一种诱导间质细胞向上皮细胞转化及重编程的诱导剂
文献发布时间:2023-06-19 19:27:02
技术领域
本发明涉及医药领域,具体涉及一组化合物,及其在诱导间质细胞向上皮细胞转化过程中,以及重编程中的应用。
背景技术
上皮细胞和间充质细胞之间的相互转化是一个高度保守和可逆的细胞过程,其中极化的、不动的上皮细胞从其基底表面延伸丝状伪足并产生迁移的间充质细胞。上皮-间充质和间充质-上皮转化是公认的生物学事件,它们不仅在正常组织和器官发育中,而且在疾病的发病机制中都具有重要作用。上皮和间充质状态之间的细胞表型变化,分为上皮-间质转化(EMT)和间充质-上皮转化(MET),这种细胞形态的转换不仅是原肠胚形成和器官发生过程中胚胎和器官结构复杂重塑的核心,它们也被公认作为许多癌症转移的关键事件(Thiery.JP.Nat Rev Cancer.2002;2:442-54)。
在果蝇的早期发育中,新形成的上皮胚层将参与复杂的形态发生运动以使胚胎发育,比如形成的外胚层细胞将保留其上皮表型(Tepass,U.Bioessays 19,1997,673–682;Tepass,U.&Hartenstein,V.Dev.Biol.161,1994:563–596)。中胚层的发育过程也观察到EMT-MET循环,这些细胞通过腹侧沟内陷形成背侧血管性腺片和马尔皮基小管(Campbell,K.et al.,Mech.Dev.2010.127,345–357)。大多数其他后生动物的发育是通过受精卵的一系列分裂和上皮样结构的逐步组装进行的(Stern,C.D.(ed.).Gastrulation:From Cellsto Embryos(Cold Spring Harbor Laboratory Press,New York,2004)。因此MET发生在正常发育过程中,包括了体细胞发生、肾脏发育、心脏发生、肝发生和体腔形成等过程(Bin Let al.,PLoS One.2011;6(2):e17092;Nakajima Y et al.,Anat Rec.2000;258:119–127)。上述发现显示,在每个器官形态发生过程中MET的机制是相似的,呈现出上皮相关基因被上调而间充质基因被下调的统一趋势,但每个过程都有独特的信号通路来诱导MET和基因表达的相关变化。
类似的,胚胎干细胞(ESC)或诱导多能干细胞(iPSCs)也经历EMT和MET分化成体细胞类型。那么在分化的体细胞也可以重新编程通过连续的EMT-MET进入多能性状态,其中MET是获得多能性的关键步骤(Shu,X.&Pei,D.Curr.Opin.Genet.Dev.28,2014,32–37)。研究表明,重编程中的MET是通过与代谢转换、表观遗传修饰协同作用实现了细胞命运变化(Wu,J.,Ocampo,A.和Belmonte,J.C.I.Cell,2016,166,1371–1385)。研究表明MET在小鼠胚胎以及人类成纤维细胞的早期体细胞重编程过程中起到非常关键的作用(Hofding,M.K.和Hyttel,P.Stem Cell Res.2015,14,39–53;Subramanyam,D.et al.Nat.Biotechnol.2011,29,443–448;Li,R.et al.Cell Stem Cell.2010,7,51–63)。在这一过程中,BMP4途径对MET的启动起到了关键性的作用,这一途径通过激活CDH1(编码E-钙粘蛋白)内含子2和启动子CLDN4(编码claudin-4)以增加MET的作用。目前,可以根据蛋白结构预测工具预测出与目标蛋白相结合的化合物,通过功能筛选得到上调目标蛋白的化合物;与此同时,多种微型核糖核酸(mirRNA)已被发现可以与MET发生呈现高度的相关性,比如mir-134,mir-145,mir-470和mir-200c,都表现出对MET负调控特征(Esther E Creemers 1,Anke J Tijsen,Yigal MPinto,Circ Res,2012Feb 3;110(3):483-95;Li,R.et al.Cell Stem Cell.2010,7,51–63)。因此,可以根据这类调控性微型核糖核酸(mirRNA)的结构设计出抑制性化合物,从而实现对MET现象的正调控。
因此,MET这一生物学现象在多种发育过程中起到重要作用,目前有多种手段在体外对发育进行调控,从而重建老化或功能损失的组织和器官,并通过多种医学手段来进行相关疾病治疗。这正是再生医学的重要研究方向,聚焦正常组织特征与功能的机理,探索创伤后修复的生物学基础,以及组织器官的再生机制及多种干细胞分化机理,从而最终获得有效的生物治疗方法。其中,胚胎干细胞(Embryonic stem cells,ESCs,简称ES、EK或ESC细胞)和诱导多能干细胞(induced pluripotent stem cell)是其中最为瞩目的研究材料。但是目前对发育的调控多涉及到基因组改造,包括病毒载体调控在内的多种手段均存在潜在的致瘤性风险。因此,使用不改变基因组序列的调控方式显得尤为重要。这其中,如果挖掘到可以调控MET的化合物,那么这种化合物可在重编程,细胞分化以及组织重建领域发挥巨大的作用,实现更安全与更灵活的调控目的。
发明内容:
基于上述原因,本发明同时根据Oct4蛋白结构,并根据结合Oct4复合物的负调控mirRNA结构,设计岀能够同时结合Oct4蛋白结构及负调控mirRNA结构的吡咯并吡啶衍生物。吡咯并吡啶衍生物能够进行Oct4化学激活,实现对其下游基因的表达调控。从而避免了使用病毒或者其它载体对Oct4的调控,进一步实现安全简便的化学小分子增强生物学表达功能。
本发明提供了吡咯并吡啶衍生物化合物的用途,可在多种细胞中实现MET这一生物学现象,引起细胞形变,同时实现上皮细胞相关基因和重编程早期基因的表达。为实现重编程的化学诱导提供了强有力的启动化合物。
其中,本发明涉及吡咯并吡啶衍生物式(I)结构的化合物或其药学上可接受的盐、溶剂化物、活性代谢物、多晶型物、酯、光学异构体或前药,包含式(I)结构的化合物的药物组合物及其作为Oct4高选择性活化剂用于细胞的重编程。
本发明提供了一种式(I)结构的化合物:
其中:
m1和m2分别为0或1;
A
A
在一些实施例中:A
在一些实施例中:A
在一些实施例中:m1是0,m2是1;A
在一些实施例中:m1是1,m2是0;A
在一些实施例中:m1是1,m2是1;A
在一些实施例中:m1是0,m2是0;A
在一些实施例中,化合物为:
本发明涉及药物组合物,药物组合物包含上述中任意一项的化合物或其药物上可接受的盐、溶剂化物、活性代谢物、多晶型物、酯、光学异构体、前药或它们中的两种以上的组合中的至少一种,以及至少一种药学上可接受的载体或赋形剂。
本发明涉及上述任意一项的所述的化合物和/或其药物上可接受的盐、溶剂化物、活性代谢物、多晶型物、酯、光学异构体、前药或其组合在制备诱导间质细胞向上皮细胞转化的用途。
本发明涉及一种活化Oct4的方法,其包括使上述任一项的化合物和/或其药物上可接受的盐、溶剂化物、活性代谢物、多晶型物、酯、光学异构体、前药或其组合与Oct4靶标蛋白接触。
附图说明
图1的上半部分显示样品池的热量变化,其中红色峰线是miR-145与A-3-6结合放热,蓝色线是不含miR-145缓冲液与A-3-6结合放热。图1的下半部分显示峰面积拟合Multiple Sites模型曲线,其中计算的nSite分别为3和1.8所对应的Kd(M)为7.017E-8和7.544E-9,空白对照组Kd(M)为1.000E-3。ITC试验结果证明了本发明得到的小分子与目标靶点能够特异性的结合;
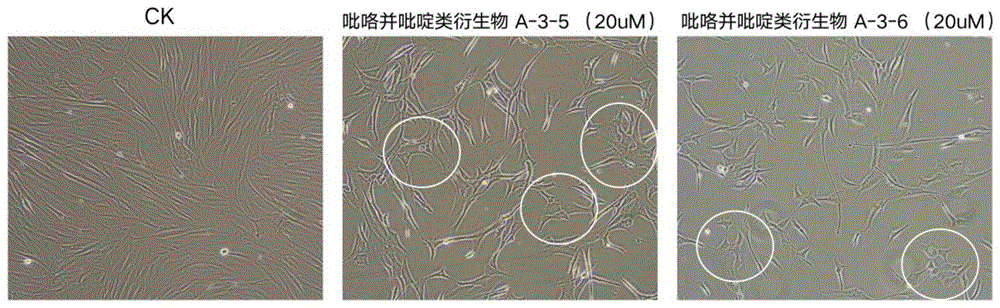
图2显示的是添加吡咯并吡啶衍生物的对照相比,在使用吡咯并吡啶类衍生物单独处理细胞24小时后就能引起MET现象发生,细胞出现典型上皮状形态(其中CK为对照组);
图3显示的是吡咯并吡啶衍生物对Oct4等基因的基础表达的结果(其中CK为对照组)。
具体实施方式
在本发明中,以下定义是可适用的:
本文的术语“烷基”是指含有1-12个碳原子的直链或支链饱和烃。(C
术语“烯基”是指含有2-12个碳原子的直链或支链不饱和烃,在链中含有至少一个C=C双键。烯基的实例包括乙烯基、丙烯基、正丁烯基、异丁烯基、戊烯基或己烯基。
术语“亚烷基”:是指二价烷基。单价烷基中的任一个可通过从烷基中夺取第二氢原子而为亚烷基。如本文所定义,亚烷基还可以是C
术语“亚烯基”:是指二价烯基。单价烯基中的任一个可通过从烯基中夺取第二氢原子而为亚烯基。如本文所定义,亚烯基可进一步是C
术语“环烷基”是指含有3-18个碳原子的单环饱和碳环。环烷基可进一步是C
术语“卤素”是指氟、氯、溴或碘。
术语“氰基”是指具有通过三键(即C≡N)与氮原子连接的碳原子的取代基。
本文所用的术语“取代”,指的是指定的原子或基团上的任意一个或多个氢原子被选自指定的范围中的基团替换,条件是不超过所述指定原子的正常化合价。
在本发明描述的某些实施方案中,本文提供了式(I)化合物,其中当A
本文所述的化合物包括但不限于:它们的光学异构体、外消旋体以及其它混合物。在这些情况下,单个对映异构体或非对映异构体,即光学活性构型,可以通过不对称合成或通过拆分外消旋体或非对映异构体混合物来获得。对于外消旋体或非对映异构体混合物的拆分,可通过常规的方法来完成,例如在拆分剂存在下结晶或使用例如手性高压液相色谱(HPLC)柱的色谱法完成。此外,这些化合物包括R-和S-构型的具有手性中心的化合物。这些化合物还包括晶型,包括多晶型物和包合物。类似地,术语“盐”也包括了所述化合物的盐的所有异构体、外消旋体、其他混合物、R-和S-构型、互变异构体和晶型。
“药物上可接受的盐”,指的是无毒的、生物学上可耐受的或其他方面在生物学上适合于给予治疗个体的式(I)、式(II)或式(III)代表的化合物的游离酸或碱的盐。一般参见:S.M.Berge,et al.,“Pharmaceutical Salts”,J.Pharm.Sci.,1977,66:1-19,以及Handbook ofPharmaceutical Salts,Properties,Selection,and Use,Stahl andWermuth,Eds.,Wiley-VCH and VHCA,Zurich,2002。优选地药学上可接受的盐是指那些药理学上有效的并且适合于接触病人组织而无异常毒性、刺激或过敏反应的盐。式(I)、式(II)或式(III)的化合物可以具有足够的酸性基团、足够的碱性基团或兼具这两种类型的功能基团,并相应地与一些无机或有机碱、及无机和有机酸反应,形成一种药学上可接受的盐。药学上可接受的盐的实例包括硫酸盐、焦硫酸盐、硫酸氢盐、亚硫酸盐、亚硫酸氢盐、磷酸盐、单氢磷酸盐、二氢磷酸盐、偏磷酸盐、焦磷酸盐、盐酸盐、氢溴酸盐、氢碘酸盐、醋酸盐、丙酸盐、癸酸盐、辛酸盐、丙烯酸盐、甲酸盐、异丁酸盐、己酸盐、庚酸盐、丙炔酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、富马酸盐、马来酸盐、丁炔-1,4-二酸盐、己炔-1,6-二酸盐、苯甲酸盐、氯代苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、羟基苯甲酸盐、甲氧基苯甲酸盐、邻苯二甲酸盐、磺酸盐、二甲苯磺酸盐、苯乙酸盐、苯丙酸盐、苯丁酸盐、柠檬酸盐、乳酸盐、γ-羟基丁酸盐、羟乙酸盐、酒石酸盐、甲烷磺酸盐、丙磺酸盐、萘-1-磺酸盐、萘-2-磺酸盐和扁桃酸盐。
“溶剂化物”例如“水合物”是通过溶剂与化合物的相互作用形成的。术语“化合物”包括化合物的溶剂化物,包括水合物。同样地,“盐”包括盐的溶剂化物,例如水合物。合适的溶剂化物是药学上可接受的溶剂化物,例如水合物,包括一水合物和半水合物。
“前药”可指指定化合物的前体,它在对受治疗者施用后,在体内经化学或生理学过程(如溶剂分解、酶分解)或在生理条件下(如在生理pH条件下将前药转化为式(I)化合物),得到该化合物。“药学上可接受的前药”是无毒的、生物学上耐受的或其它方面在生物学上适宜对受治疗者施用的前药。示例性的选择和制备合适的前药衍生物的程序在例如H.Bundgaard编着的“Design of Prodrugs”,Elsevier,1985中有描述。
“活性代谢物”指式(I)、式(II)或式(III)化合物或其盐在体内代谢的药学活性产物。可用本领域已知的或可获得的常规技术测定化合物的前药和活性代谢物。例如见Bertolini等,J.Med.Chem.1997,40,2011-2016;Shan等,J.Pharm.Sci.1997,86(7),765-767;Bagshawe,Drug Dev.Res.1995,34,220-230;Bodor,Adv.Drug Res.1984,13,224-331;Bundgaard,Design of Prodrugs(ElsevierPress,1985);及Larsen,Design andApplication of Prodrugs,Drug Design andDevelopment(Krogsgaard-Larsen et al.,eds.,Harwood Academic Publishers,1991)
“治疗有效量”是指当施用于哺乳动物(优选人)时足以实现哺乳动物(优选人)的疾病或病症的治疗(如以下定义)的本文公开的化合物的量。构成“治疗有效量”的公开的化合物的量将随着化合物、病症及其严重性和被治疗的哺乳动物的年龄而变化,但是可以由本领域普通技术人员根据他自己的知识和本文公开内容来常规确定。
术语“治疗”,指的是给予个体施用本文所述的至少一种化合物和/或至少一种其药学上可接受的盐,以减缓(减少)不希望发生的生理变化或疾病,例如炎症或癌症的发展或扩散。本发明的目的,有益的或期望的临床结果包括但不限于:减轻症状、减少疾病的严重程度、稳定(即,不恶化)疾病的状态、延迟或延缓疾病进展、改善或缓和病情、以及缓解(无论是部分还是全部)无论是检测到的还是检测不到的疾病。与那些不接受治疗的预计生存期相比,“治疗”也意味着可以延长生存期。需要治疗的个体包括具有这些疾病的症状或患有这些疾病的个体。
药物组合物
本发明提供了药物组合物,其包含一种或多种文中所述的化合物或其药学上可接受的盐或酯作为活性成分,以及一种或多种药学上可接受的赋形剂、载体,包括惰性固体稀释剂和填料、稀释剂,包括无菌水溶液和多种有机溶剂、渗透促进剂、增溶剂和助剂。该药物组合物可以单独给药或与其他治疗剂一起给药。此类组合物以药学领域公知的方式制备。
该药物组合物可以单剂量或多剂量,通过任一具有类似用途的药剂的可接受的方式给药,例如在此引入作为参考的那些专利和专利申请中所描述的,包括直肠、口含、鼻内和透皮途径、通过动脉内注射、静脉、腹膜内、肠胃外、肌内、皮下、口服、局部、作为吸入剂,或通过植入或包衣设备如支架,例如或者动脉插入柱状聚合物。
本发明还提供试剂盒,其包括包含本发明化合物或其药学上可接受的盐的药物组合物,和至少一种药学上可接受的载体。
“药学上可接受的载体或赋形剂”是指无毒的、生物学上可耐受的并且其他在生物学上适用于给予个体的物质,例如惰性物质,其被加入到药理学组合物中或者用作媒介、载体或稀释以方便活性成分的给药并与之相容。赋形剂的实例包括碳酸钙、磷酸钙、各种类型的糖或淀粉、纤维素衍生物、明胶、植物油和聚乙二醇。
“它们中的两种以上的组合”:应当理解的是“它们中的两种以上的组合”是指化合物、其药物上可接受的盐、其药物上可接受的溶剂化物、其药物上可接受的活性代谢物、其药物上可接受的多晶型物、其药物上可接受的酯、其药物上可接受的光学异构体和其药物上可接受的前药中的两种以上的组合”。
化合物及其组合物的用途
本发明提供了化合物及其组合物的用途,其主要作为Oct4高选择性活化剂,用于活化Oct4功能,通过对Oct4启动子的化学调节,实现对其下游基因的表达调控,进而将间质细胞向上皮细胞转化的用途。
在以下实施例和本文其他地方使用到的缩写列表:
CH
以下配合图式及本发明的实施例,进一步阐述本发明为达成预定发明目的所采取的技术手段。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的原料、试剂材料等,如无特殊说明,均为市售购买产品。
通用合成
本申请中所给的描述的通用合成路线可以通过用具有类似结构的其他原料替换起始原料,从而相应地得到不同的产物而进行变化。下面的合成路线描述给出了起始原料可如何变化得到相应产物的多个实例。
通用方法A-1-n
其中R为
通用方法A-3-n
其中R为C
通用方法A-4-n
其中R为
通用方法A-6-n
其中R为C
中间体合成
中间体Ⅰ-3:1H-吡咯并[2,3-c]吡啶-5-醇
合成路线:
步骤1:中间体1H-吡咯并[2,3-c]吡啶-5-重氮Ⅰ-2
向1H-吡咯并[2,3-c]吡啶-5-胺I-1(66.6mg,0.5mmol)中添加水(2mL)和20%的H
步骤2:中间体1H-吡咯并[2,3-c]吡啶-5-醇Ⅰ-3
将上述1H-吡咯并[2,3-c]吡啶-5-重氮Ⅰ-2的硫酸氢盐(48mg,0.2mmol)的水溶液(1mL),滴加到100℃的40%硫酸水溶液(5mL)中,并将混合物搅拌10分钟。向获得的反应混合物中添加NaOH至PH约为3,向反应混合物中加入乙酸乙酯,分离有机层,用盐水冲洗,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(氯仿:甲醇=50:1)纯化得到1H-吡咯并[2,3-c]吡啶-5-醇Ⅰ-3(20mg,77%),液相质谱m/z=135.1[M+H]+。
实施例1:((1H-吡咯并[2,3-c]吡啶-5-基)氧基)甲基)苯甲腈A-1-1
步骤:将1H-吡咯并[2,3-c]吡啶-5-醇Ⅰ-3(10mg,0.07mmol)与3-(溴甲基)苯甲腈(20mg,0.1mmol)与K
实施例2:5-苯乙氧基-1H-吡咯并[2,3-c]吡啶A-1-2
步骤:将1H-吡咯并[2,3-c]吡啶-5-醇Ⅰ-3(10mg,0.07mmol)与(2-溴乙基)苯(22mg,0.12mmol)与K
实施例3:(1H-吡咯并[2,3-c]吡啶-5-基)异吲哚-1-酮A-2
合成路线:
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与2-甲酰基苯甲酸(18mg,0.12mmol)混合,添加甲酸(0.2mL),三乙胺(1mL),乙醇(1mL),将混合物加热至80℃搅拌60分钟。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(二氯甲烷:乙酸乙酯=20:1)纯化得到(1H-吡咯并[2,3-c]吡啶-5-基)异吲哚-1-酮A-2(15mg,60%),液相质谱m/z=250.3[M+H]+。
实施例4:(1H-吡咯并[2,3-c]吡啶-5-基)噻吩-3-甲酰胺A-3-1
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与噻吩-2-羧酸(15mg,0.12mmol)与2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(38mg,0.1mmol)混合,添加N,N-二异丙基乙胺(0.2mL),N,N-二甲基甲酰胺(2mL),室温下搅拌18小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(乙酸乙酯:己烷=3:7)纯化得到N-(1H-吡咯并[2,3-c]吡啶-5-基)噻吩-3-甲酰胺A-3-1(10mg,41%),液相质谱m/z=244.1[M+H]+。
实施例5:(E)-2-甲基-N-(1H-吡咯并[2,3-c]吡啶-5-基)丁-2-烯酰胺A-3-2
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与(E)-2-甲基-2-烯酸(12mg,0.12mmol)与2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(38mg,0.1mmol)混合,添加N,N-二异丙基乙胺(0.2mL),N,N-二甲基甲酰胺(2mL),室温下搅拌18小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(乙酸乙酯:己烷=3:7)纯化得到(E)-2-甲基-N-(1H-吡咯并[2,3-c]吡啶-5-基)丁-2-烯酰胺A-3-2(11mg,51%),液相质谱m/z=216.1[M+H]+。
实施例6:N-(1H-吡咯并[2,3-c]吡啶-5-基)-2,3-二氢苯并呋喃-2-甲酰胺A-3-3
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与苯并呋喃-2-羧酸(19mg,0.12mmol)与2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(38mg,0.1mmol)混合,添加N,N-二异丙基乙胺(0.2mL),N,N-二甲基甲酰胺(2mL),室温下搅拌18小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(乙酸乙酯:己烷=3:7)纯化得到N-(1H-吡咯并[2,3-c]吡啶-5-基)-2,3-二氢苯并呋喃-2-甲酰胺A-3-3(13mg,46%),液相质谱m/z=280.1[M+H]+。
实施例7:3,4-二氯-N-(1H-吡咯并[2,3-c]吡啶-5-基)苯甲酰胺A-3-4
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与3,4-二氯苯甲酸(23mg,0.12mmol)与2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(38mg,0.1mmol)混合,添加N,N-二异丙基乙胺(0.2mL),N,N-二甲基甲酰胺(2mL),室温下搅拌18小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(乙酸乙酯:己烷=3:7)纯化得到3,4-二氯-N-(1H-吡咯并[2,3-c]吡啶-5-基)苯甲酰胺A-3-4(13mg,43%),液相质谱m/z=306.0[M+H]+。
实施例8:N-(1H-吡咯并[2,3-c]吡啶-5-基)苯并噻吩-2-甲酰胺A-3-5
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与苯并噻吩-2-羧酸(21mg,0.12mmol)与2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(38mg,0.1mmol)混合,添加N,N-二异丙基乙胺(0.2mL),N,N-二甲基甲酰胺(2mL),室温下搅拌18小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(乙酸乙酯:己烷=3:7)纯化得到N-(1H-吡咯并[2,3-c]吡啶-5-基)苯并噻吩-2-甲酰胺A-3-5(14mg,48%),液相质谱m/z=294.1[M+H]+。
实施例9:2-苯基-N-(1H-吡咯并[2,3-c]吡啶-5-基)乙酰胺A-3-6
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与2-苯乙酸(16mg,0.12mmol)与2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(38mg,0.1mmol)混合,添加N,N-二异丙基乙胺(0.2mL),N,N-二甲基甲酰胺(2mL),室温下搅拌18小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(乙酸乙酯:己烷=3:7)纯化得到2-苯基-N-(1H-吡咯并[2,3-c]吡啶-5-基)乙酰胺A-3-6(12mg,48%),液相质谱m/z=252.1[M+H]+。
实施例10:1-(1H-吡咯并[2,3-c]吡啶-5-基)-3-(对甲苯基)脲A-4-1
步骤1:对硝基苯基对甲苯氨基碳酸酯I-9-1
将对甲苯胺(21mg,0.2mmol)与4-硝基苯基羰酰氯(48mg,0.24mmol)混合,添加三乙醇胺(0.2mL),二氯甲烷(1mL),四氢呋喃(2mL)室温下搅拌18小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(二氯甲烷:己烷=5:1)纯化得到对硝基苯基对甲苯氨基碳酸酯I-9-1(42mg,77%),液相质谱m/z=273.1[M+H]+。
步骤2:1-(1H-吡咯并[2,3-c]吡啶-5-基)-3-(对甲苯基)脲A-4-1
将上述对硝基苯基对甲苯氨基甲酸酯I-9-1(27mg,0.1mmol)与1H-吡咯并[2,3-c]吡啶-5-胺I-1(16mg,0.12mmol)与K
实施例11:1-(3-溴苯基)-3-(1H-吡咯并[2,3-c]吡啶-5-基)脲A-4-2
步骤1:4-硝基苯基(3-溴苯基)氨基碳酸酯I-9-2
将3-溴苯胺(34mg,0.2mmol)与4-硝基苯基羰酰氯(48mg,0.24mmol)混合,添加三乙醇胺(0.2mL),二氯甲烷(1mL),四氢呋喃(2mL)室温下搅拌18小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(二氯甲烷:己烷=5:1)纯化得到4-硝基苯基(3-溴苯基)氨基碳酸酯I-9-2(46mg,68%),液相质谱m/z=338.1[M+H]+。
步骤2:1-(3-溴苯基)-3-(1H-吡咯并[2,3-c]吡啶-5-基)脲A-4-2
将上述4-硝基苯基(3-溴苯基)氨基碳酸酯I-9-2(34mg,0.1mmol)与1H-吡咯并[2,3-c]吡啶-5-胺I-1(16mg,0.12mmol)与K
实施例12:N-甲基-N-苯基-1H-吡咯并[2,3-c]吡啶-5-甲酰胺A-5
合成线路:
步骤1:5-硝基-1H-吡咯并[2,3-c]吡啶Ⅰ-10
向发烟硫酸(500μL)中的H
步骤2:1H-吡咯并[2,3-c]吡啶-5-羧酸Ⅰ-11
将上述5-硝基-1H-吡咯并[2,3-c]吡啶Ⅰ-10(39mg,0.24mmol)与KCN(195mg,3mmol)混合添加1-丁基咪唑四氟硼酸盐(3mg,0.014mmol),EtOH(2mL),水(2mL)加热至80℃搅拌19小时。然后加入10mL水,用CH
步骤3:N-甲基-N-苯基-1H-吡咯并[2,3-c]吡啶-5-甲酰胺A-5
将上述1H-吡咯并[2,3-c]吡啶-5-羧酸Ⅰ-11(16mg,0.1mmol)与N-甲基苯胺(13mg,0.12mmol)与2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(38mg,0.1mmol)混合,添加N,N-二异丙基乙胺(0.2mL),N,N-二甲基甲酰胺(2mL),室温下搅拌18小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(乙酸乙酯:己烷=3:7)纯化,减压蒸馏除去溶剂,得到N-甲基-N-苯基-1H-吡咯并[2,3-c]吡啶-5-甲酰胺A-5(18mg,72%),液相质谱m/z=252.1[M+H]+。
实施例13:N-(四氢-2H-吡喃-4-基)-1H-吡咯并[2,3-c]吡啶-5-胺A-6-1
将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与四氢-4H-吡喃-4-酮(12mg,0.12mmol)混合,添加甲醇(2mL),室温下搅拌2小时,然后添加氰基硼氢化钠(20mg,0.3mmol),室温下搅拌2小时。然后向混合物中添加NaOH水溶液(10mL,0.3mol/L)。所得混合物用DCM(20mL x 3)萃取。合并的有机相在无水Na
实施例14:N-(吡啶-4-基甲基)-1H-吡咯并[2,3-c]吡啶-5-胺A-6-2
将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与异烟醛(13mg,0.12mmol)混合,添加甲醇(2mL),室温下搅拌2小时,然后添加氰基硼氢化钠(20mg,0.3mmol),室温下搅拌2小时。然后向混合物中添加NaOH水溶液(10mL,0.3mol/L)。所得混合物用DCM(20mL x 3)萃取。合并的有机相在无水Na
实施例15:N-环丁基-1H-吡咯并[2,3-c]吡啶-5-胺A-6-3
将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与环丁酮(8mg,0.12mmol)混合,添加甲醇(2mL),室温下搅拌2小时,然后添加氰基硼氢化钠(20mg,0.3mmol),室温下搅拌2小时。然后向混合物中添加NaOH水溶液(10mL,0.3mol/L)。所得混合物用DCM(20mL x 3)萃取。合并的有机相在无水Na
实施例16:2-(1H-吡咯并[2,3-c]吡啶-5-基)异吲哚-1,3-二酮A-7
合成路线:
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与异苯并呋喃-1,3-二酮(18mg,0.12mmol)混合,添加N,N-二甲基乙酰胺(2mL),室温下搅拌24小时,然后再加入二甲苯(1mL),油浴140℃搅拌48小时。完成后(通过TLC监测),通过过滤分离不溶性催化剂,用丙酮洗涤并干燥。在减压下浓缩有机层以得到所需产物,用水洗涤,并在乙醇中重结晶,粗品经硅胶柱层析(CH
实施例17:N-苯基-1H-吡咯并[2,3-c]吡啶-5-胺A-8
合成路线:
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与碘苯(24mg,0.12mmol),(N,N-联吡啶基咪唑亚基)二溴化铜(10mg,0.023mmol),碳酸铯(100mg,0.3mmol)混合,添加1,4-二氧六环(5mL),油浴170℃搅拌12小时。然后向混合物中添加NaOH水溶液(10mL,0.3mol/L)。所得混合物用DCM(20mL x 3)萃取。合并的有机相在无水Na
实施例18:(E)-5-苯乙烯基-1H-吡咯并[2,3-c]吡啶A-9
合成路线:
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与苯乙烯(13mg,0.12mmol),双(二亚苄基丙酮)-钯(0)(5mg,0.0087mmol)混合,添加亚硝基叔丁酯(0.5mL),氯乙酸(0.5mL),乙酸(3mL)油浴50℃搅拌2小时。然后向混合物中添加NaOH水溶液(10mL,0.3mol/L)。所得混合物用DCM(20mL x 3)萃取。合并的有机相在无水Na
实施例19:N-(1H-吡咯并[2,3-c]吡啶-5-基)噻吩-2-磺酰胺A-10
合成路线:
步骤:将1H-吡咯并[2,3-c]吡啶-5-胺I-1(13mg,0.1mmol)与噻吩-2-磺酰氯(22mg,0.12mmol)混合,添加三乙胺(0.5mL),二氯甲烷(3mL),氮气保护下室温搅拌24小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(乙酸乙酯:己烷=3:7)纯化,减压蒸馏除去溶剂,得到N-(1H-吡咯并[2,3-c]吡啶-5-基)噻吩-2-磺酰胺A-10(14mg,50%),液相质谱m/z=280.0[M+H]+。
实施例20:N-(2-((1H-吡咯并[2,3-c]吡啶-5-基)氨基)乙基)甲磺酰胺A-11
合成路线:
步骤1:N1-(1H-吡咯并[2,3-c]吡啶-5-基)乙烷-1,2-二胺Ⅰ-19
将1H-吡咯并[2,3-c]吡啶-5-胺I-1(26mg,0.2mmol)与2-溴乙烷-1-胺I-18(29mg,0.24mmol)混合,加入水(5mL),油浴95℃搅拌18小时。反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。混合物在硅胶(CH
步骤2:N-(2-((1H-吡咯并[2,3-c]吡啶-5-基)氨基)乙基)甲磺酰胺A-11
将上述N1-(1H-吡咯并[2,3-c]吡啶-5-基)乙烷-1,2-二胺Ⅰ-19(24mg,0.13mmol)与甲磺酰氯Ⅰ-20(28mg,0.16mmol)混合,加入二氯甲烷(5mL),冰水浴搅拌24小时。向反应混合物中加入乙酸乙酯和水,分离有机层,用硫酸钠干燥,减压浓缩。残渣经硅胶柱层析(乙酸乙酯:己烷=3:7)纯化,减压蒸馏除去溶剂,得到N-(2-((1H-吡咯并[2,3-c]吡啶-5-基)氨基)乙基)甲磺酰胺A-11(12mg,36%),液相质谱m/z=255.1[M+H]+。
实施例21:本发明分别采用两种化合物预测方法来进行吡咯并吡啶衍生物预测,方法如下:
方法一:蛋白结构对接预测
利用AutoDock Vina和LeDock软件分别对实施例1-20中的吡咯并吡啶衍生物分子与Oct4靶标蛋白进行分子对接,分别生产10个对接构象。计算每一吡咯并吡啶分子与Oct4靶标蛋白最优对接结果的结合能与配体效率,综合对接结果进行吡咯并吡啶分子筛选,具体对接结果如表1所示:表第二、三列分别由autodock vina和LeDock分子对接软件计算获得的结合自由能(binding energy),其值越负,小分子配体与靶蛋白结合能力越强。表第四列则代表由LeDock计算产生的配体效率(ligand efficiency),其绝对值越大表示小分子活性具有更强的潜能。在本发明中根据两个软件的独立算法,预测出本发明涉及化合物的结合能水平,预测值显示本发明中的化合物其结合能均远大于本发明根据靶点特征设定的阈值3。
表1.本发明所用吡咯并吡啶衍生物分子与靶向序列的结合能预测
/>
方法二:mir-RNA结构对接预测:
mir-145与小分子亲和力的预测采用了一种新的计算工具RLDOCK软件。RLDOCK是一个基于物理形变的配体-RNA结合能力预测模型。采用全新的结合位点全局多步搜索算法,优化了搜索效率和稳定性,使该算法可以有效的预测未知结合位点。RLDOCK多步计算中,首先详尽的扫描miR-145结构上可能的结合位点和该位点小分子可能的结合姿势,采用最小Lennard-Jones势能计分函数对所有结合可能给出打分值并排序,结果如表2所示:评分结果是一个负数值,其绝对值越大,配体-RNA亲和力越强。RLDock的最终打分是综合其采纳的不同能量函数的总值,目前,经过验证的可用的RNA靶点数量有限,目前还没有成熟的计算体系,能区分实际相互作用的阈值。本方法依据预测结合能排序,优先验证高排名分子。
表2:吡咯并吡啶衍生物与靶向mir-RNA的结合能预测
/>
通过Oct4蛋白结构对接预测,以及mir-145结构对接预测,发现吡咯并吡啶衍生物在两种结构对接预测中均表现出较大的结合能,因此推测吡咯并吡啶衍生物具有Oct4的表达增强能力,因此采取等温滴定量热法及细胞学实验验证吡咯并吡啶衍生物对Oct4的表达增强作用,从而验证吡咯并吡啶衍生物的Oct4高选择性活化剂作用。
实施例22:利用等温滴定量热法验证小分子化合物与目的核酸的互作
等温滴定量热法(ITC)利用功率补偿原理,通过单次浓度扫描能够准确得到体系在较宽的浓度组成范围内发生的分子间相互作用的焓变,测量结合时的热量变化,提供核酸-配体相互作用的完整热力学特性。对复合物热力学特征和能量的量化是分析微小核酸miR-145与化合物相互作用的分子基础。本发明采用沃特世(Waters)公司的Nano-ITC滴定热量计来体外验证核酸-小分子相互作用。实验过程为:将体积300μL,浓度为50μM的单链miR-145溶液放在一个温控样品池中,通过一个热电偶回路与参比池中等体积去离子水偶联。以A-3-6小分子为例,将体积50μL,浓度500μM的小分子作为配体置于注射器中。通过测量样品池的热量变化,并通过与参比池进行平衡化计算,显示为一个吸热或放热的峰。用仪器自带的软件对两反应物相互作用模式进行拟合,可以直接计算溶液中两个或多个分子之间反应结合焓(ΔH)、定压热容(ΔCp)、结合位点数(n)、结合平衡常数(Ka),综合计算获得动力学数据。图1上半部分显示样品池的热量变化,其中红色峰线是miR-145与A-3-6结合放热,蓝色线是不含miR-145缓冲液与A-3-6结合放热。下图1是峰面积拟合Multiple Sites模型曲线,其中计算的nSite分别为3和1.8所对应的Kd(M)为7.017E-8和7.544E-9,空白对照组Kd(M)为1.000E-3。ITC试验结果证明了本发明得到的小分子与目标靶点能够特异性的结合。
实施例23:验证小分子化合物引起的MET细胞形态变化
在T25培养人间充质细胞,按照4x10
实施例24:验证小分子引起的转录表达差异验证
本发明的目的是使用特定化合物达到实现MET转化的作用,此类活化剂的重要功能为增强MET形变的产生的表达。根据前期文献的报导,在验证本发明的小分子功能时,除了与MET形成的结构蛋白KRT家族,以及调控基因MSX系列等均作为检测指标,结果表明吡咯并吡啶及其衍生物能够诱导MET现象的产生;能够Oct4基因的下游基因的表达增加也是本发明中化合物的一个功能验证的指标,本发明使用Nanog重编程早期基因的表达对吡咯并吡啶及其衍生物对Oct4的激活功能进行进一步的验证。
在T25培养人间充质细胞,按照4x10
表3.化合物效应基因QPCR引物序列
- 一种尿路上皮细胞的诱导剂和尿路上皮细胞的诱导方法
- 一种过表达SMAD3诱导体细胞重编程为乳腺上皮细胞的方法