标二抗进行1:10000稀释时,P/N值最高。确定酶标二抗最佳作用稀释度为1:10000。
八、底物显色时间的确定
1、实验方法
以最适抗原包被浓度(4μg/mL)和最佳包被时间(4℃过夜)将实施例6纯化的△S1D蛋白包被酶标版,用5%(w/v)的BSA的PBST作为封闭液封闭酶标板,封闭时间为90min。封闭后,乳清按照确定的最佳稀释度1∶160加入,37℃作用120min,将HRP-山羊抗猪IgA分别进行1:10000稀释,37℃作用60min,TMB室温避光分别作用10min、15min、20min、 25min,其他步骤同上,比较各组阴、阳性乳清的P/N值,以选择底物显色时间时间。
2、实验结果
结果如图30所示,当底物(TMB)显色时间为25min时,P/N值最高。确定底物最佳显色时间为25min。
九、临界值的确定
1、实验方法
取10份经上海酶联生物科技有限公司生产的PEDV IgA ELISA试剂盒鉴定的阴性乳清,检测乳清OD450nm值,计算样品OD450nm值的平均值
建立的检测条件为:
以最适抗原包被浓度(4μg/mL)和最佳包被时间(4℃过夜)将实施例6纯化的△S1D蛋白包被酶标版,用5%(w/v)BSA的PBST作为封闭液封闭酶标板,封闭时间为90min。封闭后,乳清按照确定的最佳稀释度1∶160加入,37℃作用120min,将HRP-山羊抗猪IgA分别进行1:10000稀释,37℃作用60min,TMB室温避光作用25min,其他步骤同上。
2、实验结果
结果如图31所示。根据计算可得10份样品的平均值为0.090,标准偏差为0.018。根据统计学原理,当
实施例10乳清IgA ELISA检测方法的重复性
一、批内重复试验
1、实验方法
将同一批次纯化的实施例6纯化得到的△S1D片段蛋白蛋白包被ELISA板,取4份阳性乳清样品和1份阴性乳清样品,每份样品重复5个孔进行ELISA检测,按照实施例9建立的检测方法进行检测,根据结果计算每份样品5次重复的平均值、标准差和变异系数。
2、实验结果
结果如表4所示,批内重复试验的变异系数均小于10%,表明建立的间接ELISA方法重复性良好。
4批内重复实验
二、批间重复试验
1、实验方法
用同一批次纯化的蛋白抗原包被5块不同批次的酶标板,取4份阳性乳清样品和1份阴性乳清样品进行ELISA检测,根据结果计算每份样品在5个不同批次酶标板上的平均值、标准差和变异系数。
2、实验结果
结果如表5所示,批间重复试验的变异系数均小于10%,表明建立的间接ELISA方法稳定性良好.
表5批间重复实验
实施例11乳清IgAELISA检测方法的敏感性
一、实验方法
取4份阳性乳清样品和1份阴性乳清样品分别做1∶40,1∶80,1∶160,1∶320,1∶ 640,1∶1280共6个梯度稀释,按实施例9建立好的间接ELISA方法进行检测,对结果进行敏感性分析。
二、实验结果
结果如图32所示。当阳性乳清稀释到160倍时,4份阳性乳清样品仍能检出阳性,证明本实验建立的ELISA检测方法敏感性良好。
实施例12乳清IgAELISA检测方法与市售产品的比较
一、实验方法
将送检的133份临床腹泻母猪的乳清样品,分别用按实施例9建立的IgA抗体检测方法和上海酶联生物科技有限公司生产的PEDV IgAELISA试剂盒进行检测,并计算两种检测方法的符合率,即两种方法同时检测为阳性或阴性的样品数之和,与样品总数的比值。
二、实验结果
检测结果如6所示。本研究建的方法敏感性为85.4%,特异性为80.0%,两种方法符合率为85.0%。
表6
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,对于本领域的普通技术人员来说,在上述说明及思路的基础上还可以做出其它不同形式的变化或变动,这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
序列表
<110> 华南农业大学
<120> 一种猪流行性腹泻病毒S1D片段蛋白的编码基因及其应用
<160> 1
<170> SIPOSequenceListing 1.0
<210> 1
<211> 438
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 1
atgaccctgg atgtgtgcac caaatatacc atttatggct ttaaaggcga aggcattatt 60
accctgacca atagcagctt tctggcgggc gtgtattata ccagcgatag cggccagctg 120
ctggcgttta aaaatgtgac cagcggcgcg gtgtatagcg tgaccccgtg cagctttagc 180
gaacaggcgg cgtatgtgga tgatgatatt gtgggcgtga ttagcagcct gagcagcagc 240
acctttaata gcacccgcga actgccgggc tttttttatc atagcaatga tggcagcaat 300
tgcaccgaac cggtgctggt gtatagcaat attggcgtgt gcaaaagcgg cagcattggc 360
tatgtgccga gccagagcgg ccaggtgaaa attgcgccga tggtgaccgg caatattagc 420
attccgacca attttagc 438
- 一种猪流行性腹泻病毒S1D片段蛋白的编码基因及其应用
- 重组蛋白、其编码基因、其应用及猪流行性腹泻病毒抗体的检测试剂盒及检测方法