微小残留病灶的检测方法、装置、设备和存储介质
文献发布时间:2023-06-19 18:32:25
技术领域
本申请属于基因检测技术领域,具体涉及微小残留病灶的检测方法、装置、设备和存储介质。
背景技术
基于循环肿瘤DNA(Circulating tumor DNA,ctDNA)指导的MRD(微小/可测量/分子残留病灶,Minimal/ Measurable/ Molecular residual disease)评估,可优于传统的临床或影像学方法鉴定出有MRD的患者,并在预测疾病复发风险等方面具有较高的灵敏度和特异性。
在相关技术中,比如公开号为CN112236535A的中国发明专利,记载了一种用于借助于循环肿瘤DNA的个人化检测的癌症检测和监测的方法,用于检测乳癌、膀胱癌或结肠直肠癌中的单核苷酸变异体,通过对核酸进行多重扩增反应来产生扩增子集合,核酸是从来自已接受乳癌、膀胱癌或结肠直肠癌治疗的患者的血液或尿液样品或其一部分分离,其中扩增子集合中的每个扩增子跨越与乳癌、膀胱癌或结肠直肠癌相关联的患者特异性单核苷酸变异体基因座集合中的至少一个单核苷酸变异体基因座;以及确定扩增子集合中的每个扩增子的至少一个区段的序列,所述至少一个区段包含患者特异性单核苷酸变异体基因座,其中一种或多种患者特异性单核苷酸变异体的检测指示乳癌、膀胱癌或结肠直肠癌的早期复发或转移。
但是上述检测方法,以血液或尿液样品中核酸作为投入样本来进行多重扩增反应,无法准确去除重复序列,且高循环数扩增可能会引入扩增错误。此外,该方法采用常规WES panel进行组织位点的确定,对于高证据级别基因及位点未进行重点监测,而这些属于通用肿瘤患者数据库中发生频率较高且带有临床证据的区域。再者,该方法仅进行了个性化panel追踪,对于血液样本中可能藏匿的第二原发变异或肿瘤进化变异等无法进行监测。
发明内容
1. 发明目的
本申请的目的在于提供微小残留病灶的检测方法、装置、设备和存储介质,来解决以上背景技术部分提到的技术问题之一。
2. 技术方案
为了解决上述问题,本申请所采用的技术方案如下:
作为本申请的第一方面,本申请提供了一种微小残留病灶的检测方法,该方法基于第二代测序技术,主要包括如下步骤:
S1,获取患者肿瘤组织DNA和血细胞DNA的WDC测序数据,即:分别构建肿瘤组织DNA文库和血细胞DNA文库;将两个文库等质量比混合,并用WDC探针进行杂交捕获获得捕获后DNA文库,其中WDC探针是全外显子测序探针(WES探针)与靶向用药基因Panel按照1:(2~8)混合后形成的混合探针;对捕获后DNA文库进行测序,获得肿瘤患者WDC测序数据。该WDC探针可以实现测序深度的差异化,即WES其他区域:肿瘤相关基因区域:靶向用药基因区域可以实现1:(1.5~3):(2~6)的有效深度比例,可以降低靶向用药核心基因及肿瘤相关基因的检测下限,提高灵敏度;
S2,获取患者基因组突变信号,将S1中获得的WDC测序数据预处理后与hg19人类参考基因组比对,获得肿瘤组织样本DNA突变信号、血细胞样本DNA突变信号,比较并保留只存在肿瘤组织样本的DNA突变信号作为基因组突变信号,DNA突变信号包括体细胞变异(SNV)、插入缺失(Indel)、融合(fusion)或其他类型突变中一种或多种;
S3,筛选追踪突变信号,将S2中的基因组突变信号按照功能及可信度进行排序,筛选排序靠前的预设个数基因组突变信号作为追踪突变信号,排序规则为:首先有重要功能的驱动突变,给予最高排序优先级排序;其次按突变频率及主克隆-亚克隆排序,针对突变频率大于5%的突变,按突变频率从大到小排序;对突变频率在1%~5%的突变,优先按主克隆>亚克隆排序,次优先按突变频率排序;
S4,制备个性化组合panel(CCP探针),根据追踪突变信号设计追踪突变信号序列探针(customized探针),并与固定突变信号序列探针(core探针)和SNP探针混合,制备个性化组合panel,其中固定突变信号序列探针(core探针)用于检测肿瘤进化或第二原发,SNP探针用于鉴别样本来源和评估样本污染程度;
S5,获取患者肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA的个性化组合panel测序数据,构建含有UMI接头的血浆cfDNA文库,并将肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:(6~12)进行不同样本类型文库的混合;通过CCP探针杂交捕获获得捕获后DNA文库,对捕获后DNA文库进行测序,获得肿瘤患者个性化组合panel测序数据,利用该质量比例混合,可以得到肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA1:1:(3~6)的数据量,可以同时兼顾测序深度与成本的平衡,在达到血浆10万×超高深度的同时,组织可以达到1万×深度以便得到更准确的组织突变谱,及血细胞的1万×以上深度可以辅助血浆排除克隆性造血的干扰;
S6,追踪突变信号校正及确定追踪突变序列和位置,利用肿瘤组织样本和血细胞样本的个性化组合panel测序数据校正追踪突变信号,去除不再被确定为体细胞小突变和融合突变的信号,去除克隆性造血来源的突变,更新追踪突变信号生成最终追踪突变信号并确定最终追踪突变信号序列和位置;
S7,获取血浆cfDNA的追踪突变信号检测结果,提取覆盖在最终追踪突变信号位置上血浆样本的reads对,提取两端的分子标签序列、基因组上的起始位置、插入片段长度和方向等信息,确定单链一致性序列(SSCS)和双链一致性序列,结合UMI序列过滤并确定追踪突变信号检测结果;
S8,结合所有追踪突变信号的检测结果得到肿瘤患者的MRD检测结果,统计S7中追踪突变信号的阳性突变个数,与预先设置的阈值相比,大于预先设置的阈值,则肿瘤患者的MRD状态为阳性,否则为阴性。
进一步地,上述S1中靶向用药基因Panel中基因包括AKT1、ALK、AR、ARAF、BRAF、BRCA1、BRCA2、CDK4、CTNNB1、DDR2、EGFR、ERBB2、ERBB3、ERRFI1、ESR1、FBXW7、FGFR1、FGFR2、FGFR3、FLT1、GNA11、GNAQ、HRAS、IDH1、IDH2、KIT、KRAS、MAP2K1、MAPK1、MET、MTOR、NF1、NF2、NOTCH1、NRAS、NTRK1、NTRK2、NTRK3、PDGFRA、PIK3CA、PTEN、RAC1、RB1、RET、RICTOR、ROS1、SMAD4、TERT、TP53、TSC1、VEGFA、AKT2、AKT3、APC、ATM、ATR、ATRX、CDK6、CDKN2A、CHEK2、FLT3、FLT4、JAK1、JAK2、KDR、KEAP1、MDM2、MYC、PALB2、VHL、ABL1、BTK、SMO、ETV6、EWSR1、NTRK、HER2和BRCA等基因中一种或多种,涉及的适应症包括肺癌、结直肠癌、乳腺癌、胃癌、胃肠间质瘤、甲状腺癌、头颈部鳞癌、卵巢癌和黑色素瘤等实体瘤中的一种或多种。肿瘤的基因状态,尤其肿瘤驱动基因的变异状态可提示肿瘤的进展、药物敏感或耐药信息,同时可用于评估预后、复发和转移风险,由此类基因所组成的Panel,即为靶向用药基因Panel。更进一步地,可以根据需要选择不同的目的基因或组合。
进一步地,上述S1中,WES探针与靶向用药基因Panel进行1:2的方式进行混合,WES其他区域:肿瘤相关基因区域:靶向用药核心基因区域可以实现1:1.5:2的去重后有效深度比例。
进一步地,上述S1中,WES探针与靶向用药基因Panel进行1:4的方式进行混合,WES其他区域:肿瘤相关基因区域:靶向用药核心基因区域可以实现1:2:3的去重后有效深度比例。
进一步地,上述S1中,WES探针与靶向用药基因Panel进行1:8的方式进行混合,WES其他区域:肿瘤相关基因区域:靶向用药核心基因区域可以实现1:3:6的去重后有效深度比例。
进一步地,上述S1中,肿瘤组织样本可以是分离好的福尔马林固定、石蜡包埋的肿瘤组织样本。
进一步地,上述S2中,WDC测序数据预处理包括去除接头和低质量碱基,推荐使用Trimmomatic软件。
进一步地,上述S2中,比对至hg19人类参考基因组序列,推荐使用BWA软件。
进一步地,上述S2中,比对至hg19人类参考基因组序列后还包括去重、重比对和质量值矫正,去重包括调用商业软件Sentieon-202112.05,采用命令“sentieon driver--algo Dedup—rmdup”对初始Bam文件进行去重复处理,生成去重后的Bam文件;重比对包括调用商业软件Sentieon-202112.05,采用命令“sentieon driver--algo Realigner”对去重后的Bam文件进行重比对处理,生成重比对后的Bam文件;质量值矫正包括调用商业软件Sentieon-202112.05,采用命令“sentieon driver--algo QualCal”对重比对后的Bam文件进行质量值矫正处理,生成校正后的Bam文件。
进一步地,上述S2中,体细胞变异(SNV)检测包括通过比较肿瘤组织样本和血细胞样本校正后的Bam文件,获得初始的体细胞突变列表。
进一步地,上述S2中,融合突变(fusion)检测包括通过比较肿瘤组织样本和血细胞样本校正后的Bam文件,获得肿瘤组织样本的融合突变检测结果。
进一步地,上述S2中,比较比较肿瘤组织样本和血细胞样本校正后数据,使用配对方法找出待测患者的体细胞变异和融合突变,推荐使用Mutect2软件。
进一步地,上述S2中,基因组突变信号还包括过滤,过滤规则如下:gnomAD、ExAC、1000g三个数据库的人群突变频率小于2%;测序深度大于40;突变频率大于1%;不在平台黑名单范围(通过大量样本,不同批次的统计,反复出现的低质量的突变,定义为黑名单突变)。
进一步地,上述S2中,基因组突变信号过滤规则还包括:支持reads>2,覆盖深度>100,正负链支持没有显著差异,所在及周围无简单重复序列,肿瘤组织突变频率/血细胞突变频率>5。
进一步地,上述S2中,还可以提供患者的其他肿瘤相关检测信息,包括TMB,MSI等。
进一步地,上述S3中,主克隆和亚克隆分类,是根据S2中的基因组突变信号和CNV检测结果,根据每个体细胞突变的支持突变reads个数及测序深度,并考虑由CNV等引入的等位基因不平衡,采用统计聚类方法,例如贝叶斯聚类方法,估计肿瘤纯度及并以此将体细胞突变做分组聚类到不同的克隆群体,并统计每个克隆群体的细胞占比,将占比最高的克隆群体定义为主克隆,其他类别定义为亚克隆。更进一步地,推荐使用factes和pyclone软件完成分类。
进一步地,上述CNV检测包括通过比较肿瘤组织样本和血细胞样本校正后的Bam文件,获得肿瘤组织样本的肿瘤纯度和肿瘤细胞等位基因拷贝数的估计值。
进一步地,上述S3中,预设个数为10~50个或全部突变信号。
进一步地,上述S4中,追踪突变信号序列探针(customized探针)设计规则如下:如果是SNV/Indel类型的突变,根据参考基因组和追踪突变列表,串联每个追踪突变信号起始位置基因组上游60 bp的参考基因组序列、追踪突变信号序列和追踪突变信号终止位置基因组下游60 bp的参考基因组序列三个序列为候选customized探针序列;如果是Fusion类型的突变,根据参考基因组和融合突变的方向,串联融合突变上游基因gene1断点1上游(沿转录本方向)60 bp的序列和融合变异下游基因gene2断点2下游(沿转录本方向)60 bp的序列为候选customized探针序列。
进一步地,上述S4中,追踪突变信号序列探针设计还包括过滤,过滤规则如下:去除在整个参考基因组范围存在“较好比对位置”个数大于20的候选探针序列,其中“较好比对位置”指匹配长度大于30 bp且比对期望值小于0.000001的位置;去除含有重复序列SSR的候选探针序列;去除GC<10%或GC>80%的异常候选序列。
进一步地,上述S4中,Core探针中固定突变信号(高证据热点)包括来自NCCN指南、专家共识等、公共数据库中靶向证据基因位点和耐药化疗证据基因位点、FDA/NMPA药品标签、结合临床试验及会议摘要等证据基因位点,同时在多癌种中筛选出一级证据基因位点和二级证据基因位点形成的集合中一种或多种。
进一步地,上述S4中,SNP探针的位点包括来源于WDC中全外显子组所覆盖的dbSNP数据库中的杂合度较高的SNPs位点集合中一种或多种。
进一步地,上述S4中,固定突变信号序列探针(core探针)的基因如表2所示,SNP探针坐标如表3所示。
进一步地,上述S4中,个性化Panel按照探针物质的量摩尔数比Customized探针:Core探针:SNP探针=8:8:1的体系混合,制备CCP杂交探针工作液,按照8:8:1配制而成,可以实现5:5:1的去重后有效深度比例,可以降低靶向用药核心基因/肿瘤相关基因的检测下限,提高灵敏度。
进一步地,上述S5中,将肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:6的混合,得到肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA 1:1:3的数据量。
进一步地,上述S5中,将肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:9的混合,得到肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA 1:1:4的数据量。
进一步地,上述S5中,将肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:12的混合,得到肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA 1:1:6的数据量。
进一步地,上述S5中,杂交捕获完成后,采用体积梯度递增的洗脱方式进行洗脱,与常规的等体积洗脱相比,得到更高Target ratio数据。杂交捕获完成后,需要将体系中或吸附在管壁上的off-target reads 清洗掉,常规操作步骤均使用相同体积的清洗液进行清洗,本申请测试出梯度体积递增的方法可以有效增加针对前一步骤吹打或斡旋清洗过程中吸附在管壁上的off-target reads 清洗掉,最终呈现出比常规操作更高的targetratio,并实现更高深度及对应的检测灵敏度。
进一步地,上述S5中,杂交捕获完成后后用100 μL预热的清洗缓冲液ⅠⅠ、145 μL预热的Stringent清洗缓冲液Ⅰ、150μL预热的Stringent清洗缓冲液Ⅰ、50μL+100 μL清洗缓冲液Ⅰ、155 μL清洗缓冲液Ⅱ、160 μL清洗缓冲液Ⅲ的体积梯度递增的洗液进行清洗,得到捕获后的文库。
进一步地,上述S6中,追踪突变信号校正包:括参照S2和S3处理个性化组合panel测序数据,获得新的追踪突变信号,并匹配S3中的追踪突变信号是否在新的追踪突变信号中,删除不存在新的追踪突变信号中的突变信号,生成最终的追踪突变信号。
进一步地,上述S6中,确定最终追踪突变序列和位置包括:获取扩展突变型序列,根据参考基因组和最终的追踪突变信号,对每个追踪突变序列,串联其起始位置到基因组上游长度a bp的参考基因组序列、追踪突变型序列及其终止位置到基因组下游a bp的参考基因组序列三段序列为候选序列;如果候选序列在包含候选序列上下游b bp范围内仅可唯一匹配,则保留候选序列为追踪突变序列,同时定义串联序列的基因组起始位置为追踪突变序列的基因组起始位置,串联序列的基因组终止位置为追踪突变序列的基因组终止位置;若不满足保留标准,则增加1 bp长度,即(a+1) bp开始重新扩展上下游序列后重复操作,直到满足保留标准或串联序列长度超过c bp。
进一步地,上述a为3~4,b为100~200,c为30~35。更进一步地,S6中,a为3,b为200,c为35。
进一步地,上述S7中,将具有相同read id号的一对reads,标记为1个fragment,并提取fragment信息:包括两端的分子标签序列、基因组上的起始位置、插入片段长度和方向等。
进一步地,上述S7中,确定单链一致性序列(SSCS)包括:将具有相匹配fragment信 息的fragment作为一组,其中相匹配fragment信息指在d bp的误差范围内的UMI序列、起始 位置或插入片段差异等,具有几乎完全相同的fragment信息;从最终追踪突变信号序列的 基因组起始位置所对应的fragment上的碱基位置开始,到追踪突变序列的基因组终止位置 所对应的fragment上的碱基位置截止,逐碱基的比较每个位置上每种碱基类型的个数,碱 基类型包含A,T,C,G;确定SSCS,如果满足
进一步地,d为1。
进一步地,f为2。
进一步地,上述S7中,结合UMI序列过滤并确定追踪突变信号检测结果包括:对每一个追踪突变,定义与追踪突变序列完全匹配的SSCS为一个simplex,具有配对分子标签序列的两个simplex定义为1个duplex(双链一致性);按照如下规则过滤并确定追踪突变:如果simplex上追踪突变边缘距离fragment边缘距离的更小值小于预设阈值(j),或simplex上与参考基因组序列不同的碱基个数大于预设阈值(n),则定义该simplex为低质量simplex;统计每个追踪突变的低质量simplex的比例,若大于预设阈值(r),认为该突变为一个低可信度突变,在后续分析中去除;统计过滤后每个追踪突变的simplex个数,和duplex个数,如果满足simplex个数大于预设阈值(s),同时duplex个数大于预设阈值(h),则报告该突变为一个阳性突变。
进一步地,上述j为5。
进一步地,上述n为5。
进一步地,上述r为0.5。
进一步地,上述s为0。
进一步地,h为1。
进一步地,上述S8中,预先设置的阈值为1~3,也可以根据需要自行设置。更进一步地,预先设置的阈值为1。
作为本申请的第二方面,本申请提供了一种微小残留病灶的检测装置,包括:
数据输入模块,用于输入患者肿瘤组织样本和术前血细胞样本的WDC测序数据,和输入患者上述肿瘤组织样本、血细胞样本和血浆的个性化组合panel测序数据;
数据处理模块,用于根据输入数据完成第一方面所述的获取基因组突变信号、筛选追踪突变信号、追踪突变信号校正及确定追踪突变序列和位置、获取血浆cfDNA的追踪突变信号检测结果;
结果输出模块,用于输出第一方面所述的肿瘤患者的MRD检测结果。
作为本申请的第三方面,本申请提供了一种电子设备,包括:一个或多个处理器;存储装置,其上存储有一个或多个程序,当一个或多个程序被一个或多个处理器执行,使得一个或多个处理器实现上述第一方面任一实现方式所描述的方法。
作为本申请的第四方面,本申请提供了一种计算机存储介质,其上存储有计算机程序,其中,程序被处理器执行时实现上述第一方面任一实现方式所描述的方法。
3. 有益效果
本申请与现有技术相比,其有益效果在于:
(1)本申请提供的微小残留病灶的检测方法,使用WDC组合测序方式,即差异化深度的WES+靶向用药基因Panel,一方面包含全外显子测序,相对其他单独固定panel,通过差异化深度全外显子/靶向用药基因Panel可在更大范围内筛选患者特异性的突变谱,显著增加可追踪位点数目,提高检测灵敏度;另一方面包含高深度的固定加强panel方式,重点检测通用肿瘤患者数据库中发生频率较高且带有临床证据的区域,可以检测出更多更低频且肿瘤高频率发生/肿瘤高证据的组织变异位点,解决了现有技术中采用常规WES检测组织样本位点可能会存在遗漏低频率位点的问题,同时可包含经典融合区间,该区间通常不在外显子区域;最后本申请方式可以同时给出其他肿瘤标志物指标,例如TMB和MSI等,这些指标或在全外显子测序上表现更优(TMB),或在高深度的固定加强panel上表现更优(MSI)。
(2)本申请提供的微小残留病灶的检测方法,通过按功能及可信度的排序方式筛选有限个数的突变信号作为追踪突变信号,可在有限检测成本控制下得到更准确的检测结果。驱动突变、高频突变和主克隆突变均是有更大概率被释放到血浆中的突变,按此排序就可选择到更大可能性在血浆中可被检测到的突变信号,提高检测灵敏度。
(3)本申请提供的微小残留病灶的检测方法,使用的个性化组合panel(CCP探针),即10万×超高深度的个性化Customized探针+高证据/高频热点Core探针+SNP探针的组合Panel,使用突变型的customized序列探针可以更高效的抓取到待测样本的突变信号,固定core序列探针可为本申请使用者提示重要肿瘤进化/第二原发突变的出现,固定SNP序列探针则用于质量控制区分待测样本是否发生污染,相对于现有通过扩增子方法扩增几十个循环中极易发生污染,本申请方法可以监控污染造成的不合格样本,可以避免假阳或假阴的出现。换言之,本申请提供的微小残留病灶的检测方法,既可以监测肿瘤来源的变异位点,又可以同时检测第二原发变异位点及监测肿瘤进化,进一步实现检测灵敏度的提升,克服了现有技术中只进行组织突变谱追踪的应用局限性。
(4)本申请提供的微小残留病灶的检测方法,获得肿瘤组织样本DNA、血细胞样本DNA和血浆样本DNA共捕获的10万×血浆超高深度个性化组合panel数据,并将其用于更新追踪突变列表,提高追踪位点变异检测的准确性。即,通过高深度的个性化组合panel的方式再次获取肿瘤组织样本DNA数据,可检查使用WDC组合测序方式确定的突变是否为真实突变,减少由于WDC组合测序方式的测序深度限制导致追踪突变并不是真实的患者特异性突变的情况出现,提高检测结果准确性。
(5)本申请提供的微小残留病灶的检测方法,在检测待测血浆样本在追踪突变信号结果时,仅对覆盖追踪位点的reads使用结合唯一分子标签(UMI-unique molecularidentifiers,UMI)的duplex信息与严格的可信度过滤模型的方式进行,通过唯一分子标签进行去除重复序列,以提升血浆游离ctDNA单点检测的准确性,解决了现有技术中数据无法准确去除重复的问题;仅对覆盖追踪位点的reads进行检测相较于全区间的变异检测有效降低了运算成本;结合分子标签技术的duplex信息与严格的可信度过滤模型,使用迭代方式找寻唯一匹配的扩展突变型序列,可有效提高Indel检测的准确性,同时使用duplex及后续严格的过滤模型提高了SNV,Indel和fusion等各种突变类型检测的准确性。
(6)本申请提供的微小残留病灶的检测方法,基于差异化深度全外显子/靶向用药测序和组织、血细胞、血浆共捕获技术,及10万×超高深度个性化/高证据热点组合Panel测序的方式,评估血浆样本中微小残留病灶和肿瘤进化/第二原发的方法,克服现有方法在组织检测下限较高或追踪位点过少,和血液中ctDNA含量较低时检测灵敏度、准确度不足或检测成本过高,个性化追踪检测与肿瘤进化/第二原发检测无法兼得等问题,在有限成本范围内显著提高患者治疗后复发风险预测的准确性。
附图说明
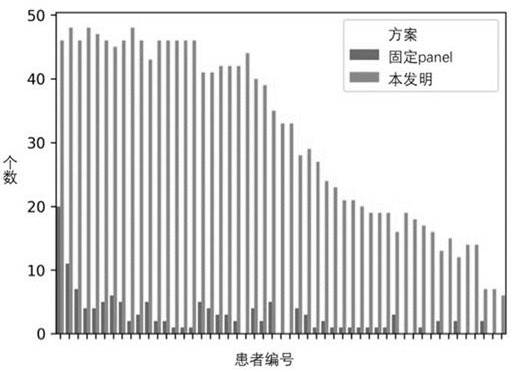
图1是实施例1及对比例1可追踪的突变个数。
图2是实施例1及对比例1检出的阳性突变。
图3是全外显子测序探针与靶向用药基因Panel按不同比例混合后形成的WDC探针的差异化测序深度。
图4是不同质量比组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库的CCP探针杂交共捕获的测序数据深度。
图5是杂交捕获体系中等体积清洗与体积梯度递增清洗的效果比较。
具体实施方式
下面结合具体实施例对本申请进一步进行描述。
需要说明的是,本说明书中所引用的如“上”、“下”、“左”、“右”、“中间”等用语,亦仅为便于叙述的明了,而非用以限定可实施的范围,其相对关系的改变或调整,在无实质变更技术内容下,当亦视为本申请可实施的范畴。
除非另有定义,本文所使用的所有的技术和科学术语与属于本申请的技术领域的技术人员通常理解的含义相同;本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
如本文所使用,术语“约”用于提供与给定术语、度量或值相关联的灵活性和不精确性。本领域技术人员可以容易地确定具体变量的灵活性程度。
如本文所使用,术语“......中的至少一个”旨在与“......中的一个或多个”同义。例如,“A、B和C中的至少一个”明确包括仅A、仅B、仅C以及它们各自的组合。
浓度、量和其他数值数据可以在本文中以范围格式呈现。应当理解,这样的范围格式仅是为了方便和简洁而使用,并且应当灵活地解释为不仅包括明确叙述为范围极限的数值,而且还包括涵盖在所述范围内的所有单独的数值或子范围,就如同每个数值和子范围都被明确叙述一样。例如,约1至约4.5的数值范围应当被解释为不仅包括明确叙述的1至约4.5的极限值,而且还包括单独的数字(诸如2、3、4)和子范围(诸如1至3、2至4等)。相同的原理适用于仅叙述一个数值的范围,诸如“小于约4.5”,应当将其解释为包括所有上述的值和范围。此外,无论所描述的范围或特征的广度如何,都应当适用这种解释。
实施例1
本实施例对51例I期肺癌患者术前血浆中MRD进行检测,由于血浆为术前血浆,可以理解的上述血浆样本为MRD阳性样本,包括如下步骤:
S1:获取患者肿瘤组织DNA和血细胞DNA的WDC测序数据,即:分别构建肿瘤组织DNA文库和血细胞DNA文库;将两个文库等质量比混合,并用WDC探针进行杂交捕获获得捕获后DNA文库,其中WDC探针是全外显子测序探针(WES探针)与靶向用药基因Panel按照1:(2~8)混合后形成的混合探针;对捕获后DNA文库进行测序,获得肿瘤患者WDC测序数据。具体包括如下步骤:
S11:DNA提取及核酸片段化,取患者肿瘤组织样本和术前全血,术前全血经密度梯度离心获得血细胞样本和血浆样本,提取肿瘤组织样本DNA并稀释至0.5 ng/μL~6 ng/μL,提取血细胞样本DNA并稀释至6 ng/μL;提取血浆中cfDNA并稀释至0.5 ng/μL~1 ng/μL,使用核酸打断仪处理肿瘤组织样本DNA和血细胞样本DNA获得片段化的肿瘤组织样本DNA和片段化的血细胞样本DNA。在实施例中,肿瘤组织可以是分离的经福尔马林固定、石蜡包埋的肿瘤组织样本。
S12:肿瘤组织样本和血细胞样本DNA文库构建,使用Roche的KAPA Hyper Prepkit(KK8504)试剂盒,对片段化的肿瘤组织样本DNA、片段化的血细胞样本DNA进行末端修复加A,使用Roche的KAPA HiFi HotStart ReadyMix(KK2602)试剂盒进行预扩增反应,通过Beckman的AMPure XP beads将预扩增产物纯化至新的EP管,即为肿瘤组织样本和血细胞样本DNA文库。在实施例中,还可以对DNA文库进行Qubit浓度检测和Agilent 2100质检,使用核酸浓度检测仪定量使得肿瘤组织样本DNA文库≥800 ng,血细胞样本DNA文库≥500 ng;并用生物分析仪对文库分析,肿瘤组织样本和血细胞样本DNA文库的主峰应位于150~500bp之间。
S13:WDC探针杂交捕获获得捕获后DNA文库(WDC 文库),利用WDC探针捕获目标区域片段,构建捕获后DNA文库。在实施例中,WDC探针为WES探针与靶向用药基因Panel按照1:(2~8)混合后形成的混合探针,利用该比例混合的探针可以实现测序深度的差异化,即WES其他区域:肿瘤相关基因区域:靶向用药基因区域可以实现1:(1.5~3):(2~6)的有效深度比例,可以降低靶向用药基因及肿瘤相关基因的检测下限,提高灵敏度。在实施例中,靶向用药的基因包括AKT1、ALK、AR、ARAF、BRAF、BRCA1、BRCA2、CDK4、CTNNB1、DDR2、EGFR、ERBB2、ERBB3、ERRFI1、ESR1、FBXW7、FGFR1、FGFR2、FGFR3、FLT1、GNA11、GNAQ、HRAS、IDH1、IDH2、KIT、KRAS、MAP2K1、MAPK1、MET、MTOR、NF1、NF2、NOTCH1、NRAS、NTRK1、NTRK2、NTRK3、PDGFRA、PIK3CA、PTEN、RAC1、RB1、RET、RICTOR、ROS1、SMAD4、TERT、TP53、TSC1、VEGFA、AKT2、AKT3、APC、ATM、ATR、ATRX、CDK6、CDKN2A、CHEK2、FLT3、FLT4、JAK1、JAK2、KDR、KEAP1、MDM2、MYC、PALB2、VHL、ABL1、BTK、SMO、ETV6、EWSR1、NTRK、HER2和BRCA。在实施例中,WDC文库构建具体为:将肿瘤组织样本DNA文库和血细胞样本DNA文库按照样本类型进行等质量比混合,并置于真空离心浓缩仪中60℃蒸干约20 min得到蒸干的文库;向蒸干后的DNA文库加入DNA杂交体系和WDC杂交探针,震荡混匀离心后室温孵育,按照95℃30s,70℃hold的16个小时杂交反应条件进行杂交;杂交后的文库使用市售试剂盒Twist Standard Hyb and Wash Kit(104447)进行目标区域杂交捕获和杂交后洗脱,洗脱后的带目标区域片段的beads再使用KAPA HiFi HotStart ReadyMix(KK2602)试剂盒进行杂交后扩增反应,最后用Beckman的AMPure XP beads 将预扩增产物纯化至新的EP管,即为WDC探针杂交捕获后DNA文库(WDC文库)。在实施例中,还可以对DNA文库进行Qubit浓度检测。在实施例中,也可以使用市售试剂盒xGen™ Hybridization and Wash Kit(1080584)行目标区域杂交捕获和杂交后洗脱,可以达到相同效果。
S14:WDC文库测序,获得WDC测序数据。在实施例中,具体为:用基因测序仪对WDC文库进行上机测序,得到肿瘤组织样本、血细胞样本10:3的数据量产出。
S2:获取患者基因组突变信号,即:将S1中获得的WDC测序数据预处理后与hg19人类参考基因组比对,获得肿瘤组织样本DNA突变信号、血细胞样本DNA突变信号,保留只存在肿瘤组织样本的DNA突变信号作为基因组突变信号,DNA突变信号包括体细胞变异(SNV)、插入缺失(Indel)、融合(fusion)或其他类型突变中一种或多种。具体包括如下步骤:
S21:WDC测序数据预处理及比对,包括去除接头和低质量碱基、比对至hg19人类参考基因组序列、去重、重比对和质量值矫正,获得校正后的Bam文件。在实施例中,WDC测序数据预处理是利用商业软件处理。在实施例中,去除接头和低质量碱基包括调用Trimmomatic-0.36将每一对FASTQ文件都作为paired reads进行去除接头和低质量碱基处理,采用“ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:8:true LEADING:3 TRAILING:3SLIDINGWINDOW:4:20 MINLEN:51”参数,生成去接头后的FASTQ文件。在实施例中,比对至hg19人类参考基因组序列包括调用商业软件Sentieon-202112.05将去接头后的FASTQ文件作为paired reads,分别使用bwa men模块比对到hg19人类参考基因组序列,util sort模块对比对结果进行排序后生成初始Bam文件。在实施例中,去重包括调用商业软件Sentieon-202112.05,采用命令“sentieon driver--algo Dedup—rmdup”对初始Bam文件进行去重复处理,生成去重后的Bam文件。在实施例中,重比对包括调用商业软件Sentieon-202112.05,采用命令“sentieon driver--algo Realigner”对去重后的Bam文件进行重比对处理,生成重比对后的Bam文件。在实施例中,质量值矫正包括调用商业软件Sentieon-202112.05,采用命令“sentieon driver--algo QualCal”对重比对后的Bam文件进行质量值矫正处理,生成校正后的Bam文件。
S22:体细胞变异(SNV)检测,包括通过比较肿瘤组织样本和血细胞样本校正后的Bam文件,获得初始的体细胞突变列表。在实施例中,比较是利用商业软件处理校正后的Bam文件完成。在实施例中,调用gatk-package-4.1.9.0的Mutect2模块的配对样本模式得到初始的体细胞突变列表。在实施例中,使用gatk-package-4.1.9.0的FilterMutectCalls模块过滤掉某些指标不满足软件默认条件的突变,具体指标包括:map_qual,base_qual,germline,fragment,normal_artifact,position和haplotype。在实施例中,还包括突变注释,获得位点信息,用于后续位点过滤和排序操作。在实施例中,突变注释通过商业软件完成。在实施例中,使用ANNOVAR软件对初始突变列表进行注释,生成注释后突变列表,使用参数:–protocol refGene,ljb26_sift,ljb2_pp2hdiv,ljb2_pp2hvar,exac03,clinvar_20220709,cadd14,gnomad_exome,cytoBand,snp138,gnomad_genome,1000g2015aug_all,1000g2015aug_chb,1000g2015aug_chs,1000g2015aug_afr,1000g2015aug_eas,1000g2015aug_eur,1000g2015aug_sas,1000g2015aug_amr,simpleRepeat,cosmic80,HGMD,rmsk,BIC,OMIM,reliability,Pro_CancerRepeat,hgmd_202004。
S23:融合突变(fusion)检测,包括通过比较肿瘤组织样本和血细胞样本校正后的Bam文件,获得肿瘤组织样本的融合突变检测结果。在实施例中,比较是利用商业软件处理校正后的Bam文件。在实施例中,调用LUMPY(V0.2.13)软件,输入配对的肿瘤组织样本和血细胞样本校正后的Bam文件,获得肿瘤组织样本的融合突变检测结果。
S24:拷贝数变异(CNV)检测,包括通过比较肿瘤组织样本和血细胞样本校正后的Bam文件,获得肿瘤组织样本的肿瘤纯度和肿瘤细胞等位基因拷贝数的估计值。在实施例中,比较是通过商业软件处理校正后的Bam文件。在实施例中,调用R包FACTES,输入配对的肿瘤组织样本和血细胞样本校正后的Bam文件,得到肿瘤组织样本的肿瘤纯度和肿瘤细胞等位基因拷贝数的估计值,该值用于后续的主克隆和亚克隆分类。
在实施例中,还包括S25:突变过滤,包括按照如下过滤规则过滤掉突变获得最终的基因组突变信号,过滤规则包括:gnomAD、ExAC、1000g三个数据库的人群突变频率小于2%;测序深度大于40;突变频率大于1%;不在平台黑名单范围(通过大量样本,不同批次的统计,反复出现的低质量的突变,定义为黑名单突变);支持reads>2;覆盖深度>100;正负链支持没有显著差异;所在及周围无简单重复序列;肿瘤组织突变频率/血细胞突变频率>5。
在实施例中,还包括TMB和MSI分析,分析方法参考公告号为CN112029861B的发明名称为基于捕获测序技术的肿瘤突变负荷检测装置及方法和公告号为CN112365922B的发明名称为用于检测MSI的微卫星位点、其筛选方法及应用的中国发明专利。
S3:筛选追踪突变信号,即:将S2中的基因组突变信号按照功能及可信度进行排序,首先有重要功能的驱动突变,给予最高排序优先级排序;其次按突变频率及主克隆-亚克隆排序,针对突变频率大于5%的突变,按突变频率从大到小排序;对突变频率在1%~5%的突变,优先按主克隆>亚克隆排序,次优先按突变频率排序,排序后筛选排序靠前的预设个数基因组突变信号作为追踪突变信号,具体包括如下步骤:
S31:主克隆和亚克隆分类,根据S2中的基因组突变信号和CNV检测结果,根据每个体细胞突变的支持突变reads个数及测序深度,并考虑由CNV等引入的等位基因不平衡,采用统计聚类方法,例如贝叶斯聚类方法,估计肿瘤纯度及并以此将体细胞突变做分组聚类到不同的克隆群体,并统计每个克隆群体的细胞占比,将占比最高的克隆群体定义为主克隆,其他类别定义为亚克隆。在实施例中,分类是通过商业软件处理完成。在实施例中,调用PyClone-0.13.1软件的run_analysis_pipeline模块,使用参数“--num_iters 10000--burnin 1000--prior major_copy_number--max_clusters 2”,根据基因组突变信号和CNV检测结果,判断出每个突变的分类,即属于主克隆还是亚克隆。
S32:排序,根据如下排序规则进行排序,排序规则为:根据预先归纳的有重要功能的驱动突变数据库,筛选在数据库中的突变,给予最高排序优先级排序;按突变频率及主克隆-亚克隆排序,针对突变频率大于5%的突变,按突变频率从大到小排序;对突变频率在1%~5%的突变,优先按主克隆>亚克隆排序,然后按突变频率排序。
S33:筛选追踪突变信号,包括选择S32中排序在前列的基因组突变信号作为追踪突变信号。在实施例中,选择排序在前列的50个基因组突变信号作为追踪突变信号。在实施例中,选择全部的基因组突变信号作为追踪突变信号。
S4:制备个性化组合panel(CCP探针工作液),即:根据追踪突变信号设计追踪突变信号序列探针(customized探针),并与固定突变信号序列探针(core探针)和SNP探针混合,制备个性化组合panel,其中固定突变信号序列探针(core探针)用于检测肿瘤进化或第二原发,SNP探针用于鉴别样本来源和评估样本污染程度,具体包括如下步骤:
S41:筛选候选customized探针序列,筛选规则如下:如果是SNV/Indel类型的突变,根据参考基因组和追踪突变信号,串联每个追踪突变信号序列起始位置基因组上游60bp的参考基因组序列、追踪突变信号序列和追踪突变信号序列终止位置基因组下游60 bp的参考基因组序列三个序列为候选customized探针序列;如果是Fusion类型的突变,根据参考基因组和融合突变的方向,串联融合突变上游基因gene1断点1上游(沿转录本方向)60bp的序列和融合变异下游基因gene2断点2下游(沿转录本方向)60 bp的序列为候选customized探针序列。本方法使用针对特定追踪突变信号的探针序列,可以更有效的抓取特定追踪突变的序列,提高检测的灵敏度,而传统的基于参考基因组的探针序列,由于带特定追踪突变的序列的片段与探针的匹配性降低,抓取这些特定追踪突变的序列能力也将减弱。
S42,候选customized探针序列过滤,过滤规则如下:去除在整个参考基因组范围存在“较好比对位置”个数大于20的候选探针序列,其中“较好比对位置”指匹配长度大于30bp且比对期望值小于0.000001的位置;去除含有SSR的候选探针序列;去除GC<10%或GC>80%的异常候选序列。在实施例中,上述过滤可以通过商业软件完成。在实施例中,调用blat(V.35)软件去除在整个参考基因组范围存在“较好比对位置”个数大于20的探针序列。在实施例中,调用软件MISA检测重复序列SSR,去除含有SSR的候选序列。在实施例中,调用MFEprimer(v.3.2.6)软件对候选探针序列做质量控制(GC,Tm和Dg),去除GC<10%或GC>80%的异常候选序列。
S43,制备CCP探针工作液,按照探针摩尔数Customized探针:Core探针:SNP探针=8:8:1的体系混合,Customized探针如表1所示,固定突变信号序列探针(core探针)的基因如表2所示,SNP探针坐标如表3所示,按照8:8:1配制而成的CCP探针,测序后可以实现5:5:1的有效深度比例,可以降低靶向用药基因/肿瘤相关基因的检测下限,提高灵敏度。CCP探针工作液制备所需要的Core探针和SNP探针,承担各自不同的功能,由于Core探针需要承担检测肿瘤进化或第二原发等功能,因此也需要血浆10万×数据深度才能增加检测灵敏度,而SNP探针只需要用于鉴别样本来源和评估样本污染程度即可,因此仅需要较低数据深度即可。在实施例中,Core探针来自于臻和肿瘤精准医学证据库,其中证据基因位点均来自NCCN指南、专家共识等、公共数据库中靶向证据基因位点和耐药化疗证据基因位点、FDA/NMPA药品标签、结合临床试验及会议摘要等证据基因位点,同时在多癌种中筛选出一级证据基因位点和二级证据基因位点,形成的集合为固定突变信号panel(core panel)。在实施例中,SNP探针用于鉴别样本来源和评估样本污染程度,是确保样本检测的准确性的不可或缺的一部分。SNP探针主要来源于WDC中全外显子组所覆盖的dbSNP数据库中的杂合度较高的SNPs位点集合。在实施例中,Core探针、SNP探针与Customized探针按照探针摩尔数比例进行混合,体系还包括IDTE。
表1 Customized探针
表2 Core探针基因
表3 SNP探针坐标
S5:获取患者肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA的个性化组合panel测序数据,即:构建血浆cfDNA文库,并将肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:(6~12)进行不同样本类型文库的混合;通过CCP探针杂交捕获获得捕获后DNA文库,对捕获后DNA文库进行测序,获得肿瘤患者个性化组合panel测序数据。具体包括如下步骤:
S51:血浆cfDNA预文库构建,本申请使用Roche的KAPA Hyper Prep kit(KK8504)试剂盒,对血浆cfDNA进行末端修复加A和接头连接的反应,使用Roche的KAPA HiFiHotStart ReadyMix (KK2602)试剂盒进行预扩增反应,通过Beckman的AMPure XP beads将预扩增产物纯化至新的EP管,即为血浆cfDNA预文库。在实施例中,还包括对末端修复加A处理后的血浆cfDNA进行唯一分子标签(UMI-unique molecular identifiers,UMI)接头连接处理,通过唯一分子标签进行去除重复序列,可以提升血浆游离ctDNA单点检测的准确性,解决了现有技术中数据无法准确去除重复的问题。在实施例中,具体为:末端修复加A的PCR反应结束后瞬离并加入稀释好的5 μL UMI接头溶液,再加入45 μL连接混合液(5 μL 超纯水+30 μL 连接缓冲液+10 μL DNA连接酶),震荡混匀、瞬离并置于PCR仪20℃孵育30min。然后通过Beckman的AMPure XP beads 将连接反应后的DNA产物纯化至新的EP管,用于下一步预扩增。在实施例中,还可以对DNA文库进行Qubit浓度检测和Agilent 2100质检,使用核酸浓度检测仪定量使得血浆cfDNA文库≥1000 ng;并用生物分析仪对文库分析,血浆cfDNA文库的主峰应位于150~400 bp之间。
S52:CCP探针杂交捕获获得捕获后DNA文库(CCP 文库),利用CCP探针捕获目标区域片段,构建捕获后DNA文库。在实施例中,具体为:将肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:(6~12)进行不同样本类型文库的混合,利用该比例混合,可以得到肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA 1:1:(3~6)的数据量,可以同时兼顾测序深度与成本的平衡,在达到血浆10万×超高深度的同时,组织可以达到1万×深度以便得到更准确的组织突变谱,及血细胞的1万×以上深度可以辅助血浆排除克隆性造血的干扰,混合后置于真空离心浓缩仪中60℃蒸干约20 min得到蒸干的文库,向蒸干后的DNA文库加入DNA杂交体系和CCP杂交探针,震荡混匀离心后室温孵育,按照95℃ 30 s,65℃ 4 h,65℃ hold的16个小时杂交反应条件进行杂交。杂交后的DNA文库使用市售试剂盒xGen™ Hybridization and Wash Kit(1080584)进行目标区域杂交捕获和杂交后洗脱,洗脱后的带目标区域片段的beads再使用KAPA HiFi HotStart ReadyMix (KK2602)试剂盒进行杂交后扩增反应,最后用Beckman的AMPure XP beads将预扩增产物纯化至新的EP管,即为CCP探针杂交捕获后DNA文库(CCP文库)。在实施例中,还可以对最终捕获文库文库进行Qubit浓度检测。在实施例中,目标区域杂交后的洗脱采用体积梯度递增的洗脱方式进行,与常规的等体积洗脱相比,得到更高Target ratio数据。在实施例中,体积梯度递增的洗脱方式包括如下步骤:孵育完成后,加入100 μL 65℃预热的清洗缓冲液I,混匀后置于磁力架上1 min至液体澄清,弃上清、瞬离后置于磁力架上弃残液;加入145 μL 65℃预热的Stringent清洗缓冲液,吹打混匀后65℃孵育5 min,置于磁力架上1 min至液体澄清,弃上清;加入150 μL 65℃预热的Stringent清洗缓冲液,吹打混匀后65℃孵育5 min,置于磁力架上1min至液体澄清,弃上清,瞬离后置于磁力架上弃残液;加入50μL室温放置的清清洗缓冲液I,轻轻吹打磁珠至重悬,转移重悬磁珠至新的PCR管内,补充100 μL清洗缓冲液Ⅰ,振荡瞬离,置于磁力架上1min至液体澄清,弃上清;从磁力架取下PCR管,瞬离后置于磁力架上,用10 μL移液器彻底弃去离心管底部的残留液体;加入155 μL室温放置的清洗缓冲液Ⅱ,持续振荡中间瞬离两次,置于磁力架上1min至液体澄清,弃上清,瞬离后置于磁力架上弃残液;加入160μL室温放置的清洗缓冲液Ⅲ,持续振荡瞬离,置于磁力架上1 min至液体澄清,弃去上清,瞬离后置于磁力架上弃残液;向PCR管中加入20 μL超纯水洗脱,转移至新的PCR管内,获得捕获文库,进行下一步扩增。杂交完成后,需要将体系中或吸附在管壁上的off-target reads清洗掉,常规操作步骤均使用相同体积的清洗液进行清洗,本申请测试出梯度体积递增的方法可以有效增加针对前一步骤吹打或斡旋清洗过程中吸附在管壁上的off-target reads的清洗,最终呈现出比常规操作高7.18%的target ratio,并实现更高深度及对应的检测灵敏度。
S53:CCP文库测序,获得个性化组合panel测序数据。在实施例中,具体为:用基因测序仪对扩增后捕获CCP DNA文库进行上机测序,得到肿瘤组织样本、血细胞样本和血浆cfDNA样本的测序数据。在实施例中,将肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:6的混合,得到肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA1:1:3的数据量。在实施例中,将肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:9的混合,得到肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA 1:1:4的数据量。在实施例中,将肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:12的混合,得到肿瘤组织样本DNA、血细胞样本DNA和血浆cfDNA 1:1:6的数据量。常规的基因检测手段均采用几百、几千×测序深度进行检测,随着MRD组织先验策略深入临床研究,各个研究机构均预期采用固定panel较高深度测序来提升MRD检测灵敏度,由于成本压力,目前应用较多的3万×血浆测序深度。本发明采取患者个性化组织Panel(相对较小panel)进行个性化追踪检测,因此可以在有效控制成本的情况下可以采用10万×超高深度进行MRD个性化突变谱的追踪检测。可以同时兼顾测序深度与成本的平衡。
S6:追踪突变信号校正及确定追踪突变序列和位置,即:利用肿瘤组织样本和血细胞样本的个性化组合panel测序数据校正追踪突变信号,去除不再被确定为体细胞小突变和融合突变的信号,去除克隆性造血来源的突变,更新追踪突变信号生成最终追踪突变信号并确定最终追踪突变信号序列和位置,具体包括如下步骤:
S61:参考步骤S2和S3处理个性化组合panel测序数据,获得新的追踪突变信号,并匹配S3中的追踪突变信号是否在新的追踪突变信号中,删除不存在新的追踪突变信号中的突变信号,生成最终追踪突变信号。如上所述,WDC组合测序中的组织和血细胞仅200×测序深度,而CCP组合测序数据中组织和血细胞在1万×以上测序深度,高深度下可以将组织中位点频率定位更准确,同时高深度血细胞检测到的克隆性造血可以一并排除,即经过个性化组合panel测序数据将组织突变谱进行了精筛,使样本检测更准确。
S64:确定最终追踪突变序列和位置,按照如下方法确定:获取扩展突变型序列,即首先根据参考基因组和最终追踪突变信号,对每个追踪突变序列,串联其起始位置到基因组上游3 bp的参考基因组序列、追踪突变序列及其终止位置到基因组下游3 bp的参考基因组序列三段序列为候选序列;如果候选序列在包含候选序列上下游200 bp范围内仅可唯一匹配,则保留候选序列为追踪突变序列,同时定义串联序列的基因组起始位置为追踪突变序列的基因组起始位置,串联序列的基因组终止位置为追踪突变序列的基因组终止位置;若不满足保留标准,则增加1 bp长度,即4 bp开始重新扩展上下游序列后重复操作,直到满足保留标准或串联序列长度超过35 bp。确定在追踪突变信号附近唯一的一段包含追踪突变的序列,有效避免了匹配到附近其他位置的可能性。而上下游扩展提高了这种唯一性片段存在的可能性,同时更长的片段匹配及定位更准确。另一方面直接使用突变后的序列做上下游扩展,可以更直接确定每个测序序列(read)或单链一致性序列(SSCS)是否支持该追踪突变信号,而传统的与参考基因序列做比对的方法,尤其在长片段的插入缺失时无法准确比对及定位。对于Indel,尤其是长片段的插入缺失,本申请可以有效提高匹配及定位准确率。
S7:获取血浆cfDNA的最终追踪突变信号检测结果,即:提取覆盖在最终追踪突变信号位置上血浆样本的reads对,提取两端的分子标签序列、基因组上的起始位置、插入片段长度和方向等信息,确定单链一致性序列和双链一致性序列,并确定追踪突变信号检测结果,具体包括如下步骤:
S71:去除接头,提取UMI序列并比对,提取覆盖在最终追踪突变信号位置上血浆样本的reads。在实施例中,去除接头和提取UMI序列,调用fastp(0.23.2)将每一对FASTQ文件都作为paired reads进行去除接头和UMI序列处理,采用“--trim_poly_g --poly_g_min_len 10 --cut_right --cut_window_size 4 --cut_mean_quality 20 --overlap_len_require 30 --overlap_diff_limit 5 --overlap_diff_percent_limit 20 --length_required 51 --adapter_fasta adapters/TruSeq3-PE.f”参数,生成去接头和提取UMI序列后的FASTQ文件,提取的UMI序列存在于对应read的ID中。比对并提取UMI序列,调用商业软件Sentieon-202112.05将去接头后的FASTQ文件作为paired reads,分别使用umiextract模块提取UMI序列、bwa men模块比对到hg19人类参考基因组序列,和util sort模块对比对结果进行排序后生成初始Bam文件。在实施例中,如果是双端测序,则将具有相同read id号的一对reads,标记为1个fragment,提取fragment的包括两端UMI序列、基因组上的起始位置、插入片段长度和方向的信息。
S72:确定单链一致性序列(SSCS),将具有相匹配fragment信息的fragment作为一 组,其中相匹配fragment信息指在1 bp的误差范围内的UMI序列、起始位置或插入片段差异 等,具有几乎完全相同的fragment信息,从最终追踪突变信号序列的基因组起始位置所对 应的fragment上的碱基位置开始,到追踪突变序列的基因组终止位置所对应的fragment上 的碱基位置截止,逐碱基的比较每个位置上每种碱基类型的个数,碱基类型包含A,T,C,G,; 确定SSCS,如果满足
S73:确定支持追踪突变的类型:对每一个追踪突变,定义与追踪突变信号序列完全匹配的SSCS为一个simplex,具有配对UMI序列的两个simplex定义为1个duplex。
S74,追踪突变过滤及确定:按照如下规则过滤追踪突变:如果simplex上追踪突变边缘距离fragment边缘距离的最小值小于5,或simplex上与参考基因组序列不同的碱基个数大于5,则定义该simplex为低质量simplex,统计每个追踪突变的低质量simplex的比例,若大于0.5,则认为该突变为一个低可信度突变,在后续分析中去除;统计过滤后每个追踪突变的simplex个数,和duplex个数,如果满足simplex个数大于0,同时duplex个数大于1,则报告该突变为一个阳性突变。
S8:获取MRD检测结果,结合所有追踪突变信号的检测结果得到待测患者的MRD检测结果,即经过上述严格过滤后仍然有大于预先设置阈值个数的阳性突变,则定义该患者MRD状态为阳性,否则为阴性。在实施例中,预先设置阈值=1。
实施例2
本实施例提供不同比例全外显子测序探针(WES探针)与靶向用药基因Panel混合制备的WDC探针进行WDC探针杂交捕获并测序的各区域测序深度,其他步骤参考实施例1。
结果如图3所示,WDC探针可以实现测序深度的差异化。WDC组合Panel相较于只有WES探针杂交捕获的测序数据,可以实现WES其他区域:肿瘤相关基因区域:靶向用药核心基因区域可以实现1:(1.5~3):(2~6)的有效深度比例,可以降低靶向用药核心基因及肿瘤相关基因的检测下限,从而提高组织检测的灵敏度。
实施例3
本实施例提供不同比例肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库混合后CCP探针杂交捕获后的测序深度,其他步骤参考实施例1。
结果如图4所示,低于2:1:(6-12)的比例需要增加测序数据量及测序成本才能实现血浆高深度的等效性,而高于该比例则需要更高数据量实现组织和血细胞的深度等效性。由于组织、血细胞和血浆共捕获体系中,组织片段损伤程度比血细胞大,因此投入量需比血细胞更多。同时结合组织和血细胞投入量(30ng-300ng)比血浆(10ng-50ng)高的情况,因此血浆需要更高的数据量才能达到更高的测序深度。本发明综合成本因素和超高深度需求,最终确定肿瘤组织样本DNA文库、血细胞样本DNA文库和血浆cfDNA文库按照质量比2:1:(6-12)进行CCP探针杂交捕获时,既可以控制测序成本又可以血浆样本中位深度达到10万×数据深度,组织和血细胞1万×数据深度。
实施例4
本实施例提供CCP探针杂交捕获过程中目标区域杂交后的洗脱采用体积梯度递增的洗脱方式与常规的等体积洗脱方式的比较结果,其他步骤参考实施例1。
结果如图5所示,相交于常规操作步骤均使用相同体积的清洗液进行清洗,本实施例测试出梯度体积递增的方法可以有效增加针对前一步骤吹打或斡旋清洗过程中吸附在管壁上的off-target reads清洗掉,最终呈现出比常规操作的target ratio能高7.18%,并实现更高深度及对应的检测灵敏度。
实施例5
本是实施例提供了一种微小残留病灶的检测装置,包括:
数据输入模块,用于输入患者实施例1中患者肿瘤组织样本和术前血细胞样本的WDC测序数据,和输入患者上述肿瘤组织样本、血细胞样本和血浆的个性化组合panel测序数据;
数据处理模块,用于根据输入数据完成实施例1中所述的获取患者基因组突变信号、筛选追踪突变信号、追踪突变信号校正及确定追踪突变序列和位置及获取血浆cfDNA的追踪突变信号检测结果;
结果输出模块,用于输出实施例1中所述的肿瘤患者的MRD检测结果。
对比例1
本对比例提供专利号为CN109477138A,发明名称为肺癌检测方法的对51例I期肺癌患者带瘤状态的术前血血浆样本进行检测,具体方法参考CN109477138A。
结果分析:
本申请实施例1及对比例1检测的可追踪的突变个数如图1所示,本申请51例样本可追踪1794个突变,平均每个样本可追踪35个突变(中位值39),而对比例1的51例样本可追踪168个突变,平均每个样本可追踪3个突变(中位值2)。由此可见,本申请方案有更多的可追踪的突变个数。
本申请实施例1及对比例1检测的阳性突变如图2所示,本申请51例样本中检测到22个样本的37个阳性突变,而对比例1中仅检测到2个样本的2个阳性突变。由此可见,本申请方案检出更多的阳性突变。
本申请实施例1及对比例1的阳性检测率通过如下公式计算,
本申请阳性检出率为22/51=43.13%, 而对比例1阳性检出率为2/51=3.9%,提高极显著。同时对比所有可公开查询的其他机构结果,平均阳性检出率大部分低于10%,本申请具有显著的效果。
- 存储设备在线检测方法、装置、设备及可读存储介质
- 医学图像的病灶定位识别方法、装置、设备及存储介质
- 一种液位检测装置和包含其的设备以及液位检测方法、电子设备及计算机可读存储介质
- 一种存储系统的状态检测方法、装置、设备及存储介质
- 设备的检测方法、装置、计算机设备和存储介质
- 确定关于实体瘤微小残留病灶的检测参数的方法、设备和存储介质
- 检测实体瘤微小残留病灶的方法、计算设备和存储介质