使用无循环mRNA谱分析检测阿尔茨海默病风险的系统和方法
文献发布时间:2023-06-19 18:34:06
本申请要求2020年3月18日提交的美国临时专利申请系列第62/991,513号和2020年3月20日提交的美国临时专利申请系列第62/992,723号的优先权。上述专利申请的全部内容通过引用并入本文。
背景技术
阿尔茨海默病(AD)是一种神经变性病症(neurodegenerative disorder),特点在于认知和行为损害,显著干扰患者的日常正常功能。它是一种临床前期长、病程渐进的不可治愈的疾病。
阿尔茨海默病是影响全球大部分老年人群的痴呆的最常见原因,并且预计到2050年将增加两倍。阿尔茨海默病是一种神经变性病症,其特征通常在于淀粉样-β肽的积累、tau蛋白和神经原纤维缠结的沉积、突触和神经元功能障碍的发作、由小胶质细胞引起的炎症反应的激活以及线粒体功能障碍。目前临床前阿尔茨海默病的诊断指南利用心理测试来确定认知损害的存在,随后使用成像和脑脊液(CSF)生物标志物来确定该损害是否由阿尔茨海默病引起。尽管尸检组织学仍然是建立阿尔茨海默病病理学的金标准,但CSF Aβ1-42和淀粉样蛋白正电子发射断层扫描(PET)的评估可用作替代物。此外,脑中的变化在具有已知的症状前变化的临床症状之前数年显现,包括皮质变薄和淀粉样蛋白-β、tau蛋白和神经原纤维缠结的沉积。虽然这些病理变化可以通过成像测试和CSF蛋白标志物来测量,但是成像方式昂贵并且CSF收集是侵入性的。因此,需要用于阿尔茨海默病诊断的高度可行的非侵入性检验。
发明内容
本文公开了一种检测对象中的阿尔茨海默病(AD)的方法,该方法包括:(a)定量生物样品中多种无细胞信使RNA(cf-mRNA)的cf-mRNA水平;和(b)处理所述多种cf-mRNA水平中的一种或多种以鉴定对象的组织的疾病状态和对象的年龄,其中处理包括将对象的cf-mRNA水平与多种cf-mRNA的阈值进行比较。生物样品可以包括对象的血液。处理可以包括对所述多种cf-mRNA的水平中的一种或多种应用机器学习分类器。机器学习分类器可以包括LASSO回归模型。该方法还可包括(c)定量第二生物样品中多种cf-mRNA的cf-mRNA水平,以及(d)处理第二生物样品中多种cf-mRNA的所述水平中的一种或多种以鉴定所述对象的所述组织的第二疾病状态。第二生物样品可以在对象已经接受神经变性病症的治疗或疗法之后获得。治疗或疗法可包括胆碱酯酶抑制剂或美金刚胺中的一种或多种。定量可以包括使多种cf-mRNA经历逆转录、多核苷酸扩增、测序、探针杂交、微阵列杂交或其组合中的至少一种。
该方法还可包括形成包含来源于多种cf-mRNA的多种cDNA的下一代测序(NGS)文库。定量还可以包括检测对不是来自血液的生物样品有贡献的多种cf-mRNA的比例。定量还可以包括检测对来自对象的大脑的生物样品有贡献的多种cf-mRNA的比例。多种cf-mRNA可以对应于选自KIAA0100、MAGl1、NNMT、MXD1、ZNF75A、SELL、ASS1、MNDA和AC132217.4的两种或更多种基因。该方法还可包括将对象鉴定为具有患阿尔茨海默病的高风险并推荐治疗。该方法还可包括治疗患者的阿尔茨海默病。治疗可包括胆碱酯酶抑制剂或美金刚胺中的一种或多种。
本文公开了一种检测对象中阿尔茨海默病(AD)阶段的方法,该方法包括:(a)从对象获得生物样品;以及(b)检测生物样品中多种无细胞信使RNA(cf-mRNA)的cf-mRNA水平,其中多种cf-mRNA对应于选自KIAA0100、MAGl1、NNMT、MXD1、ZNF75A、SELL、ASS1、MNDA和AC132217.4的两种或更多种基因。该方法还可包括使用机器学习分类器处理多种cf-mRNA的水平。机器学习分类器可以包括LASSO回归模型。该方法还可包括(c)从对象获得第二生物样品;以及(d)检测第二生物样品中多种无细胞信使RNA(cf-mRNA)的cf-mRNA水平。第二生物样品可以在对象已经接受神经变性病症的治疗或疗法之后获得。治疗或疗法可包括胆碱酯酶抑制剂或美金刚胺中的一种或多种。该方法还可包括鉴定对象患阿尔茨海默病的阶段的风险。阿尔茨海默病的阶段可选自:临床前阿尔茨海默病、由阿尔茨海默病引起的轻度认知损害、由阿尔茨海默病引起的轻度痴呆、由阿尔茨海默病引起的中度痴呆或由阿尔茨海默病引起的重度痴呆。该方法还可以包括将多种cf-mRNA的cf-mRNA水平与多种cf-mRNA的cf-mRNA水平的阈值进行比较。
该方法还可包括将cf-mRNA水平输入分类器以获得风险评分,其中风险评分指示对象患有AD的可能性。分类器可以是经过训练的机器学习算法。经过训练的机器学习算法可以包括LASSO回归模型。可以使用来自被诊断患有阿尔茨海默病的对象的生物样品来训练经过训练的机器学习算法。风险评分可确定为至少80%的灵敏度。风险评分可确定为至少90%的灵敏度。风险评分可具有0.44的截止值。风险评分可指示对象的阿尔茨海默病的特定发展状态。在确定对象的风险评分之前,对象可能没有被诊断为患有阿尔茨海默病。该方法还可以包括基于风险评分生成报告。该方法还可包括将报告发送给健康从业者。该报告可包括施用胆碱酯酶抑制剂和/或美金刚胺的建议。
该方法还可包括给对象分配临床痴呆评定量表(CDR)评分或简易精神状态检查量表(MMSE)评分。分配还可包括(a)定量生物样品中第二多种cf-mRNA的cf-mRNA水平,其中第二多种cf-mRNA对应于选自SLU7、HNRNPA2B1、GGCT、NDUFA12、HSPB11、ATP6V1B2、SASS6、SUMO1、KRCC1和LSM6的两种或更多种基因;以及(b)将对象中的第二多种cf-mRNA水平与第二多种cf-mRNA的阈值进行比较。定量可以包括使第二多种cf-mRNA经历逆转录、多核苷酸扩增、测序、探针杂交、微阵列杂交或其组合中的至少一种。生物样品可以是血浆或血清。生物样品可以是脑脊液。第一多种cf-mRNA和第二多种cf-mRNA可以来自端脑(cerebrum)、小脑、背根神经节、颈上神经节、松果腺、杏仁核、三叉神经节、大脑皮质和下丘脑中的至少两种。该方法还可包括监测AD进展。监测可包括磁共振成像(MRI)脑扫描或计算机断层摄影(CT)脑扫描。该方法还可包括对对象进行精神敏锐度测试。
本文公开了一种检测对象中的阿尔茨海默病(AD)的方法,该方法包括:(a)定量生物样品中多种无细胞信使RNA(cf-mRNA)的cf-mRNA水平,其中多种无细胞mRNA对应于编码转录因子的基因,该转录因子涉及sirtuin信号转导通路、IL-8信号转导通路、蛋白质泛素化通路、氧化磷酸化通路、sumoylation通路、线粒体功能障碍通路、炎性体通路、GABA受体信号转导通路、netrin信号转导通路、突触长期抑制信号转导通路、阿片样物质信号转导通路或其组合中的至少一种;以及(b)将对象的cf-mRNA水平与多种cf-mRNA的阈值进行比较。
本文公开了一种用于定量生物样品中多种无细胞信使RNA(cf-mRNA)的cf-mRNA水平的组合物,其中多种无细胞mRNA对应于包含KIAA0100、MAGl1、NNMT、MXD1、ZNF75A、SELL、ASS1、MNDA和AC132217.4的多种基因,该组合物包含具有与从多种cf-mRNA转录的cDNA序列杂交的序列的多种寡核苷酸引物。
本文公开了一种用于检测对象中阿尔茨海默病(AD)阶段的可能性的方法,该方法包括:(a)从对象获得生物样品;以及(b)检测生物样品中多种无细胞信使RNA(cf-mRNA)的cf-mRNA水平,其中多种cf-mRNA对应于包含KIAA0100、MAGI1、NNMT、MXD1、ZNF75A、SELL、ASS1、MNDA和AC132217.4的多种基因,其中该方法的准确度大于85%。该方法可具有至少80%的灵敏度。该方法可具有至少90%的灵敏度。该方法可具有至少80%的特异性。生物样品可以是血液。生物样品可以是血清。
本文公开了一种测定活性剂的方法,该方法包括(a)在第一时间点评估对象的第一无细胞表达谱;(b)向对象施用活性剂;和(c)在第二时间点评估对象的第二无细胞表达谱。该方法还可包括将第一无细胞表达谱与第二无细胞表达谱进行比较。第一表达谱和第二表达谱之间的差异可以指示治疗的效果。活性剂可以是治疗阿尔茨海默病的药物化合物。该方法还可包括在第三时间点评估对象的第三无细胞表达谱。评估可包括测序、阵列杂交或核酸扩增中的一种或多种。第二时间点可以是第一时间点之后的四周。该方法还可以包括在18个月的时间段内,在第一时间点之后每四周评估一次时间点。该方法可包括追踪和/或检测一种或多种无细胞表达谱,以测量用于治疗和/或药物发现和/或开发的一种或多种感兴趣的靶标。该方法还可包括在治疗和/或药物发现和开发期间测量用于先导优化和/或临床开发的药效学。该方法还可包括产生基因表达谱以表征与用于治疗和/或药物发现和/或开发的特异性靶标的参与相关的一种或多种药效学效应。该方法可以包括检测用于治疗和/或药物发现和开发的药效学靶标参与的变化。对象可能患有或疑似患有阿尔茨海默病。
在本说明书中提到的所有出版物、专利和专利申请都通过引用并入本文,其程度如同每个单独的出版物、专利或专利申请被具体且单独地指出通过引用而并入。
附图说明
本发明的新颖特征在所附权利要求中具体阐述。通过参考阐述说明性实施方案的以下详细描述和附图,将获得对本发明的特征和优点的更好理解,在说明性实施方案中利用了本发明的原理,并且附图中:
图1A-1D显示了RNA浓度和基因表达谱样品分布。图1A说明了从血浆中提取的RNA的典型生物分析仪图谱(上图)。从AD和NCI血浆中提取的RNA的RNA浓度。图1B显示了两个重复之间的皮尔逊相关系数的直方图。图1C显示了所有测序样品的主成分分析。图1D显示了校正后所有测序样品的主成分分析。
图2A-2D显示了无细胞信使核糖核酸(cf-mRNA)测序是表征cf-mRNA转录组的全面和准确的方法。图2A显示了每个样品检测到的转录本的直方图。图2B显示了具有掺入(spiked-in)的内源对照的皮尔逊相关系数的直方图。图2C显示了使用皮尔逊相关性分析的单个转录本的重复之间的相关性的实例。图2D显示了一致检测的基因的所有外显子-内含子连接处的聚集覆盖(在所有NCI对照中TPM>5,总共3490个基因)。
图3A-3C显示了AD患者中cf-mRNA的转录景观和基于基因集分析(gene-setanalysis)和功能注释的功能暗示。图3A显示了研究设计的示意图。图3B显示了AD(n=126)和NCI对照(n=115)之间cf-mRNA中差异表达的基因的火山图。FDR<0.05用作截止标准。图3C显示了使用基因集富集分析鉴定的最重要的通路(上图,上调基因;下图,下调基因)。黑色垂直虚线代表显著性阈值(p<0.05)。
图4A-4C显示了与AD相关的生物学过程和信号转导通路。图4A显示了通过IPA分析确定的AD的cf-mRNA中上调的基因作为输入的生物学过程(左)。通过IPA分析确定AD的cf-mRNA中下调的基因作为输入的最突出的生物学过程(右)。图4B显示了AD的cf-mRNA中下调的基因作为输入在神经系统发育和功能(IPA)中的子分类(subcategories)。图4C显示了由基因本体确定的AD的cf-mRNA中上调的基因作为输入的生物学过程(左)和由基因本体确定的AD的cf-mRNA中下调的基因作为输入的最突出的生物学过程(右)。
图5A-5C显示了cf-mRNA转录本与脑组织转录本和AD中失调的转录本显著重叠。图5A显示了AD的cf-mRNA中基因型-组织表达(GTEx)限定的大脑富集基因和下调基因之间的重叠(左),以及AD的cf-mRNA中GTEx限定的肝脏富集基因和下调基因之间的重叠(右)。P值显示了重叠基因数目与预期数目之间的比较。图5B显示了与NCI相比在AD的cf-mRNA中上调的基因与在AD患者的脑组织中上调的基因之间的重叠(左)。图5C显示了与NCI相比在AD的cf-mRNA中下调的基因与在AD患者的脑组织中下调的基因之间的重叠(左)。
图6A-6E说明了cf-mRNA分类器鲁棒性地区分AD与NCI。图6A示出了分类器建立的示意图。图6B示出了使用训练队列对分类准确度的评价。y轴描绘了各个算法的AUROC。图6C显示了用于鉴别AD与NCI的cf-mRNA分类器的ROC曲线(左)和鉴别AD与NCI的瀑布图(右)。图6D显示了用于区分AD与NCI的9基因微型分类器的ROC曲线。图6E显示了9个微型分类器基因的总队列(123个AD和114个NCI)中AD和NCI之间的读段计数。
图7A说明了CDR≤1的AD患者中1,496个失调基因的表达水平(FDR<0.05)。图7B显示了在“早期”AD患者中下调的基因主要在神经系统功能和发育过程(例如,Netrin信号转导、神经元中的CREB信号转导、钙转运和神经发生的调节)中富集和在免疫应答和蛋白稳态(例如,蛋白质泛素化、炎性体通路和免疫应答的激活)中上调的基因。
图8A-8G显示了cf-mRNA基因与认知损害的严重程度相关。图8A显示了一致性矩阵NMF聚类鉴定大小生物学上不同的聚类(size biologically distinct cluster)。来自2591个差异表达基因的无监督NMF聚类。图8B显示了按CDR等级分类的“突触传递”和“免疫和炎症应答”聚类的表达。图8C显示了基因的CDR和TPM的FDR(表示为-log)与皮尔逊相关系数之间的图。红色虚线代表FDR=0.05。图8D显示了使用与CDR评分相关的706个基因在IPA通路分析中鉴定的最重要的经典通路(top canonical pathway)。红色虚线代表FDR=0.05。图8E显示了基于CDR和MMSE评分(CDR评分(顶部)和MMSE(底部))的SLU7的表达。图8F显示了用于区分NCI(CDR=0)与CDR评分为0.5-1的cf-mRNA分类器的平均ROC曲线。进行15次交叉验证的迭代,并且曲线代表那些15条ROC曲线的平均值。图8G显示了使用基于图8A中鉴定的NMF聚类的AD患者的cf-mRNA谱的AD患者的无监督聚类。
图9A-9C显示了cf-mRNA基因针对认知损害评分的表达。图9A说明了使用ANOVA分析-图基事后检验(ANOVA analysis-Tukey’s post-hoc test)鉴定的5个患者组中5个AD患者子分类、年龄和MMSE分布中每一个的聚类值。图9B显示了基因的MMSE和TPM的FDR(表示为-log)与皮尔逊相关系数之间的图。红色虚线代表FDR=0.05。图9C显示了使用与MMSE评分相关的520个基因在IPA通路分析中鉴定的最重要的经典通路。红色虚线代表FDR=0.05。图9D显示了与MMSE和CDR评分相关的基因之间的重叠基因。
图10描述了与本文公开一致的计算机系统。
图11显示了每百万转录本(TPM)中TCF7按年龄组的差异表达。
图12显示了TPM中PTK2(衰老细胞中的粘着斑激酶)按年龄组的差异表达。
图13显示了TPM中FER按年龄组的差异表达。
图14显示了TPM中CD36按年龄组的差异表达。CD36是与年龄相关的GO0000302组合(panel)“对活性氧物质的应答”功能的18个基因之一。
图15显示了TPM中WWTR1按年龄组的差异表达。WWTR1在与YAP/TAZ复合物相关的Hippo通路中表达。WWTR1是40个与年龄相关的非血液基因之一。
图16显示了TPM中CAV1按年龄组的差异表达。CAV1是涉及细胞膜穴样内陷(caveolae)形成的小窝蛋白1(Caveolin 1)。CAV1是40个与年龄相关的非血液基因之一。
图17显示了年龄相关基因与其他数据集的比较。两个基因NELL2和LTB始终与年龄高度相关。
图18显示了与非血液基因重叠的41个年龄相关基因的表达的热图,p值为3.93e-11。
图19显示了使用GTEx数据的多种组织的年龄相关基因的图表。
具体实施方式
本文所述的方法、系统和试剂盒涉及使用标志物类型的组合来快速、非侵入性地检测病症,以便考虑到由个体自然衰老引起的基因表达的变化,同时确定可能的病症和可能的受胁迫组织。在一些实施方案中,将包含已知在对象年龄的个体中差异表达的基因的基因组合(gene panel)应用于对象的无细胞RNA(cfRNA)表达谱。通过本文公开的实践,可以预测疾病身份(disease identity)及其对一种或多种组织的影响程度,而无需对怀疑受影响的一种或多种组织进行侵入性研究。
需要开发一种可靠且无创的检测,以在早期准确诊断阿尔茨海默病。医师通常使用数字量表——临床痴呆评定量表(CDR)来量化神经变性病症的严重程度。此外,在临床和研究环境中使用简易精神状态检查量表(MMSE)或Folstein检验来测量认知损害。
在循环中,例如在血液样品中,疾病标志物的鉴定可以是允许鉴定患病组织而不需要侵入性操作例如活组织检查的有用工具。这对于对这种侵入性的、痛苦的过程弹性较小的老年人群是有用的。也可以考虑可能影响基因表达的疾病以外的因素。一些组织的基因表达随个体年龄而改变。鉴定与年龄相关的基因标志物以及它们如何差异表达可能是重要的,以便在诊断患病组织时考虑它们。
在此,通过在年龄匹配的AD患者和对照个体之间进行血浆cf-mRNA谱的转录组范围比较,概念验证(proof-of-concept)显示循环转录组具有以非侵入性方式揭示神经变性疾病如AD的分子和功能信息的潜力。本文公开了测定的技术性能,以及循环中数千个基因的检测和定量,以显示AD患者血浆中失调的基因可反映已知与认知损害和神经变性病症相关的生物过程和通路。例如,本文公开了AD患者中与神经系统功能和发育关联的多种通路(例如,突触损失、GABA信号转导和神经传递)的总体下降,伴随着炎症、线粒体功能障碍、氧化和蛋白稳态涉及的基因的水平升高。此外,发现AD患者血浆中失调的基因和生物过程基本上与死后脑活检标本的RNA-seq数据集中鉴定的那些重叠。血浆中的无细胞mRNA可以为AD患者中脑内稳态(brain homeostasis)的非侵入性分子评价的替代物。
受益于对AD中涉及的分子机制的更好理解的一个潜在应用是开发新的治疗策略。cf-mRNA测序可提供AD患者的循环转录组的颗粒表征,包括AD患者中失调的或与AD严重程度相关的数千个基因。除了在已知与AD相关的生物过程(例如,参与GABA信号转导的26个失调基因)上显示高分辨率之外,还观察到与AD患者中的神经发生相关的基因的水平降低,不受任何一种特定理论的束缚,这可以支持AD中成人神经发生被破坏的假设。此外,在AD患者中涉及RNA剪接的许多因子(例如SLU7)被鉴定为失调,其水平与疾病严重程度强烈相关。证据表明了可变RNA剪接在衰老和神经变性中的作用。观察到AD患者中Netrin信号转导的显著降低,包括NETRIN-1水平的显著降低,NETRIN-1结合APP并且已被提议作为Aβ水平的主要调节剂。NETRIN-1表达降低与Aβ浓度增加有关。整合的cf-mRNA技术方案可以提供更好地理解AD的异质性病因学的方法,并且可以帮助鉴定具有治疗潜力的新分子实体,并且增加它们在临床前和临床阶段的技术成功的可能性。
实际上,作为在其发作和进展期间影响多种生物通路和过程的复杂神经变性疾病,AD的异质性代表了AD药物开发的一个主要困难。迄今为止,靶向β-淀粉样蛋白和tau蛋白的治疗药物已经显示出适度的结果,因此靶向AD中通常受影响的通路(例如,炎症、线粒体功能障碍)的多种化合物和神经保护化合物目前正被开发并检验为AD治疗的替代物。用于异质性AD群体的治疗剂的成功开发可依赖于适当地丰集可能应答候选药物的AD患者的试验组的能力。由于基于脑活检的患者的分子表征通常是不可行的,因此能够预先选择最适合于每种治疗的患者的非侵入性工具可用于临床试验。本公开指示了由循环转录组揭示的分子信息可以为疾病相关过程的个性化表征铺平道路,因此使得能够更有效地管理患者并且提高干预成功的概率。此外,考虑到cf-mRNA能够“实时”监测器官健康和器官系统对治疗干预的应答,以及在循环中鉴定的AD相关过程的所有组成成分,cf-mRNA测序和临床信息的整合还可以允许监测AD患者中的治疗应答。
尽管尸检组织学仍然是建立AD病理学的金标准,但目前CSF、PET和MRI可用于诊断AD患者。然而,成像方式可能是昂贵的,并且CSF收集可能是侵入性的。因此,对于AD患者的管理,需要可扩展的、可访问的和有成本效益的基于血液的测试。迄今为止,几种基于蛋白质的血液生物标志物,包括测量Aβ肽的循环水平的那些,似乎是作为AD的诊断生物标志物的有希望的候选物,尽管考虑到Aβ也存在于无痴呆的个体中并且其水平不一致地预测认知衰退的速率,但并非没有限制。对cf-mRNA转录组进行图谱绘制代表了一种用于开发鉴定AD患者的分子分类器的非侵入性方法,如基于cf-mRNA的分类器鉴别对照个体与AD患者的性能所示。因此,cf-mRNA图谱绘制可以提供一种用于更个性化的患者管理的新方法,该方法将疾病状态的临床信息与对患者特异性分子特征的了解整合在一起以产生用于改善患者管理的解决方案。cf-mRNA图谱绘制可例如作为潜在工具辅助临床试验,用于鉴别患有或不患有AD的患者,减少需要Aβ-PET进行AD诊断的患者的数量,以及基于其分子特征对具有增加的应答治疗的可能性的患者进行分级。
本文提供了用于例如使用对象的生物样品评估或检测对象中的阿尔茨海默病(AD)的非侵入性方法、系统、组合物和试剂盒。该方法包括从生物样品中分离无细胞信使RNA(cf-mRNA)。在一些实施方案中,生物样品是血浆或血清。在其他实施方案中,生物样品是脑脊液(CSF)。
本文公开了AD和NCI之间血浆cf-mRNA谱的第一转录组范围比较,并鉴定了与AD不同的cf-mRNA特征。基因集富集分析显示了AD的cf-mRNA谱反映了AD中常失调的信号通路和生物学过程。此外,本文公开了与认知损害的严重程度相关的“免疫与炎症应答(immune&inflammatory response)”和“突触传递(synaptic transmission)”基因聚类。此外,与AD的另一属性神经元功能相关的基因在AD患者的cf-mRNA转录组中减弱。本文公开了与CDR和MMSE认知损害评分相关的一组基因,其中一些甚至在具有非常轻度至轻度认知损害的AD患者中与没有认知损害的那些相比具有显著的基因表达改变。本文还公开了一种分类器,其可以将具有适度认知损害的AD患者与不具有认知损害的正常对照区分开,指示循环中的转录变化可能适合作为AD的早期诊断工具。
该方法还可以使用预先离心以减少来自cf-mRNA测序数据的不需要的“血液”转录本的污染。本文的方法可以降低来自组织特异性cf-mRNA信号的“血液组分”血细胞内的背景噪声。这样的噪声可以增加测序深度要求并且稀释来自组织特异性cf-mRNA的信号。通过该纯化步骤,可以说cf-mRNA转录本很可能来源于对象的大脑。通过用“血液组分”转录本降低背景噪声,检测到的cf-mRNA转录本可能源自大脑。
通常,从对象收集血清、血浆或其他生物样品,并通过去除细胞碎片优化样品。在一些实施方案中,从远程位置处的对象收集样品并经由递送服务运送到测试点。一些对象是健康的,一些经历认知损害,以及一些被诊断患有AD。在某些情况下,样品可以富集非血液转录本。包括来自不同基因组来源,如端脑、小脑、背根神经节、颈上神经节、松果腺、杏仁核、三叉神经节、大脑皮质和下丘脑的遗传物质的混合物的cf-mRNA可以从优化的样品中分离。
可以使用宽范围的离心范围来优化样品,以便去除血液转录本。在某些情况下,范围可以包括1,500g至20,000g、1,900g至16,000g、4,000g至16,000g、8,000g至16,000g、10,000g至14,000g、11,000g至13,000g、11,500g至12,500g或合适的更低或更高的范围。在一些情况下,样品可以是约12,000g、大体上12,000g、基本上12,000g或12,000g的离心机。一些范围跨越约12,000g。一些范围在100g至12,000g之内。一些离心方案与12,000g没有显著差异,例如在12,000g离心。还考虑了具有在以上列出的低数字处的起点或在以上列出的高数字处结束的替代范围。这样的离心方案有助于用于加工的RNA文库的多样性的2.5x的改善。在各种情况下,离心方案可有助于用于加工的RNA文库的多样性的1.1x、1.2x、1.3x、1.4x、1.5x、1.6x、1.7x、1.8x、1.9x、2.0x、2.1x、2.2x、2.3x、2.4x、2.5x、2.6x、2.7x、2.8x、2.9x、3.0x、3.1x、3.2x、3.3x、3.4x、3.5x、3.6x、3.7x、3.8x、3.9x、4.0x或大于4.0x的改善。
此外,可基于分离的cf-mRNA转化cDNA以形成包括NGS文库的cDNA文库。例如,cDNA可以由cf-mRNA样品的逆转录产生。此外,可以富集cDNA用于定量。
构建cDNA文库后,许多方法可用于定量不同cDNA的水平。例如,多核苷酸扩增、测序、探针杂交、RT-PCR和微阵列杂交以及其他合适的方法可用于定量cDNA的水平。可以使用各种方法富集cDNA。例如,这些方法中的一些是基于与设计用于与不同cDNA杂交的寡核苷酸的杂交。杂交可以是与固定在高密度或低密度微阵列上的寡核苷酸的杂交或与用配体修饰的寡核苷酸的液相杂交,该配体随后可用于将杂交体固定到固体表面如珠子上。其他方法可以使用序列特异性扩增(例如,PCR)来扩增液滴中的特异性cDNA,允许扩增特异性cDNA用于下游测序。基于液滴的扩增可以实现高度多重PCR,而没有大量PCR引物对的潜在非特异性相互作用和随后的非特异性扩增产物的产生以及降低的cDNA的扩增效率。
此外,还可以使用微阵列技术鉴定或证实差异基因表达。在该方法中,可以将感兴趣的多核苷酸序列(包括cDNA和寡核苷酸)铺板或排列在微芯片基底上。然后可将阵列序列与来自感兴趣的细胞或组织的特异性DNA探针杂交。
此外,还可以使用测序技术鉴定或证实差异基因表达。感兴趣的多核苷酸序列(包括cDNA和寡核苷酸)可用作合成测序文库的模板。可以对文库进行测序,并将读段映射到适当的参考。示例性的测序技术可以包括,例如,乳化PCR、来自Roche 454的焦磷酸测序、来自Ion Torrent的半导体测序、来自Life Technologies的通过连接的SOLiD测序,来自Intelligent Biosystems的通过合成的测序、流动池上的桥式扩增(例如,Solexa/Illumina)、通过Wildfire技术的等温扩增(Life Technologies)或通过滚环扩增产生的卷状物/纳米球(完整基因组,Intelligent Biosystems,Polonator)。诸如Heliscope(Helicos)、SMRT技术(Pacific Biosciences)或纳米孔测序(Oxford Nanopore)的测序技术可以是合适的测序平台,其可以允许单个分子的直接测序而无需先前的克隆扩增。其他测序方法也在本公开的范围内。可以在有或没有靶标富集的情况下进行测序。此外,RT-PCR可用于定量不同的基因表达水平。通常,逆转录反应步骤可以使用特异性引物、随机六聚体或寡dT引物引发,这取决于表达图谱绘制的目标。逆转录酶可以是禽成髓细胞瘤病毒逆转录酶(AMV-RT)、莫洛尼鼠白血病病毒逆转录酶(MLV-RT)或其他合适的逆转录酶。
尽管PCR步骤可使用多种热稳定的DNA依赖性DNA聚合酶,但其通常使用Taq DNA聚合酶,其可具有5′-3′核酸酶活性但缺乏3′-5′校正核酸内切酶活性。因此,TaqManTM PCR通常利用Taq或Tth聚合酶的5'-核酸酶活性来水解与其靶标扩增子结合的杂交探针,但可使用具有等效5'核酸酶活性的任何合适的酶。两种寡核苷酸引物可用于产生PCR反应的典型扩增子。可以设计第三寡核苷酸或探针来检测位于两个PCR引物之间的核苷酸序列。探针可以是不能被Taq DNA聚合酶延伸的,并且可以用报告荧光染料和猝灭荧光染料(quencherfluorescent dye)标记。当两种染料靠近在一起时,例如当它们在探针上时,来自报告染料的任何激光诱导的发射都可以被淬灭染料淬灭。在扩增反应期间,Taq DNA聚合酶可以模板依赖性方式切割探针。所得探针片段可以在溶液中解离,并且来自释放的报告染料的信号可以从第二荧光团的猝灭效应中释放。对于合成的每个新分子,可以释放一个报告染料分子,并且未淬灭的报告染料的检测可以为数据的定量解释提供基础。
TaqManTM RT-PCR可使用市售设备进行,例如ABI PRISM7700TM序列检测系统TM(Perkin-Elmer-Applied Biosystems,Foster City,Calif.,USA)或Lightcycler(RocheMolecular Biochemicals,Mannheim,德国)。在某些实施方案中,5'核酸酶程序在实时定量PCR装置如ABI PRISM 7700TM序列检测系统TM上运行。该系统包括热循环仪、激光器、电荷耦合器件(CCD)、照相机和计算机。该系统包括用于运行仪器和分析数据的软件。5'-核酸酶测定数据(5'-nuclease assay data)最初可表示为Ct(阈值循环)。可以在每个循环期间记录荧光值,并且荧光值代表了扩增反应中该点的产物的量。当荧光信号首次记录为统计学显著时的点可以是阈值循环(Ct)。
差异表达基因组合
包含本文所述的多个差异表达的蛋白质编码基因的生物标志物组合(biomarkerpanel)可促进灵敏和非侵入性检验以检测对象是否患有AD或确定AD的临床发展阶段。阿尔茨海默病的临床发展阶段包括(1)临床前阿尔茨海默病、(2)由阿尔茨海默病引起的轻度认知损害、(3)由阿尔茨海默病引起的轻度痴呆、(4)由阿尔茨海默病引起的中度痴呆和(5)由阿尔茨海默病引起的重度痴呆。包含多个差异表达的蛋白质编码基因的生物标志物组合通常容易通过从个体抽血获得。使用本文公开的生物标志物组合的益处可以包括快速和方便地检测AD而无需繁琐和不可靠的检验。
可以选择如本文公开的生物标志物组合,使得它们作为组合的预测值显著大于它们单独成员的预测值。组合成员通常不会彼此共同变化,使得组合成员对组合的总体健康信号提供独立的贡献。生物标志物组合可包含AD患者血浆中失调的基因,以及与疾病严重程度相关的基因,其富含与AD相关的生物过程,例如,突触功能障碍、线粒体功能障碍和炎症。循环失调的基因可用于鉴定异质性群体患者中的AD患者子类型(subtype),并构建基于cf-mRNA的分类器,其从AD患者中鉴别(例如,鲁棒性地鉴别)年龄匹配的对照。无细胞mRNA生物标志物组合可以非侵入性地揭示与神经变性和AD相关的分子特征,并且支持将cf-mRNA与临床信息整合以潜在地改善AD患者管理的潜力,鉴定新的治疗靶标,并且对患者进行分级以增加治疗剂的研究和开发的技术成功的可能性。因此,一个组合能够基本上胜过指示个体AD状态的任何个体成分的表现,从而获得商业和医学上相关的置信度(例如,灵敏度、特异性或灵敏度和特异性)。
在一些情况下,组合成员彼此独立地变化。因此,尽管事实上如果单独测量,组合的一个或多个个体成员将不指示存在健康风险,但是本文的组合通常指示了健康风险。在其他情况下,本文的组合以显著的置信水平指示了健康风险,尽管事实上没有单独的组合成员自己以显著的置信水平指示了健康风险。在另外的情况下,本文的组合可以以显著的置信度水平指示了健康风险,尽管事实上至少一个个体成员以显著的置信度水平指示不存在健康风险。
一些生物标志物组合包含本文所述的差异表达的蛋白质编码基因中的一些或全部(参见表1A)。在一些情况下,生物标志物组合可以包含至少九种蛋白质编码基因。在一些情况下,生物标志物组合可以包含来自表1A的任意两种基因。在一些情况下,生物标志物组合可以包含来自表1A的任意三种基因。在一些情况下,生物标志物组合可以包含来自表1A的任意四种基因。在一些情况下,生物标志物组合可以包含来自表1A的任意五种基因。在一些情况下,生物标志物组合可以包含来自表1A的任意六种基因。在一些情况下,生物标志物组合可以包含来自表1A的任意七种基因。在一些情况下,生物标志物组合可以包含来自表1A的任意八种基因。在一些情况下,生物标志物组合可以包含来自表1A的九种基因。
表1A:差异表达的基因列表
此外,一些生物标志物组合可以包含本文所述的一些或全部差异表达的蛋白质编码基因(参见表1B)。在一些情况下,生物标志物组合可以包含至少14种蛋白质编码基因。在一些情况下,生物标志物组合可以包含来自表1B的任意两种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意三种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意四种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意五种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意六种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意七种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意八种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意九种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意十种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意十一种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意十二种基因。在一些情况下,生物标志物组合可以包含来自表1B的任意十三种基因。在一些情况下,生物标志物组合可以包含来自表1B的十四种基因。
表1B:额外的差异表达的基因列表
在构建各种生物标志物组合之后,如本文提供的非侵入性诊断方法中所述,生物标志物组合可用于确定对象是否患有AD。此外,生物标志物组合也可用于确定AD的特定发展阶段。通常,AD的不同发展阶段被分配CDR评分或MMSE评分。本文的一些方法包括将对象中生物标志物组合的水平与相同生物标志物组合的阈值水平进行比较。在一些情况下,生物标志物组合的阈值水平等于对照对象的生物标志物组合的水平。在一些情况下,对照对象是具有已知诊断的人。例如,对照对象可以是阴性对照对象。阴性对照对象可以是未患有AD的对象。对于其他实例,对照对象可以是阳性对照对象。阳性对照对象可以是确诊为AD的对象。阳性对照对象可以是确诊为AD的对象。此外,阳性对照对象可以是确诊AD的任何阶段的对象。例如,阳性对照对象的CDR评分可为0.5、1、2或3。阳性对照对象的MMSE评分可为1-6、6-12、12-18、18-24或24-30。阈值可以是生物标志物的预定水平,其中预定水平基于对照对象中生物标志物的测量量来设定。
本文描述的用于检测对象中的AD的诊断方法可以以大于75%、大于80%、大于85%、大于90%、大于95%、大于96%、大于97%、大于98%、大于99%或约100%的灵敏度检测AD。这类诊断方法可以以70%至100%、80%至100%或90%至100%的灵敏度检测阿尔茨海默病(AD)。这类诊断方法可以以大于70%、大于75%、大于80%、大于85%、大于90%、大于95%、大于96%、大于97%、大于98%、大于99%或约100%的特异性检测AD。这类诊断方法可以以50%至100%、60%至100%、70%至100%、80%至100%或90%至100%的特异性检测AD。在各种实施方案中,这类诊断方法可以以50%或更高、60%或更高、70%或更高、75%或更高、80%或更高、85%或更高、或90%或更高的灵敏度和特异性检测AD。在某些实施方案中,这类诊断方法可以以50%至100%、60%至100%、70%至100%、80%至100%或90%至100%的灵敏度和特异性检测AD。
分类器
可以使用许多不同的技术来开发分类器。例如,计算机系统可用于开发和生成分类器。从多个差异表达的蛋白质编码基因收集的数据(例如cf-mRNA水平)可用于训练机器学习算法以获得分类器。
机器学习可以概括为学习机器在经历了学习数据集之后对新的、未见过的实例/任务精确执行的能力。机器学习可以包括本文提供的概念和方法。监督学习概念可以包括:AODE;人工神经网络,例如,反向传播算法(Backpropagation)、自编码算法(Autoencoders)、霍普菲尔德网络(Hopfield networks)、波尔兹曼机(BoltzmannMachine)、有限波尔兹曼机(Restricted Boltzmann Machines)和Spiking神经网络;贝叶斯统计,例如,贝叶斯网络和贝叶斯知识库;案例推理;高斯过程回归;基因表达编程;成组数据处理法(GMDH);归纳逻辑编程;基于实例的学习;懒惰学习;学习自动机;学习向量量化;逻辑模型树;最小消息长度(决策树、决策图等),例如,最近邻算法和模拟建模;概率近似正确学习(PAC,Probably approximately correct learning)学习;涟波下降规则(Ripple down rule),一种知识获取方法;符号机器学习算法;支持向量机(SVM);随机森林;分类器集成,例如,自举汇聚法(装袋法)和提升法(元算法);有序分类(Ordinalclassification);信息模糊网络(IFN);条件随机场(Conditional Random Field);ANOVA;线性分类器,例如,Fisher线性判别式、线性回归、逻辑回归、多项式逻辑回归、朴素贝叶斯分类器、Perceptron、支持向量机;二次分类器;k最近邻(k-nearest neighbors);提升法(Boosting);具有L1正则化(LASSO)的逻辑回归;具有L2正则化的逻辑回归(岭回归分类器);决策树,例如,C4.5、随机森林、ID3、CART、SLIQ、SPRINT;贝叶斯网络,例如,朴素贝叶斯;以及隐马尔可夫模型(Hidden Markov models)。无监督学习概念可以包括:期望最大化算法;矢量量化;生成地形图(Generative topographic map);信息瓶颈方法;人工神经网络,例如,自组织映射(Self-organizing map);关联规则学习,例如,Apriori算法、Eclat算法和FP-growth算法;层次聚类(Hierarchical clustering),例如,单链接聚类和概念聚类;聚类分析,例如,K均值算法、模糊聚类DBSCAN和OPTICS算法;以及离群值检测,例如,局部离群因子(例如,)。半监督学习概念可以包括:生成模型、低密度分离、Graphbased方法和共训练。强化学习概念可以包括:时间差分学习(Temporal difference learning)、Q学习、学习自动机机(Learning Automata)和SARSA。深度学习概念可以包括:深度信念网络、深度玻尔兹曼机、深度卷积神经网络、深度递归神经网络和分层时间记忆。
在一些情况下,在一些情况下通过ROC的AUC评估分类器的性能。ROC考虑了分类器在所有可能的模型评分截止点处的性能。然而,当需要做出分类决定时(例如,该患者是否生病或健康?),使用截止点定义两组。在各种实施方案中,在截止点或高于截止点的分类评分被评估为阳性(或患病),而低于截止点的分类评分被评估为阴性(或健康)。
对于本文公开的一些分类模型,通过选择验证ROC上的最大准确度点来建立分类评分截止点。ROC上的最大准确度点是正确分类呼叫的总数被最大化的一个或多个截止点。这里,正和负分类调用(positive and negative classification calls)被相等地加权。在给定ROC上存在多个最大准确度点的情况下,可以选择具有相关最大灵敏度的点。
临床结果评分
用于子集选择(sub-selecting)鉴别生物标志物和/或对象特征以及用于构建分类模型的机器学习算法在本文的一些方法和系统中用于确定临床结果评分。这些算法包括但不限于弹性网络、随机森林、支持向量机和逻辑回归。这些算法可以帮助选择重要的生物标志物特征,并将潜在的测量转换为与例如临床结果、疾病风险、疾病可能性、疾病存在与否、治疗响应和/或疾病状态分类相关的评分或概率。
可通过将定量的cf-mRNA水平输入本文所述的分类器来产生临床结果评分。此外,通过将对应于从对象获得的生物样品中至少两种差异表达的基因的cf-mRNA水平与这两种基因的参考cf-mRNA水平进行比较来确定临床结果评分。可替代地或组合地,通过将一组对应于差异表达的基因的cf-mRNA水平的对象特异性谱与差异表达的基因的参考谱进行比较来确定临床结果评分。通常,参考水平或参考谱代表了已知的诊断。例如,参考水平或参考谱代表了AD的阳性诊断。作为另一个实例,参考水平或参考谱代表了AD的阴性诊断。类似地,参考水平或参考谱代表了与CDR或MMSE相关的特定评分。
在一些情况下,评分的增加指示了以下中的一项或多项的可能性增加:不良的临床结果、良好的临床结果、高疾病风险、低疾病风险、完全应答、部分应答、稳定的疾病、无应答和用于疾病管理的推荐治疗(或多种治疗)。在一些情况下,定量评分的降低指示了以下一项或多项的可能性增加:不良的临床结果、良好的临床结果、高疾病风险、低疾病风险、完全应答、部分应答、稳定的疾病、无应答和用于疾病管理的推荐治疗(或多种治疗)。此外,在一些实施方案中,评分的增加指示了更高的CDR或MMSE评分。
从患者到参考谱的类似谱通常指示了以下一项或多项的可能性增加:不良的临床结果、良好的临床结果、高疾病风险、低疾病风险、完全应答、部分应答、稳定的疾病、无应答和用于疾病管理的推荐治疗(或多种治疗)。在一些应用中,从患者到参考谱的不同生物标志物谱可以指示以下的一项或多项:不良的临床结果、良好的临床结果、高疾病风险、低疾病风险、完全应答、部分应答、稳定的疾病、无应答和用于疾病管理的推荐治疗(或多种治疗)。
对应于一种或多种差异表达的基因的cf-mRNA水平的阈值增加通常指示了以下一种或多种的可能性增加:不良的临床结果、良好的临床结果、高疾病风险、低疾病风险、完全应答、部分应答、稳定的疾病、无应答和用于疾病管理的推荐治疗(或多种治疗)。在一些应用中,一种或多种生物标志物阈值的降低可以指示以下一种或多种的可能性增加:不良的临床结果、良好的临床结果、高疾病风险、低疾病风险、完全应答、部分应答、稳定的疾病、无应答和用于疾病管理的推荐治疗(或多种治疗)。
定量评分、一个或多个阈值或类似生物标志物谱值中的至少一个的增加指示了以下一项或多项的可能性增加:不良的临床结果、良好的临床结果、高疾病风险、低疾病风险、完全应答、部分应答、稳定的疾病、无应答和用于疾病管理的推荐治疗(或多种治疗)。类似地,定量评分、一个或多个生物标志物阈值、类似的生物标志物谱值或其组合中的至少一个的降低指示了以下一项或多项的可能性增加:不良的临床结果、良好的临床结果、高疾病风险、低疾病风险、完全应答、部分应答、稳定的疾病、无应答和用于疾病管理的推荐治疗(或多种治疗)。
治疗和监测方案
本文提供了用于实施本文所述的用于检测AD的存在或不存在和/或AD的治疗的任何方法的诊断、监测和治疗方案。
例如,可以执行简易精神状态检查量表(MMSE)来评估对象大脑中涉及学习、记忆、思考或计划技能的区域是否存在问题。可替代地或另外地,计算机断层扫描(CT)可用于监测阿尔茨海默病后期常见的大脑变化。类似地,磁共振成像(MRI)、CSF和PET可有助于测量淀粉样标志物以监测与AD相关的大脑变化。可替代地或另外地,可以进行神经心理学测试以监测大脑和行为之间的关系。神经心理学测试可以帮助诊断影响思维、情绪和行为的病症,包括AD。
这里也考虑了许多治疗方法。不同类型的药物可治疗记忆丧失、行为改变、睡眠问题和其他AD症状。例如,西酞普兰、氟西汀、帕罗西汀和舍曲林可用于治疗AD患者经历的情绪、抑郁和易怒问题。阿普唑仑、丁螺环酮、iorazepam和奥沙西泮可用于治疗与AD相关的焦虑或躁动。可替代地或另外地,可施用胆碱酯酶抑制剂和/或美金刚胺以减轻与AD相关的症状。此外,非常规疗法,例如激素替代疗法、艺术和音乐疗法以及补充剂(例如,维生素E)可替代地或另外地用于治疗AD。
本文公开的方法、系统和试剂盒可旨在非侵入性地检测受胁迫的对象中的组织或器官,以及确定哪些疾病或病症影响受胁迫的组织或器官。在一些情况下,该方法、系统和试剂盒可提供用于治疗对象的疾病或病症。本文公开的一些方法可包括选择用于治疗对象的疾病或病症的方法或疗法。本文公开的一些试剂盒和系统可以提供选择用于治疗对象的疾病或病症的方法或疗法。本文公开的一些方法包括监测对象中的疾病或病症或进行疾病或病症的测试。本文公开的一些试剂盒和系统提供了监测对象的疾病或病症或进行疾病或病症的测试。本文公开的一些方法包括治疗对象的疾病或病症、监测对象的疾病或病症或进行疾病或病症的测试。在一些情况下,本文公开的方法包括确定对象患有疾病或病症,从而告知对象或其健康护理提供者治疗或测试将对对象是适当的、合适的或有益的。在一些情况下,本文公开的方法包括确定对象患有疾病或病症并推荐对该疾病或病症的治疗。在一些情况下,本文公开的方法包括确定对象患有疾病或病症并治疗对象的疾病或病症。在一些情况下,本文公开的方法包括确定对象患有疾病或病症并监测对象的疾病或病症。在一些情况下,本文公开的方法包括确定对象相对于不具有疾病或病症的相同年龄范围内的个体具有增加的患有疾病或病症的风险或可能性,以及进行针对对象的疾病或病症的测试。在一些情况下,本文公开的方法包括确定对象相对于不具有疾病或病症的相同年龄范围内的个体具有疾病或病症的增加的风险或可能性,以及推荐针对对象的疾病或病症的测试。
本文提供了用于治疗疾病和病症的治疗剂、组合物、化合物和药剂。即使没有明确描述每种组合和类似物,这些药剂的组合和类似物也是本文设想和预期的。本文所用的“类似物(analog)”通常是指类似于天然存在的化合物的修饰的或合成的化合物,其中至少50%的类似物结构与至少50%的天然存在的化合物相同。
可以在疾病的早期以更高的准确度确定对象中的疾病存在和位置,因为本文所述的系统和方法提供了快速结果,考虑了随年龄的基因表达变化,并且是非侵入性的和廉价的。因此,在疾病进展至与早期相比相对更难以控制或治疗的晚期之前,可以有利地治疗对象。例如,本文公开的系统和方法可以允许确定哪些组织或器官在症状发作之前显示出神经变性的迹象。这样,本文公开的方法和系统可以在疾病的早期阶段提供重点分析(focused analysis)和靶向治疗。
该方法和系统可以提供用对于组织损伤的程度合适或最佳的疗法来治疗对象。在一些情况下,该方法可包括检测标志物和/或组织特异性多核苷酸以评估治疗的有效性或毒性。在某些情况下,该方法可包括定量标志物和/或组织特异性多核苷酸以评估治疗的有效性或毒性。在一些情况下,继续该治疗。在各种情况下,中断该治疗。在某些情况下,用另一种治疗代替该治疗。无论如何,由于方法和系统的快速和非侵入性性质,相对于常规治疗优化,可以更频繁地评估和优化治疗效果。
在一些方面,本公开提供了本文公开的系统、样品、标志物和组织特异性多核苷酸的用途。在一些情况下,本文公开了体外样品用于非侵入性检测受胁迫的对象的组织或器官以及导致胁迫的疾病或病症的用途。在一些情况下,本文公开了离体样品用于通过将基因表达数据与年龄依赖性表达对照进行比较来非侵入性地检测受胁迫的对象的组织或器官以及导致胁迫的疾病或病症的用途。通常,本文公开的用途包括定量样品中的标志物和组织特异性多核苷酸,样品包括离体样品和体外样品。本文公开的一些用途包括比较第一样品中标志物的量和组织特异性多核苷酸的量,并将该量与第二样品中的相应量进行比较。在一些情况下,第一样品来自第一对象,第二样品来自对照对象(例如,健康对象或患有其中对象与第一对象年龄范围相同的病症的对象)。在一些情况下,第一样品在第一时间点来自对象,第二样品在第二时间点来自同一对象。第一时间点可以在对对象进行治疗之前获得,而第二时间点可以在治疗之后获得。因此,本文还提供了本文公开的样品、标志物、组织特异性多核苷酸、试剂盒和系统以监测或评价对象的病症、对象的组织健康状态或治疗剂的效果的用途。
在一些方面,本公开提供了监测患有慢性病症的人类对象的至少一种组织的至少一种并发症的存在的方法。在一些方面,本公开提供了监测患有慢性病症的人类对象的至少一种组织的至少一种并发症的增加的风险的方法。
一些方法包括监测人类对象的至少三种组织中的任一种的并发症。一些方法包括监测人类对象在至少三种组织的任一种中的并发症的增加的风险。
本文公开的基因表达组合可具有这样的性质,即,使用来源于循环血液的cfRNA表达水平信息结合个体年龄的知识得出关于个体组织疾病状态的敏感性、特异性结论。本发明的基因标志物组合的益处是它们使用方便地、非侵入性获得的样品提供灵敏的、特异性的组织健康评估。可能不需要依赖于从侵入式活检获得的额外数据。因此,顺应性率可以显著更高,并且组织健康问题在其进展早期更容易被识别,使得它们可以被更有效地治疗。
细胞类型和组织类型特异性多核苷酸
本文提供了使用本文公开的细胞类型特异性基因表达、细胞类型特异性核酸(例如,RNA)和细胞类型特异性核酸修饰(例如,甲基化模式)的试剂盒、装置、系统和方法。术语“细胞类型特异性核酸”、“细胞类型特异性多核苷酸”、“组织特异性核酸”和“组织特异性多核苷酸”在本文中可互换使用。术语“细胞类型特异性”可用于表征在对象的单个组织中表达的核酸。可选地,术语“细胞类型特异性”可用于表征主要在本文公开的特定细胞功能或信号转导通路中表达的核酸。细胞功能或通路可包括神经炎症、免疫应答、缺氧信号转导、一氧化氮产生、系统性红斑狼疮信号转导、toll样受体信号转导、NG-kappaB信号转导、炎性体通路、线粒体功能障碍、蛋白质泛素化等。为了本申请的目的,主要表达可意指组织特异性核酸在特异性的组织中以比组织特异性核酸在对象的任何其他组织中的RNA水平高至少50%的RNA水平表达。然而,在一些情况下,在特异性的组织中以比任何其他组织高至少30%的RNA水平表达的组织特异性核酸可足以用于本文公开的方法。在其他情况下,本文公开的方法可能需要在特异性的组织中以比任何其他组织高至少80%的RNA水平表达的组织特异性核酸。主要表达可以指组织特异性核酸在感兴趣的特异性的组织中的RNA水平比组织特异性核酸在对象的任何其他组织中的RNA水平高至少2倍。主要表达可以指组织特异性核酸在感兴趣的特异性的组织中的RNA水平比组织特异性核酸在对象的任何其他组织中的RNA水平高至少5倍。主要表达可以指组织特异性核酸在感兴趣的特异性的组织中的RNA水平比组织特异性核酸在对象的任何其他组织中的RNA水平高至少10倍。主要表达可以指仅当对组织特异性核酸主要表达的特异性的组织发生损伤时,可检测量的组织特异性核酸将出现在对象的生物流体(例如,血浆)中。
本文提供了用于检测或定量来自对象的样品中的生物分子的试剂盒、系统和方法,包括作为非限制性实例的多核苷酸、肽/蛋白质、脂质和甾醇。本文公开的生物分子可以是组织特异性的。本文所用的术语“组织特异性(tissue-specific)”通常是指生物分子或其修饰物,其在对象的单个组织中比在任何其他组织中以更高水平表达。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少10%。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少20%。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少30%。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少40%。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少50%。因此,可以认为组织特异性生物分子主要存在于或主要表达于单个组织中。本文公开的组织特异性生物分子可以是组织特异性多核苷酸。组织特异性多核苷酸是以组织特异性方式表达或修饰的核酸。例如,可以仅存在单个组织或器官或一小组组织或器官主要负责特定基因的表达(例如,对象中基因总表达的至少60%、70%、80%、90%、95%或更多)。
本文提供了用于检测或定量样品中的组织特异性多核苷酸的试剂盒、系统和方法。至少一个遗传信息数据库可用于鉴定组织特异性多核苷酸或一组组织特异性多核苷酸。因此,本公开的各方面提供了用于使用和开发数据库的系统和方法。本公开的方法可以利用包含跨组织类型产生的现有数据的数据库来鉴定组织特异性基因。这种数据库可用于鉴定组织特异性基因。数据库可以是基于网络的基因表达谱。基于网络的基因表达库的非限制性实例是公众可获得的,例如www_proteinatlas_org的人蛋白质图谱,biogps_org的BioGPS和www_ebi_ac_uk/gxa/的欧洲生物信息学研究所表达图谱,ncbi_nlm_nih_gov/geo/的高通量基因表达(Gene Expression Omnnibus,GEO),所有这些文献的内容通过引用并入本文。这样的数据库也可作为印刷和在线期刊中的出版文章公开可用。数据库还可以包括图谱,例如,人133A/GNF1H基因图谱(参见,Su等人,Proc Natl Acad Sci U S A,2004,第101卷,第6062-7页,原始出版物)和RNA-seq图谱(参见,Krupp等人,Bioinformatics,2012,第15卷,第1184-5页,原始出版物),两者均通过引用并入本文。这些数据库和网站结合了来自许多独立研究的数据,并且常常确证物种之间的组织特异性基因表达模式。这种交叉验证可以为本文公开的方法、系统和试剂盒提供有用的组织特异性多核苷酸。在一些情况下,本文公开的组织特异性多核苷酸通过至少两个公开的数据集被鉴定为具有组织特异性表达。在一些情况下,本文公开的组织特异性多核苷酸通过至少三个公开的数据集被鉴定为具有组织特异性表达。在一些情况下,本文公开的组织特异性多核苷酸通过至少四个公开的数据集被鉴定为具有组织特异性表达。在一些情况下,本文公开的组织特异性多核苷酸通过至少五个公开的数据集被鉴定为具有组织特异性表达。为了从至少一个数据库中鉴定组织特异性转录本,某些实施方案对数据库使用模板匹配算法。可以使用用于过滤数据的模板匹配算法,参见例如Pavlidis P,Noble WS(2001)Analysis of strain andregional variation in gene expression in mouse brain.Genome Biol 2:research0042.1-0042.15。组织特异性基因的实例包括在US20130252835的图18中出现的那些,其通过引用并入本文。
本文提供了用于检测或定量样品中的组织特异性多核苷酸的试剂盒、系统和方法。组织特异性核酸可以指在对象群体中每个对象的单个组织中表达的核酸。组织特异性核酸可以指主要在对象群体中的每个对象的特异性的组织中表达的核酸。对象群体可能是健康的。对象群体可能患有常见的疾病或病症。对象群体可以包括两名对象。对象群体可以包括五名对象。对象群体可以包括十名对象。对象群体可以包括二十名对象。对象群体可以具有共同的种族、共同的遗传背景、共同的性别、共同的年龄或其组合。组织特异性核酸可指在单一组织中表达或主要在特异性的组织中表达的核酸,如公开的研究或数据库所示。公开的研究可采用微阵列技术或RNA-seq分布来测量组织特异性核酸水平。在一些情况下,特异性的组织的损伤由导致特异性的组织中的细胞凋亡的疾病或病症引起,从而将无细胞组织特异性核酸释放到对象的循环流体中。组织特异性核酸可以是在特异性的组织中足够高表达的核酸,当发生对特异性的组织的损伤时,可以在循环生物流体(例如,血液、血浆)中检测到该核酸。组织特异性核酸可以是在特异性的组织中足够高表达的核酸,当至少10%、至少20%、至少30%、至少40%或至少50%的特异性的组织发生损伤时,可以在循环生物流体(例如,血液、血浆)中检测到该核酸。
本文公开了用于检测、定量和/或分析组织特异性多核苷酸的方法、试剂盒和系统。通常,组织特异性多核苷酸是在细胞、组织或器官损伤或伤害后释放到生物流体(例如,血液、脑脊液、淋巴液和尿液)中的无细胞多核苷酸。如本文所用,对细胞、组织或器官的损伤或伤害可归因于导致细胞膜破坏或细胞或组织或器官表面内或表面上的至少一种细胞的细胞膜完整性丧失的疾病或病症。细胞膜的破坏或细胞膜完整性的丧失可导致细胞内多核苷酸的释放。细胞膜的破坏可能是由于例如坏死、自溶或凋亡。组织特异性多核苷酸的非限制性实例包括组织特异性RNA和包含组织特异性甲基化模式的DNA。组织特异性RNA可包括但不限于信使RNA(mRNA)、微小RNA(miRNA)、前体miRNA(pre-miRNA)、初级miRNA(pri-miRNA)、前体mRNA(pre-mRNA)、环状RNA(circRNA)、长非编码RNA(lncRNA)和外泌体RNA(exosomal RNA)。本文提供了具有组织特异性表达的基因的实例。
本文提供了用于检测或定量来自对象的样品中的生物分子的试剂盒、系统和方法。本文公开的生物分子可以是组织特异性的。本文所用的术语“组织特异性(tissue-specific)”通常是指生物分子或其修饰物,其在对象的单个组织中比在任何其他组织中以更高水平表达。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少10%。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少20%。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少30%。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少40%。在一些情况下,其在单个组织中的表达比在对象的任何其他组织中的表达高至少50%。因此,可以认为组织特异性生物分子主要存在于或主要表达于单个组织中。本文公开的组织特异性生物分子可以是组织特异性多核苷酸。组织特异性多核苷酸是以组织特异性方式表达或修饰的核酸。例如,可以仅存在单个组织或器官或一小组组织或器官主要负责特定基因的表达(例如,对象中基因总表达的至少60%、70%、80%、90%、95%或更多)。
在一些情况下,本文公开的方法包括单个组织特异性多核苷酸的水平与组织特异性多核苷酸的相应参考水平的比较足以确定组织是否已被疾病或病症损伤。在其他情况下,可将多种组织特异性多核苷酸的水平与组织特异性多核苷酸的相应参考水平进行比较,以确定组织是否已被疾病或病症损伤。本文公开的方法可以包括将少至1、2、3、4、5、6、7、8、9或10个组织特异性多核苷酸的水平与相应的参考水平进行比较以确定组织是否已被疾病或病症损伤。将少至1、2或3个组织特异性多核苷酸与相应的参考水平进行比较可能是有利的。
在一些情况下,本文公开的将组织特异性多核苷酸的水平与组织特异性多核苷酸的相应参考水平进行比较的方法可确定组织特异性多核苷酸的水平高于相应参考水平。在一些情况下,相应的参考水平是健康个体中组织特异性多核苷酸的水平,并且组织特异性多核苷酸的水平高于相应的参考水平指示对象中特异性的组织、器官或细胞的损伤或伤害。组织特异性多核苷酸的水平可以比相应的参考水平高至少5%、至少10%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%、至少80%、至少90%、至少100%、至少150%或至少200%。
在一些情况下,本文公开的将组织特异性多核苷酸的水平与组织特异性多核苷酸的相应参考水平进行比较的方法可确定组织特异性多核苷酸的水平低于相应参考水平。在一些情况下,相应的参考水平是患有疾病或病症的个体或群体中组织特异性多核苷酸的水平,并且组织特异性多核苷酸的水平低于相应的参考水平指示对对象中的特异性的组织、器官或细胞不存在损伤或伤害的最小量。组织特异性多核苷酸的水平可以比相应的参考水平低至少5%、至少10%、至少20%、至少30%、至少40%、至少50%、至少60%、至少70%、至少80%、至少90%或至少95%。
本文公开的组织特异性多核苷酸可描述为“对应于基因”。在一些情况下,短语“对应于基因”意指组织特异性多核苷酸从基因转录。因此,在一些情况下,组织特异性多核苷酸是组织特异性RNA转录本。组织特异性RNA转录本包括全长转录本、转录本片段、转录本剪接变体、酶或化学切割的转录本、来自两个或多个融合基因的转录本和来自突变基因的转录本。片段和切割的转录本必须保留足够的全长多核苷酸以被识别为对应于该基因。在一些情况下,5%的全长多核苷酸是足够的全长多核苷酸。在一些情况下,10%的全长多核苷酸是足够的全长多核苷酸。在一些情况下,15%的全长多核苷酸是足够的全长多核苷酸。在一些情况下,20%的全长多核苷酸是足够的全长多核苷酸。在一些情况下,25%的全长多核苷酸是足够的全长多核苷酸。在一些情况下,30%的全长多核苷酸是足够的全长多核苷酸。在一些情况下,40%的全长多核苷酸是足够的全长多核苷酸。在一些情况下,50%的全长多核苷酸是足够的全长多核苷酸。在一些情况下,短语“对应于基因”意指组织特异性多核苷酸是基因的修饰形式(例如,组织特异性DNA修饰模式)。
分离、定量和检测
通常,本文公开的方法包括检测或定量本文公开的疾病或病症的标志物的量以确定对象受相应疾病或病症影响或对象处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少1个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少5个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少10个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少15个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少20个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少25个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少30个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少40个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少50个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。在一些情况下,检测或定量至少100个拷贝/ml的标志物足以确定对象受相应疾病或病症影响或处于受相应疾病或病症影响的风险中。
通常,本文公开的方法包括检测或定量本文公开的组织特异性多核苷酸的量,以确定相应的组织正受疾病或病症影响。在一些情况下,方法包括检测或定量至少1个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少5个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少10个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少15个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少20个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少25个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少30个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少35个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少40个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少45个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少50个拷贝/ml的组织特异性多核苷酸。在一些情况下,方法包括检测或定量至少100个拷贝/ml的组织特异性多核苷酸。
本文公开的一些方法包括检测或定量至少一定量的标志物或组织特异性多核苷酸,以确定疾病或病症正在影响相应的组织。在一些情况下,其中标志物是多核苷酸或组织特异性多核苷酸的标志物的量为至少1个拷贝/mL、至少10个拷贝/mL、至少20个拷贝/mL、至少30个拷贝/mL、至少40个拷贝/mL或至少50个拷贝/mL、至少80个拷贝/细胞、至少100个拷贝/细胞、至少120个拷贝/细胞、至少150个拷贝/细胞或至少200个拷贝/细胞。在一些情况下,标志物是蛋白质、脂质或其他非多核苷酸生物分子的标志物的量为至少5pg/mL、至少10pg/mL、至少20pg/mL、至少30pg/mL、至少50pg/mL、至少60pg/mL、至少80pg/mL、至少100pg/mL、至少150pg/mL、至少200pg/mL或至少500pg/mL。
如以上和以下描述中所讨论的,本文公开的方法和系统旨在通过检测、定量或以其他方式分析本文公开的至少一种标志物和至少一种组织特异性多核苷酸来非侵入性地检测受胁迫的对象中的组织或器官,以及确定哪些疾病或病症正在影响受胁迫的组织或器官。在一些情况下,至少一种标志物包含多核苷酸(例如,无细胞多核苷酸)或多肽。一些方法包括通过使多核苷酸或多肽与至少一种探针接触来检测该多核苷酸或多肽。在一些情况下,至少一种探针仅能够结合野生型多核苷酸或多肽。在一些情况下,至少一种探针仅能够结合多核苷酸或多肽的突变形式。在一些情况下,例如,其中标志物是多核苷酸,检测包括测序。
本文公开的一些方法包括分离至少一种标志物和/或至少一种组织特异性多核苷酸。在一些情况下,至少一种标志物和/或至少一种组织特异性多核苷酸包含无细胞多核苷酸。在一些情况下,分离无细胞多核苷酸包括将来自对象的样品分级分离。一些方法包括从样品中去除完整细胞。例如,一些方法包括离心血液样品并收集上清液即血清或血浆或过滤样品以去除细胞。在一些实施方案中,分析无细胞多核苷酸而不分级分离来自对象的样品。例如,尿液、脑脊髓液或其他含有很少或不含细胞的液体可能不需要分级分离。一些方法包括充分纯化无细胞多核苷酸以检测、定量和/或分析无细胞多核苷酸。各种试剂、方法和试剂盒可用于纯化无细胞多核苷酸。试剂可以包括但不限于Trizol、苯酚-氯仿、糖原、碘化钠和胍树脂。试剂盒包括但不限于Thermo Fisher
本文公开的一些方法包括富集样品中的无细胞多核苷酸。例如,感兴趣的样品可以含有来自细菌的RNA/DNA。一些方法包括exomal捕获,从而消除不需要的序列并富集样品中的感兴趣的多核苷酸。在一些情况下,exomal捕获包括基于阵列的捕获或溶液中捕获,分别对应于拴系到表面或珠子上的感兴趣的RNA的DNA片段。一些方法还包括从样品中过滤或去除其他生物分子或细胞如蛋白质或血小板。在一些情况下,富集样品中的无细胞多核苷酸包括防止血浆样品的血细胞RNA污染。在一些情况下,使用不含EDTA的管子防止或减少了血浆/血清样品中血细胞RNA的存在。
通常,本文公开的方法包括检测或定量至少一种标志物和/或至少一种组织特异性多核苷酸。在一些情况下,定量和/或检测至少一种标志物和/或至少一种组织特异性多核苷酸包括扩增至少一种标志物和/或至少一种组织特异性多核苷酸。在涉及无细胞RNA的一些情况下,定量和/或检测至少一种标志物和/或至少一种组织特异性多核苷酸包括逆转录无细胞RNA。可使用多种方法中的任一种来检测和/或定量样品中的标志物或组织特异性多核苷酸。在涉及无细胞、组织特异性RNA的一些情况下,从样品中分离RNA,并在进一步操作如扩增和/或测序之前逆转录产生cDNA。在一些实施方案中,扩增在3'端开始,并且随机地遍及样品中的整个转录组以允许mRNA和非多腺苷酸化转录本两者的扩增。用于扩增cDNA的合适试剂盒包括例如
本文公开的一些方法包括定量本文所述的至少一种标志物和/或至少一种组织特异性多核苷酸。在一些情况下,定量可用于确定病症的严重程度。例如,一些方法包括将标志物和/或组织特异性多核苷酸的量与对象中第一时间的第一样品中标志物和/或组织特异性多核苷酸的量进行比较,并在第二时间定量第二样品中的标志物和/或组织特异性多核苷酸,其中对象在第一时间和第二时间之间接受治疗。一些方法包括基于由量化产生的信息来维持治疗或改变治疗(例如,类型、剂量)。一些方法包括在额外时间定量额外样品中的标志物和/或组织特异性多核苷酸,在该额外时间之间调整治疗。
本文公开的定量核酸的一些方法包括对至少一种核酸进行测序。测序可以是靶向测序。在一些情况下,靶向测序包括特异性扩增本文公开的选定标志物或选定组织特异性多核苷酸并对扩增产物进行测序。在一些情况下,靶向测序包括特异性扩增本文公开的选定标志物的子集或选定组织特异性多核苷酸的子集,并对扩增产物进行测序。可选地,包括靶向测序的一些方法不包括扩增标志物或组织特异性多核苷酸。一些方法包括非靶向测序。在一些情况下,非靶向测序包括对扩增产物进行测序,其中一部分无细胞核酸不是标志物或组织特异性多核苷酸。在一些情况下,非靶向测序包括扩增来自对象的样品中的无细胞核酸并对扩增产物进行测序,其中一部分无细胞核酸不是标志物或组织特异性多核苷酸。在一些情况下,非靶向测序包括扩增包含本文所述的标志物或组织特异性多核苷酸的无细胞核酸。测序可提供对应于标志物或组织特异性多核苷酸的相对数量的多个读段(read)。在一些情况下,测序提供了对应于标志物或组织特异性多核苷酸的绝对数量的多个读段。在一些实施方案中,通过全转录组鸟枪法测序(也称为“RNA-seq”)对扩增的cDNA进行测序。全转录组鸟枪法测序(RNA-seq)可以使用多种下一代测序平台来完成,例如Illumina基因组分析平台(Illumina Genome Analyzer platform)、ABI Solid测序平台或Life Science的454测序平台。在一些情况下,通过微阵列,例如肽阵列或寡核苷酸阵列进行特异性靶标的鉴定,其中可寻址结合元件(addressable binding element)的阵列特异性结合相应的靶标,并且与结合程度成比例的信号用于确定样品中靶标的量。在一些情况下,定量方法可包括测序。在一些情况下,测序允许平行查询(parallel interrogation)数千个基因而没有扩增子干扰。在一些实例中,定量方法可包括定量PCR(qPCR)。在一些情况下,通过qPCR精确定量基因表达需要如此多的对照基因,所以用qPCR定量效率低。在其他情况下,测序效率和通过测序的精确定量可能不受分析的(对照)基因的数目的影响。至少出于上述原因,测序可用于本文公开的一些方法,其中评估多个器官(例如,大脑、心脏、肾、肝等)的健康状态。
本文公开的定量核酸的一些方法包括定量PCR(qPCR)。在一些情况下,qPCR包括本文所述的无细胞RNA的逆转录反应以产生相应的cDNA。在一些情况下,无细胞RNA包含标志物、组织特异性多核苷酸和既不是标志物也不是组织特异性多核苷酸的无细胞RNA。一些无细胞RNA包含本文所述的标志物、本文所述的组织特异性多核苷酸和既不是本文所述的标志物也不是组织特异性多核苷酸的无细胞RNA。在一些情况下,qPCR包括使对应于标志物、组织特异性多核苷酸或管家基因(例如,ACTB、ALB、GAPDH)的cDNA与对标志物、组织特异性多核苷酸或管家基因具有特异性的PCR引物接触。
本文公开的一些方法包括定量血细胞特异性多核苷酸。本文公开的包括qPCR的方法可包括使cDNA与对应于血细胞特异性多核苷酸的引物接触。本文公开的一些血细胞特异性多核苷酸是核酸,该核酸主要由一种或多种类型的血细胞表达或甚至仅由一种或多种类型的血细胞表达。血细胞的类型通常可分为白细胞(也称为白血球)、红细胞(也称为红血球)和血小板。在一些情况下,血细胞特异性多核苷酸在包括定量本文公开的组织特异性多核苷酸和疾病标志物的方法中用作对照。在一些情况下,利用对应于血细胞特异性多核苷酸的引物扩增的产物的缺失可用于证实该方法是检测血液、血浆或血清样品中的无细胞RNA而不是血细胞中表达的RNA。作为非限制性实例,血细胞特异性多核苷酸包括在白细胞、血小板或红细胞及其组合中表达的多核苷酸。白细胞包括但不限于淋巴细胞、T细胞、B细胞、树突细胞、粒细胞、单核细胞和巨噬细胞。作为非限制性实例,血液特异性多核苷酸可以由选自CD4、TMSB4X、MPO、SOX6、HBA1、HBA2、HBB、DEFA4、GP1BA、CD19、AHSP和ALAS2的基因编码。血细胞特异性多核苷酸可由CD4编码并主要由白细胞表达。血细胞特异性多核苷酸可由TMSB4X编码并由多种血细胞类型(全血)表达。血细胞特异性多核苷酸可由MPO编码并主要由中性粒细胞表达。血细胞特异性多核苷酸可由DEFA4编码并主要由嗜中性粒细胞表达。血细胞特异性多核苷酸可由GP1BA编码并主要由血小板表达。血细胞特异性多核苷酸可由CD19编码并主要由B细胞表达。血细胞特异性多核苷酸可由ALAS2、SOX6、HBA1、HBA2或HBB编码并主要由红细胞表达。
在一些情况下,定量方法可以是qPCR。qPCR可以是更灵敏的方法,因此更准确地定量以非常低的水平存在的RNA。在一些情况下,定量方法可以是测序。在一些情况下,测序需要更复杂的RNA样品制备并且需要核酸的消耗或富集以提供精确的定量。
通常,本文公开的方法包括检测或定量标志物的组合或组织特异性多核苷酸的组合。在一些情况下,如果检测到多个组织特异性多核苷酸,则可以对对象进行更决定性的诊断或评估。在一些情况下,对象的血液样品中每种组织特异性多核苷酸的存在将不指示对感兴趣的组织或来源的损伤。然而,它们的存在可以共同指示对感兴趣的组织或来源的损伤。类似地,如果检测到多个标志物,则可以对对象进行更决定性的诊断或评估。在一些情况下,对象血液样品中每种标志物的存在将不指示对感兴趣的组织或来源的损伤。然而,它们的存在可以共同指示感兴趣的组织或来源的病症。该方法可包括检测或定量2、3、4、5、6、7、8、9或10个组织特异性多核苷酸。该方法可包括检测或定量2、3、4、5、6、7、8、9或10个标志物。可能已知两种或更多种标志物在共同的遗传通路或共同的分子信号转导通路中相互作用。共同的分子信号转导通路可以是相互作用以启动细胞功能的几种蛋白质的网络,例如,作为非限制性实例,炎症应答、细胞凋亡、胆固醇摄取等。
类似地,在无细胞DNA的情况下,本文公开的一些方法使用DNA或染色质的组织特异性修饰来鉴定样品中的组织特异性多核苷酸。例如,组织特异性无细胞DNA可包含组织特异性甲基化模式。组织特异性无细胞DNA可与指示特异性来源组织的蛋白质(例如,已知在特定组织中转录该基因的转录因子)复合。无细胞或循环染色质或染色质片段可具有组织特异性组蛋白修饰(例如,甲基化、乙酰化和磷酸化)。在这些情况中的一些中,诸如染色质免疫沉淀等方法可适用于检测/定量组织特异性多核苷酸。无细胞组织特异性DNA可以是单链或双链DNA。
本文公开的一些方法包括使用多种检测甲基化模式的方法。通常,DNA将经历选择性修饰甲基化或未甲基化核苷酸的化学转化过程。例如,可用亚硫酸氢盐(bisulfite)处理DNA,其将胞嘧啶残基转化为尿嘧啶(尿嘧啶在PCR后转化为胸腺嘧啶),但不影响5-甲基胞嘧啶残基。因此,亚硫酸氢盐处理在DNA序列中引入了依赖于单个胞嘧啶残基的甲基化状态的特定变化(“甲基化特异性修饰”),生成了关于DNA片段的甲基化状态的单核苷酸解析信息。可以对改变的序列进行各种分析以检索该信息。
本文公开的一些方法包括在亚硫酸氢盐处理之前,将DNA置于氧化或还原条件下,以便鉴定其他表观遗传标记(epigenetic mark)的模式。例如,可以进行氧化性亚硫酸氢盐反应。5-甲基胞嘧啶和5-羟甲基胞嘧啶在亚硫酸氢盐测序中均读作C。氧化性亚硫酸氢盐反应允许在单碱基分辨率下鉴别5-甲基胞嘧啶和5-羟甲基胞嘧啶。通常,该方法采用5-羟甲基胞嘧啶到5-甲酰基胞嘧啶的特定化学氧化,其随后在亚硫酸氢盐处理期间转化为尿嘧啶。然后读作C的唯一碱基是5-甲基胞嘧啶,给出了DNA样品中真实甲基化状态的图谱。5-羟甲基胞嘧啶的水平也可以通过测量亚硫酸氢盐和氧化性亚硫酸氢盐测序之间的差异来定量。在亚硫酸氢盐处理之前,也可将DNA置于还原条件下。还原将样品核苷酸序列中的5-甲酰基胞嘧啶残基转化为5-羟甲基胞嘧啶。如上所述,在亚硫酸氢盐处理后,5-甲酰基胞嘧啶转化为尿嘧啶,但5-羟甲基胞嘧啶不转化。通过将经历还原性亚硫酸氢盐处理的样品的第一部分与仅经历亚硫酸氢盐处理的样品的第二部分进行比较,可以鉴定5-甲酰基胞嘧啶标记的位置。
作为基于甲基化诱导序列改变的替代方案,本文公开的方法可以包括通过分离或富集包含甲基化的多核苷酸并基于甲基化的多核苷酸的序列(例如,通过测序或探针杂交)鉴定甲基化的多核苷酸来推断甲基化状态。富集甲基化序列的一种方法包括以甲基化特异性方式修饰碱基、富集包含修饰的多核苷酸(例如,通过纯化)、和/或扩增富集的多核苷酸、然后鉴定多核苷酸。例如,5-羟甲基修饰的胞嘧啶(5hmC)可以在UDP-葡萄糖分子和β-葡糖基转移酶的存在下被选择性糖基化。UDP-葡萄糖分子可以包含标记,使得标记在与UDP-葡萄糖反应时与含5hmC的多核苷酸缀合。标记可以是结合对的成员(例如,链霉抗生物素蛋白/生物素或抗原/抗体),其允许在与结合对的相应成员结合时分离修饰的片段。分离的多核苷酸可以在鉴定之前例如在扩增反应(例如,PCR)中进一步富集。
可以使用本文公开的任何合适的序列检测方法检测多核苷酸的存在和/或数量(相对或绝对)以及亚硫酸氢盐处理导致的序列变化。实例包括但不限于探针杂交、引物指导的扩增和测序。可以使用任何方便的低通量或高通量测序技术或平台对多核苷酸进行测序,包括Sanger测序、Solexa-Illumina测序、基于连接的测序(SOLiD)、焦磷酸测序;strobe测序(SMR);以及半导体阵列测序(Ion Torrent)。Illumina或Solexa测序基于可逆染料终止剂(reversible dye-terminator)。DNA分子通常附着在载玻片上的引物上并扩增,从而形成局部克隆集落。随后,可以一次性加入一种类型的核苷酸,并洗去未掺入的核苷酸。随后,可以拍摄荧光标记的核苷酸的图像,并且从DNA中化学去除染料,允许下一个循环。Applied Biosystems的SOLiD技术采用通过连接进行的测序。该方法基于使用所有可能的固定长度的寡核苷酸的库,该寡核苷酸根据测序位置进行标记。这类寡核苷酸被退火和连接。随后,用于匹配序列的DNA连接酶的优先连接通常导致在该位置的核苷酸的信号信息。由于DNA通常通过乳化PCR扩增,因此所得珠子(每个仅含有相同DNA分子的拷贝)可沉积在载玻片上,得到数量和长度与Illumina测序相当的序列。设想的测序方法的另一个实例是焦磷酸测序,特别是例如基于Roche 454基因组测序仪的454焦磷酸测序。该方法在油溶液中放大水滴内的DNA,每个液滴含有附着于单个引物包被的珠子上的单个DNA模板,然后形成克隆集落。焦磷酸测序使用荧光素酶产生光,用于检测添加到新生DNA中的单个核苷酸,并且组合数据用于产生序列读出(read-out)。另一种方法是基于Helicos的Heliscope技术,其中片段被束缚于阵列的polyT寡聚物捕获。在每个测序循环中,加入聚合酶和单个荧光标记的核苷酸并对阵列成像。随后去除荧光标记,并重复该循环。合适的测序技术的其他实例是通过杂交的测序、通过使用纳米孔的测序、基于显微镜的测序技术、微流体Sanger测序或基于微芯片的测序方法。高通量测序平台允许在单个反应容器中产生多个不同的测序读段,例如10
计算机控制系统
本公开提供了计算机控制系统,其被编程以实现本公开的方法。图10示出了计算机系统1001,其被编程或以其他方式配置为评估或检测对象中的AD。计算机系统1001可以调节本公开的各个方面,例如,接收或获得生物样品;定量生物样品中多种无细胞信使RNA(cf-mRNA)的cf-mRNA水平,其中所述多种无细胞mRNA对应于包含KIAA0100、MAGI1、NNMT、MXD1、ZNF75A、SELL、ASS1、MNDA和AC132217.4的第一多种基因或包含SLU7、HNRNPA2B1、GGCt、NDUFA12、HSPB11、ATP6V1B2、SASS6、SUMO1、KRCC1和LSM6的第二多种基因;将所述cf-mRNA水平输入分类器以获得风险评分;基于所述风险评分生成报告等。计算机系统1001可以是用户的电子装置或相对于该电子装置远程定位的计算机系统。电子装置可以是移动电子装置。
计算机系统1001包括中央处理器(CPU,本文也称为“处理器”和“计算机处理器”)1005,其可以是单核或多核处理器或用于并行处理的多个处理器。计算机系统1001还包括存储器或存储位置1010(例如,随机存取存储器、只读存储器、闪存)、电子存储单元1015(例如,硬盘)、用于与一个或多个其他系统通信的通信接口1020(例如,网络适配器)以及外围装置1025如高速缓存、其他存储器、数据存储和/或电子显示适配器。存储器1010、存储单元1015、接口1020和外围装置1025通过诸如主板等通信总线(实线)与CPU 1005通信。存储单元1015可以是用于存储数据的数据存储单元(或数据储存库)。计算机系统1001可以借助于通信接口1020可操作地耦合到计算机网络(“网络”)1030。网络1030可以是因特网(Internet)、互联网(internet)和/或外联网或与因特网通信的内联网和/或外联网。在一些情况下,网络1030是电信和/或数据网络。网络1030可以包括一个或多个计算机服务器,其可以实现诸如云计算等分布式计算。在一些情况下,借助于计算机系统1001,网络1030可以实现对等网络(peer-to-peer network),该对等网络可以使耦合到计算机系统1001的设备能够表现为客户机或服务器。
CPU 1005可以执行机器可读指令序列,其可以包含在程序或软件中。该指令可存储在存储器位置(例如,存储器1010)中。指令可以被引导到CPU 1005,其可以随后对CPU1005进行编程或以其他方式配置以实现本公开的方法。由CPU 1005执行的操作的实例可以包括提取、解码、执行和回写。
CPU 1005可以是诸如集成电路等电路的一部分。系统1001的一个或多个其他组件可以包括在电路中。在一些情况下,该电路是专用集成电路(ASIC)。
存储单元1015可以存储文件,例如驱动程序、库和保存的程序。存储单元1015可以存储用户数据,例如用户偏好和用户程序。在一些情况下,计算机系统1001可以包括在计算机系统1001外部的一个或多个附加数据存储单元,例如位于通过内联网或因特网与计算机系统1001通信的远程服务器上。
计算机系统1001可以通过网络1030与一个或多个远程计算机系统通信。例如,计算机系统1001可以与用户(例如,正在查询风险评分的医务人员)的远程计算机系统通信。远程计算机系统的实例包括个人计算机(例如,便携式PC),平板计算机或平板电脑(例如,
本文所述的方法可以通过存储在计算机系统1001的电子存储位置(例如,存储器1010或电子存储单元1015)上的机器(例如,计算机处理器)可执行代码来实现。可以以软件的形式提供机器可执行或机器可读代码。在使用期间,代码可由处理器1005执行。在一些情况下,可从存储单元1015中检索代码并将其存储在存储器1010上以供处理器1005随时访问。在一些情况下,可以排除电子存储单元1015,并且将机器可执行指令存储在存储器1010上。
代码可以被预编译并被配置成与具有适于执行该代码的处理器的机器一起使用,或者可以在运行时期间被编译。代码可以以编程语言提供,该编程语言可以被选择为使得该代码能够以预编译(pre-compiled)或编译时(as-compiled)的方式执行。
本文提供的系统和方法的各方面诸如计算机系统1001可以在编程中实现。技术的各个方面可以被认为是通常为机器(或处理器)可执行代码和/或相关联的数据的形式的“产品”或“制品”,其被承载在或体现在一种类型的机器可读介质中。机器可执行代码可以存储在电子存储单元如存储器(例如,只读存储器、随机存取存储器、闪存)或硬盘上。“存储”型介质可包括计算机、处理器等的任何或所有有形存储器或其相关联的模块,例如各种半导体存储器、磁带驱动器、磁盘驱动器等,其可在任何时间为软件编程提供非暂时性存储。软件的全部或部分有时可以通过因特网或各种其他电信网络进行通信。这种通信例如可以使得能够将软件从一个计算机或处理器加载到另一个计算机或处理器中,例如从管理服务器或主机计算机加载到应用服务器的计算机平台中。因此,可以承载软件元件的另一类型的介质包括例如通过有线和光陆线网络(optical landline network)以及通过各种空中链路在本地设备之间的物理接口上使用的光、电和电磁波。承载这种波的物理元件,例如有线或无线链路、光链路等,也可以被认为是承载软件的介质。如本文所用,除非限制为非暂时性的、有形的“存储”介质,诸如计算机或机器等术语“可读介质”是指参与向处理器提供指令以供执行的任何介质。
因此,诸如计算机可执行代码等机器可读介质可以采取多种形式,包括但不限于有形存储介质、载波介质或物理传输介质。非易失性存储介质包括例如光盘或磁盘(例如任何计算机中的任何存储设备)等,例如可用于实现图中所示的数据库等。易失性存储介质包括动态存储器,例如这种计算机平台的主存储器。有形传输介质包括同轴电缆;铜线和光纤,包括包含计算机系统内的总线的线。载波传输介质可以采用电信号或电磁信号或者声波或光波的形式,例如在射频(RF)和红外(IR)数据通信期间产生的那些。计算机可读介质的常见形式因此包括例如:软盘(floppy disk)、柔性盘(flexible disk)、硬盘、磁带、任何其他磁性介质、CD-ROM、DVD或DVD-ROM、任何其他光学介质、穿孔卡纸带(punch cardspaper tape)、具有孔图案的任何其他物理存储介质、RAM、ROM、PROM和EPROM、FLASH-EPROM、任何其他存储器芯片或盒、传输数据或指令的载波、传输这种载波的电缆或链路或计算机可从其读取编程代码和/或数据的任何其他介质。许多这些形式的计算机可读介质可涉及将一个或多个指令的一个或多个序列携带到处理器以供执行。
计算机系统1001可以包括电子显示器1035或与电子显示器1035通信,该电子显示器1035包括用户界面(UI)1140,用于提供例如基于风险评分的报告,该报告包含直接用于监测和/或治疗AD进展的信息。UI的实例包括但不限于图形用户界面(GUI)和基于web的用户界面。
本公开的方法和系统可以通过一个或多个算法来实现。算法可以在中央处理器1005执行时通过软件来实现。算法可以例如用于生成分类器以计算具有AD或认知损害的风险评分。
试剂盒
本公开还提供了试剂盒。在一些情况下,本文所述的试剂盒包含用于测量和/或检测对应于本文所述的一种或多种基因的cf-mRNA的一种或多种组合物、试剂和/或装置组件。本文所述的试剂盒还可包含用于实施本文提供的任何方法的说明书。试剂盒还可包含能够通过各种测定类型如逆转录、多核苷酸扩增、测序、探针杂交和微阵列杂交检测cf-mRNA的试剂。试剂盒还可以包含计算机可读介质,该计算机可读介质包括用于实现本文所述方法的计算机可执行代码。
在一些实施方案中,本文提供的试剂盒包含与从对应于本文公开的差异表达基因列表的cf-mRNA转录的cDNA序列杂交的多种寡核苷酸引物。
在一些实施方案中,本文所述的试剂盒包含包装材料。如本文所用,术语“包装材料”可指容纳试剂盒的组分的物理结构。包装材料可以保持试剂盒组分的无菌性,并且可以由通常用于这种目的的材料(例如,纸、波纹纤维板、玻璃、塑料、箔、安瓿等)制成。试剂盒还可以包含缓冲剂、防腐剂或蛋白质/核酸稳定剂。试剂盒可以包含用于从患者获得生物样品的组件。此类组件的非限制性实例可以是手套、皮下注射针或注射器、用于容纳生物样品的管状物(tubing)、管子或器皿、灭菌组件(例如,异丙醇擦拭物或无菌纱布)和/或冷却材料(例如,冷却袋、干冰或冰)。在一些情况下,本文公开的试剂盒根据任何公开的方法使用。
本文可以提供系统和试剂盒,以非侵入性地检测受胁迫的对象中的组织或器官,以及确定哪些疾病或病症影响受胁迫的组织或器官,同时考虑衰老过程导致的基因表达的变化。本文公开了用于检测对象中的疾病或病症的试剂盒,该试剂盒包含用于检测至少一种标志物的至少一种试剂和用于检测至少一种组织特异性多核苷酸的至少一种试剂。另外地或可选地,本文公开的试剂盒可用于确定对象中疾病或病症的位置(例如,组织)和/或进展。另外地或可选地,本文公开的试剂盒可用于确定施用于对象的疗法是否影响疾病或病症的进展或阶段。另外地或可选地,本文公开的试剂盒可用于确定施用于对象的疗法是否已导致任何非预期的毒性或副作用。
本文提供了包含至少一种本文公开的试剂的试剂盒。至少一种用于检测组织特异性多核苷酸的试剂可以包括至少一种用于检测无细胞多核苷酸的试剂。用于检测至少一种标志物的至少一种试剂可以包括用于检测无细胞多核苷酸的至少一种试剂。至少一种无细胞多核苷酸可包含无细胞DNA或无细胞RNA。无细胞DNA可具有组织特异性甲基化模式。无细胞多核苷酸可以是组织特异性基因转录本。用于检测至少一种标志物的至少一种试剂和/或用于检测组织特异性多核苷酸的至少一种试剂可包含多核苷酸探针。多核苷酸探针可与无细胞多核苷酸结合。多核苷酸探针可以以序列依赖性方式与无细胞多核苷酸结合。多核苷酸探针可与对应于基因的野生型形式而非基因的突变形式无细胞多核苷酸结合。可选地,多核苷酸探针可与对应于基因的突变形式而非基因的野生型形式的无细胞多核苷酸结合。多核苷酸探针可以附着于信号转导部分。作为非限制性实例,信号转导部分可选自半抗原、荧光分子和放射性同位素。试剂盒对一种疾病或病症可以是特异性的。试剂盒可包含少至1、2、3、4或5个多核苷酸探针以检测对象中的疾病或病症。试剂盒对多种疾病或病症可以是特异性的。试剂盒可包含5至10、10至20、10至100、10至1000、100至1000、100至10,000或更多个多核苷酸探针。
本文提供了包含至少一种本文公开的试剂的试剂盒。用于检测至少一种标志物的至少一种试剂和/或用于检测组织特异性多核苷酸的至少一种试剂可包含引物。引物可以是逆转录酶引物。引物可以是PCR引物。引物可以扩增至少一种标志物、至少一种组织特异性多核苷酸或其部分。引物可以以序列依赖性方式扩增无细胞多核苷酸。引物可以扩增对应于基因的野生型形式而非基因的突变形式的无细胞多核苷酸或其部分。可选地,引物可扩增对应于基因的突变形式而非基因的野生型形式的无细胞多核苷酸或其部分。试剂盒还可以包含扩增报告分子,其向试剂盒的使用者提供用于检测组织特异性多核苷酸的至少一种标志物和/或至少一种试剂的量。通常,该量是基于参考样品的相对量。扩增信号转导试剂可以选自插入荧光染料或染料。扩增信号转导试剂可以是SYBR Green。
本文提供了包含至少一种本文公开的试剂的试剂盒。用于检测至少一种标志物的至少一种试剂和/或用于检测组织特异性多核苷酸的至少一种试剂可以包含与至少一种标志物或组织特异性多核苷酸结合的肽。肽可以是抗体或多核苷酸结合蛋白(例如,转录因子、组蛋白)的一部分。用于检测至少一种标志物的至少一种试剂和/或用于检测组织特异性多核苷酸的至少一种试剂可包含发射信号的信号转导部分,其中发射或丢失的信号指示标志物或组织特异性多核苷酸的存在或量。信号转导部分的实例包括但不限于染料、荧光团、酶和放射性粒子。至少一种试剂还可以包含用于检测信号或其不存在的信号转导部分检测器。
本文公开了用于检测组织或器官是否受病症影响的试剂盒,其中该试剂盒包含用于病症的标志物的至少一种探针或引物。本文还公开了用于检测肿瘤、病原体或疾病的位置的试剂盒,其中该试剂盒包含用于病症的标志物的至少一种探针或引物。在一些情况下,试剂盒包含至少一种探针和至少一种引物。在一些情况下,标志物是多核苷酸、引物或探针是与感兴趣的靶标杂交的多核苷酸。在一些情况下,标志物是肽或蛋白质,并且探针是能够结合肽或蛋白质的抗体或抗体片段。在一些情况下,探针是与标志物结合的小分子。在一些情况下,探针与可用于检索标志物、定量标志物或检测标志物的标签缀合。至少一种病症或疾病可以是以下中的至少一种:炎症、凋亡、坏死、纤维化、感染、自身免疫性疾病、关节炎、肝病、神经变性疾病和癌症。
本文公开了用于检测对象中的疾病或病症的试剂盒,该试剂盒包含用于检测至少一种标志物的至少一种试剂和用于检测至少一种组织特异性多核苷酸的至少一种试剂。试剂盒还包含固体支持物,其中多核苷酸探针、引物和/或肽附着于该固体支持物。固体支持物可以选自珠子、芯片、凝胶、颗粒、孔、柱、管子、探针、载玻片、膜和基质。
本文公开了用于检测对象中的疾病或病症的试剂盒,该试剂盒包含用于检测至少一种标志物的至少一种试剂和用于检测至少一种组织特异性多核苷酸的至少一种试剂。本文公开的试剂盒的两种或更多种组分可以是分开的。本文公开的试剂盒的两种或更多种组分可以是集成的。本文公开的试剂盒的两种或更多种组分可以集成到装置中。装置可以允许使用者简单地将来自对象的至少一个样品添加到该装置中并且接收指示对象是否患有疾病或病症和/或对象的哪个或哪些组织受疾病或病症影响的结果。在一些情况下,使用者可以将至少一种试剂添加到装置中。在其他情况下,用户不必向装置中添加任何试剂。
本文公开了用于检测对象中的疾病或病症的试剂盒,该试剂盒包含用于检测至少一种标志物的至少一种试剂和用于检测至少一种组织特异性多核苷酸的至少一种试剂。至少一种组织特异性多核苷酸或标志物可包含无细胞多核苷酸。至少一种标志物可包含RNA。至少一种组织特异性多核苷酸可包含至少一种组织特异性RNA,其中组织特异性RNA是仅在特异性的组织中表达的RNA,或者是在特异性的组织中表达的水平显著高于在其他组织中表达的水平的RNA。例如,组织特异性基因可以是这样的基因,其在特定组织或组织群中的表达比任何其他组织或组织群(例如,任何单独的或组合的所有其他组织或组织群)高至少2倍、5倍、10倍或25倍。至少一种组织特异性多核苷酸或标志物可包含至少一种组织特异性甲基化DNA,其中组织特异性甲基化DNA包含组织特异性甲基化模式。可选地或另外地,组织特异性甲基化DNA可包含具有甲基化模式的DNA,该甲基化模式仅在一种组织中发生或在组织中的水平显著高于其在其他组织中发生的水平。可以通过以下状况确定组织被损伤:(a)是否至少一种标志物的水平高于至少一种标志物的参考水平,和(b)是否至少一种组织特异性多核苷酸的水平高于至少一种组织特异性多核苷酸的参考水平。至少一种组织特异性多核苷酸可包含两种或更多种多核苷酸,每种多核苷酸对不同组织(例如,2、3、4、5、10、15、25或更多种不同组织)是特异性的。组织可以是以下中的至少一种:全血、骨、上皮、下丘脑、平滑肌、肺、胸腺、淋巴结、甲状腺、心脏、肾、大脑、小脑、肝和皮肤。标志物和/或组织特异性多核苷酸可对应于基因。通常,如果标志物或组织特异性多核苷酸是包含基因(或其可识别部分)的DNA分子,或是该基因的表达产物(例如,RNA转录本或蛋白质产物),则该标志物或组织特异性多核苷酸“对应于基因”。
本文还公开了用于执行本公开的方法的系统。通常,系统可以包括能够执行本文公开的方法的步骤的各种单元,例如,样品处理单元、扩增单元、测序单元、检测单元、定量单元、比较单元和/或报告单元。在一些实施方案中,系统包括:存储器单元,该存储器单元被配置成用于存储以下各项的结果:(i)用于检测对象的第一样品中的至少一种病症的至少一种标志物的测定,和(ii)用于检测对象的第二样品中的至少一种组织特异性RNA的测定,其中至少一种组织特异性RNA是对组织特异性的无细胞RNA;至少一个处理器,其被编程为:(i)定量至少一种标志物的水平;(ii)定量至少一种组织特异性多核苷酸的水平;(iii)将至少一种标志物的水平与标志物的相应参考水平进行比较;(iv)将至少一种组织特异性多核苷酸的水平与组织特异性多核苷酸的相应年龄依赖性参考水平进行比较;以及(v)基于上述比较确定组织损伤的存在或组织损伤的相对变化;以及输出单元,其向接收者递送报告,其中报告提供步骤(b)的结果。该系统可以基于步骤(b)的结果提供对医疗行动的推荐。医疗行动可以包括治疗。第一样品和第二样品可以相同。第一样品和第二样品可以不同。第一样品和第二样品可以是不同的,因为它们是在不同的时间获得的。第一样品和第二样品可以是不同的,因为它们是不同的流体。第一和/或第二样品可以是选自以下的流体:血液、血液级分、唾液、痰液、尿液、精液、经阴道的液体、脑脊液、汗液或乳汁。第一和/或第二样品可以是血浆。
本文公开的系统可与本文公开的试剂盒或装置中的任一种一起使用。系统可以与本文公开的试剂盒或装置中的任一种集成。本文公开的装置可以包括本文公开的系统中的任何一个。在一些实施方案中,系统包括计算机系统。在系统中使用的计算机可以包括至少一个处理器。处理器可与计算机系统的至少一个控制器、计算单元和/或其他单元相关联,或根据需要植入固件中。如果以软件实现,则例程可以存储在任何计算机可读存储器中,例如RAM、ROM、闪存、磁盘、激光盘或其他合适的存储介质中。同样地,软件可以经由任何已知的递送方法被递送到计算装置,该递送方法包括例如经由诸如电话线、因特网、无线连接等通信信道,或者经由诸如计算机可读盘、闪存驱动器等可传输介质。各个步骤可以被实现为各个块、操作、工具、模块和技术,其进而可以在硬件、固件、软件或硬件、固件和/或软件的任何组合中实现。当以硬件实现时,一些或所有块、操作、技术等可以以例如定制集成电路(IC)、专用集成电路(ASIC)、现场可编程逻辑阵列(FPGA)、可编程逻辑阵列(PLA)等来实现。客户机-服务器关系数据库体系结构可用于系统的实施方案中。客户机-服务器体系结构是网络体系结构,其中网络上的每个计算机或进程是客户机或服务器。服务器计算机通常是专用于管理磁盘驱动器(文件服务器)、打印机(打印服务器)或网络流量(网络服务器)的强大计算机。客户计算机包括用户在其上运行应用程序的PC(个人计算机)或工作站,以及本文公开的示例性输出装置。客户端计算机可以依赖于服务器计算机来获取资源,例如文件、装置、甚至处理能力。在一些实施方案中,服务器计算机处理所有数据库功能。客户计算机可以具有处理所有前端数据管理的软件,并且还可以接收来自用户的数据输入。
本文公开的系统可以被配置成接收对样品进行检测反应的用户请求。用户请求可以是直接的或间接的。直接请求的实例包括经由诸如键盘、鼠标或触摸屏的输入装置发送的那些请求。间接请求的实例包括经由诸如互联网(有线或无线)上的通信介质的传输。
本文公开的系统还可以包括向接收者发送报告的报告生成器,其中该报告包含本文描述的方法的结果。可以实时生成报告,例如在测序读段期间或在分析测序数据时,随着过程的进行而定期更新。此外,或可选地,可以在分析结束时生成报告。在一些实施方案中,响应于来自用户的指令生成报告。除了检测或比较的结果之外,报告还可以包含基于这些结果的分析、结论或建议。例如,检测与疾病或病症相关的标志物并且组织特异性多核苷酸的水平高于正常范围,报告可包括关于这种关联的信息,例如对象患有疾病或病症的可能性、哪些组织受到或未受到影响以及任选地基于这种信息的建议(例如,额外的测试、监测或补救措施)。报告可以采取多种形式中的任一种。可以预见,与本公开相关的数据可以通过这样的网络或连接(或用于传输信息的任何其他合适的装置,包括但不限于邮寄物理报告,诸如打印输出)来传输,以便由接收器接收和/或检查。接收器可以是但不限于个人或电子系统(例如,至少一台计算机和/或至少一个服务器)。
本公开提供了一种包括代码的计算机可读介质,该代码在由至少一个处理器执行时实现本公开的方法。包括计算机可执行代码的机器可读介质可以采取多种形式,包括但不限于有形存储介质、载波介质或物理传输介质。非易失性存储介质包括例如光盘或磁盘(例如,任何计算机中的任何存储设备)等,例如可用于实现数据库等。易失性存储介质包括动态存储器,例如这种计算机平台的主存储器。有形传输介质包括同轴电缆;铜线和光纤,包括包含计算机系统内的总线的线。载波传输介质可以采用电信号或电磁信号或者声波或光波的形式,例如在射频(RF)和红外(IR)数据通信期间产生的那些。计算机可读介质的常见形式因此包括例如:软盘、柔性盘、硬盘、磁带、任何其他磁性介质、CD-ROM、DVD或DVD-ROM、任何其他光学介质、穿孔卡纸带、具有孔图案的任何其他物理存储介质、RAM、ROM、PROM和EPROM、FLASH-EPROM、任何其他存储器芯片或盒、传输数据或指令的载波、传输这种载波的电缆或链路或计算机可从其读取编程代码和/或数据的任何其他介质。许多这些形式的计算机可读介质可涉及将至少一个指令的至少一个序列携带到处理器以供执行。
除非另外定义,否则本文使用的所有技术术语具有与本发明所属领域的普通技术人员通常理解的含义相同的含义。
如本文所用,除非上下文另外明确指出,否则单数形式“一种(a/an)”和“所述(the)”包括复数引用。本文中对“或”的任何引用旨在涵盖“和/或”,除非另有说明。
如本文所用,术语“约”在数字的上下文中是指从大于该数字10%至小于该数字10%的范围。
如本文所用,短语“至少一种”、“一种或多种”和“和/或”是开放式表达,其在操作中既是联合的也是分离的。例如,每种表述“A、B和C中的至少一种”、“A、B或C中的至少一种”、“A、B、和C中的一种或多种”、“A、B或C中的一种或多种”和“A、B、和/或C”是指单独的A、单独的B、单独的C、一起的A和B、一起的A和C、一起的B和C或一起的A、B和C。
术语“确定(determining)”、“测量(measuring)”、“评价(evaluating)”、“评估(assessing)”、“测定(assaying)”和“分析(analyzing)”在本文中通常可互换使用以指代测量形式,并包括确定是否元素存在与否(例如,检测)。这些术语可包括定量、定性或定量和定性确定。可选地评估也可以是相对的或绝对的。“检测……的存在(detecting thepresence of)”包括确定存在的某物的量以及确定它是否存在。
术语“组合(panel)”、“生物标志物组合”、“蛋白质组合”、“分类器模型”和“模型”在本文中可互换使用,是指一组生物标志物,其中该组生物标志物包含至少两种生物标志物。示例性生物标志物是映射到本文公开的差异表达基因列表的cf-mRNA。然而,还考虑了另外的生物标志物,例如提供样品的个体的年龄或性别。生物标志物组合通常预测和/或提供对象健康状况、疾病或病症的信息。
生物标志物组合的“水平”是指组合的组成标志物的绝对和相对水平以及组合的组成生物标志物的相对模式。
术语“对象”、“个体”或“患者”在本文中通常可互换使用。“对象”可以是含有表达的遗传物质的生物实体。生物实体可以是植物、动物或微生物,包括例如细菌、病毒、真菌和原生动物。对象可以是体内获得或体外培养的生物实体的组织、细胞及其后代。对象可以是哺乳动物。哺乳动物可以是人。对象可能被诊断或怀疑处于疾病的高风险中。疾病可以是认知损害。认知损害可以是AD的症状。在一些情况下,对象不一定被诊断或怀疑处于疾病的高风险中。
术语灵敏度或真阳性率可以指测试的正确鉴定病症的能力。例如,在诊断测试中,测试的灵敏度是已知患有疾病的患者的比例,该患者将对该疾病测试为阳性。在一些情况下,这通过确定真阳性(即,患有疾病的测试为阳性的患者)与患有病症的群体中的个体总数(即,测试为阳性且患有病症的患者与测试为阴性且患有病症的患者的总和)的比例来计算。
当选择不同的诊断截止值(cut-off)时,灵敏度和特异性之间的定量关系可以改变。可以用ROC曲线代表这种变化。ROC曲线的x轴显示测定的假阳性率,其可计算为(1-特异性)。ROC曲线的y轴报告测定的灵敏度。这允许容易地确定对于给定特异性的测定的灵敏度,反之亦然。
如本文所用,术语“治疗(treatment)”或“治疗(treating)”用于指用于在接受者中获得有益或期望结果的药物或其他干预方案。有益的或期望的结果包括但不限于治疗益处和/或预防益处。治疗益处可指消除或改善所治疗的症状或潜在病症。此外,通过根除或改善与潜在病症相关的一种或多种生理症状可实现治疗益处,使得在对象中观察到改善,尽管对象可能仍然患有潜在病症。预防作用包括延迟、预防或消除疾病或病症的出现;延迟或消除疾病或病症的症状的发作;减缓、停止或逆转疾病或病症的进展或其任何组合。为了预防性益处,处于发展特定疾病的风险中的对象或报告疾病的一种或多种生理症状的对象可以经历治疗,即使可能尚未做出该疾病的诊断。
如本文所用,术语“机器学习”、“机器学习过程”、“机器学习操作”和“机器学习算法”一般是指可以逐步提高任务的计算机性能的任何系统或分析和/或统计过程。机器学习可以包括机器学习算法。机器学习算法可以是经过训练的算法。机器学习(ML)可以包括一种或多种监督的、半监督的或无监督的机器学习技术。例如,ML算法可以是通过监督学习训练的训练算法(例如,各种参数被确定为权重或缩放因子)。ML可以包括回归分析、正则化、分类、降维、整体学习、元学习、关联规则学习、聚类分析、异常检测、深度学习或超深度学习中的一个或多个。ML可以包括但不限于:k均值、k均值聚类、k最近邻、学习向量量化、线性回归、非线性回归、最小二乘回归、偏最小二乘回归、逻辑回归、逐步回归、多元自适应回归样条(multivariate adaptive regression splines)、岭回归、主成分回归、最小绝对收缩和选择操作、最小角回归、正则相关分析、因子分析、独立成分分析、线性判别分析、多维标度、非负矩阵分解、主成分分析、主坐标分析、投影寻踪、Sammon映射、t-分布随机邻居嵌入、AdaBoosting、提升法(boosting)、梯度提升(gradient boosting)、自助聚集(bootstrapaggregation)、集平均(ensemble averaging)、决策树、条件决策树、提升决策树、梯度增强决策树、随机森林、堆叠泛化、贝叶斯网络、贝叶斯信念网络、朴素贝叶斯、高斯朴素贝叶斯、多项式朴素贝叶斯、隐马尔可夫模型、分层隐马尔可夫模型、支持向量机、编码器、解码器、自动编码器、堆叠自动编码器、感知器、多层感知器、人工神经网络、前馈神经网络、卷积神经网络、递归神经网络、长短期记忆、深度信念网络、深度玻尔兹曼机、深度卷积神经网络、深度递归神经网络或生成式对手网络。
实施例
以下说明性实施例是本文所述的组合物和方法的实施方案的代表,并不意味着以任何方式进行限制。
实施例1-临床标本
检查了来自AD和NCI的五个独立患者队列的总共242份血浆标本,包括126名阿尔茨海默病患者和116名年龄匹配的对照。这些队列包括:加利福尼亚大学圣地亚哥分校、肯塔基大学、圣路易斯华盛顿大学、GEMS(印第安纳州)和BioIVT。详细的患者人口统计学资料和临床病理学特征如表2所示。从所有患者获得书面知情同意书,且研究获得所有参与机构的机构审查委员会批准。
表2:总体患者特征
所有临床诊断均根据NINCDS-ADRDA(美国国立神经病语言障碍卒中研究所和阿尔茨海默病及相关疾病学会的标准)以及根据来自美国国家衰老研究所-阿尔茨海默病协会工作组关于阿尔茨海默病诊断指南的建议进行。
实施例2-RNA提取、文库制备和全转录组RNA-seq
使用QIA amp循环核酸试剂盒(Qiagen)从至多1mL血浆中提取RNA,并以15μl体积洗脱。ERCC RNA Spike-In Mix(Thermo Fisher Scientific,货号4456740)根据制造商的说明(Ambion)添加到RNA中作为外源性掺入对照。Agilent RNA 6000Pico芯片(AgilentTechnologies,货号5067-1513)用于评估提取的RNA的完整性。将RNA样品转化为测序文库。使用基于芯片的电泳进行NGS文库制备过程的定性和定量分析,并使用基于qPCR的定量试剂盒对文库进行定量。使用Illumina NextSeq500平台(Illumina Inc.)、使用配对末端测序、75-循环测序进行测序。使用FASTQ生成应用程序在Illumina BaseSpace平台(IlluminaInc)上执行碱基识别。对于测序数据分析,去除衔接子序列,并使用cutadapt(v1.11)修剪低质量碱基。从后续分析中排除短于15个碱基对的读段。使用具有GENCODE v24基因模型的STAR(v2.5.2b)将大于15个碱基对的读段序列与人参考基因组GRCh38进行比较。使用samtools(v1.3.1)rmdup命令删除重复读段。使用RSEM(v1.3.0)由重复数据删除(de-duplicated)的BAM文件计算基因表达水平。
使用读段作为输入,用DESeq2(v1.12.4)实施差异表达分析。在整个队列中具有少于250个总读段的基因从随后的分析中被排除。在DE分析之前对技术重复进行平均和合并。
从表3中描述的五个不同来源获得样品。为了校正与样品来源相关的分批效应,实施了多因素模型“~来源+疾病状态”,包括作为潜在混杂因素的样品来源。分批校正是有效的,如校正后的PCA图所示。使用Benjamin-Hochberg校正来校正多重检验并获得调节的p值(使用0.05的FDR截止值来选择失调的基因)。
使用Ingenuity通路分析(IPA)软件版本47547484进行通路富集分析。将差异表达和与MMSE和CDR相关的基因的完整列表上传到IPA,并使用表达分析确定高度富集的通路。检查了包括经典通路和“最重要的疾病和生物功能(Top diseases and bio functions)”的IPA分类。
实施例3-脑特异性基因建立
与其他组织类型(细胞类型)相比,在特定组织(细胞类型)中显示显著更高表达的基因被认为是组织(细胞类型)特异性基因,例如脑特异性基因。从以下两个公共数据库获得组织(细胞类型)转录组表达水平:用于在51种人类组织中基因表达的GTEx(www_gtexportal_org/home/)和用于在56种人类造血细胞类型中基因表达的蓝图表观基因组(www_blueprint-epigenome_eu/)。对于每个单独的基因,根据其特定基因的表达对组织(细胞类型)进行分级,并且如果在最重要的组织(top tissue)(细胞类型)中的表达比所有其他组织(细胞类型)高约20倍以上,则认为该基因对最重要的组织(细胞类型)是特异性的。
实施例4-生物信息学分析/分类器开发
为了构建基因表达分类器,将队列分为65%和35%,前65%被指定为“训练队列”,且后35%被指定为“验证队列”。应用这些基因表达值和具有岭正则化的逻辑回归模型鉴定AD样品。使用在scikit-learn Python库中具有L1正则化的逻辑回归分析来实施分类。通过进行15次交叉验证、随机保留“训练队列”中用于验证的样品的40%来确定Meta参数。
为了确保分类器性能的无偏性评价,将来自肯塔基大学的样品用作“训练队列”,将来自所有其他来源的样品用作“验证队列”。在模型训练期间,验证队列中没有以任何方式使用样品。在特征选择步骤中,在训练队列上运行DESeq2,并选择AD和NCI样品之间差异表达的前1,476个基因。然后将这些1,658个基因的表达水平(TPM)用于分类器的后续训练。使用Python文库scikit-learn(scikit-learn_org/stable/,v0.20.1)实施分类器的训练。用分类sklearn.linear_model.LogisticRegression、sklearn.ensemble.RandomForestClassifier、sklearn.svm.SVC和sklearn.neighbors.KNeighborsClassifier分别实现了逻辑回归、随机森林、支持向量机(SVM)、K最近邻分类器。通过对训练队列进行15倍交叉验证来确定Meta参数。接下来,将经过训练的分类器应用于验证队列,并获得验证队列中每个样品的预测风险评分。通过将风险评分与样品的真实疾病状态进行比较,能够绘制接收器操作特征(ROC)曲线并计算曲线下面积(AUC)。根据DeLong计算ROC曲线的置信区间。
首先进行标准化,由此将每个基因的表达水平除以其在样品中的最大值。该步骤被设计成重新调节不同基因之间的表达水平,以避免少数高度表达的基因控制分解过程。然后使用Python文库scikit-learn(//scikit-learn_org/stable/)中的sklearn.decomposition.NMF对归一化的表达矩阵进行NMF分解。NMF分解通过将表达式矩阵分解为两个矩阵X=WH的乘积来实现数据的更简洁的表示。X是具有n行(n个样品)和m列(m个基因)的表达矩阵;W是具有n行(n个样品)和p列(p个分量)的系数矩阵;H是具有p行(p个分量)和m列(m个基因)的载荷矩阵(loading matrix)。W在某种意义上是具有减少的维数的原始矩阵H的概括。H包含关于每个基因对组分贡献多少的信息。通过对每种成分贡献最多的热门基因(top genes)进行通路分析,实现衍生成分的生物学解释。通过对系数矩阵W进行分级聚类来进行患者分组。使用Python library SciPy(v1.3.0)classscipy.cluster.hierarchy.linkage以参数方法=“平均值(average)”和度量=“相关性(correlation)”来实现分级聚类。
为了确保无偏性地评估分类器性能,首先仅使用来自肯塔基大学的样品(对照n=24,AD n=66)构建分类器(图6A)。选择仅在本肯塔基大学(UKy)队列中鉴定的差异表达基因(1,658个FDR<0.05的基因)作为分类器的输入特征。该基因集与使用整个队列鉴定的2,591个失调基因显著重叠(即,使用UKy队列鉴定的1,094个下调基因中的942个与使用整个队列鉴定的那些基因重叠,p值<10e-8;在使用UKy群组鉴定的564个上调基因中的451个与使用整个群组鉴定的那些基因重叠,p值<10e-8;超几何分布检验)。然后在由来源于四个独立来源的剩余AD(n=60)和对照样品(n=92)组成的检验集上检验分类器模型。通过计算检验队列中的AUROC(接收器操作特征下的面积)评价的分类性能为AUROC:0.83(95%CI:0.77-0.89)(图6B)。使用约登指数(Youden Index)建立0.868的截止值,灵敏度为83.3(95%CI:71.5-91.7%),特异性为68.5(95%CI:58.0-77.8%)。
实施例5-统计分析
使用来源于基因分类器多变量逻辑回归模型的风险评分绘制接收器操作特征(ROC)曲线并计算曲线下面积(AUC)。计算交叉验证的15次迭代中的每次迭代的ROC曲线下面积(AUC)。由这15个交叉验证计算平均ROC曲线。使用DeLong方法计算ROC曲线的置信区间。用皮尔逊相关性分析检验两个变量之间的相关性。使用学生t检验评价两个变量之间的差异。使用R(3.3.3,R Development Core Team,//cran_r-project_org/)和MedCalc统计软件版本19(MedCalc Software bvba,Ostend,Belgium)进行所有统计分析。
实施例6-使用低输入血浆RNA的cf-RNA转录组的鲁棒表征
对从126名AD患者和116名年龄匹配的对照获得的400μm至1ml血浆中提取的RNA进行测序。AD和NCI对照之间的平均血浆cf-RNA产量没有差异(分别为8.55和9.55ng)(图1A)。测序运行后,鉴定的平均蛋白编码基因为11,714(在>5TPM检测到转录本)(图2A)。利用外部RNA掺入混合物对照ERCC(外部RNA对照聚生体),用与预期掺入拷贝数相关的ERCC转录本的观察水平证实了本方案的准确性(平均值r=0.92,图2B)。此外,96个样品中技术重复之间转录本水平的比较紧密相关(平均值r=0.87),突出了方案的鲁棒技术再现性(图2C和1B)。最后,跨外显子-内含子剪接点的读段分布显示DNA污染可忽略不计(图2D)。这些结果一起证明了cf-mRNA测序方案用于产生多样的、定量的和可再现的测序数据的可靠的技术性能,而与患者的AD状态无关。
实施例7-与cf-mRNA基因表达谱相关的阿尔茨海默病的鉴定
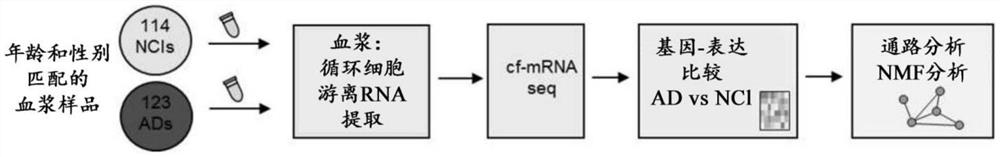
为了鉴定AD患者和对照之间循环转录组的差异,对从241份血浆样品中分离的cf-mRNA进行测序,该血浆样品由来自五个独立来源的126名AD患者和115名年龄匹配的NCI对照组成(图3A;参与者特征参见表2和3)。
表3:队列特征
在AD和NCI之间鉴定了2,591个差异表达的基因(FDR<0.05,图3B),其中在AD患者的循环中,2,057个转录本下调,而534个转录本上调。术语“上调”和“下调”用于描述与NCI对照相比AD患者循环中转录本数量的变化。为了评价这些差异表达的基因的功能作用,使用IPA通路分析来确定最受AD影响的通路和生物过程。IPA分析揭示由AD患者的下调转录本鉴定的许多经典通路与神经元信号转导通路有关,包括:GABA受体信号转导、netrin信号转导、突触长期抑制和阿片样物质信号转导通路,而上调的转录本在与免疫应答(例如,IL-8信号转导、炎性体和神经炎症信号转导通路)、线粒体活性(例如,sirtuin信号转导通路和线粒体功能障碍)和蛋白稳态(例如,sumoylation)相关的经典通路中富集。使用AD患者中下调的转录本鉴定的最重要的经典通路与神经元功能有关,包括GABA受体信号转导、神经元中的CREB信号转导、netrin信号转导和突触发生信号转导通路等(图3C)。
此外,使用IPA分析来检查AD中失调的生物过程。与经典通路分析一致,AD患者中上调的转录本在与免疫应答激活(例如,IL-8信号转导和炎性体通路)、线粒体活性(例如,线粒体功能障碍、氧化磷酸化和sirtuin信号转导通路)和蛋白稳态(例如,sumoylation、蛋白质泛素化和解折叠蛋白应答)相关的通路中显著富集(图3C)。关于生物学过程,AD患者中下调的基因富含“神经系统发育和功能”类别。与神经元和突触丧失相关的生物过程,包括“神经元发育”、“神经传递”和“突触传递”,是最显著富集的术语,指示了AD患者cf-mRNA转录组中神经元和突触连接相关转录本的总体下降(图4B)。一致地,观察到在AD患者的cf-mRNA中下调的基因的显著部分是脑特异性基因(p=6.17x 10
此外,一部分脑特异性基因在AD患者的cf-mRNA中下调(p=6.17x 10-10,图5A)。为了进一步确定AD相关的cf-mRNA中的转录变化对应于脑组织中的基因表达变化,将ADcf-mRNA中鉴定的差异表达基因与先前的RNA-seq数据集进行比较,该数据集检查了海马解剖组织中的转录变化(图5B)。对于上调和下调基因,在脑组织中观察到AD和NCI的差异表达基因与cf-mRNA的重叠(p<10-5)。此外,在cf-mRNA和脑组织之间存在鉴定的通路的重叠(图5C)。这些数据共同支持cf-mRNA转录组捕获与AD相关的转录变化。
实施例8-基于cf-mRNA谱的阿尔茨海默病患者相对于非认知损害对照的鲁棒分类
使用机器学习算法构建可区分AD患者与NCI个体的基于cf-mRNA的分类器。为了确保无偏性地评估分类器性能,首先将队列随机分成训练集(队列的65%)和检验集(队列的35%)(图6A)。随后,对训练组进行差异表达分析,并选择所有差异表达的基因(1,476个基因,FDR<0.05)作为输入特征。使用以下算法训练分类模型:具有L1正则化的逻辑回归(LASSO)、随机森林、具有L2正则化的逻辑回归(岭分类器)、最近邻分类器和支持向量机(SVM)(图6B)。然后将在训练数据集上训练的模型应用于检验集,并通过计算AUROC(接收器操作特征下的面积)评价其性能。在所有评价的算法中,岭分类器提供最佳分类性能,AUROC为0.902(图6C),平均AUROC为0.844(图6D)。使用0.44的疾病风险评分截止值,分类器的灵敏度为0.81且特异性为0.85。通过调整LASSO逻辑回归分类器中的正则化参数,减少了合并到分类器中的特征的数量。在分类器中掺入不同数量的基因并在检验集上评估其性能后,分类器中使用的基因数量减少至9,同时保持高分类性能(AUROC=0.861)。患者总队列中9个基因(KIAA0100、MAGI1、NNMT、MXD1、ZNF75A、SELL、ASS1、MNDA和AC132217.4(非编码RNA))中每一个的表达示于图6E中。
实施例9-与AD严重程度相关的cf-mRNA特征的鉴定
使用非负矩阵分解(NMF)对本文公开的基因进行无监督聚类,以鉴定与不同生物过程相关的六个基因聚类(图8A和9A)。
两个聚类,突触传递以及免疫和炎症应答的标准化表达值显示与CDR评分显著相关(图8B和9A)。突触传递聚类基因的表达随着CDR评分的增加而降低(r=-0.48,p<0.0001),并且在CDR评分0和0.5之间(p=0.001)。相反,免疫和炎症应答聚类的表达水平随着CDR评分增加至1,但对于CDR较高的患者,表达值没有增加(r=0.54,p<0.0001)。
使用非负矩阵分解(NMF)的无监督分解鉴定了六个基因聚类(图8A)。IPA通路分析揭示与涉及AD发作和进展的过程相关(图8A)。例如,聚类3富集了与突触传递通路相关的基因,而聚类5富集了与免疫应答和神经炎症相关的基因(图8A)。基于这6个基因聚类的分子分布将异质性AD患者群体分级为子类型。特别地,基于六个基因聚类的大小的所有126名AD患者的无监督分级聚类揭示了五个不同的组(图8G)。例如,“D组”患者的特征在于聚类5基因(例如,免疫应答和神经炎症)的水平升高。观察到的患者分组不是由于样品来源、年龄差异或认知损害的严重程度(图9A),并且表明cf-mRNA谱分析可用于AD患者的非侵入性子类型化。
接下来,为了更好地理解这些通路/过程中的变化与AD进展之间的关系,研究了这些聚类中的任一个是否与患者临床痴呆评定量表(CDR)评分相关。分析揭示了两个基因聚类,聚类3(“突触传递”)和聚类5(“免疫应答、神经炎症”)的标准化表达值与CDR评分显著相关(图3D)。特别地,“突触传递”基因聚类显示表达随着CDR评分的增加而降低(r=-0.48,相关p值p<0.0001),并且甚至在无痴呆的个体(CDR=0)和患有非常轻度痴呆的患者(CDR=0.5)之间观察到显著差异(p=0.001)。相反,“免疫应答和神经炎症”聚类的表达水平随着CDR评分而增加(r=0.54,相关p值p<0.0001),在CDR阶段0至1之间发生最剧烈的变化。
基于这些观察结果,寻找其表达水平与疾病严重程度显著相关的单个基因。鉴定出与CDR评分相关的707个基因(FDR<0.05,图9B)。基因本体分析揭示这些基因主要参与蛋白稳态、氧化磷酸化和线粒体功能紊乱(图9C),所有已知的都与AD相关。为了确保基因始终与认知损害相关,使用MMSE评分重复相同的分析,MMSE评分是认知损害评估的另一种广泛使用的临床度量。519个基因与MMSE评分相关(图9B)。与CDR和MMSE评分相关的鉴定的基因以及使用这些基因鉴定的分子通路明显重叠(图9C和9D)。有趣的是,已显示一种参与前mRNA剪接的基因SLU7在衰老个体和患有神经变性病症的患者(26)的脑组织中失调,与CDR和MMSE评分高度相关(图9D和8E)。
实施例10-基于cfRNA的衰老研究
收集294名个体的基因表达、cfRNA数据,每名个体具有先前测量的cfRNA表达数据。记录收集表达数据的对象的年龄,并基于年龄范围将数据分成5个库(bin):20-35、35-50、50-66、66-81和81-96。计算表达数据和个体年龄之间的斯皮尔曼相关性(spearmancorrelation)。应用0.05的错误发现率(FDR)截止值,得到774个发现与年龄相关的基因。在这些774个基因中,660个与年龄正相关(上调),且114个与年龄负相关(下调)。图11-16显示了发现与年龄相关的六个基因的差异表达:TCF7、PTK2、FER、CD36、WWTR1和CAV1。
实施例11-基因表达与对氧物质反应的蛋白质的关系
将实施例10中鉴定的774个基因与基因集GO0000302“对活性氧物质的应答”进行比较。有趣的是,这些基因中有18个与年龄相关。如果显著高于单独偶然预期的,则这在基因集之间形成重叠,P值为4.99e-。
实施例12-与年龄相关的非血液基因
对三个个体的全血、血沉棕黄层和单独的cf-RNA进行测序。在测序的512个非血液基因中,意味着它们仅在cf-RNA部分中发现,40个通过实施例10中使用的方法显示与年龄相关。
实施例13-年龄相关基因与其他数据集的比较
将实施例10中鉴定的774个年龄相关基因与其他数据集中鉴定的年龄相关基因进行比较,总结在图17中。甚至在调整混杂临床参数之前,鉴定的774基因与基因集重叠良好。两个基因NELL2和TLB在所有数据集中始终与年龄高度相关,因为它们位于所有数据集中前30个差异表达的基因中。
实施例14-混杂因素影响(confounder effect)的校正
应用多变量回归来校正混杂因素对收集的774个年龄相关基因的表达数据的影响。示例性混杂因素包括但不限于预处理方案(旋转、过滤等)、生物流体的类型(血清与血浆)和样品来源(哪个中心/大学/医院)。回归的结果是120个基因与年龄显著相关(FDR<0.1)。
在与年龄相关的120个基因中,15个基因显示出随年龄降低的表达。这15个基因包括:LEF1、TCF7和BCL11B。
在与年龄相关的120个基因中,105个基因显示出随年龄增加的表达。这105个基因包括:ID1、CDKN1C、CDH5和PPARG。
实施例15-显示随年龄和非血液基因表达增加的120个基因的重叠
在显示与年龄相关的来自实施例14的120个基因中,41个与实施例12中测序的非血液基因重叠。图18显示了41个基因的热图。关系的p值为3.93e-11。41个基因包括HMGN5、PPARG、FABP4、C1orf115、RAPGEF3、AFAP1L1、RAPGEF5、ERG、LIMCH1、ID1、LMCD1、NNMT、PALM、PRKCDBP、PTRF、FAM167B、RAMP2、TINAGL1、SNCG、RBPi、MGP、IL33、S100A16、NRN1、TEAD4、RAI14、MPDZ、CDH5、LAMA4、C8orf4、PALMD、SHROOM4、CALCRL和CYYR1。
实施例16-年龄相关基因与GTEx数据的重叠
将来自实施例15的120个基因与来自Yang等人报道的GTEx数据的年龄相关基因进行比较,总结在图19中。
虽然本文已经示出和描述了本发明的优选实施方案,但是对于本领域技术人员显而易见的是,这些实施方案仅作为示例提供。在不脱离本发明的情况下,本领域技术人员将想到许多变化、改变和替换。应当理解,在实施本发明时可以采用本文所述的本发明实施方案的各种替代方案。以下权利要求书旨在限定本发明的范围,并且由此涵盖这些权利要求书范围内的方法和结构及其等同物。
- 用于无创血糖检测的光谱分析设备和相关使用方法
- 用于无创血糖检测的光谱分析设备和相关使用方法