一种在酵母基因中定点敲入外源基因的方法
文献发布时间:2023-06-19 18:37:28
技术领域
本发明属于基因工程技术领域,具体涉及一种在酵母基因中定点敲入外源基因的方法。
背景技术
酿酒酵母(Saccharomyces cerevisiae)是一种典型的真核生物系统,其具有优良的鲁棒性、对苛刻发酵条件的耐受性、基因工程操作的高效性和公认的安全性,是代谢工程中最重要的宿主之一,在学术研究和工业发酵中已经得到了广泛应用。酿酒酵母中ADE2基因被破坏后,酵母的腺嘌呤(Adenine)的合成会受到抑制,当平板上的腺嘌呤被消耗完毕时,酵母试图通过自身代谢合成腺嘌呤以供应用,但是ADE2基因被破坏,合成受阻,又因为ADE4、ADE5、ADE6、ADE7、ADE8基因均正常,从而造成中间产物P-ribosylamino imidazole(AIR)在细胞中积累使得菌落变成粉色。因此,可以将ADE2基因作为靶向基因,进行基因编辑,包括敲除、敲入、替换和点突变等,例如,在ADE2基因的特定位点,引入外源基因片段,以实现外源基因片段功能的研究和应用,这展现了ADE2基因的重要用途。
然而,随着基因工程的进一步拓展,细胞工厂的构建与途径优化过程也暴露出了缺陷和局限,例如,需要在单一位点进行多次操作等,从而使其成为基因和代谢工程的限速步骤。CRISPR/Cas9基因编辑技术能方便地对细胞基因组进行定点突变、缺失、插入、多基因敲除以及染色体重排等,具有低成本、高效率和操作简单等特点,因而该项技术被广泛应用于生物医药、基因治疗以及作物育种等领域。
Di Carlo等首次报道了利用CRISPR/Cas系统对酿酒酵母基因进行敲除;在没有筛选标签的情况下,通过90bp同源序列实现了CAN1基因的敲除(Nucleic Acids Res,2013,41(7):4336-4343)。
本发明致力于通过同源重组,在利用较短同源臂的情况下,实现酿酒酵母ADE2基因特定位点的基因编辑,尤其是在特定位点精确引入外源基因片段,为酵母的研究提供新思路与新方法,从而使该技术和方案在今后的农业、工业中具有更广阔的应用前景。
发明内容
本发明提供了一种在酵母基因中定点敲入外源基因的方法,该方法选自如下步骤:
将携带有Cas9基因和sgRNA转录基因的载体,与外源基因片段,共同转化至酵母体内,转录后的sgRNA引导Cas9蛋白至酵母靶向基因的靶点处,切割靶点,并在靶点处引入DNA双链断裂,然后利用同源重组,将外源基因敲入靶点中,从而实现外源基因在酵母基因中的定点敲入。
在上述方法中,sgRNA转录基因是根据酵母基因的靶点序列设计的。在本发明中,所述靶点指的是,基因中任意想被本领域技术人员敲入外源基因的核苷酸位点,例如,酵母ADE2基因第640~641位碱基之间的核苷酸位点。
在上述方法中,外源基因片段的5'端和3'端均含有同源臂序列,所述同源臂序列与酵母靶向基因互为同源臂。同源臂序列能够介导外源基因与酵母靶向基因之间发生同源重组,从而实现外源基因的敲入。在本发明中,外源基因可选自任意想被本领域技术人员敲入酵母基因的基因片段,其可以是单基因片段,也可以是双基因片段、三基因片段以及四基因片段等多基因片段。
本发明提供了一种携带有Cas9基因和sgRNA转录基因的载体,即pACT2-Cas9-ADE2载体,由如下方法制备而成:
将pACT2-AD载体利用HindIII限制性内切酶进行酶切处理,去除ADH1启动子与ADH1终止子之间的部分;以CPB载体为模板,使用引物Cas9 F、引物Cas9 R,通过KOD-Plus高保真酶进行Cas9及核定位信号的扩增,获得Cas9基因片段;将酶切后的pACT2-AD载体与Cas9基因片段利用连接酶进行连接,获得pACT2-Cas9载体;将pACT2-Cas9载体使用HpaI限制性内切酶进行酶切处理,获得线性化pACT2-Cas9载体,将线性化载体和sgRNA转录基因片段利用NEB连接酶进行连接,获得pACT2-Cas9-ADE2载体。
上述pACT2-Cas9-ADE2载体制备方法中,引物Cas9 F的核酸序列选自SEQ ID NO:14,引物Cas9 R的核酸序列选自SEQ ID NO:15。
上述pACT2-Cas9-ADE2载体制备方法中,可在sgRNA转录基因片段中添加启动子,用于sgRNA的转录启动。所述启动子选自SNR52启动子。
在一个具体实施方案中:
采用酿酒酵母实施本发明的上述技术方案,靶向基因选自ADE2基因,靶点选自ADE2基因第640~641位碱基之间的核苷酸位点,靶点序列选自aattgtagagactatccaca,sgRNA转录基因片段可根据该靶点序列进行设计,以使转录形成的sgRNA能够识别ADE2基因中的该靶点序列,引导Cas9蛋白切割靶点,为同源重组创造有利条件。
本发明的有益效果为:
酿酒酵母中ADE2基因被破坏后,酵母的腺嘌呤(Adenine)的合成会受到抑制,当平板上的腺嘌呤被消耗完毕时,酵母试图通过自身代谢合成腺嘌呤以供应用,但是ADE2基因被破坏,合成受阻,又因为ADE4、ADE5、ADE6、ADE7、ADE8基因均正常,从而造成中间产物P-ribosylamino imidazole(AIR)在细胞中积累使得菌落变成粉色。因此,可以将ADE2基因作为靶向基因,在特定位点引入外源基因片段,以实现外源基因片段功能的研究和应用,这展现了ADE2基因的重要用途。
本发明取得了至少如下技术效果:首次在酿酒酵母ADE2基因第640~641位碱基之间敲入外源基因;并证明了在该位点进行操作,能以更短的同源臂序列完成同源重组。这在现有技术的基础上具有突出贡献;因为,同源臂序列越长,引物合成的时间越长,实验成本越高;同时,同源臂越长,出现突变的几率将大幅增加,从而引起碱基的替换、缺失等问题,造成同源重组的效率和效果变差。因此,当人们研究外源基因片段的功能时,可根据本发明提供的靶点序列(aattgtagagactatccaca)设计sgRNA,通过CRISPR/Cas9技术,以最短的同源臂序列实现外源基因在ADE2基因第640~641位碱基之间的插入,从而能够减少突变几率,保证了外源基因功能研究的准确性。
附图说明
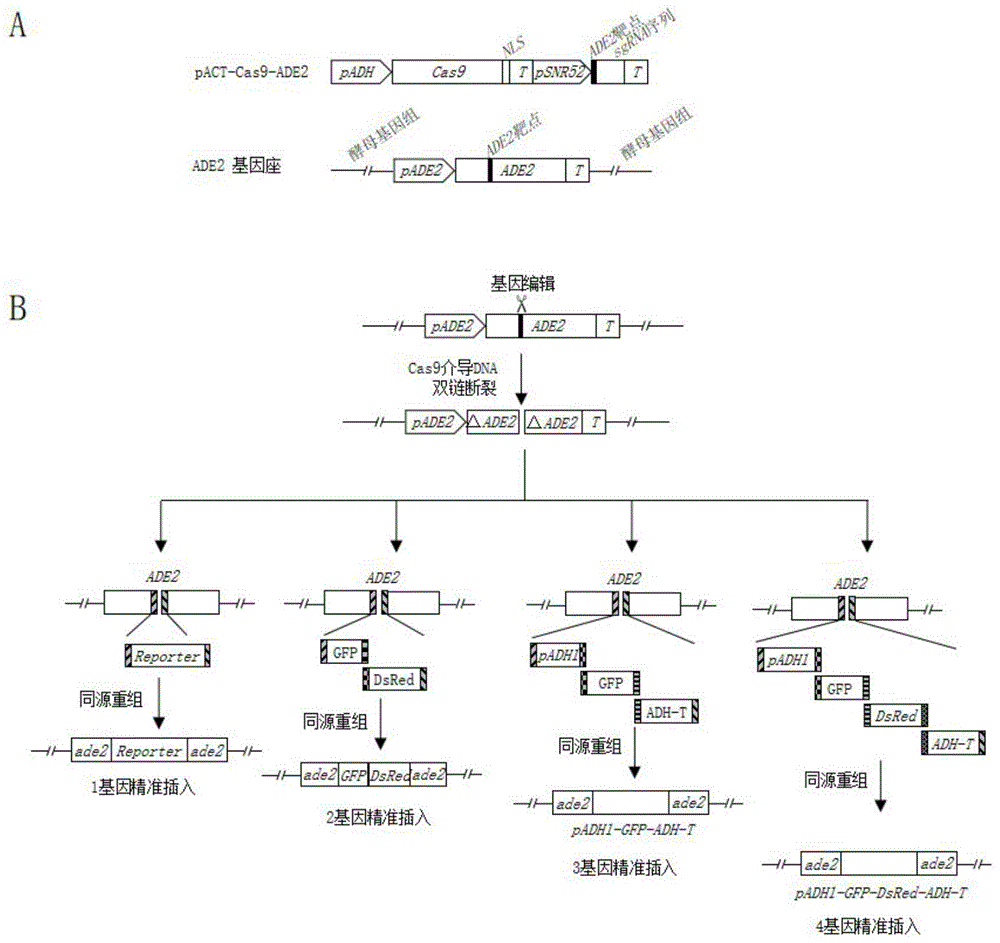
图1为CRISPR-Cas9基因编辑技术介导酿酒酵母基因组精准插入相关载体及技术原理;其中,A图为CRISPR-Cas9介导的酵母内源基因ADE2基因编辑载体pACT-Cas9-ADE2图谱及ADE2基因座图谱;B图为CRISPR-Cas9基因编辑技术介导外源基因1~4个片段精准插入ADE2基因座原理图。
图2为CRISPR-Cas9介导单个基因片段精准插入;其中,A图为ADE2基因座精准插入GFP菌液及菌落表型,Bar=100μm;B图为ADE2基因座精准插入GFP序列测序峰图;C图为ADE2基因座精准插入DsRed菌液及菌落表型,Bar=100μm;D图为ADE2基因座精准插入DsRed序列测序峰图;
图3为CRISPR-Cas9介导双基因片段精准插入;其中,A图为ADE2基因座精准插入GFP基因及DsRed基因菌液及菌落表型,Bar=100μm;B图为ADE2基因座精准插入GFP基因及DsRed基因序列测序峰图;
图4为CRISPR-Cas9介导酿酒酵母基因组ADE2基因座多基因片段精准插入,ADE2基因座精准插入pADH1启动子、GFP基因及ADH1终止子序列测序峰图;
图5为CRISPR-Cas9介导酿酒酵母基因组ADE2基因座多基因片段精准插入,ADE2基因座精准插入pADH1启动子、GFP基因、DsRed基因及ADH1终止子序列测序峰图;
图6为目的质粒用量优化试验图;
图7为Carrier DNA用量优化试验图;
图8为同源重组片段用量优化试验图。
具体实施方式
本发明所使用的材料、方法,如下所示:
商业化载体pACT2-AD:购自北京华越洋生物科技有限公司,VECT2400。
酵母BY4741感受态细胞:购自北京酷来搏科技有限公司,CC301。
Carrier DNA:购自北京酷来搏科技有限公司,YT0003。
CPB载体:由实验室保存,参考文献为:Li C,et.al.RNA-guided Cas9 as an invivo desired-target mutator in maize.Plant Biotechnology Journal.2017,15(12):1566-1576.doi:10.1111/pbi.12739。
DFP双荧光载体:由实验室保存,参考文献为:Le D,et.al.Genome Editing andDouble Fluorescence Proteins Enable Robust Maternal Haploid Induction andIdentification in Maize.Molecular Plant,2018:S1674205218302181.
基因靶点设计网站:http://staff.biosustain.dtu.dk/laeb/crispy_cenpk/。
KOD-Plus反应程序如下:
94℃2min,循环1次;94℃15s,59℃30s,68℃1kb/min,循环35次;68℃7min,循环1次;12℃,循环∞次。
PCR扩增体系:
10×Buffer,5μL;2mM dNTP,5μL;25mM MgSO
本发明在具体实施方式中选择以酵母ADE2基因为靶向基因。酵母中ADE2基因被破坏后,酵母的腺嘌呤(Adenine)的合成会受到抑制,当平板上的腺嘌呤被消耗完毕时,酵母试图通过自身代谢合成腺嘌呤以供应用,但是ADE2基因被破坏,合成受阻,又因为ADE4、ADE5、ADE6、ADE7、ADE8基因均正常,从而造成中间产物P-ribosylamino imidazole(AIR)在细胞中积累使得菌落变成粉色。
本发明中所使用的其它术语,除非有另外说明,一般具有本领域普通技术人员通常理解的含义。下面结合具体实施例,并参照数据进一步详细的描述本发明。以下实施例只是为了举例说明本发明,而非以任何方式限制本发明的范围。
1、pACT2-Cas9-ADE2载体的构建
以商业化载体pACT2-AD为基础载体,使用HindⅢ限制性内切酶将ADH1启动子与ADH1终止子之间的GAL4激活域(activation domain)及一段载体骨架进行切除,通过琼脂糖凝胶电泳分离目标条带,并进行目的条带的切胶回收。
以CPB载体为Cas9模板,使用引物Cas9 F、引物Cas9 R,通过KOD-Plus高保真酶进行Cas9及核定位信号(NLS)的扩增,通过琼脂糖凝胶电泳分离目的条带,获得大小为4180bp的Cas9基因片段,切胶回收。
将酶切后的pACT2-AD载体与Cas9基因片段进行连接,连接时使用
以酵母ADE2基因为靶标基因设计靶点,经过比较分析,最终确定ADE2基因的靶点为AATTGTAGAGACTATCCACA(SEQ ID NO:37)。靶点确定后可用于设计sgRNA。sgRNA的作用在于,引导Cas9蛋白切割靶向基因ADE2,使DNA双链产生断裂(DSB)。
以CPB载体为sgRNA模板,使用ADE2+sgRNA F、ADE2+sgRNA R引物,进行gRNAscaffold的扩增,获得长度为124bp的目的片段,切胶回收,获得sgRNA片段。
以酵母基因组为模板,使用ADE2+SNR52 F、ADE2+SNR52 R引物,通过KOD-Plus反应程序,进行SNR52启动子的扩增,获得长度为312bp的目的片段,切胶回收,获得SNR52启动子片段。
将SNR52启动子片段、sgRNA片段与酶切后的pACT2-Cas9载体片段使用
将pACT2-Cas9载体使用HpaI(NEB)限制性内切酶进行酶切处理,获得线性化pACT2-Cas9载体,将线性化载体和SNR52-sgRNA片段使用NEB连接酶进行连接,转化大肠杆菌,待菌斑长出大小均匀且适宜的单菌落时,挑取培养基上生长的单克隆进行菌落PCR,选择条带大小正确的菌落进行sanger测序,测序结果如SEQ ID NO:1所示。将验证正确的进行质粒的提取,获得pACT2-Cas9-ADE2载体。该载体的靶向基因为酵母ADE2基因,其靶点在酵母ADE2基因上。
构建完成的pACT2-Cas9-ADE2载体包含两个组成成分:一个是Cas9表达框,由酵母pADH1启动子启动的Cas9基因并以酵母ADH1-T终止子结束;另外一个是靶向酵母内源基因ADE2的sgRNA表达盒单元,由酵母SNR52启动子启动的ADE2基因靶点以及sgRNA骨架组成;如图1A所示,即pADH-Cas9-NLS-ADH终止子-SNR52-sgRNA。
当pACT2-Cas9-ADE2载体通过遗传转化步骤转入酵母细胞中后,pADH1启动子启动Cas9基因表达,同时酵母内源SNR52启动子启动以ADE2基因为靶点的sgRNA转录,随后由sgRNA引导Cas9蛋白对酵母内源的ADE2基因的640-641位碱基之间引入切点,从而使DNA双链产生断裂。
2、同源重组DNA片段的制备
由于酵母体内缺少DNA非同源末端修复方式,而同源重组修复途径比较活跃,因此,为完成外源DNA片段在酵母体内的同源重组,需同时向胞内引入相应的同源重组DNA片段,即同源臂。同源重组片段在制备时已经包含同源臂序列,同源臂序列是通过引物的方式添加在片段上的。各同源重组DNA片段的制备,如下文所示:
(1)单基因同源重组片段制备:
以DFP双荧光载体为GFP及DsRed基因片段模板。使用引物ADE2+EGFP F、引物ADE2+EGFP R进行EGFP单片段的扩增。使用引物ADE2+DsRed F、引物ADE2+DsRed R进行DsRed单片段的扩增。
(2)双基因同源重组片段制备:
以DFP双荧光载体为GFP及DsRed基因片段模板。使用引物ADE2+EGFP F、引物EGFP+DsRed R进行EGFP+DsRed双片段定点插入中EGFP片段的扩增。使用引物EGFP+DsRed F、引物ADE2+DsRed R进行EGFP+DsRed双片段定点插入中DsRed片段的扩增。
(3)三基因同源重组片段制备:
以商业化载体pACT2-AD为pADH1启动子以及ADH1-T终止子基因片段的扩增模板,以DFP双荧光载体为GFP基因片段模板。使用引物ADE2+ADH1启F、引物EGFP+ADH启R进行pADH1启动子片段的扩增。使用引物ADH启+EGFP F、引物ADH终+EGFP R进行EGFP片段的扩增。使用引物EGFP+ADH终F、引物ADH终+ADE2 R进行ADH1-T终止子片段的扩增。
(4)四基因同源重组片段制备:
以商业化载体pACT2-AD为pADH1启动子以及ADH1-T终止子基因片段的扩增模板,以DFP双荧光载体为GFP基因以及DsRed基因片段为模板。使用引物ADE2+ADH1启F、引物EGFP+ADH启R进行pADH1启动子片段的扩增。使用引物ADH启+EGFP F、引物DsRed+EGFP R进行EGFP片段的扩增。使用引物EGFP+DsRed F、引物ADH终+DsRed R进行DsRed片段的扩增。使用引物DsRed+ADH终F、引物ADH终+ADE2 R进行ADH1-T终止子片段的扩增。
上述同源重组片段在制备时所采用的PCR扩增体系及扩增反应程序,与载体构建时使用的程序相同。扩增后PCR产物通过琼脂糖凝胶电泳分离目标条带,并进行目的条带的切胶回收,使用聚和美胶回收试剂盒进行回收。
3、酵母遗传转化
将pACT2-Cas9-ADE2载体和同源重组片段共转入酵母中,使用SD-Leu固体培养基进行涂板并置于30℃下进行培养;
转化步骤如下:
(1)取100μL冰上融化的酵母BY4741感受态细胞,依次加入1~5μL预冷的目的质粒(pACT2-Cas9-ADE2载体),2~10μL同源重组片段,10μL Carrier DNA(10ng/μL),500μLPEG/LiAc,并吸打几次混匀。上述Carrier DNA在加入前需进行变性处理:95-100℃,5min,快速冰浴,重复一次;然后置于冰上,5min之内使用。
(2)30℃水浴30min,其中,在15min时翻转6~8次混匀。
(3)42℃水浴15min,其中,在7min时翻转6~8次混匀。
(4)12000rpm离心15s弃上清,使用800~1000μL YPDA重悬,置于30℃摇床中220rpm/min摇60~90min。
(5)12000rpm离心15s弃上清,使用800μL ddH
上述pACT2-Cas9-ADE2载体和同源重组片段的添加量,如下所示:
当定点插入单片段时,添加3μL pACT2-Cas9-ADE2载体质粒,浓度为120ng/μL;添加加2μL同源重组片段,浓度为70ng/μL。
当定点插入双片段时,添加3μL pACT2-Cas9-ADE2载体质粒,浓度为120ng/μL;添加加2μL同源重组片段,浓度为60ng/μL。
当定点插入三片段时,添加4μL pACT2-Cas9-ADE2载体质粒,浓度为120ng/μL;添加加2μL同源重组片段,浓度为70ng/μL。
当定点插入四片段时,添加4.5μL pACT2-Cas9-ADE2载体质粒,浓度为120ng/μL;添加加2.5μL同源重组片段,浓度为70ng/μL。
4、单菌株及菌落荧光表型筛选
单菌株的观察采用莱卡DM1000荧光显微镜。
将菌液滴加在玻片上,利用488nm荧光观察GFP荧光蛋白表达情况,利用530nm激发光源观察DsRed蛋白表达状态。菌落的观察使用美国路阳LUYOR-3415RG便携式荧光蛋白激发光源,佩戴黄色眼镜,使用蓝色激发光照射培养皿中菌落,观察定点插入GFP片段的菌落发光情况。佩戴红色观察眼镜使用绿光激发光照射培养皿中菌落,观察定点插入DsRed片段的菌落发光情况。
将pACT2-Cas9-ADE2载体与单基因片段、双基因片段、三基因片段或四基因片段转入酵母细胞中后,Cas9蛋白在sgRNA引导下切割ADE2基因第640-641位碱基位点,引入DNA双链断裂(DSB),产生缺口,利用酿酒酵母细胞内HR DNA损伤修复途径,通过同源重组,从而实现带有同源臂的外源片段的定点插入,如图1B所示。
当GFP片段3’端与5’端分别含有27bp同源臂序列(SEQ ID NO:2),且与ADE2基因互为同源臂;此时,胞内通过同源重组作用在ADE2基因第214位氨基酸处引入GFP基因序列,并使得ADE2蛋白的N端第214位氨基酸与GFP蛋白融合表达,此时菌落或单菌株在488nm下激发出绿色荧光。
当DsRed片段3’端与5’端分别含有27bp同源臂序列(SEQ ID NO:3),且与ADE2基因互为同源臂;此时,胞内通过同源重组作用在ADE2基因第214位氨基酸处引入DsRed基因序列,并使得ADE2蛋白的N端第214位氨基酸与DsRed蛋白融合表达,此时菌落或单菌株在530nm下激发出红色荧光。
当GFP片段3’端含有27bp同源臂序列(SEQ ID NO:4),且与ADE2基因互为同源臂;GFP片段5’端含有27bp同源臂序列(SEQ ID NO:4),且与DsRed基因3’端含有的27bp同源臂序列(SEQ ID NO:5)互为同源臂;DsRed片段5’端含有27bp同源臂序列(SEQ ID NO:5),且与ADE2基因互为同源臂;此时,胞内通过同源重组作用在ADE2基因第214位氨基酸处引入GFP基因序列与DsRed基因序列,并使得ADE2蛋白的N端第214位氨基酸与GFP蛋白及DsRed蛋白融合表达,此时菌落或单菌株在488nm下激发出绿色荧光,而在530nm下激发出红色荧光。
当ADH1启动子3’端含有27bp同源臂序列(SEQ ID NO:6),且与ADE2基因互为同源臂;ADH1启动子5’端含有27bp同源臂序列(SEQ ID NO:6),且与GFP片段3’端含有的27bp同源臂序列(SEQ ID NO:7)互为同源臂;GFP片段5’端含有27bp同源臂序列(SEQ ID NO:7),且与ADH1终止子3’端含有的27bp同源臂序列(SEQ ID NO:8)互为同源臂;ADH1终止子5’端含有27bp同源臂序列(SEQ ID NO:8),且与ADE2基因5’互为同源臂序列;此时,胞内通过同源重组作用在ADE2基因第214位氨基酸处引入ADH1启动子序列、GFP基因及ADH1终止子序列,并使得ADE2蛋白的N端第214位氨基酸与ADH1启动子、GFP蛋白及ADH1终止子融合表达,此时菌落或单菌株在488nm下激发出绿色荧光。
当ADH1启动子3’端含有27bp同源臂序列(SEQ ID NO:9),且与ADE2基因互为同源臂;ADH1启动子5’端含有27bp同源臂序列(SEQ ID NO:9),且与DsRed片段3’端含有的27bp同源臂序列(SEQ ID NO:10)互为同源臂;DsRed片段5’端含有27bp同源臂序列(SEQ ID NO:10),且与ADH1终止子3’端含有的27bp同源臂序列(SEQ ID NO:11)互为同源臂;ADH1终止子5’端含有27bp同源臂序列(SEQ ID NO:11),且与ADE2基因5’互为同源臂序列;此时,胞内通过同源重组作用在ADE2基因第214位氨基酸处引入ADH1启动子序列、DsRed基因及ADH1终止子序列,并使得ADE2蛋白的N端第214位氨基酸与ADH1启动子、DsRed蛋白及ADH1终止子融合表达,此时菌落或单菌株在530nm下激发出红色荧光。
当ADH1启动子3’端含有27bp同源臂序列(SEQ ID NO:6),且与ADE2基因互为同源臂;ADH1启动子5’端含有27bp同源臂序列(SEQ ID NO:6),且与GFP片段3’端含有的27bp同源臂序列(SEQ ID NO:12)互为同源臂;GFP片段5’端含有27bp同源臂序列(SEQ ID NO:12),且与DsRed片段3’端含有的27bp同源臂序列(SEQ ID NO:13)互为同源臂;DsRed片段5’端含有27bp同源臂序列(SEQ ID NO:13),且与ADH1终止子3’端含有的27bp同源臂序列(SEQ IDNO:11)互为同源臂;ADH1终止子5’端含有27bp同源臂序列(SEQ ID NO:11),且与ADE2基因5’互为同源臂序列;此时,胞内通过同源重组作用在ADE2基因第214位氨基酸处引入ADH1启动子序列、GFP基因、DsRed基因及ADH1终止子序列,并使得ADE2蛋白的N端第214位氨基酸与ADH1启动子、GFP蛋白、DsRed蛋白及ADH1终止子融合表达,此时菌落或单菌株在488nm下激发出绿色荧光,而在530nm下激发出红色荧光。
5、同源重组测序验证
为判断ADE2基因定点插入单基因片段、双基因片段、三基因片段或四基因片段同源重组准确性,将转化获得的阳性菌落进行基因组提取,通过在ADE2基因插入位点上下游设计了引物ADE2-1 F、引物ADE2-1 R,用于扩增ADE2靶点处插入的片段,获得PCR产物经过Sanger测序并进行比对,鉴定ADE2基因座内单基因片段、双基因片段、三基因片段及四基因片段同源重组的准确性。
测序结果表明:
ADE2基因第214位氨基酸处精准插入GFP核酸序列,GFP片段3’端与5’端均与ADE2基因衔接正确(图2B)。
ADE2基因第214位氨基酸处精准插入DsRed核酸序列,DsRed片段3’端与5’端均与ADE2基因衔接正确(图2D)。
ADE2基因第214位氨基酸处精准插入GFP、DsRed核酸序列。GFP片段3’端与ADE2衔接正确、GFP片段5’端与DsRed片段3’端衔接正确、DsRed片段5’端与ADE2基因衔接正确(图3B)。
ADE2基因第214位氨基酸处精准插入ADH1启动子、GFP、ADH1终止子核酸序列,ADH1启动子3’端与ADE2基因衔接正确、ADH1启动子5’端与GFP片段3’端衔接正确、GFP片段5’端与ADH1终止子3’端衔接正确、ADH1终止子5’端与ADE2基因衔接正确(图4)。
ADE2基因第214位氨基酸处精准插入ADH1启动子、DsRed、ADH1终止子核酸序列,ADH1启动子3’端与ADE2基因衔接正确、ADH1启动子5’端与DsRed片段3’端衔接正确、DsRed片段5’端与ADH1终止子3’端衔接正确、ADH1终止子5’端与ADE2基因衔接正确。
ADE2基因第214位氨基酸处精准插入ADH1启动子、GFP、DsRed、ADH1终止子核酸序列,ADH1启动子3’端与ADE2基因衔接正确、ADH1启动子5’端与GFP片段3’端衔接正确、GFP5’端与与DsRed片段3’端衔接正确、DsRed片段5’端与ADH1终止子3’端衔接正确、ADH1终止子5’端与ADE2基因衔接正确(图5)。
本发明所涉及的引物序列,如下述表1所示:
表1
使用上述引物扩增目标产物,均按常规的PCR扩增程序进行。
优化例1转化条件的优化
上述试验内容已对本发明的技术方案进行了完整地阐述和实施。但为了优化影响转化效率的各种因素,按照单一变量原则,进行以下试验:
1、优化目的质粒的量
设计不同的质粒浓度梯度,在转化时加入不同的质粒量,统计相同菌液量中菌的数目,以获得最佳转化效果时质粒的量。试验结果如图6所示,在一定范围内,增加质粒的量,可使得转化时菌落数目增加,但当质粒的量达到一定数值时,随着质粒的增加,菌落数目没有明显改变。综合来看,当质粒的量为160ng时,转化效果较好。
2、优化Carrier DNA的量
在确定好最佳质粒量的条件下,设计不同梯度的carrier DNA,确定转化时最佳Carrier DNA的量。在相同的质粒量条件下,探索Carrier DNA的量,三次重复,取平均值,试验结果如图7所示,在Carrier DNA的量为80ng时,菌落数目最多。
3、优化同源重组片段的量
在确定质粒与Carrier DNA的量的条件下,探索转化时同源重组片段的量,设置8个梯度,三次重复。试验结果如图8所示,在片段的量为500ng时,转化的效果较好。
本发明具体实施方式所涉及的其它核酸序列,如下所示,其中,下划线处所示的序列为同源臂序列:
SEQ ID NO:1:
SEQ ID NO:2:
SEQ ID NO:3:
SEQ ID NO:4:
SEQ ID NO:5:
SEQ ID NO:6:
SEQ ID NO:7:
SEQ ID NO:8:
SEQ ID NO:9:
SEQ ID NO:10:
SEQ ID NO:11:
SEQ ID NO:12:
SEQ ID NO:13:
以上所述,仅是本发明的较佳实施例而已,并非是对本发明作其它形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更或改型为等同变化的等效实施例。但是凡是未脱离本发明技术方案内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与改型,仍属于本发明技术方案的保护范围。