用微生物核酸鉴定转移性癌症的存在及起源组织
文献发布时间:2023-06-19 19:28:50
相关申请的交叉引用
本申请要求于2020年9月21日和2020年10月26日提交的美国临时申请第63/081,075号和第63/105,624号的优先权益,上述申请通过引用方式并入本文。
政府赞助
本发明是在美国国立卫生研究院授予的政府资助号F30 CA243480下完成的。政府在本发明中具有某些权利。
技术领域
本发明涉及用存在于组织和液体活检中的非人微生物核酸鉴定转移性癌症的存在和/或其起源组织。在本发明的至少一个实施例中,机器学习(ML)模型被训练为诊断模型,以在转移性癌症的类型之间和类型内进行区分。
背景技术
越来越多的证据表明,细菌、病毒、真菌、古细菌和噬菌体微生物群在癌变中起着关键作用。事实上,据估计,多达20%的全球癌症负担是由微生物制剂直接引起的。许多研究人员认为,潜在的机制是通过微生物对免疫系统的影响,其能够增加或抑制炎症,以及操纵受试者的免疫细胞的能力以及其他机制。
基于使用定殖有一种或多种特定细菌的悉生小鼠模型的研究数据,似乎微生物群可以通过多种机制改变癌症易感性和进展,如调节炎症、诱导DNA损伤和产生参与肿瘤发生或肿瘤抑制的代谢物。除了癌变和癌症进展外,新出现的证据表明,微生物群可以预测对癌症治疗的应答或被操纵以改善癌症治疗,包括“传统”化疗(例如吉西他滨)和更“创新”的免疫疗法(例如PD-1阻断)。
虽然大部分文献集中于检查宿主肠道微生物组的组成或功能及其对癌症的影响,但文献中的最近实例已经探索了原发性肿瘤组织内或患有原发性肿瘤的患者血液内的癌症相关微生物群(PMID:32214244、32467386、29567829、31578522)。由于原发性肿瘤相关微生物群与肿瘤形成的潜在因果关系,以及与其多个转移性对应物相比,容易获得单个原发性肿瘤,因此,原发性肿瘤相关微生物群常常是研究的热点。
然而,大多数癌症死亡并非源于原发性肿瘤,而是源于转移,并且关于癌症相关微生物群和转移性癌症之间的关系仍然知之甚少。如果该领域中的这一空白能够得到解决,它可以导致新类型的癌症诊断,即通过早期检测转移性癌症的存在和/或起源组织来预防大量患者的发病率和死亡率。此外,转移性癌症的起源组织的准确鉴定对于确定应该给予患者何种临床治疗是至关重要的。作为一个人为的实例,在患者脑中发现的转移性肺癌将具有与起源于患者的脑中的脑癌(即原发性肿瘤)不同的临床治疗。因此,改善转移性癌症的起源组织诊断的方法也影响给定治疗的最佳类型或剂量以及患者的预后。
历史上,鉴定转移性癌症的起源组织的过程依赖于从转移组织活检获得人类分子信息:免疫组织化学(IHC)蛋白染色、人类DNA测序(例如,以鉴定已知与特定原发性肿瘤类型相关的突变)、DNA(例如,表观基因组)的测序修饰或人类RNA测序(例如,以鉴定与特定原发性肿瘤类型相关的基因表达模式)。然而,这些方法用于定位转移性肿瘤起源组织的准确性是有限的。例如,Weiss等人(PMID:23287002)报告了使用IHC方法的准确率仅为69%,而在相同样本上使用92基因表达特征时仅为79%。这些结果表明,在鉴定>20%的患者转移性癌症的起源组织时,失败率是惊人的,因为所有癌症死亡的绝大多数是由于转移。这些低准确率反映了有多少转移性肿瘤失去了其原发性肿瘤组织的原始细胞标志物,使其来源难以用人类信息自信且快速地识别,这可能刺激临床上对患者原发性肿瘤的侵入性、昂贵且紧急的搜寻。
就目前有关癌症相关微生物领域的科学现状而言,以下是已知的:(i)许多癌症相关微生物位于原发性肿瘤癌症细胞和邻近免疫细胞的细胞内(PMID:32467386),(ii)几乎所有原发性肿瘤都含有癌症类型特异性微生物群(PMID:32214244),以及(iii)在结肠癌的情况下,当胞内微生物从原发性肿瘤转移时,它们可以在癌细胞内移动(PMID:29170280)。
然而,未知但至关重要的是:(i)转移瘤的微生物群是否忠实地反映了其起源组织,或者转移瘤的新身体部位(与原发性肿瘤相比)是否破坏了微生物的组成或功能;(ii)是否所有癌症类型,特别是那些不包括结肠癌的癌症类型,在原发性肿瘤和其转移瘤之间共享胞内(或胞外)微生物,这将影响依赖于微生物信息的转移瘤泛癌诊断方法的可行性;(iii)是否可以在血液中检测到转移瘤的微生物群,如果可以,此类信息是否可以提供癌症起源组织的信息。
此前,WO2020093040A1专注于使用患者组织和血液中的非人微生物核酸开发用于原发性肿瘤的新癌症诊断法。此外,US20180291463A1、WO2018200813A1和WO2018031545A1描述了一种基于微阵列的技术,用于检测原发性肿瘤样本(不是转移瘤,也不是血液或其他体液)中预选的(“偏向的”)微生物群体。US20180223338描述了使用原发性肿瘤组织微生物组或唾液微生物组来鉴定和诊断头颈癌。US20180258495A1描述了使用原发性肿瘤组织微生物组或粪便微生物组来检测结肠癌、与结肠癌相关的某些种类的突变,以及收集和扩增相应微生物的试剂盒。
发明内容
根据至少一个实施例,本发明的公开内容提供了一种仅使用来自人组织活检或血液来源样本的非人来源的核酸来准确诊断或确定是否存在转移性癌症、其起源组织及其对某些疗法的应答的可能性的方法。
在实施例中,本发明提供了一种使用血液来源组织广泛创建与转移性癌症的存在和/或类型相关的微生物存在或丰度模式(“特征”)的方法。然后,这些特征可以用于诊断人类中转移性癌症的存在和/或起源组织。
在实施例中,本发明提供了一种使用转移性肿瘤组织广泛创建与转移性癌症的起源组织相关的微生物存在或丰度模式的方法。然后,这些特征可以用于诊断人类中转移性癌症的存在和/或起源组织。
在实施例中,本发明提供了一种用于确定受试者是否存在转移性癌症的方法,其包括:检测患有癌症的受试者的生物样本中的微生物存在;从微生物存在中去除污染的微生物特征,从而产生净化的微生物存在;将净化的微生物存在与来自一个或多个患有癌症的受试者的一个或多个生物样本的微生物存在进行比较,从而产生微生物-癌症比较数据集;以及从微生物癌症比较数据集中确定受试者是否存在转移性癌症。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中从微生物癌症比较数据集中确定受试者是否存在转移性癌症包括鉴定转移性癌症的起源组织。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中微生物存在进一步包括微生物丰度。微生物存在或丰度可以例如包括以下非哺乳类生命域:细菌、真菌、病毒、古细菌、原生动物、噬菌体或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中通过以下测量微生物存在或丰度:生态学鸟枪法测序、定量聚合酶链反应、免疫组织化学、原位杂交、流式细胞术、宿主全基因组测序、宿主转录组测序、癌症全基因组测序、癌症转录组测序或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中通过扩增以下微生物来源的核酸区域来测量微生物存在或丰度:16SrRNA的V1、V2、V3、V4、V5、V6、V7、V8、V9可变结构域,18S rRNA的内部转录间隔区(ITS)区域或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中通过靶向微生物DNA、RNA或其任意组合的核酸测量来检测微生物存在或丰度,其中靶向微生物DNA、RNA或其任意组合的核酸测量与受试者的哺乳类DNA、RNA或其任意组合的测量同时发生。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中转移性癌症包括:急性髓系白血病、肾上腺皮质癌、膀胱尿路上皮癌、脑低级别胶质瘤、乳腺浸润性癌、宫颈鳞状细胞癌和宫颈内膜腺癌、胆管癌、结肠腺癌、淋巴样肿瘤弥漫性大B细胞淋巴瘤、食管癌、多形性胶质母细胞瘤、头颈部鳞状细胞癌、肾嫌色细胞癌、肾透明细胞癌、肾乳头状细胞癌、肝细胞癌、肺腺癌、肺鳞状细胞癌、间皮瘤、卵巢浆液性囊腺癌、胰腺癌、嗜铬细胞瘤和副神经节瘤、前列腺癌、直肠腺癌、肉瘤、皮肤恶性黑色素瘤、胃腺癌、睾丸生殖细胞肿瘤、甲状腺癌、胸腺瘤、子宫癌肉瘤、子宫内膜癌、葡萄膜黑色素瘤或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中转移性癌症包括癌症类型,其中该癌症类型包括:肺癌、前列腺癌、黑色素瘤癌、乳腺癌、甲状腺癌或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中污染的微生物特征包括微生物存在的分类分配。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中去除污染的微生物特征是任选的,而不是必需的。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中用于形成微生物-癌症比较数据集的比较生物样本来自患有一种或多种原发性肿瘤、转移性肿瘤或其任意组合的受试者。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中微生物-癌症比较数据集进一步包括哺乳类特征,其中该哺乳类特征包括:肿瘤组织的免疫组织化学蛋白标志物、肿瘤组织DNA、肿瘤组织RNA、肿瘤组织甲基化模式、无细胞肿瘤DNA、无细胞肿瘤RNA、外泌体来源肿瘤DNA、外泌体来源肿瘤RNA、循环肿瘤细胞来源DNA、循环肿瘤细胞来源RNA、无细胞肿瘤DNA的甲基化模式、无细胞肿瘤RNA的甲基化模式、循环肿瘤细胞来源DNA的甲基化模式、循环肿瘤细胞来源RNA的甲基化模式、循环肿瘤细胞来源RNA的甲基化模式或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的用于确定受试者是否存在转移性癌症的方法,其中生物样本包括组织样本、液体活检、全血活检或其任意组合。生物样本可以进一步包含全血的一种或多种成分,包括:血浆、白细胞、红细胞、血小板或其任意组合。
在实施例中,本发明提供了一种基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其包括:检测来自患有癌症的受试者的生物样本中的微生物存在;从微生物存在中去除污染的微生物特征,从而产生净化的微生物存在;在净化的微生物存在和存在于受试者中的转移性癌症之间生成关联;以及向受试者施用由净化的微生物存在和转移性癌症之间的关联确定的治疗。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中微生物存在进一步包括微生物丰度,其中微生物存在或丰度包括以下非哺乳类生命域:细菌、真菌、病毒、古细菌、原生动物、噬菌体或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中污染的微生物特征包括微生物存在的分类分配。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中从微生物存在中去除污染的微生物特征是任选的步骤,并且关联可以在所检测到的微生物存在和存在于受试者中的转移性癌症之间生成。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中生物样本包括组织样本、液体活检、全血活检或其任意组合。生物样本可以进一步包含全血的一种或多种成分,包括:血浆、白细胞、红细胞、血小板或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中治疗不会因净化的微生物存在而代谢或变得失活。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中治疗包括:小分子、激素疗法、生物制剂、工程化宿主来源细胞类型、益生菌、工程化细菌、天然但选择性的病毒、工程化病毒、噬菌体或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中转移性癌症包括:急性髓系白血病、肾上腺皮质癌、膀胱尿路上皮癌、脑低级别胶质瘤、乳腺浸润性癌、宫颈鳞状细胞癌和宫颈内膜腺癌、胆管癌、结肠腺癌、淋巴样肿瘤弥漫性大B细胞淋巴瘤、食管癌、多形性胶质母细胞瘤、头颈部鳞状细胞癌、肾嫌色细胞癌、肾透明细胞癌、肾乳头状细胞癌、肝细胞癌、肺腺癌、肺鳞状细胞癌、间皮瘤、卵巢浆液性囊腺癌、胰腺癌、嗜铬细胞瘤和副神经节瘤、前列腺癌、直肠腺癌、肉瘤、皮肤恶性黑色素瘤、胃腺癌、睾丸生殖细胞肿瘤、甲状腺癌、胸腺瘤、子宫癌肉瘤、子宫内膜癌、葡萄膜黑色素瘤或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中治疗包括与针对转移性癌症的主要治疗联合给予的佐剂,以提高主要治疗的疗效。佐剂可以是例如抗生素或抗微生物剂。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中治疗基于与转移性癌症或转移性癌症的环境相关的微生物成分或抗原。治疗可以包括靶向微生物抗原的过继细胞转移、针对微生物抗原的癌症疫苗、针对微生物抗原的单克隆抗体、设计成至少部分靶向微生物抗原的抗体-药物缀合物、设计成至少部分靶向一种或多种微生物抗原的多价抗体、抗体片段、其抗体衍生物,或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中治疗包括靶向微生物存在的一类功能上或生物学上相似的微生物的抗生素。治疗可以进一步包括两种或更多种治疗类型,其中该两种或更多种治疗类型被组合,使得该两种或更多种治疗类型中的至少一种类型利用与转移性癌症或转移性癌症环境相关的微生物存在或丰度来增强治疗效果。
在实施例中,本发明提供了一种如上文/下文所述的基于微生物存在施用治疗以治疗受试者的转移性癌症的方法,其中净化的微生物存在和转移性癌症之间的关联进一步包括转移性癌症的起源、类型或其任意组合。
在实施例中,本发明提供了一种被配置为确定受试者是否存在转移性癌症的系统,其包括:一个或多个处理器;以及包括软件的非瞬态计算机可读存储介质,其中该软件包含可执行指令,作为执行的结果,该可执行指令使得计算机系统的一个或多个处理器:获得与来自患有癌症的受试者的生物样本的一个或多个核酸分子相关的第一数据;从与生物样本的一种或多种核酸相关的第一数据的非微生物核酸中分离微生物核酸,从而确定第二数据;基于第二数据,鉴定微生物核酸的微生物存在;从第二数据中去除微生物存在的污染的微生物特征,从而产生净化的微生物存在的表;将净化的微生物存在的表输入到机器学习模型中;并且从机器学习模型接收指示转移性癌症的存在或不存在的输出。在实施例中,本发明提供了一种被配置为确定受试者是否存在转移性癌症的系统,其中该系统包括Illumina NovaSeq 6000仪器。Illumina NovaSeq 6000仪器可以通信耦合(例如,经由网络连接)到一个或多个计算机系统可访问的网络存储位置,该计算机系统能够访问和处理由Illumina NovaSeq 6000仪器生成的数据。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中微生物存在进一步包括微生物丰度,其中微生物存在或丰度包括以下非哺乳类生命域:细菌、真菌、病毒、古细菌、原生动物、噬菌体或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中该系统进一步确定转移性癌症的起源组织。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中净化的微生物特征包括微生物存在的分类分配。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中去除污染的微生物特征是任选的。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中通过将一个或多个核酸分子与微生物和非微生物基因组的参考数据库进行比对来分离微生物核酸和非微生物核酸。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中在不将一个或多个核酸分子与微生物和非微生物基因组的参考数据库进行比对的情况下来分离微生物核酸和非微生物核酸。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中净化的微生物存在的表进一步包括哺乳类特征,其中该哺乳类特征包括:肿瘤组织的免疫组织化学蛋白标志物、肿瘤组织DNA、肿瘤组织RNA、肿瘤组织甲基化模式、无细胞肿瘤DNA、无细胞肿瘤RNA、外泌体来源肿瘤DNA、外泌体来源肿瘤RNA、循环肿瘤细胞来源DNA、循环肿瘤细胞来源RNA、无细胞肿瘤DNA的甲基化模式、无细胞肿瘤RNA的甲基化模式、循环肿瘤细胞来源DNA的甲基化模式、循环肿瘤细胞来源RNA的甲基化模式、循环肿瘤细胞来源RNA的甲基化模式或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中转移性癌症包括:急性髓系白血病、肾上腺皮质癌、膀胱尿路上皮癌、脑低级别胶质瘤、乳腺浸润性癌、宫颈鳞状细胞癌和宫颈内膜腺癌、胆管癌、结肠腺癌、淋巴样肿瘤弥漫性大B细胞淋巴瘤、食管癌、多形性胶质母细胞瘤、头颈部鳞状细胞癌、肾嫌色细胞癌、肾透明细胞癌、肾乳头状细胞癌、肝细胞癌、肺腺癌、肺鳞状细胞癌、间皮瘤、卵巢浆液性囊腺癌、胰腺癌、嗜铬细胞瘤和副神经节瘤、前列腺癌、直肠腺癌、肉瘤、皮肤恶性黑色素瘤、胃腺癌、睾丸生殖细胞肿瘤、甲状腺癌、胸腺瘤、子宫癌肉瘤、子宫内膜癌、葡萄膜黑色素瘤或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中转移性癌症包括癌症类型,其中该癌症类型包括:肺癌、前列腺癌、黑色素瘤癌、乳腺癌、甲状腺癌或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中生物样本包括组织样本、液体活检、全血活检或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中生物样本包含全血的成分,包括:血浆、白细胞、红细胞、血小板或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中该机器学习模型经训练以鉴别非转移性和转移性癌组织或血液样本。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中机器学习模型经训练以辨别一种或多种癌症类型。该一种或多种癌症类型可以包括:急性髓系白血病、肾上腺皮质癌、膀胱尿路上皮癌、脑低级别胶质瘤、乳腺浸润性癌、宫颈鳞状细胞癌和宫颈内膜腺癌、胆管癌、结肠腺癌、淋巴样肿瘤弥漫性大B细胞淋巴瘤、食管癌、多形性胶质母细胞瘤、头颈部鳞状细胞癌、肾嫌色细胞癌、肾透明细胞癌、肾乳头状细胞癌、肝细胞癌、肺腺癌、肺鳞状细胞癌、间皮瘤、卵巢浆液性囊腺癌、胰腺癌、嗜铬细胞瘤和副神经节瘤、前列腺癌、直肠腺癌、肉瘤、皮肤恶性黑色素瘤、胃腺癌、睾丸生殖细胞肿瘤、甲状腺癌、胸腺瘤、子宫癌肉瘤、子宫内膜癌、葡萄膜黑色素瘤或其任意组合。
在实施例中,本发明提供了一种如上文/下文所述的被配置为确定受试者是否存在转移性癌症的系统,其中输出进一步包含转移性癌症的类型、起源组织或其任意组合的指示。
在实施例中,本发明提供了一种广泛诊断受试者转移性癌症的方法,其包括:检测来自受试者的组织或血液样本中的微生物存在或丰度;确定所检测到的微生物存在或丰度不同于在不存在转移的情况下取得的一个或多个正常组织样本中的微生物存在或丰度;以及将所检测到的微生物存在或丰度与转移性癌症的已知微生物存在或丰度相关联,从而诊断转移性癌症。
在实施例中,本发明提供了一种广泛诊断受试者转移性癌症的起源组织的方法,其包括:检测来自患有转移性癌症的受试者的组织或血液样本中的微生物存在或丰度;确定所检测到的微生物存在或丰度与先前研究的患有原发性肿瘤的受试者群体中的微生物存在或丰度相似或不同;以及将所检测到的转移性癌症的微生物存在或丰度与最相似的原发性肿瘤类型相关联,从而诊断转移性癌症的起源组织。
在实施例中,本发明提供了一种诊断受试者转移性癌症的起源组织的方法,其包括:检测来自受试者的液体活检中的微生物存在或丰度;确定所检测到的微生物存在或丰度与来自健康受试者和/或患有原发性肿瘤的受试者群体的一个或多个液体活检中的微生物存在或丰度相似或不同;以及将所检测到的微生物存在或丰度与该群体中最相似的液体活检相关联,从而诊断转移性癌症的存在或不存在,并且如果存在,诊断其起源组织。
在实施例中,本发明提供了一种诊断转移性癌症的身体部位的方法,其中起源位置是骨骼(肉瘤)、肾上腺、膀胱、脑、乳房、子宫颈、胆囊、结肠、食道、颈部(头颈部鳞状细胞癌)、肾脏、肝脏、肺、淋巴结(弥漫性大B细胞淋巴瘤)、皮肤、卵巢、前列腺、直肠、胃、甲状腺和子宫,并且其中受试者是人。
在实施例中,本发明提供了一种诊断转移性癌症的方法,其中癌症为肾上腺皮质癌、膀胱癌、脑癌(低级别胶质瘤;胶质母细胞瘤)、乳腺癌、宫颈癌、胆管癌、结肠癌、食管癌、头颈癌、肾癌(嫌色细胞;肾透明细胞癌;乳头状细胞癌)、肝癌、肺癌(腺癌;鳞状细胞癌)、淋巴样肿瘤弥漫性大B细胞淋巴瘤、黑色素瘤(皮肤黑色素瘤、葡萄膜黑色素瘤)、卵巢癌、前列腺癌、直肠癌、肉瘤、胃癌、甲状腺癌(甲状腺癌、胸腺瘤)和子宫癌,并且其中受试者是人。
在实施例中,本发明提供了一种使用非人特征预测人转移性癌症的分子特征的方法,其中分子特征是人突变,其中非人特征是微生物存在或丰度。
在实施例中,本发明提供了一种预测哪些受试者对转移性癌症的特定治疗应答或不应答的方法,其中受试者是人,其中治疗是免疫疗法,其中免疫疗法是PD-1阻断(例如纳武利尤单抗、帕普利珠单抗)。
在实施例中,本发明提供了一种诊断转移性癌症的方法,其进一步包括基于所鉴定的疾病的非人特征或所鉴定的转移性癌症的起源组织来治疗受试者的转移性癌症,其中受试者是人,其中非人特征是微生物存在或丰度。
在实施例中,本发明提供了一种诊断转移性癌症的方法,其进一步包括基于受试者的非人特征设计一种新治疗以治疗受试者的转移性癌症,其中非人特征是微生物特征,其中受试者是人。
在实施例中,本发明提供了一种诊断转移性癌症的方法,其进一步包括基于受试者的非人特征将其与受试者的早期癌症相区分,其中非人特征是微生物特征,其中受试者是人。
在实施例中,可以使用以下方式中的一种或多种设计新的疗法来靶向和利用与转移性癌症相关的非人特征:小分子、激素疗法、生物制剂、工程化宿主来源细胞类型、益生菌、工程化细菌、天然但选择性的病毒、工程化病毒和噬菌体。
在实施例中,本发明提供了一种诊断转移性癌症的方法,其进一步包括对其非人特征的纵向监测,以指示原发性肿瘤何时转移和/或疾病何时对治疗有应答,其中受试者是人。
在实施例中,本发明提供了一种试剂盒来测量转移性癌症组织或血液样本中的微生物存在或丰度,从而允许诊断转移性癌症和/或其起源组织。
在实施例中,本发明提供了一种计算机系统来分析转移性癌症组织或血液样本中的微生物存在或丰度,并将机器学习应用于该微生物存在或丰度,从而诊断转移性癌症和/或其起源组织。
在实施例中,本发明利用基于机器学习架构的诊断模型。
在实施例中,本发明利用基于规则化机器学习架构的诊断模型。
在实施例中,本发明利用基于机器学习架构集合的诊断模型。
在实施例中,本发明鉴定并选择性地去除作为污染物(“噪声”)的某些非人特征,同时选择性地保留其他非人特征作为非污染物(“信号”),其中非人特征是微生物特征。
在实施例中,本发明提供了一种诊断转移性癌症的方法,其中微生物来自细菌、真菌、病毒、古细菌、原生动物和/或噬菌体来源,或其任意组合。
在实施例中,本发明提供了一种诊断转移性癌症的方法,其中将微生物存在或丰度信息与关于受试者和/或受试者转移性癌症的信息结合,以产生比仅具有单独的微生物存在或丰度信息具有更好预测性能的诊断模型,其中受试者是人。
在实施例中,诊断模型利用受试者信息与来自以下来源的一个或多个的微生物存在或丰度信息的组合:肿瘤组织的免疫组织化学蛋白标志物、肿瘤组织DNA、肿瘤组织RNA、肿瘤组织甲基化模式、无细胞肿瘤DNA、无细胞肿瘤RNA、外泌体来源肿瘤DNA、外泌体来源肿瘤RNA、循环肿瘤细胞来源DNA、循环肿瘤细胞来源RNA、无细胞肿瘤DNA的甲基化模式、无细胞肿瘤RNA的甲基化模式、循环肿瘤细胞来源DNA的甲基化模式或循环肿瘤细胞来源RNA的甲基化模式、循环肿瘤细胞来源RNA的甲基化模式。
在实施例中,微生物存在或丰度通过以下检测:生态鸟枪法测序、定量聚合酶链式反应、免疫组织化学、原位杂交、流式细胞术、宿主全基因组测序、宿主转录组测序、癌症全基因组测序、癌症转录组测序或其任意组合,和/或其中使用扩增以下微生物来源的核酸区域中的一个或多个来检测微生物存在或丰度:16S rRNA的V1、V2、V3、V4、V5、V6、V7、V8或V9可变结构域区域;或18S rRNA的内部转录间隔区(ITS)区域,和/或其中通过靶向微生物DNA、RNA或其任意组合的核酸测量来检测微生物存在或丰度,其中靶向微生物DNA、RNA或其任意组合的测量与宿主DNA、RNA或其任意组合的测量同时发生。
在实施例中,通过以下方法中的一种或多种测量宿主的转移性癌症组织中微生物存在或不存在的地理空间分布:肿瘤组织和/或其微环境的多重取样、免疫组织化学、原位杂交、数字空间基因组学、数字空间转录组学或其任意组合。
在实施例中,微生物核酸与来自宿主的核酸同时被检测并随后进行区分。
在实施例中,在测量(例如测序)组合的核酸池之前,选择性地耗尽受试者的核酸并选择性地保留微生物核酸,其中受试者是人。
在实施例中,在测量(例如测序)受试者的组合核酸池之前,选择性地微生物核酸,其中受试者是人。
在实施例中,通过将核酸与微生物和非微生物基因组的参考数据库进行比对来分离微生物核酸和非微生物核酸。
在实施例中,通过将核酸与参考基因组数据库进行比对来分离微生物核酸和非微生物核酸。
在实施例中,本发明提供了生物样本是血液、血液成分(例如血浆)或组织活检,其中转移性组织活检是恶性或非恶性的,或其任意组合。
在实施例中,本发明提供了生物样本是液体活检,包括但不限于血浆、尿液、唾液或眼泪,或其任意组合。
在实施例中,通过测量受试者微生物组的其他身体部位中的微生物存在或丰度来推断转移性癌症的微生物存在或丰度,其中受试者是人。
在实施例中,受试者生物样本中的微生物存在或丰度同时告知转移性癌症的存在和起源组织的信息。
在一些实施例中,本公开描述了一种确定对治疗受试者的转移性癌症具有至少70%治疗效果的治疗的方法,其包括:(a)检测来自患有转移性癌症的受试者的生物样本中的微生物存在;(b)从微生物存在中去除污染的微生物特征,从而产生净化的微生物存在;(c)在净化的微生物存在和受试者的转移性癌症之间生成关联;以及(d)基于净化的微生物存在和转移性癌症之间的关联,确定对治疗受试者的转移性癌症具有至少70%治疗效果的治疗。在一些实施例中,治疗包括至少80%或至少90%的治疗效果。在一些实施例中,治疗应答包括积极应答者、无应答者、不良应答者或其任意组合。在一些实施例中,微生物存在进一步包括微生物丰度,其中微生物存在或丰度包括以下非哺乳类生命域:细菌、真菌、病毒、古细菌、原生动物、噬菌体或其任意组合。在一些实施例中,污染的微生物特征包括微生物存在的分类分配。在一些实施例中,省略了步骤(b)。在一些实施例中,生物样本包括组织样本、液体活检、全血活检或其任意组合。在一些实施例中,生物样本包含全血的一种或多种成分,包括:血浆、白细胞、红细胞、血小板或其任意组合。在一些实施例中,治疗不会因净化的微生物存在而代谢或变得失活。在一些实施例中,治疗包括:小分子、激素疗法、生物制剂、工程化宿主来源细胞类型、益生菌、工程化细菌、天然但选择性的病毒、工程化病毒、噬菌体或其任意组合。在一些实施例中,转移性癌症包括:急性髓系白血病、肾上腺皮质癌、膀胱尿路上皮癌、脑低级别胶质瘤、乳腺浸润性癌、宫颈鳞状细胞癌和宫颈内膜腺癌、胆管癌、结肠腺癌、淋巴样肿瘤弥漫性大B细胞淋巴瘤、食管癌、多形性胶质母细胞瘤、头颈部鳞状细胞癌、肾嫌色细胞癌、肾透明细胞癌、肾乳头状细胞癌、肝细胞癌、肺腺癌、肺鳞状细胞癌、间皮瘤、卵巢浆液性囊腺癌、胰腺癌、嗜铬细胞瘤和副神经节瘤、前列腺癌、直肠腺癌、肉瘤、皮肤恶性黑色素瘤、胃腺癌、睾丸生殖细胞肿瘤、甲状腺癌、胸腺瘤、子宫癌肉瘤、子宫内膜癌、葡萄膜黑色素瘤或其任意组合。在一些实施例中,治疗包括与针对转移性癌症的主要治疗联合给予的佐剂,以提高主要治疗的疗效。在一些实施例中,佐剂是抗生素或抗微生物剂。在一些实施例中,治疗基于与转移性癌症或转移性癌症的环境相关的微生物成分或抗原。在一些实施例中,治疗包括靶向微生物抗原的过继细胞转移、针对微生物抗原的癌症疫苗、针对微生物抗原的单克隆抗体、设计成至少部分靶向微生物抗原的抗体-药物缀合物、设计成至少部分靶向一种或多种微生物抗原的多价抗体、抗体片段、其抗体衍生物,或其任意组合。在一些实施例中,治疗包括靶向微生物存在的一类功能上或生物学上相似的微生物的抗生素。在一些实施例中,治疗包括两种或更多种治疗类型,其中该两种或更多种治疗类型被组合,使得该两种或更多种治疗类型中的至少一种类型利用与转移性癌症或转移性癌症环境相关的微生物存在或丰度来增强治疗效果。在一些实施例中,净化的微生物存在与转移性癌症之间的关联进一步包括转移性癌症的起源、类型或其任意组合。
在一些实施例中,本公开描述了一种预测受试者转移性癌症的治疗应答的方法,其包括:(a)检测来自患有转移性癌症的受试者的生物样本中的微生物存在;(b)从微生物存在中去除污染的微生物特征,从而产生净化的微生物存在;(c)在净化的微生物存在和受试者的转移性癌症之间生成关联;以及(d)基于净化的微生物存在和转移性癌症之间的关联,预测受试者转移性癌症的治疗应答。在一些实施例中,治疗应答包括积极应答者、无应答者、不良应答者或其任意组合。在一些实施例中,微生物存在进一步包括微生物丰度,其中微生物存在或丰度包括以下非哺乳类生命域:细菌、真菌、病毒、古细菌、原生动物、噬菌体或其任意组合。在一些实施例中,污染的微生物特征包括微生物存在的分类分配。在一些实施例中,省略了步骤(b)。在一些实施例中,生物样本包括组织样本、液体活检、全血活检或其任意组合。在一些实施例中,生物样本包含全血的一种或多种成分,包括:血浆、白细胞、红细胞、血小板或其任意组合。在一些实施例中,治疗不会因净化的微生物存在而代谢或变得失活。在一些实施例中,治疗包括:小分子、激素疗法、生物制剂、工程化宿主来源细胞类型、益生菌、工程化细菌、天然但选择性的病毒、工程化病毒、噬菌体或其任意组合。在一些实施例中,转移性癌症包括:急性髓系白血病、肾上腺皮质癌、膀胱尿路上皮癌、脑低级别胶质瘤、乳腺浸润性癌、宫颈鳞状细胞癌和宫颈内膜腺癌、胆管癌、结肠腺癌、淋巴样肿瘤弥漫性大B细胞淋巴瘤、食管癌、多形性胶质母细胞瘤、头颈部鳞状细胞癌、肾嫌色细胞癌、肾透明细胞癌、肾乳头状细胞癌、肝细胞癌、肺腺癌、肺鳞状细胞癌、间皮瘤、卵巢浆液性囊腺癌、胰腺癌、嗜铬细胞瘤和副神经节瘤、前列腺癌、直肠腺癌、肉瘤、皮肤恶性黑色素瘤、胃腺癌、睾丸生殖细胞肿瘤、甲状腺癌、胸腺瘤、子宫癌肉瘤、子宫内膜癌、葡萄膜黑色素瘤或其任意组合。在一些实施例中,治疗包括与针对转移性癌症的主要治疗联合给予的佐剂,以提高主要治疗的疗效。在一些实施例中,佐剂是抗生素或抗微生物剂。在一些实施例中,治疗基于与转移性癌症或转移性癌症的环境相关的微生物成分或抗原。在一些实施例中,治疗包括靶向微生物抗原的过继细胞转移、针对微生物抗原的癌症疫苗、针对微生物抗原的单克隆抗体、设计成至少部分靶向微生物抗原的抗体-药物缀合物、设计成至少部分靶向一种或多种微生物抗原的多价抗体、抗体片段、其抗体衍生物,或其任意组合。在一些实施例中,治疗包括靶向微生物存在的一类功能上或生物学上相似的微生物的抗生素。在一些实施例中,治疗包括两种或更多种治疗类型,其中该两种或更多种治疗类型被组合,使得该两种或更多种治疗类型中的至少一种类型利用与转移性癌症或转移性癌症环境相关的微生物存在或丰度来增强治疗效果。在一些实施例中,净化的微生物存在与转移性癌症之间的关联进一步包括转移性癌症的起源、类型或其任意组合。
在一些实施例中,本公开描述了一种确定在受试者的受试者转移性癌症的治疗过程中的动作的方法,其包括:(a)检测来自患有转移性癌症的受试者的生物样本中的微生物存在;(b)从微生物存在中去除污染的微生物特征,从而产生净化的微生物存在;(c)在净化的微生物存在和受试者的转移性癌症之间生成关联;以及(d)基于净化的微生物存在和转移性癌症之间的关联,确定在受试者转移性癌症的治疗过程中的动作。在一些实施例中,动作包括停止、开始或暂停受试者转移性癌症的治疗。在一些实施例中,微生物存在进一步包括微生物丰度,其中微生物存在或丰度包括以下非哺乳类生命域:细菌、真菌、病毒、古细菌、原生动物、噬菌体或其任意组合。在一些实施例中,污染的微生物特征包括微生物存在的分类分配。在一些实施例中,省略了步骤(b)。在一些实施例中,生物样本包括组织样本、液体活检、全血活检或其任意组合。在一些实施例中,生物样本包含全血的一种或多种成分,包括:血浆、白细胞、红细胞、血小板或其任意组合。在一些实施例中,治疗不会因净化的微生物存在而代谢或变得失活。在一些实施例中,治疗包括:小分子、激素疗法、生物制剂、工程化宿主来源细胞类型、益生菌、工程化细菌、天然但选择性的病毒、工程化病毒、噬菌体或其任意组合。在一些实施例中,转移性癌症包括:急性髓系白血病、肾上腺皮质癌、膀胱尿路上皮癌、脑低级别胶质瘤、乳腺浸润性癌、宫颈鳞状细胞癌和宫颈内膜腺癌、胆管癌、结肠腺癌、淋巴样肿瘤弥漫性大B细胞淋巴瘤、食管癌、多形性胶质母细胞瘤、头颈部鳞状细胞癌、肾嫌色细胞癌、肾透明细胞癌、肾乳头状细胞癌、肝细胞癌、肺腺癌、肺鳞状细胞癌、间皮瘤、卵巢浆液性囊腺癌、胰腺癌、嗜铬细胞瘤和副神经节瘤、前列腺癌、直肠腺癌、肉瘤、皮肤恶性黑色素瘤、胃腺癌、睾丸生殖细胞肿瘤、甲状腺癌、胸腺瘤、子宫癌肉瘤、子宫内膜癌、葡萄膜黑色素瘤或其任意组合。在一些实施例中,治疗包括与针对转移性癌症的主要治疗联合给予的佐剂,以提高主要治疗的疗效。在一些实施例中,佐剂是抗生素或抗微生物剂。在一些实施例中,治疗基于与转移性癌症或转移性癌症的环境相关的微生物成分或抗原。在一些实施例中,治疗包括靶向微生物抗原的过继细胞转移、针对微生物抗原的癌症疫苗、针对微生物抗原的单克隆抗体、设计成至少部分靶向微生物抗原的抗体-药物缀合物、设计成至少部分靶向一种或多种微生物抗原的多价抗体、抗体片段、其抗体衍生物,或其任意组合。在一些实施例中,治疗包括靶向微生物存在的一类功能上或生物学上相似的微生物的抗生素。在一些实施例中,治疗包括两种或更多种治疗类型,其中该两种或更多种治疗类型被组合,使得该两种或更多种治疗类型中的至少一种类型利用与转移性癌症或转移性癌症环境相关的微生物存在或丰度来增强治疗效果。在一些实施例中,净化的微生物存在与转移性癌症之间的关联进一步包括转移性癌症的起源、类型或其任意组合。
在一些实施例中,本公开描述了一种产生治疗受试者转移性癌症的治疗的方法,其包括:(a)检测来自患有转移性癌症的受试者的生物样本中的微生物存在;(b)从微生物存在中去除污染的微生物特征,从而产生净化的微生物存在;(c)在净化的微生物存在和受试者的转移性癌症之间生成关联;以及(d)基于净化的微生物存在和转移性癌症之间的关联,产生治疗受试者转移性癌症的治疗。在一些实施例中,微生物存在进一步包括微生物丰度,其中微生物存在或丰度包括以下非哺乳类生命域:细菌、真菌、病毒、古细菌、原生动物、噬菌体或其任意组合。在一些实施例中,污染的微生物特征包括微生物存在的分类分配。在一些实施例中,省略了步骤(b)。在一些实施例中,生物样本包括组织样本、液体活检、全血活检或其任意组合。在一些实施例中,生物样本包含全血的一种或多种成分,包括:血浆、白细胞、红细胞、血小板或其任意组合。在一些实施例中,治疗不会因净化的微生物存在而代谢或变得失活。在一些实施例中,治疗包括:小分子、激素疗法、生物制剂、工程化宿主来源细胞类型、益生菌、工程化细菌、天然但选择性的病毒、工程化病毒、噬菌体或其任意组合。在一些实施例中,转移性癌症包括:急性髓系白血病、肾上腺皮质癌、膀胱尿路上皮癌、脑低级别胶质瘤、乳腺浸润性癌、宫颈鳞状细胞癌和宫颈内膜腺癌、胆管癌、结肠腺癌、淋巴样肿瘤弥漫性大B细胞淋巴瘤、食管癌、多形性胶质母细胞瘤、头颈部鳞状细胞癌、肾嫌色细胞癌、肾透明细胞癌、肾乳头状细胞癌、肝细胞癌、肺腺癌、肺鳞状细胞癌、间皮瘤、卵巢浆液性囊腺癌、胰腺癌、嗜铬细胞瘤和副神经节瘤、前列腺癌、直肠腺癌、肉瘤、皮肤恶性黑色素瘤、胃腺癌、睾丸生殖细胞肿瘤、甲状腺癌、胸腺瘤、子宫癌肉瘤、子宫内膜癌、葡萄膜黑色素瘤或其任意组合。在一些实施例中,治疗包括与针对转移性癌症的主要治疗联合给予的佐剂,以提高主要治疗的疗效。在一些实施例中,佐剂是抗生素或抗微生物剂。在一些实施例中,治疗基于与转移性癌症或转移性癌症的环境相关的微生物成分或抗原。在一些实施例中,治疗包括靶向微生物抗原的过继细胞转移、针对微生物抗原的癌症疫苗、针对微生物抗原的单克隆抗体、设计成至少部分靶向微生物抗原的抗体-药物缀合物、设计成至少部分靶向一种或多种微生物抗原的多价抗体、抗体片段、其抗体衍生物,或其任意组合。在一些实施例中,治疗包括靶向微生物存在的一类功能上或生物学上相似的微生物的抗生素。在一些实施例中,治疗包括两种或更多种治疗类型,其中该两种或更多种治疗类型被组合,使得该两种或更多种治疗类型中的至少一种类型利用与转移性癌症或转移性癌症环境相关的微生物存在或丰度来增强治疗效果。在一些实施例中,净化的微生物存在与转移性癌症之间的关联进一步包括转移性癌症的起源、类型或其任意组合。
附图说明
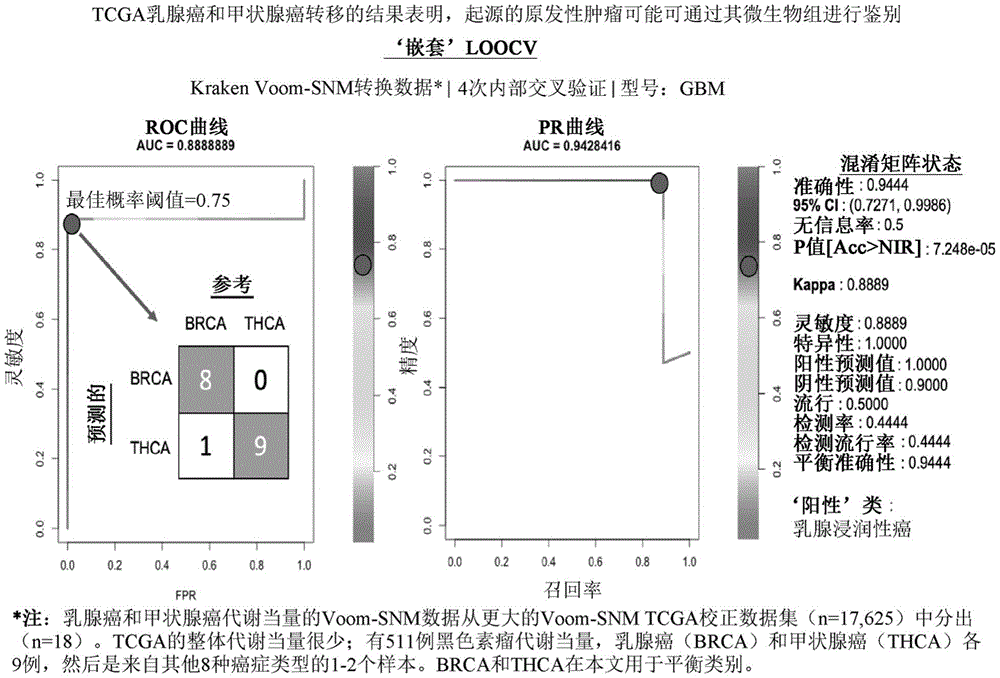
图1示出了留一交叉验证(LOOCV)机器学习结果,该结果鉴别转移性乳腺癌和转移性甲状腺癌组织样本,从而通过18名受试者的组织微生物组诊断起源原发性肿瘤(因为转移性癌症是根据其起源组织命名的)。
图2示出了使用来自559名受试者的血源性微生物DNA预测转移性癌症与非转移性癌症的分析。
图3示出了使用来自15名受试者的基于血液的微生物DNA对转移性黑色素瘤和其他转移性癌症类型的鉴别。图3中描绘的标有“其他转移性癌症类型”的样本包括乳腺癌(2个样本)、转移性甲状腺癌(2个样本)和转移性食管癌(1个样本)。在不同的实施例中,这些癌症类型和/或其他组合可以组合以提供足够的数量进行测试。
图4a图示了示出由微生物检测管线鉴定的总测序读数百分比,以及Kraken在TCGA数据集中在属层次解析的那些读数百分比的棒棒糖图(Lollipop plot)。LAML,急性髓系白血病;PAAD,胰腺癌;GBM,多形性胶质母细胞瘤;PRAD,前列腺癌;ESCA,食管癌;TCGT,睾丸生殖细胞肿瘤;BRCA,乳腺浸润性癌;THCA,甲状腺癌;KICH,肾嫌色细胞癌;THYM,胸腺瘤;READ,直肠腺癌;SARC,肉瘤;UVM,葡萄膜黑色素瘤;CHOL,胆管癌;ACC,肾上腺皮质癌;UCEC,子宫内膜癌;LUSC,肺鳞状细胞癌;PCPG,嗜铬细胞瘤和副神经节瘤;BLCA,膀胱尿路上皮癌;UCS,子宫癌肉瘤;LGG,脑低级别胶质瘤(图4a)。所有癌症类型中包括的样本总数为17,625。图4b图示了示出质量控制处理和剩余样本数量的CONSORT式图表。FFPE,固定福尔马林石蜡包埋。图4c图示了Voom归一化数据的主成分分析(PCA),其中癌症微生物组样本按测序中心着色。图4d图示了Voom-SNM数据的PCA。图4e图示了原始分类计数数据、Voom归一化数据和Voom-SNM数据的主方差分量分析。图4f-h图示了用于区分TCGA原发性肿瘤(图4f)、肿瘤和正常样本(图4g)以及I期和IV期癌症(图4h)的从灰度红色(高)到灰度蓝色(低)的分类器性能指标(AUROC(ROC)和AUPR(PR))的热图。“NA”可以指示在任何ML类中没有足够的样本(例如,少于20个)可用于模型训练。
图5a-g图示了根据至少一个实施例的TCGA癌症微生物组数据集内病毒和细菌读数的生态验证。图5a图示了使用在HMP2数据集上训练的SourceTracker2对COAD(n=70)患者的实体组织正常样本的平均身体部位归属。图5b图示了梭杆菌(Fusobacterium)属在与梭杆菌属BDN相关的常见胃肠道(GI)癌症中的不同丰度,BDN:血液来源,正常;STN:实体组织,正常;PT:原发性肿瘤。图5c图示了分组GI癌症(n=8:COAD、READ、CHOL、LIHC、PAAD、HNSC、ESCA、STAD;缩写见图8a)和非GI癌症(n=24)中梭杆菌丰度的差异(见方法)。图5d-e图示了患有CESC(图5d)或HNSC(图5e)的HPV感染患者的归一化HPV丰度,如TCGA中的临床所示。ISH,原位杂交;IHC,免疫组织化学。图5f图示了具有临床判定危险因素的LIHC患者的归一化正肝病毒丰度:HepB,既往乙型肝炎感染;EtOH,重度饮酒;HepC,既往丙型肝炎感染。图5g图示了STAD整合分子亚型中的归一化EBV丰度:CIN,染色体不稳定;GS,基因组稳定;MSI,微卫星不稳定;EBV,EBV感染的样本。在所有图中,血液来源正常和/或实体组织正常数据示出为比较阴性对照;对于两个以上的比较,双边曼-惠特尼U检验(Mann-Whitney U-test)与多重检验校正一起使用;箱形图示出了中间值(直线)、第25和第75个百分位数(箱形)以及1.5倍的四分位数间距(IQR,晶须)。灰度蓝色数字示出每组的样本数量。
图6a-d图示了使用血液中的mbDNA进行癌症鉴别的分类器性能,并作为癌症“液体”活检的补充诊断方法。图6a图示了类似于图4f-h的模型性能热图,以使用血液mbDNA预测一种癌症类型与所有其他癌症类型,TCGA研究ID在右侧(图8a);每个ML少数类别至少需要20个样本才有资格。图6b图示了使用Ia-IIc期癌症的血液mbDNA预测一种癌症类型与所有其他癌症类型的ML模型性能。图6c-d图示了根据Guardant360(图6c)和FoundationOneLiquid(图6d)ctDNA测定,使用来自没有可检测的原发性肿瘤基因组改变的患者的血液mbDNA的ML模型性能。FD,完整数据;LCR,可能由测序中心去除的污染物;APCR,由测序中心去除的所有假定的污染物;PCCR,去除的板中心污染物;MSF,由测序中心最严格的过滤。所包括的用于评估每次比较性能的样本数量可以在cancermicrobiome.ucsd.edu/CancerMicrobiome_DataBrowser的数据浏览器混淆矩阵中找到。
图7a-k图示了使用血浆来源的无细胞mbDNA鉴别癌症类型和健康对照的ML模型的性能。图7a,验证研究中分析的样本的人口统计学。所有患者均患有多种亚型的高分级(III-IV期)癌症,并被汇总为PC、LC和SKCM组。图7b图示了用于区分分组的癌症样本(n=100)和非癌症健康对照(n=69)的自举性能估计值。利用不同的训练-测试分割(70%训练-30%测试),来自500次迭代的ROC(顶图)和PR(底图)曲线数据的栅格化密度图。图7c–h图示了两类之间的留一(LOO)迭代ML性能:前列腺癌(PC)与对照(Ctrl;图7c),肺癌(LC)与对照(图7d),黑色素瘤(SKCM)与对照(图7e),PC与LC(图7f),LC与SKCM(图7g)以及PC与SKCM(图7h)。图7i-k图示了多类别(n=3或4)、LOO迭代ML性能,以区分癌症类型(图7i)以及患有癌症的混合患者与健康对照个体(分别为图7j和图7k)。将LOO ML总体性能计算为将一个与所有其他性能进行比较时的性能平均值(示出在混淆矩阵下方)。
图8a-g图示了TCGA癌症微生物组的持续概述。图8a图示了TCGA研究缩写表。图8b图示了Voom归一化数据的PCA,其中灰度颜色表示样本的测序平台,每个点表示一个癌症微生物组样本。图8c图示了连续Voom-SNM监督归一化后数据的PCA,如测序平台所标记的。图8d图示了Voom归一化数据的PCA,其中灰度颜色表示样本的实验策略,每个点表示一个癌症微生物组样本。图8e图示了连续Voom-SNM监督归一化后数据的PCA,如实验策略所标记的。图8f-g图示了元数据质量控制后TCGA中所有类型癌症的给定样本类型中的样本数量归一化的微生物读数计数(图4b),包括论文中分析的三种主要样本类型(图8f)和其余样本类型(图8g)。ANP,另外的,新的主要;AM,另外的转移;MM,转移性的;RT,复发性肿瘤。对于原始数据和归一化数据的PCA,n=17,625。
图9a-h图示了使用微生物丰度鉴别TCGA癌症类型之间和之内的性能度量。图9a–f图示了图4f-h中热图的实例。灰度-颜色梯度(顶图)表示ROC和PR曲线上任意点的概率阈值。使用50%概率阈值截止值示出了插入的混淆矩阵,该阈值截止值可用于计算ROC和PR曲线上相应点处的灵敏度、特异性、精度、召回率、阳性预测值、阴性预测值等。图9g-h图示了模型性能的线性回归,特别是AUROC(图9g)和AUPR(图9h),用于以一种癌症类型与所有其他类型的方式鉴别癌症类型,作为少数类大小的函数。示出了使用在原发性肿瘤中检测到的微生物的模型的性能,其中比较的样本数量(n=13,883)和癌症类型(n=32)最多。由于AUROC和AUPR的域为[0,1],并且少数类大小从20到1,238个样本不等,后者在log
图10a-i图示了ML模型管线的内部验证。图10a图示了TCGA原始微生物计数数据的两个独立部分被归一化并用于模型训练,以使用肿瘤微生物DNA和RNA预测一种癌症类型与所有其他癌症类型;然后将每个模型应用于另一半的归一化数据。与完整数据集的50–50%分割的训练和测试(分割1:n=8,814个样本;分割2:n=8,811个样本;总样本:n=17,625)相比,该热图比较了这些模型的性能。图10b-c图示了当通过多个测序中心的原发性肿瘤RNA样本(n=11,741)对完整Voom-SNM数据进行子集化以预测一种癌症类型与所有其他癌症类型(图10b,AUROC;图10c,AUPR)时的模型性能比较。图10d-e示出了当通过多个测序中心的原发性肿瘤RNA样本(n=2,142)对完整Voom-SNM数据进行子集化以预测一种癌症类型与所有其他癌症类型(图10d,AUROC;图10e,AUPR)时的模型性能比较。图10f-g图示了当通过来自UNC(n=9,726,仅进行RNA-seq)的样本对完整Voom-SNM数据进行子集化以使用原发肿瘤RNA样本(图10f,AUROC;图10g,AUPR)预测一种癌症类型与所有其他癌症类型时的模型性能比较。图10h-i图示了当通过来自HMS(n=898,仅进行WGS)的样本对完整Voom-SNM数据进行子集化以使用原发肿瘤DNA样本(图10h,AUROC;图10i,AUPR)预测一种癌症类型与所有其他癌症类型时的模型性能比较。图10b-i图示了广义线性模型,s.e.以灰色示出;虚线对角线表示完美的线性关系;对于样本量比较,完整的Voom-SNM数据集包含13,883个原发性肿瘤样本。
图11a-t图示了Kraken来源的TCGA癌症微生物组谱及其ML性能的正交验证。图11a-h图示了在使用肿瘤微生物DNA和RNA经由直接基因组比对(BWA)进行基于Kraken的分类分配后,对四种TCGA癌症类型(CESC,n=142(DNA)和n=309(RNA);STAD,n=322(DNA)和n=770(RNA);LUAD,n=351(DNA)和n=600(RNA);和OV,n=189(DNA)和n=850(RNA))进行了另外的筛选。针对以下对归一化、BWA过滤数据和匹配的、独立归一化的Kraken数据之间的ML性能进行比较:使用原发性肿瘤微生物比较一种癌症类型与所有其他癌症类型(图11a,AUROC;图11b,AUPR)、肿瘤与正常鉴别(图11c,AUROC;图11d,AUPR)、使用原发性肿瘤微生物进行I期与IV期肿瘤鉴别(图11e,AUROC;图11f,AUPR)、以及使用血液来源微生物比较一种癌症类型与所有其他癌症类型(图11g,AUROC;图11h,AUPR)(见方法)。图11i图示了BWA过滤数据和Kraken完整数据之间的分类群计数的维恩图。图11j-t图示了名为SHOGUN的正交微生物检测管线,并在TCGA样本的子集(n=13,517个总样本)上运行单独的数据库,经由Voom-SNM(类似于其Kraken对应物)归一化,并用于下游ML分析。图11j,SHOGUN来源的微生物分类群(S)和Kraken来源的微生物分类群(K)的维恩图。注意,SHOGUN的数据库不包括病毒,而Kraken数据库包括病毒。图11k-l图示了Voom(图11k)的PCA和Voom-SNM(图11l)归一化的SHOGUN数据,由测序中心灰度着色。图11m-t图示了使用相同的70%–30%分割,针对以下对在SHOGUN数据上训练和测试的模型与匹配的Kraken数据之间的ML性能进行比较:使用原发性肿瘤微生物比较一种癌症类型与所有其他癌症类型(图11m,AUROC;图11n,AUPR)、肿瘤与正常鉴别(图11o,AUROC;图11p,AUPR)、使用原发性肿瘤微生物进行I期与IV期肿瘤鉴别(图11q,AUROC;图11r,AUPR)、以及使用血液来源微生物比较一种癌症类型与所有其他癌症类型(图11s,AUROC;图11t,AUPR)。为了公平比较,匹配的Kraken数据是通过去除原始Kraken计数数据中的所有病毒分配并子集化到SHOGUN分析的相同的13,517个TCGA样本获得的;然后,这些匹配的Kraken数据以与SHOGUN数据相同的方式通过Voom-SNM独立地归一化(见方法),并输入下游ML管线。对于所有ML性能,要求每类中≥20个样本合格。对于回归子图,虚斜线表示完美的性能对应关系;示出了具有s.e.带的广义线性模型。
图12a-e图示了泛癌微生物丰度和用于TCGA癌症微生物组分析和ML模型检测的交互式网站。图12a示出了梭杆菌的泛癌归一化丰度,其中对每种样本类型的各种癌症类型的微生物丰度进行了单向ANOVA(Kruskal-Wallis)检验。样本量以灰度-蓝色插入,箱形图示出中值(直线)、第25和第75个百分位数(箱形)和1.5×IQR(晶须);TCGA研究缩写如下所列并定义于图8a。图12b图示了基于HMP2数据的TCGA-COAD实体组织正常样本(n=70)和TCGASKCM原发性肿瘤样本(n=122)的粪便贡献的SourceTracker2结果。只有一个实体组织正常样本可用于TCGA-SKCM(补充表4),因此原发性肿瘤被用作预期皮肤菌群的最佳代表。预计结肠样本的粪便贡献应高于皮肤,因此使用了单侧曼-惠特尼U检验。由于SourceTracker2输出每个源(即HMP2)对每个汇(即COAD、SKCM样本)的平均分数贡献,因此每个条形图的中心值是这些值的平均值,误差线表示s.e.m。样本量以灰度蓝色示出如下。图12c图示了阿尔法乳头瘤病毒属的泛癌归一化丰度,其中对每种样本类型的各种癌症类型的微生物丰度进行了单向ANOVA(Kruskal-Wallis)检验。样本量以灰度-蓝色插入,箱形图示出中值(直线)、第25和第75个百分位数(箱形)和1.5×IQR(晶须);TCGA研究缩写如下所列并定义于图8a。TCGA研究将临床测试的HPV感染的患者分为阴性和阳性组。图12d图示了示出使用Kraken来源的数据绘制的阿尔法乳头瘤病毒属归一化微生物丰度的交互式网站的屏幕截图。使用SHOGUN来源的归一化微生物丰度绘制可在网站的另一个选项卡(左侧)上获得。图12e图示了ML模型检查的交互式网站的屏幕截图。选择数据类型(例如,去除所有可能的污染物)、癌症类型(例如浸润性乳腺癌)和感兴趣的比较(例如,肿瘤与正常)将自动更新ROC和PR曲线,以及混淆矩阵(使用50%的概率截止阈值)和分级模型特征列表。网站可在cancermicrobiome.ucsd.edu/CancerMicrobiome_DataBrowser访问。
图13a-l图示了净化方法及其对癌症微生物组数据的结果、益处和局限性。图13a图示了用于评估、减轻、去除和/或模拟污染源的各种方法。图13b图示了在不同程度的净化后,TCGA剩余分类群或微生物读数的比例。测序中心的净化去除了在任何一个测序中心被鉴定为污染物的所有分类群(n=8批次);通过平板-中心组合净化去除了任何具有十个以上TCGA样本的单个测序板上被鉴定为污染物的所有分类群(n=351批次)。图13c-f图示了对可能的污染物去除数据集(图13c)、板中心净化数据集(图13d)、所有假定污染物去除数据集(图13e)和最严格的过滤数据集(图13f)的身体部位归属预测。图13g-l图示了使用上述四个净化数据集(如上所示,每个数据集标有不同的灰度-颜色)重新生成的模型和伴随性能值(AUROC和AUPR)。从净化数据集上训练和测试的模型获得的AUROC和AUPR值相对于完整数据集(图4f-h)中的AUROC或AUPR值作图。虚线对角线表示完美的线性关系。广义线性模型已拟合到相应数据集的AUROC和AUPR值;线性拟合的s.e.由关联的阴影区域示出。COAD(n=1,006个总样本)模型性能在所有图中均有标识。
图14a-c图示了净化对每种样本类型的平均读数比例的影响。将每种主要样本类型(原发性肿瘤(图14a)、实体组织正常(图14b)、血液来源正常(图14c))的总读数计数(DNA和RNA)相加并除以每种样本类型中的样本总数。然后将该归一化读数计数(每种样本类型)除以每种癌症类型的所有样本类型的归一化读数总和,从而提供对每种癌症类型每种样本类型的平均读数比例的估计。如图例所示,对所有五个数据集重复该过程,以评估净化是否对某些类型的样本和/或癌症有不同的影响;所示百分比的相对稳定性将表明缺乏差异污染。本文中未通过净化或ML进一步分析的次要样本类型(例如,另外的转移性病变;n=4种样本类型;图8g)未在此处示出。注意,在特殊情况下,对于给定的癌症类型(ACC、MESO、UCS中的原发性肿瘤),仅存在一种样本类型,则所有条形将示出100%的归一化读数来自该样本类型。检查的癌症样本总数为17,625。
图15a-e图示了在下游ML模型中测量加标伪污染物的贡献,以及在TCGA患者中测量市售的基于宿主的ctDNA测定的理论灵敏度。图15a-b图示了使用原发性肿瘤微生物DNA或RNA(图15a)或使用血液来源的mbDNA(图15b),在所有四个净化数据集(图13b)中,针对经训练以鉴别一种癌症类型与所有其他癌症类型的模型中使用的所有分类群计算特征重要性评分。这些净化数据集在净化和归一化管线之前加标了伪污染物,以评估其性能(见方法),所示模型的测试集性能分别在图13g-h和图6a中给出。模型使用的任何加标伪污染物的特征重要性分数除以该模型中所有特征重要性分数的总和,以估计它们对做出准确预测的百分比贡献;分数越高(满分100分),模型在生物学上就越不可靠。注意,零表示模型没有使用加标的伪污染物进行预测;在板-中心净化数据上生成的模型均未包括加标的伪污染物作为特征。所包括的用于评估每次比较性能的样本数量可以在cancermicrobiome.ucsd.edu/CancerMicrobiome_DataBrowser的数据浏览器混淆矩阵中找到。图15c-d图示了在FoundationOne Liquid ctDNA编码基因(图15c)或Guardant360ctDNA编码基因(图15d)上具有一个或多个基因组改变的患者的TCGA研究中的百分比分布。检查的样本数量和原始数据可在cbioportal.org获得。图15e图示了包含FoundationOne和Guardant360 ctDNA测定的编码基因列表及其检查的改变(方法中列出的来源)的表格。
图16a-k图示了健康个体和多种癌症类型之间的真实世界、血浆来源、无细胞微生物DNA分析的分析。图16a图示了用于为真实世界的验证研究提供经验支持的TCGA中的鉴别性模拟(图7;见方法)。每个分层样本量的中心值是十次迭代中性能的平均值;误差条指代s.e.m。图16b图示了使用Kraken和SHOGUN来源数据评估阳性对照细菌(Aliivibrio)单一培养物、阴性对照空白和人样本类型中的Aliivibrio属丰度值(原始读数计数)。图16c图示了细菌单一培养稀释液中的Aliivibrio属丰度(原始读数计数)。图16d图示了无癌症健康对照个体(Ctrl)和患有肺癌(LC)、前列腺癌(PC)或黑色素瘤(SKCM)分组患者的年龄分布。图16e图示了插入皮尔逊χ
图17a-j图示了SHOGUN来源的ML性能,以使用无细胞微生物DNA鉴别癌症类型和健康、无癌症的个体。图17a图示了区分分组癌症患者(n=100)和无癌症健康对照个体(n=69)的自举性能估计。来自具有不同训练-测试分割(70%训练-30%测试)的500次迭代的ROC和PR曲线数据示出在栅格化密度图上;示出了平均值和95%置信区间估计值。图17b-g图示了两类之间的LOO迭代ML性能:前列腺癌(PC)与对照(图17b),肺癌(LC)与对照(图17c),黑色素瘤(SKCM)与对照(图17d),PC与LC(图17e),LC与SKCM(图17f)以及PC与SKCM(图17g)。图17h-j图示了多类别(n=3或4)、留一法(LOO)迭代ML性能,以区分癌症类型,以及癌症患者和健康的无癌症对照个体。平均AUROC和AUPR,由一对所有其他AUROC和AUPR值计算得出,示出在混淆矩阵下方。图17h图示了所研究的三种癌症类型之间的LOO ML性能。图17i示出了至少有20个样本属于少数类的三种样本类型之间的LOO ML性能(即TCGA分析中使用的截止值,图4f-h)。图17j图示了所研究的所有四种样本类型之间的LOO ML性能。对于具有混淆矩阵图的所有子图:由于样本量小,使用LOO ML代替单一或自举训练-测试分割;这些混淆矩阵还反映了用于每次比较的样本数量。
图18是图示根据本公开的一个或多个示例性实施例可以执行一种或多种技术(例如,方法)的计算设备或计算机系统的实例的框图。
具体实施方式
本说明书中提及的所有出版物、专利和专利申请均以引用方式并入本文,其程度与每个单独的出版物、专利或专利申请被明确且单独地指出为通过引用并入的程度相同。
除非另外定义,否则本文使用的所有技术和科学术语和任何首字母缩写具有与本发明所属领域的普通技术人员通常理解的含义相同的含义。虽然可以在本发明的实践中使用与本文所描述的那些类似或等效的任何方法和材料,但是本文中描述了示例性方法、设备和材料。
除非另有说明,各种实施例的实践将采用分子生物学(包括诊断技术)、微生物学、细胞生物学、生物化学和免疫学的常规技术,这些技术属于本领域技术人员的技术范围内。此类技术在以下文献中有充分解释:如《分子克隆:实验室手册(Molecular Cloning:ALaboratory Manual)》,第2版(Sambrook等人,1989);《寡核苷酸合成(OligonucleotideSynthesis)》(M.J.Gait编辑,1984);《动物细胞培养(Animal Cell Culture)》(R.I.Freshney编辑,1987);《酶学方法(Methods in Enzymology)》(学术出版社(AcademicPress,Inc.));《分子生物学最新进展(Current Protocols in Molecular Biology)》(F.M.Ausubel等人编辑,1987,并且定期更新);《PCR:聚合酶链式反应(PCR:ThePolymerase Chain Reaction)》(Mullis等人编辑,1994);《雷明顿:药学科学与实践(Remington,The Science and Practice of Pharmacy)》,第20版,(利平科特·威廉斯·威尔金斯出版公司(Lippincott,Williams&Wilkins)2003),和《雷明顿:药学科学与实践》,第22版,(英国医药出版社和科学大学费城药学院(Pharmaceutical Press andPhiladelphia College of Pharmacy at University of the Sciences)2012)。
至少一个实施例提供了基于患有转移性癌症的受试者的组织或血液中的微生物群来检测和测定起源的转移组织的方法。在实施例中,本发明提供了一种用于使用微生物核酸基于组织或血液中的微生物群来测定转移的起源组织的方法,其包括:
(a)从患者活检获得转移性癌症组织的样本,包括实体组织或血液;
(b)从癌症组织的样本中提取核酸,例如用ZymoBIOMICS DNA Miniprep试剂盒;
(c)从提取的核酸制备核酸测序文库,如使用KAPA HyperPlus试剂盒;
(d)使用下一代测序(NGS)对核酸文库进行测序,如在Illumina NovaSeq 6000仪器上;
(e)将输出的核酸测序读数与已知的微生物基因组进行比对,以获得样本的微生物丰度表;如使用SHOGUN算法(PMID:30443602);以及
(f)将微生物丰度表输入到机器学习算法中,以确定或预测转移性癌症的起源组织,如使用梯度提升分类树。
至少一个实施例提供了核酸可以是DNA或RNA。在实施例中,这些步骤可以用于关注微生物DNA或RNA。其他替代方法包括微生物DNA和RNA与宿主DNA和RNA的组合,以对转移的起源组织进行更准确的诊断。
至少一个实施例提供了在将核酸测序读数与已知微生物基因组进行比对之前,去除非微生物核酸。
至少一个实施例提供了在将核酸测序读数与已知微生物基因组进行比对之前,去除污染的微生物核酸。
至少一个实施例提供了在将核酸测序读数与已知微生物基因组进行比对之后但在将微生物丰度表输入机器学习算法之前,去除污染的微生物核酸。
当将输出的核酸测序读数与已知微生物基因组进行比对时,至少一个实施例产生微生物存在或不存在信息,其中微生物存在或不存在信息稍后用于机器学习。
至少一个实施例提供了可以从受试者的任何组织中提取核酸,包括实体组织、肿瘤、血液、液体活检或其任意组合。因此,核酸可以从循环血液、循环血液的成分(例如血浆、白细胞、血小板)或其任意组合中提取。
至少一个实施例进一步提供了基于转移性癌症的起源组织的确定来预测、预防规程和/或治疗受试者的方法,其包括向受试者施用指示转移的有效量的治疗组合物或治疗方案。
定义
为了促进理解本发明,如本文中所使用的多个术语和缩写如下定义在下文中:
如本文所用,术语“包括(comprises)”、“包括(comprising)”、“包含(includes)”、“包含(including)”、“具有(has)”、“具有(having)”“含有(contains)”、“含有(containing)”、“特征在于”或其任何其他变体旨在涵盖所列举的组分的非排他性包含,经受以其他方式明确指出的任何限制。例如,“包括”要素列表(例如,组分、特征或步骤)的融合蛋白、药物组合物和/或方法不一定仅限于那些元素(或组分或步骤),而是可以包含融合蛋白、药物组成和/或方法未明确列出或固有的其他元素(或组分或步骤)。
如本文所用,过渡短语“由……组成(consists of)”和“由……组成(consistingof)”不包括未指定的任何要素、步骤或组分。例如,权利要求中使用的“由……组成(consists of)”或“由……组成(consisting of)”将权利要求限制到权利要求中具体列举的组分、材料或步骤,除了通常与其相关的杂质(即,给定组分内的杂质)之外。当短语“由……组成(consists of)”或“由……组成(consisting of)”出现在权利要求主体的条款中,而不是紧接在序言之后时,短语“由……组成(consists of)”或“由……组成(consisting of)”仅限制所述条款中阐述的要素(或组分或步骤);其他要素(或组分)作为整体未被排除在权利要求之外。
如本文所用,过渡短语“基本上由……组成(consists essentially of)”和“基本上由……组成(consisting essentially of)”用于定义融合蛋白、药物组合物和/或方法,除了那些字面上公开的之外,所述融合蛋白、药物组合物和/或方法包括材料、步骤、特征、组分或要素,条件是这些另外的材料、步骤、特征、组分或要素不会实质性地影响所要求保护的发明的基本和新颖特性。术语“基本上由……组成”占据介于“包括”与“由……组成”之间的中间地带。
当介绍本发明或其优选的实施例的元素时,冠词“一个(a)”、“一种(an)”、“该(the)”和“所述(said)”旨在表示存在元素中的一个或多个元素。术语“包括”、“包含”和“具有”旨在是包含性的,并且意味着除了所列出的元素之外,还可以有另外的元素。
当用于两个或更多个项的列表中时,术语“和/或”意指所列项中的任何一项可以单独使用或与所列项中的任何一项或多项组合使用。例如,表达“A和/或B”意在意指A和B中的一者或两者,即单独的A、单独的B或A和B的组合。表达“A、B和/或C”旨在意指单独的A、单独的B、单独的C、A和B的组合、A和C的组合、B和C的组合,或A、B和C的组合。
应理解,本文所述的本发明的方面和实施例包含“由方面和实施例组成”和/或“基本上由方面和实施例组成”。
应当理解的是,采用范围格式的描述仅仅是为了方便和清楚,而不应被理解为对本发明范围的僵化限制。因此,范围的描述应该被认为已经具体公开了所有可能的子范围以及该范围内的单个数值。举例来说,如1到6的范围的描述应该认为是已经确切地公开了子范围,如1到3、1到4、1到5、2到4、2到6、3到6等,以及所述范围内的单个数值,例如1、2、3、4、5和6。无论范围的广度如何,这都适用。在本文中,值或范围可以表达为“约”、从“约”一个特定值和/或到“约”另一个特定值。当表达这类值或范围时,公开的其他实施例包括所列举的特定值、从一个特定值和/或到另一特定值。类似地,当通过使用先行词“约”将值表示为近似值时,将理解该特定值形成了另一个实施例。将进一步理解,其中公开了多个值,并且每个值在本文还被公开为除了值本身之外的“约”所述特定值。在实施例中,“约”可以用于表示,例如,所述值的10%以内、所述值的5%以内,或所述值的2%以内。
如本文所用,“患者”或“受试者”意指待诊断或治疗的人或动物受试者。
如本文所用,术语“药物组合物”是指药学上可接受的组合物,其中该组合物包含药学活性剂,并且在一些实施例中进一步包含药学上可接受的载体。在一些实施例中,药物组合物可以是药物活性剂和载体的组合。
如本文所用,术语“药学上可接受的”意指除可安全用于动物并且更具体地可用于人和/或非人哺乳动物的其他调配物之外由联邦或州政府的监管机构批准或列于美国药典、其他公认的药典中。
如本文所用,术语“药学上可接受的载体”是指与去甲基化化合物一起施用的赋形剂、稀释剂、防腐剂、增溶剂、乳化剂、佐剂和/或媒剂。此类载体可以是无菌液体,如水和油,包括石油、动物、植物或合成来源的那些油,如花生油、大豆油、矿物油、芝麻油等,聚乙二醇、甘油、丙二醇或其他合成溶剂。抗菌剂,如苯甲醇或对羟基苯甲酸甲酯;抗氧化剂,如抗坏血酸或亚硫酸氢钠;螯合剂,如乙二胺四乙酸;以及如氯化钠或右旋糖等用于调节张力的药剂也可以是载体。本领域技术人员已知用于产生与载体组合的组合物的方法。在一些实施例中,语言“药学上可接受的载体”旨在包含与药物施用相容的任何和所有溶剂、分散介质、包衣、等渗剂以及吸收延迟剂等。此类介质和药剂用于药学活性物质的用途是所属领域熟知的。参见,例如《雷明顿:药学科学与实践》,第20版,(利平科特·威廉斯·威尔金斯出版公司2003)。除了任何常规介质或药剂与活性化合物不相容的情况之外,设想此类介质或药剂在组合物中的用途。
如本文所用,“治疗有效量”是指足以治疗或改善,或以某种方式减轻与疾病和医疗病症相关的症状的药物活性化合物的量。当参考方法使用时,所述方法足以有效治疗或改善,或以某种方式减轻与疾病或病症相关的症状。例如,关于疾病的有效量是足以阻断或预防发作的量;或者如果疾病病理已经开始,则缓解、改善、稳定、逆转或减缓疾病的进展,或以其他方式减少疾病的病理后果。在任何情况下,有效量可以单剂量或分开的剂量给予。
如本文所用,术语“治疗(treat)”、“治疗(treatment)”或“治疗(treating)”至少包括与患者疾病相关的症状的改善,其中改善在广义上用于指至少减少参数的大小,例如与所治疗的疾病或病症相关的症状。因此,“治疗”还包括疾病、障碍或病理状况,或至少与之相关的症状被完全抑制(例如阻止发生)或停止(例如终止)的情况,使得患者不再患有该病症,或者至少不再患有表征该病症的症状。
如本文所用,除非另有说明,术语“预防(prevent)”、“预防(preventing)”和“预防(prevention)”是指预防疾病或障碍或其一种或多种症状发作、复发或扩散。在某些实施例中,所述术语是指在症状发作之前,使用具有或不具有一种或多种其他另外的活性剂的本文所提供的化合物或剂型进行治疗或具体地向有风险患有本文所提供的疾病或障碍的受试者施用所述化合物或剂型。这些术语涵盖抑制或减少特定疾病的症状。在某些实施例中,具有家族疾病史的受试者是预防方案的潜在候选者。在某些实施例中,具有复发症状史的受试者也是预防的潜在候选者。在这方面,术语“预防”与术语“预防性治疗”可互换使用。
如本文所用,除非另有说明,否则化合物的“预防有效量”是足以预防疾病或障碍或防止其复发的量。预防有效量的化合物是指在疾病预防中提供预防性益处的治疗剂单独或与一种或多种其他药剂组合的量。术语“预防有效量”可以包括改善总体预防或增强另一种预防剂的预防效果的量。
“扩增”是指用于获得靶核酸或其互补序列或其片段的多个拷贝的任何已知规程。所述多个拷贝可以被称为扩增子或扩增产物。在片段的情况下,扩增是指产生含有少于完整靶核酸或其互补序列的扩增核酸,例如,通过使用与靶核酸的内部位置杂交并从该位置引发聚合的扩增寡核苷酸产生的扩增核酸。已知的扩增方法包括,例如,复制酶介导的扩增、聚合酶链反应(PCR)、逆转录聚合酶链反应(RT-PCR)、连接酶链反应(LCR)、链置换扩增(SDA)和转录介导或转录相关扩增。扩增不限于起始分子的严格复制。例如,使用逆转录(RT)-PCR从样本中的RNA生成多个cDNA分子是一种扩增形式。此外,在转录过程中从单个DNA分子生成多个RNA分子也是一种扩增形式。在扩增过程中,可以使用标记的引物或掺入标记的核苷酸等方法标记扩增的产物。
“扩增子”或“扩增产物”是指在扩增规程中生成的与靶核酸或其区域互补或同源的核酸分子。扩增子可以是双链或单链,并且可以包括DNA、RNA或两者。生成扩增子的方法为本领域技术人员已知的。
“密码子”是指在核酸中共同形成遗传密码单元的三个核苷酸的序列。
“感兴趣的密码子”是指靶核酸中具有诊断或治疗意义的特定密码子(例如与病毒基因型/亚型或耐药性相关的等位基因)。
“互补”或“其互补序列”是指连续的核酸碱基序列能够通过一系列互补碱基之间的标准碱基配对(氢键)与另一个碱基序列杂交。通过使用标准碱基配对(例如,G:C、A:T或A:U配对),互补序列可以在寡聚物序列中相对于其靶序列的每个位置完全互补(即核酸双链体中没有错配),或者序列可以含有一个或多个不通过碱基配对互补的位置(例如,核酸双链体中至少存在一个错配或不匹配的碱基),但此类序列是充分互补的,因为整个低聚物序列能够在适当的杂交条件下与其靶序列特异性杂交(即部分互补)。低聚物中的连续碱基通常与预期的靶序列至少80%,优选至少90%互补,并且更优选与预期的靶序列完全互补。
“配置为”或“设计为”表示参考寡核苷酸的核酸序列配置的实际排列。例如,配置为从靶核酸生成特定扩增子的引物具有与靶核酸或其区域杂交的核酸序列,并且可以用于扩增反应以生成扩增子。同样作为实例,配置为特异性杂交到靶核酸或其区域的寡核苷酸具有在严格的杂交条件下特异性杂交到参考序列的核酸序列。
“下游”是指进一步沿着核酸序列在序列转录或读出的方向。
“上游”是指进一步沿着核酸序列在与序列转录或读出方向相反的方向。
“聚合酶链反应”(PCR)通常是指使用核酸变性、引物对退火到相反链(正向和反向)以及引物延伸的多个循环以指数地增加靶核酸序列拷贝数的过程。在称为RT-PCR的变体中,逆转录酶(RT)用于从mRNA制造互补DNA(cDNA),然后通过PCR扩增cDNA以产生多个DNA拷贝。PCR有许多本领域普通技术人员已知的排列。
“位置”是指核酸序列中的一个或多个特定氨基酸。
“引物”是指酶促可扩展的寡核苷酸,通常具有确定的序列,被设计为以反平行方式与靶核酸的互补性引物特异性部分杂交。当置于合适的核酸合成条件下时,引物可以以模板依赖的方式引发核苷酸的聚合,以产生与靶核酸互补的核酸(例如,在合适的温度和pH下,退火到靶的引物可以在核苷酸和DNA/RNA聚合酶的存在下延伸)。合适的反应条件和试剂是本领域普通技术人员所熟知的。引物通常是单链的,以获得最大的扩增效率,但也可以是双链的。如果是双链,引物通常首先处理以分离其链,然后再用于制备延伸产物。引物通常足够长,以在诱导剂(例如聚合酶)存在下引发延伸产物的合成。具体长度和序列将取决于所需DNA或RNA靶的复杂性,以及引物使用条件,如温度和离子强度。优选地,引物是约5-100个核苷酸。因此,引物的长度可以是例如5、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、40、45、50、55、60、65、70、75、80、85、90、95或100个核苷酸。引物不需要与其模板具有100%的互补性即可发生引物延伸;具有小于100%互补性的引物足以发生杂交和聚合酶延伸。如果需要,可以标记引物。引物上使用的标记可以是任何合适的标记,并且可以通过例如光谱、光化学、生物化学、免疫化学、化学或其他检测手段来检测。因此,标记引物是指在促进杂交以允许选择性检测靶序列的条件下与核酸或扩增核酸中的靶序列特异性杂交的低聚物。
如果需要,可以通过掺入可通过例如光谱、光化学、生物化学、免疫化学、化学或其他技术检测的标记来标记引物核酸。举例来说,有用的标记包括放射性同位素、荧光染料、电子致密试剂、酶(如ELISA中通常使用的)、生物素或可获得抗血清或单克隆抗体的半抗原和蛋白质。许多这些和其他标记在本文中进一步描述和/或以其他方式在本领域已知。本领域技术人员将认识到,在某些实施例中,引物核酸也可以用作探针核酸。
“区域”是指核酸的一部分,其中所述部分小于整个核酸。
“感兴趣区域”是指靶核酸的特定序列,其包括具有与待扩增和检测的基因型和/或亚型相关的至少一个单核苷酸取代突变的所有密码子位置,以及待扩增和检测的所有标记物位置(如果有的话)。
“RNA依赖性DNA聚合酶”或“逆转录酶”(“RT”)是指从RNA模板合成互补DNA拷贝的酶。所有已知的逆转录酶也具有从DNA模板制作互补DNA拷贝的能力;因此,它们都是RNA和DNA依赖性DNA聚合酶。RT也可具有RNA酶H活性。需要引物来启动RNA和DNA模板的合成。
“DNA依赖性DNA聚合酶”是一种从DNA模板合成互补DNA拷贝的酶。实例为自大肠杆菌的DNA聚合酶I、噬菌体T7 DNA聚合酶或来自噬菌体T4、Phi-29、M2或T5的DNA聚合酶。DNA依赖性DNA聚合酶可以是从细菌或噬菌体中分离的天然存在的酶或重组表达的酶,或者可以是修饰或“进化”的形式,这些形式已被工程化以具有某些理想的特征,例如,热稳定性,或从各种修饰的模板识别或合成DNA链的能力。所有已知的DNA依赖性DNA聚合酶都需要互补引物来起始合成。已知在合适的条件下,DNA依赖性DNA聚合酶可以从RNA模板合成互补DNA拷贝。RNA依赖性DNA聚合酶通常还具有DNA依赖性DNA聚合酶活性。
“DNA依赖性RNA聚合酶”或“转录酶”是一种从双链或部分双链DNA分子合成多个RNA拷贝的酶,该双链或部分双链DNA分子具有通常为双链的启动子序列。RNA分子(“转录子”)从启动子下游的特定位置开始在5'至3'方向合成。转录酶的实例是来自大肠杆菌和噬菌体T7、T3和SP6的DNA依赖性RNA聚合酶。
核酸的“序列”是指核酸中核苷酸的顺序和同一性。序列通常在5'至3'方向上读取。在两种或更多种核酸或多肽序列的情况下,术语“相同”或“同一性”百分比是指两种或更多种序列或子序列是相同的,或具有相同的特定百分比的氨基酸残基或核苷酸,当进行最大对应性的比较和比对时,例如,使用技术人员可获得的序列比较算法中的一种或通过目视检查来测量。适用于确定序列同一性百分比和序列相似性的示例性算法是BLAST规程,其描述于:例如,Altschul等人(1990)“基本局部比对搜索工具(Basic local AlignSearch Tool)”《分子生物学杂志(J.Mol.Biol.)》215:403-410,Gish等人(1993)“通过数据库相似性搜索识别蛋白质编码区域(Identification of protein coding regions bydatabase similarity search)”《自然-遗传学(Nature Genet.)》3:266-272,Madden等人(1996)“网络BLAST服务器应用(Applications of network BLAST server)”《酶学方法(Meth.Enzymol.)》266:131-141,Altschul等人(1997)“Gapped BLAST和PSI-BLAST:新一代蛋白质数据库搜索程序(Gapped BLAST and PSI-BLAST:a new generation of proteindatabase search programs))”《核酸研究(Nucleic Acids Res.)》25:3389-3402,以及Zhang等人(1997)“PowerBLAST:用于交互式或自动序列分析和注释的新型网络BLAST应用(PowerBLAST:A new network BLAST application for interinteractive or automatedsequence analysis and annotation)”《基因组研究(Genome Res.)》7:649-656,每个都通过引用并入。许多其他的最佳比对算法也是本领域已知的,并且任选地用于确定序列同一性百分比。
“标记”是指连接(共价或非共价)或能够连接至分子的部分,该部分提供或能够提供关于分子的信息(例如,关于分子的描述性、识别性等信息)或与经标记分子相互作用(例如杂交等)的另一分子。示例性标记包括荧光标记(包括例如猝灭剂或吸收剂)、弱荧光标记、非荧光标记、比色标记、化学发光标记、生物发光标记、放射性标记、质量修饰基团、抗体、抗原、生物素、半抗原、酶(包括例如过氧化物酶、磷酸酶等)等。
“接头”是指将化合物或取代基共价或非共价连接到另一部分的化学部分,例如核酸、寡核苷酸探针、引物核酸、扩增子、固体载体等。例如,接头任选地用于将寡核苷酸探针连接到固体载体上(例如,在线性或其他逻辑探针阵列中)。为了进一步说明,接头任选地将标记(例如,荧光染料、放射性同位素等)连接到寡核苷酸探针、引物核酸等上。接头通常至少是双功能化学部分,并且在某些实施例中,它们包含可裂解的连接物,其可以通过例如热、酶、化学试剂、电磁辐射等裂解以从例如固体载体中释放材料或化合物。仔细选择接头允许在与化合物的稳定性和测定方法相容的适当条件下进行切割。通常,除了例如将化学物种连接在一起或保持这些物种之间的某种最小距离或其他空间关系之外,接头没有特定的生物活性。然而,可以选择接头的成分来影响连接的化学物质的某些性质,如三维构象、净电荷、疏水性等。示例性接头包括例如寡肽、寡核苷酸、寡聚酰胺、低聚乙烯甘油、低聚丙烯酰胺、烷基链等。接头分子的另外描述提供于:例如,Hermanson,《生物共轭体技术(Bioconjugate Techniques)》,爱思唯尔科学(Elsevier Science)(1996),Lyttle等人(1996)《核酸研究》24(14):2793,Shchepino等人(2001)《核苷、核苷酸和核酸(Nucleosides,Nucleotides,&Nucleic Acids)》20:369,Doronina等人(2001)《核苷、核苷酸和核酸》20:1007,Trawick等人(2001)《生物共轭化学(Bioconjugate Chem.)》12:900,Olejnik等人(1998)《酶学方法》291:135,以及Pljevaljcic等人(2003)《美国化学会志(J.Am.Chem.Soc.)》125(12):3486,所有这些都通过引用并入。
“片段”是指含有少于完整核酸的核苷酸的一段连续核酸。
“杂交”、“退火”、“选择性结合(selective bind)”或“选择性结合(selectivebinding)”是指一种核酸与另一种核酸(通常是反平行核酸)的碱基配对相互作用,其导致双链体或其他高阶结构(即杂交复合物)的形成。反平行核酸分子之间的主要相互作用通常是碱基特异性的,例如A/T和G/C。不要求两个核酸在其全长上具有100%互补性来实现杂交。核酸杂交归因于各种充分表征的理化力,如氢键、溶剂排斥、碱基堆积等。核酸杂交的详细指南见于Tijssen(1993)《生物化学和分子生物学中的实验室技术-与核酸探针杂交(Laboratory Techniques in Biochemistry and Molecular Biology-Hybridizationwith Nucleic Acid Probes)》第一部分第2章,“杂交原理概述和核酸探针测定策略(Overview of principles of hybridization and the strategy of nucleic acidprobe assays)”(爱思唯尔,纽约),以及Ausubel(编辑)《分子生物学当前协议(CurrentProtocols in Molecular Biology)》,第一、二和三卷,1997,其通过引用并入本文。
实例
本研究基于对来自11种癌症类型的500多个转移性癌症组织样本的初步分析。图1示出了通过其组织微生物组来鉴别转移性乳腺癌和转移性甲状腺癌的机器学习结果,表明可以通过微生物特征来鉴别起源的原发性肿瘤(因为转移性癌症是根据其起源组织来命名的)。在至少一个实施例中,乳腺癌和甲状腺癌转移的Kraken Voom-SNM转换数据从较大的TCGA Voom-SNM校正数据集(n=17,625)中子集化(n=18)。TCGA包含511个黑色素瘤转移瘤,然后乳腺癌(BRCA)和甲状腺癌(THCA)各9个,然后是来自其他8种癌症类型的1-2个样本。BRCA和THCA在本文被用作具有平衡类别的说明性实例。
在至少一个实施例中,不需要本文所述的机器学习模型或算法来确定微生物丰度;相反,该步骤是在使用分类分配算法之前完成的。然后,在此类实施例中,机器学习算法对微生物的重要性进行排序,以确定哪个样本属于某种癌症类型。在各种实施例中,Kraken是分类分配算法(PMID:24580807),而机器学习算法是梯度提升(Friedman,Jerome H.“随机梯度提升(Stochastic gradient boosting)”《计算统计与数据分析(Computationalstatistics&data analysis)》38.4(2002):367-378),其中每个通过引用以其全文并入本文。
癌症微生物组的系统表征提供了开发在主要人类疾病的诊断中利用非人微生物来源的分子的技术的机会。最近的研究表明,一些类型的癌症表现出大量的微生物贡献,对来自初治患者的33种癌症类型(总共18,116个样本)的TCGA进行了全基因组和全转录组测序研究,重新检查了其微生物读数,并且使用本文所述的技术在主要类型的癌症内和之间的组织和血液中发现了独特的微生物特征。当应用于患有Ia-IIc期癌症以及患有目前在两个商业级无细胞肿瘤DNA平台上测量的缺乏任何基因组改变的癌症的患者时,这些TCGA血液特征仍然保留有预测性,尽管使用了非常严格的净化分析,丢弃了高达92.3%的总序列数据。此外,使用本文所述技术,仅使用血浆来源的无细胞微生物核酸可以鉴别来自健康、无癌症个体(n=69)的样本和来自患有多种类型癌症(前列腺癌、肺癌和黑色素瘤;总共100个样本)的患者的样本。这种潜在的基于微生物组的肿瘤学诊断工具值得进一步探索。
癌症通常被认为是人类基因组的疾病。然而,最近的研究表明,微生物组对一些类型的癌症有实质性的贡献。具体地,粪便微生物组对胃肠癌的贡献。然而,微生物对不同类型癌症的贡献程度和诊断意义仍然未知。在收集、处理和测序过程中样本污染的可能性限制了这些研究,因为很少在癌症基因组学项目中实施规程控制。在各种实施例中,可以使用最近开发的工具来最小化污染物对微生物特征的贡献,以便能够合理开发基于微生物组的诊断。
为了表征癌症相关微生物组,对TCGA全基因组测序(WGS;n=4,831)和全转录组测序(RNA-seq;n=13,285)研究中来自1万名患者和33种癌症类型的18,116个样本的微生物读数进行了检查。可以使用其他合适的数据集,并且可以在本公开的范围内设想这些数据集。微生物读数以前在即席分析中经鉴定(包括胃腺癌中的EBV和宫颈癌中的HPV),并且已经在小样本子集中进行系统地研究(例如,来自19种癌症类型中4,433个TCGA样本的病毒组和9种癌症类型中1,880个TCGA样本的细菌组)。大多数TCGA测序数据仍未探索微生物。如本文所述,使用两个正交微生物检测管线创建了综合的癌症微生物组数据集,系统地测量和减轻技术变化和污染。机器学习(ML)技术用于鉴定鉴别癌症类型和/或分期的微生物特征,并比较其性能。
可以使用本文描述的机器学习模型鉴定的癌症类型和/或分期的非详尽列表包括以下:急性髓系白血病(LAML)、肾上腺皮质癌(ACC)、膀胱尿路上皮癌(BLCA)、脑低级别胶质瘤(LGG)、乳腺浸润性癌(BRCA)、宫颈鳞状细胞癌和宫颈内膜腺癌(CESC)、胆管癌(CHOL)、结肠腺癌(COAD)、淋巴样肿瘤弥漫性大B细胞淋巴瘤(DLBC)、食管癌(ESCA)、多形性胶质母细胞瘤(GBM)、头颈部鳞状细胞癌(HNSC)、肾嫌色细胞癌(KICH)、肾透明细胞癌(KIRC)、肾乳头状细胞癌(KIRP)、肝细胞癌(LIHC)、肺腺癌(LUAD)、肺鳞状细胞癌(LUSC)、间皮瘤(MESO)、卵巢浆液性囊腺癌(OV)、胰腺癌(PAAD)、嗜铬细胞瘤和副神经节瘤(PCPG)、前列腺癌(PRAD)、直肠腺癌(READ)、肉瘤(SARC)、皮肤恶性黑色素瘤(SKCM)、胃腺癌(STAD)、睾丸生殖细胞肿瘤(TGCT)、甲状腺癌(THCA)、胸腺瘤(THYM)、子宫癌肉瘤(UCEC)、子宫内膜癌(UCS)、葡萄膜黑色素瘤(UVM)。
由于TCGA处理未控制微生物污染,且排除了健康个体,因此使用金标准微生物学方案对血液进行了另外的分析,该TCGA样本类型最有可能含有外来微生物污染。各种实施例集中于将血浆来源的微生物DNA的特征与临床上可用的无细胞肿瘤DNA(ctDNA)测定进行相应的基准测试。对来自患有前列腺癌、肺癌或皮肤癌的个体(总共n=100)以及健康、无癌症和HIV对照参与者(n=69)的血浆样本的深度宏基因组测序表明,无细胞微生物谱可用于实现健康与癌症以及癌症与癌症的鉴别。这些发现表明了一类新的基于微生物组的癌症诊断工具,可以补充现有的用于检测和监测癌症的ctDNA测定。
根据各种实施例,使用归一化数据,训练随机梯度提升ML模型,以鉴别癌症的类型和分期。这些模型的性能对于鉴别(i)一种癌症类型与所有其他癌症类型(n=32种癌症类型)和(ii)肿瘤与正常(n=15种癌症类型)是很强的(图4f-g,图9a-f;所有性能指标见cancermicrobiome.ucsd.edu/CancerMicrobiome_DataBrowser)。癌症类型之间的敏感性和特异性的差异可能部分是由于类别大小的差异,因为在少数类别和AUROC(受试者操作特征曲线下的面积;P=0.0231)值(斜率(slop)的双侧假设检验;图9g-h)之间的一种癌症类型与所有其他类型的比较中,存在显著的线性关系。癌症微生物的异质性也可能有助于该不同的表现。基于组织的微生物模型在鉴别结肠腺癌(COAD)、STAD和肾透明细胞癌(KIRC)的I期和IV期肿瘤(n=8种癌症类型)方面表现良好,但在鉴别其他五种测试的癌症方面表现不佳(图4h),在鉴别中间期方面表现也不佳(数据未显示)。这些结果表明,对于所有类型的癌症,微生物群落结构动态可能与宿主组织定义的癌症分期无关。
为了评估此类技术在数据集中的普遍性,将随机原始TCGA微生物计数分为两批,对每一批独立重复所有规程,对另一半数据测试每个独立训练的模型,并发现高度相似的性能(图10a)。在检查执行WGS或RNA-seq或仅使用基因组比对过滤Kraken数据的单一方法(WGS或RNA-seq)或测序中心时,具有鉴别性微生物特征。
为了进一步验证,应用了SHOGUN,这是一种基于比对的微生物分类管线,使用精简的、基于系统发育、仅含细菌的针对13,517个TCGA样本(WGS,n=3,434;RNA-seq,n=10,083)的数据库,这些样本涵盖了基于Kraken分析中的每种癌症分析类型(n=32)、样本类型(n=7)、测序平台(n=6)和测序中心(n=8)。尽管使用了较小的、不相同的底层数据库,但SHOGUN来源的数据复制了在Kraken来源的数据中鉴定的批次效应(图11j-l)。该数据和Kraken来源的数据的相应子集(见方法)被独立输入到归一化和ML管线中,发现数据集之间的鉴别性能没有重大差异(图11m-t)。总之,这些结果表明微生物群落对于每种癌症类型都是独特的,并且可以更广泛地应用仅基于微生物谱来区分癌症的归一化和模型训练方法。
微生物谱的生物学相关性
鉴于微生物特征的强烈鉴别,使用生态学预期和/或临床试验结果检查了其生物学相关性的证据。为了评估癌症相关微生物是否是生态预期的(即,“天然”器官特异性共生群落的一部分),对来自人体微生物组计划2(HMP2)项目中8个身体部位的217个样本的数据训练了贝叶斯微生物源跟踪算法,这些数据已通过本文所述的微生物检测和归一化管线进行处理,以估计COAD队列中70个实体组织正常样本和122个皮肤恶性黑色素瘤(SKCM)原发性肿瘤的身体部位贡献(见方法)。粪便是已知的主要身体部位因素,仅对COAD谱有作用(平均±s.e.m.分数贡献,20.17±2.55%;图5a),但在SKCM谱中没有作用(单尾曼-惠特尼U检验,P=0.0014,图12b),这表明部分群落有本地来源。
梭杆菌属在胃肠道肿瘤的发生和进展中很重要,与实体组织正常样本(所有P≤8.5×10
确认了临床注释的TCGA病毒感染,并将微生物检测管线与使用两种不同生物信息学管线检查TCGA病毒组的研究进行比较:(i)新的基因组组装方法和(ii)基于读取的方法(PathSeq算法)。在CESC和头颈部鳞状细胞癌(HNSC)样本中,经临床检测为HPV感染“阳性”或“阴性”的个体的原发性肿瘤之间的阿尔法乳头瘤病毒属丰度存在差异(所有P≤3×10
这些数据与模型在一种癌症类型与所有其他癌症类型的区别中提供的关于特征重要性的信息一致。也就是说,具有已知微生物“驱动者”或“共生体”的癌症提供了初步证据,证明这些模型是生态相关的;例如,阿尔法乳头瘤病毒属是鉴定CESC肿瘤的最重要特征;对于COAD肿瘤,粪杆菌(Faecalibacterium)属;对于LIHC肿瘤,正肝病毒属是第二重要的特征(仅次于肝毒性微囊藻属)。总的来说,这些发现为病毒和细菌数据的生物信息学和归一化方法提供了生态学验证,同时将结果扩展到更多的样本和微生物。
测量并减轻污染
在各种实施例中,测量并减轻污染的潜在影响可能是重要的,以便最好地表征推定的癌症相关微生物。基于不同类型癌症中常见的低读取丰度,先前的工作仅鉴定了TCGA中的六种污染物(表皮葡萄球菌(Staphylococcus epidermidis)、痤疮丙酸杆菌(Propionibacterium acnes)、罗尔斯通菌属(Ralstonia)、分枝杆菌(Mycobacterium)、假单胞菌(Pseudomonas)和不动杆菌(Acinetobacter)),但最近的研究表明,外部污染物的频率与样本分析物浓度呈负相关,可以使用强大的统计框架进行检测。
基于后一种方法,在TCGA样本处理(n=17,625)和分类群读取分数(n=1,993)期间计算的DNA和RNA浓度用于鉴定推定的污染物,并且还去除了通常在“阴性空白”试剂中发现的属(n=94个属;见方法)。图13a概述了从手术切除到生物信息处理所采取的方法;在原始数据集中加标了五种类型的伪污染物,以通过净化、监督归一化和ML进行跟踪。鉴于已知的技术变化(图4c-e),样本由测序中心(n=8)分批处理,并去除在任何中心发现为污染物的分类群。这鉴定了283种推定污染物,包括试剂“黑名单”中的19.1%(n=18个属)。在合并这两个列表(n=377个属)后,手动审查文献以重新允许致病属或混合证据属(病原体和常见污染物;例如分枝杆菌(Mycobacterium))。这产生了两个数据集,一个去除了可能的污染物,另一个去除了所有推定污染物。创建了第三个“最严格过滤”数据集,该数据集使用更严格的过滤模式丢弃了约92%的总读取(见方法;图13b)。最后,除了上述试剂黑名单(总共497个属)之外,将样本分组到每个中心的单独测序板中,并去除任何一个“板-中心”批次(n=351;见方法)中鉴定的所有推定污染物。净化似乎对所研究的样本类型或癌症类型没有差异影响(图14a-c)。
在至少一些实施例中,计算机模拟净化方法不能替代对癌症样本实施金标准微生物学实践,包括无菌处理、无菌认证试剂、从头到尾处理的试剂的阴性空白、以及作为“阳性”对照的多样本汇集。本文描述的计算机模拟工具反映了现有技术,但并非设计用于检测污染物或交叉污染物的大量“峰值”。这些后者污染物不应在许多中心和多年收集的癌症类型之间和内部产生统一的鉴别信号,但如果不加以控制,可能会限制生物学结论,特别是在小型研究中。
在至少一些实施例中,严格净化的风险在于反映共生、组织特异性微生物群落和伴随的癌症预测微生物谱的真实信号可能被丢弃。为了评估该问题,可以重新计算COAD实体组织正常样本的身体部位归属百分比(n=70),并发现连续的严格净化提高了在伴随组织变得无法识别之前对伴随组织的识别(图13c-f)。
重新计算图4f-h所示的ML模型,并比较它们在每种净化方法之前和之后的性能(图13g–l)。尽管淋巴瘤弥漫性大B细胞淋巴瘤(DLBC)和间皮瘤(MESO)模型(可用样本很少)似乎是例外,可能不可靠,但大多数模型不依赖于加标伪污染物(图15a)。正如预期的那样,关于组织类型的知识是信息性的比较(例如,COAD与所有其他癌症类型)在严格净化下通常表现不佳,但组织内比较(例如,肿瘤与正常)通常表现同样好或更好。这些结果表明,在某些比较中,严格过滤可能是可取的,但通用的净化方法可能会排除生物学信息结果。
利用血液中的微生物DNA进行预测
越来越多的证据表明,基于血液的微生物DNA(mbDNA)可以在癌症中提供临床信息,包括那些以血屏障或淋巴破坏为特征的癌症(例如COAD),但基于目前的技术水平,尚不清楚该应用有多广泛。使用来自TCGA血液样本的WGS数据,将ML策略应用于完整数据集和四个净化数据集,发现血源性mbDNA可以鉴别多种类型的癌症(图6a),无论用于分类的微生物分类算法和数据库如何,或者仅使用基因组比对过滤的Kraken数据时(图11g、图11h、图11s和图11t)。回顾性分析表明,很少有模型包括加标伪污染物进行预测(图15b);这样做的模型(CESC、KIRP、LIHC)可能不太值得信赖。
在这些发现的刺激下,ML模型以现有的ctDNA测定为基准,集中于ctDNA测定失败的情况:Ia-IIc期癌症和没有可检测的基因组改变的肿瘤。在从患有III期或IV期癌症的患者身上去除所有血液来源的正常样本后,建立了新的ML模型,发现它们能够使用血液mbDNA很好地鉴别癌症类型(图6b)。来自Guardant360和FoundationOne液体测定的基因列表进一步用于过滤具有一种或多种靶向修饰的TCGA患者(约70%;图15c-e),发现相同的ML方法显示出对大多数剩余的癌症类型的良好鉴别(图6c-d)。
这些分析受到以下事实的限制:ctDNA测定使用血浆而不是全血,并且mbDNA在血室中的分布是未知的。由于无法获得RNA数据,因此无法判断mbDNA是来自活微生物还是死微生物,或者由于TCGA标准操作规程(SOP)允许全血或血沉棕黄层提取,因此无法判断mbDNA是否在宿主白细胞中为无细胞(参见方法)。如果不检查主要标本和可能匹配的肠道上皮,也不可能知道血液mbDNA的来源,因为某些类型的癌症可能会以意想不到的方式泄漏mbDNA(例如,白血病中的肠道细菌翻译)。理想的净化可能是连续的,因为净化对模型性能的影响因癌症类型而异,但过滤受到以下因素的限制:(i)无法获得主要标本,(ii)属级分类分辨率,以及(iii)不知道同时处理了哪些非TCGA样本。
验证血液中的微生物特征
为了证明这些结果的实际效用,同时对基于血浆的ctDNA测定进行基准测试,在验证研究中使用血浆来源的无细胞mbDNA特征来鉴别健康个体和多种类型的癌症,同时对低生物量研究实施金标准微生物学控制。尽管血浆代表未在TCGA中研究的全血的独特子集,限制了直接的可比性,但它在档案稳定性(例如,可冻性)、生物储存库可用性和生物学解释(即非活体材料)方面具有主要优势。该队列包括69名无癌症和无HIV的个体和100名患有三种高级别(III-IV期)癌症中的一种的患者:前列腺癌(n=59;PC);肺癌(n=25;LC)和黑色素瘤(n=16;SKCM)(图7a)。在没有先前文献估计效应大小的情况下,布罗德研究所(TheBroad Institute)和HMS对来自匹配癌症类型的TCGA血液样本进行独立模拟,以估计最小样本量(图16;见方法)。从这些血浆样本中提取无细胞DNA,进行广泛的控制(图16b-c),并由有限的一组用户使用单一文库制备方法,在单个批次中,在一次深度测序运行中处理用于全宏基因组测序。在各种实施例中,技术涉及进行人读数去除、通过Kraken对剩余读数分类、使用DNA浓度和阴性空白进行严格的去污染以及Voom-SNM。人口统计比较和排列分析建议对年龄和性别进行必要的归一化(图16d-e、h-j;见方法),直接年龄回归性能表现出平均绝对误差类似于肠道微生物组(图16g)。在TCGA分析中使用的相同ML方案的“引导”示出健康对照个体和分组的癌症患者之间的强的、可概括的区别(图7b;见方法)。由于使用的样本量较小,因此对归一化数据进行了留一(LOO)迭代ML,发现除了最小的SKCM队列外,健康样本和癌症类型之间的成对和多类别比较具有较高的鉴别性表现(图7c-k)。因此,迭代地对PC和LC组进行二次抽样以匹配SKCM队列大小,并对每种类型的癌症与二次抽样的健康对照进行成对LOO鉴别(图16k;见方法)。PC和LC队列在与SKCM相同的队列大小下仍然可分离(平均(95%置信区间(CI))AUROC=0.891(0.879-0.903);平均(95%CI)AUPR=0.827(0.815-0.839);100次迭代),揭示了SKCM性能的普遍缺陷。该缺陷可能有生物学基础,因为SKCM在测试的五个数据集中的四个数据集中TCGA血液鉴别中表现第二差(图6a),尽管这需要进一步证实。为了确保Kraken的微生物分配有效,使用来自SHOGUN及其单独数据库的细菌分配重复了所有生物信息学、归一化和ML步骤,这些步骤显示出高度一致的性能(图17)。随着微生物数据库的改进,cfDNA特征的分类分配的改进也在考虑之中。可以在cancermicrobiome.ucsd.edu/CancerMicrobiome_DataBrowser探索检测到的血浆微生物丰度(图12d-e)。
总体而言,数据表明不同类型的癌症和特定微生物群之间存在广泛的关联。这些微生物谱似乎在大多数类型的癌症内部和之间有所区分,包括在低级别肿瘤阶段以及在商业ctDNA测定中没有任何可检测到的基因组改变的患者中使用基于血液的mbDNA时。即使在广泛的内部验证检查和净化后,这些结果通常仍然有效,有时会丢弃超过90%的总数据。仅使用血浆中的无细胞mbDNA,同时采用比TCGA更广泛的内部和外部污染控制,在健康对照个体和多种类型癌症患者中的高鉴别性能表明,使用广泛可用的样本进行临床相关和回顾性测试是可行的且可推广的。尽管如此,结果表明,一类新的基于微生物组的癌症诊断工具可以为患者提供实质性的未来价值。
与依靠人信息以69-79%的准确度(PMID:23287002)诊断转移的起源组织的现有技术方法相比,本发明基于微生物信息提供了至少约94%的准确度。据设想,通过将微生物信息与宿主信息相结合,可以进一步提高该准确性,如95%至100%的准确性。准确性是使用发明人先前发表的数据集(PMID:32214244)确定的,其中探索了是否可以根据肿瘤内或血液来源的微生物群分离转移性癌症类型。由于该数据集具有来自若干种已知转移性癌症类型(例如,乳腺癌、甲状腺癌、黑色素瘤)的样本,这些样本已被收集用于微生物DNA和RNA,因此机器学习被用于仅使用微生物DNA和RNA来表征区分癌症类型的性能。
本文所述机器学习方法已由发明人开发并在之前公布(PMID:32214244),并公布在PCT申请WO2020093040A1中(每个通过引用整体并入本文)。作为实例,将乳腺癌转移与仅使用肿瘤内微生物核酸的甲状腺癌转移进行比较,显示出高度的鉴别性能(ROC曲线下面积=0.889,PR曲线下面积=0.943,准确性=94.4%)。有效地,本发明的一个实施例提供了使用微生物信息诊断转移性癌症的起源组织。在其他实施例中,本发明提供了分析以区分患有原发性肿瘤的宿主和患有转移性肿瘤的宿主,从而诊断转移的存在。
方法
TCGA数据访问
所有TCGA序列数据均经由SevenBridges赞助的癌症基因组学云(CGC)访问。TCGA的SOP经由NCI生物样本研究数据库访问。匹配的患者元数据,包括分子亚型,经由以下获得:通过SevenBridges和系统生物学研究所(ISB)经由CGC访问、经由TCGA-Mutations R软件包访问,或者直接从相应TCGA出版物的补充数据中获取。所有TCGA患者的基因组改变状态均经由cBioPortal查询和下载。用于商业ctDNA测定的基因组合可从Guardant360测定和FoundationOne Liquid测定的公司白皮书中访问。对于TCGA元数据的访问和从分层格式到平面表格的转换,查询了SevenBridges的元数据本体,并在可能的情况下组织数据;对于未存储在该本体中的信息,使用ISB CGC R编程语言API来访问其最近的元数据发布。
生物信息学工具要么直接从CGC平台加载(例如,samtools、BWA),要么作为单独的Docker容器上传并运行,以创建定制的工作流程。这些工作流程将样本BAM文件作为输入,并标记每个样本内哪些DNA或RNA读数是微生物。
使用Kraken算法将与已知人参考基因组(基于原始BAM文件中的映射信息)不一致的序列读数映射到所有已知的细菌、古细菌和病毒微生物基因组。使用RepoPhlan下载了总共71,782个微生物基因组,其中5,503个是病毒基因组,66,279个是细菌或古细菌基因组。基于先前的文献,对细菌和古细菌基因组进行过滤,以获得0.8或更高的质量分数,从而留下其中的54,471个用于后续分析,或总共59,974个微生物基因组。
如前所述,Kraken算法将每个测序读数分解为k-mers(例如默认的31-mers),并将每个k-mer与微生物k-mers数据库精确匹配,该数据库在运行算法之前由上述59,974个微生物基因组构建。给定读数的精确k-mer匹配集反过来提供了该读数的最低共同祖先的推定分类分配,最准确到属级,在本文提供的数据中对此进行了总结。匹配和分类操作比执行直接基因组比对快几个数量级。为了防止假阳性并正确对管线进行基准测试,选择了四种类型的癌症(STAD、CESC、OV和LUAD),并使用BWA将Kraken分类为微生物的读数与59,974个微生物基因组进行比对,这在计算上更昂贵,但产生具有更高特异性和分类分辨率(即物种和菌株水平)的结果。基于文献中的微生物特征和/或可用的质谱蛋白质组学信息(数据未示出),直接比对的四种类型的癌症包括CESC作为推定的阳性病毒对照(针对HPV)、STAD作为推定的阳性细菌对照(针对幽门螺杆菌)、以及另外两种(LUAD、OV)。确定由Kraken(各种发现基于此)分类为属级或更低级的98.91%的读数也与BWA的微生物数据(细菌、古细菌、病毒)一致,或假阳性率为1.09%,这表明属级、Kraken标记的泛癌微生物读数足以用于未来的分析。
SHOGUN TCGA生物信息处理
为了评估使用不同的分类识别算法进行癌症类型鉴别的稳健性,使用了先前公布的浅鸟枪分类分配方法,并对TCGA样本使用单独的、以系统发育为中心的数据库,称为生命网(WoL;PMID:31792218;n=10,575细菌和古细菌基因组)。SHOGUN使用计算密集型的直接基因组比对进行分类分配,而不是Kraken使用的超快速的基于k-mer的方法。为了减少TCGA样本的处理时间,由Kraken分类为微生物来源的读数用作SHOGUN比对功能的输入,其使用Bowtie2将读数映射到WoL数据库以生成分类谱。总共处理了13,517个样本(WGS:n=3,434;RNA-seq:n=10,083),涵盖Kraken分析中研究的所有TCGA类型的癌症(n=32)、样本类型(n=7)、测序中心(n=8)和测序平台(n=6),包括21种TCGA类型癌症(n=9,444个样本),所有Kraken分析中的样本都由SHOGUN重新分析。然后使用QIIME 2将图谱折叠到属级。分析在由1,024个英特尔常春藤桥计算内核、384个AMD计算内核和12TB总RAM组成的本地计算集群上运行,计算周期约为5个月。所使用的单一癌症类型的通常任务提交是约30个内核和约250GB的RAM。
TCGA技术变化的定量测量和归一化
认识到TCGA测序中心(n=8)、测序平台(n=6)、实验策略(WGS与RNA-seq)和可能的污染之间的技术差异如何混淆结果,开发了一种管线来量化和消除批次效应,同时保持或增加归因于生物变量的信号。简而言之,过滤掉元数据质量差的样本(即缺少种族或民族、ICD10代码、DNA/RNA分析物量或FFPE状态信息);使用Voom算法将离散分类计数数据转换为近似正态分布的每百万对数计数(log-cpm)数据,该算法对数据的异方差性进行建模并消除;最后,对数据进行监督归一化(SNM),以消除所有显著的批次效应,同时保留生物效应。Voom传统上与limma结合使用,用于离散计数数据的差异表达(或丰度)分析,但用于算法转换为“类似微阵列”的数据,这允许随后的SNM。Voom和SNM模型矩阵是等效的,并且使用样本类型作为目标生物变量(n=7;例如,原发性肿瘤组织)构建,因为它们之间存在预期的生物学差异,应使用SNM保留信号;相反,以下因素被建模为在SNM期间待缓解的技术协变量:测序中心(n=8),测序平台(n=6),实验策略(n=2),组织源位点(n=191)和FFPE状态(n=2;是或否)。由于某些类型的癌症和测序中心之间完全混淆(即某些类型的癌症仅在一个TCGA位点测序),因此不可能将疾病类型建模为目标生物变量。在Voom变换期间,来自edgeR软件包的加权的M值的修剪平均值(TMM)用于大多数数据(“完整数据集”、“可能污染物去除”数据、“板-中心净化”数据和“所有假定污染物去除”数据),同时丢弃不变的特征(filterByExpr()函数;edgeR),如limma的用户指南所示。在其他情况下(“最严格过滤的”数据、“SHOGUN TCGA数据”、“与SHOGUN TCGA数据匹配的Kraken TCGA数据”以及两个血浆微生物组数据集),使用分位数归一化,因为下游SNM校正与严格过滤的TMM归一化、特征删除的数据不兼容,因为这些数据集已经具有显著减少或较低特征计数。除了“最严格过滤”的数据外,所有分位数归一化数据集仅与其他分位数归一化数据集进行比较。在Voom调整数据的SNM校正之前和之后计算主成分,主方差成分分析(PVCA)量化原始计数数据、Voom调整数据和Voom-SNM归一化数据之间的这些变化。NIEHS很好地描述了PVCA的数学基础,并基于其建议的60-90%将一个可调参数设置为80%。
使用SourceTracker2作为验证分析来解决污染问题
下载了来自NIH的HMP2项目的鸟枪测序数据,其擦拭了总计217个样本中的八个身体部位,并针对上述相同的TCGA Kraken微生物检测管线运行,包括针对相同的微生物数据库(n=59,974个细菌、古细菌和病毒宏基因组)进行分类分配。根据TCGA癌症微生物组数据,在属级上汇总HMP2数据,然后用于训练贝叶斯源跟踪模型(SourceTracker2)。使用SourceTracker的说法,这些HMP2样本充当“源”,而Voom-SNM归一化样本充当“汇”,并且SourceTracker算法用于计算每个源归属于每个汇的比例。通俗地说,使用贝叶斯模型估计了HMP2数据中归属于每个Voom-SNM归一化癌症微生物组样本的身体部位比例。在(i)将癌症微生物组数据集中的属与HMP2中的主题相交,(ii)将log
TCGA ML基准测试和泛化性
作为基准测试和泛化评估,TCGA被分成两个分层数据半(跨测序中心、样本类型和疾病类型)原始Kraken来源的属级微生物计数数据(分割#1:n=8,814;分割#2,n=8,811),通过Voom-SNM方案分别运行它们,在每个归一化的一半上构建单独的ML模型,然后在彼此的归一化数据上测试这些调整的ML模型。然后将这些模型性能与第三个ML模型进行比较,该模型基于完整的Voom-SNM归一化数据集(n=17,635个样本)构建,并使用50-50%的训练和测试分割。使用各自的50%维持测试集AUROC和AUPR比较所有三种方法的最终性能。针对另外的内部验证,仅使用(i)RNA样本或(ii)DNA样本以及(iii)来自一个测序中心的仅进行RNA-seq(UNC)或(iv)DNA-seq(HMS)的样本,建立模型来预测一种癌症类型相对于所有其他癌症类型(图10)。
TCGA净化分析
从广义上讲,有两类可能的污染会影响下一代测序数据:外部污染(例如,试剂、研究者或受试者的身体、环境因素)和内部污染(即,样本在处理或测序过程中的交叉污染)。在至少一个实施例中,整体净化方法试图(i)模拟污染以估计其对预测性能和/或模型不可靠性的贡献,(ii)尽可能减轻外部污染,以及(iii)使用合理的阳性和阴性对照测量内部污染的程度。如最近所述,使用所有TCGA样本(n=17,625)的样本分析物浓度并通过使用从测序试剂盒(类似于TCGA中使用的试剂盒)中的试剂鉴定的微生物黑名单来鉴定并去除外部污染物。如果没有接触原始样本或不知道同时运行哪些其他样本(尤其是非癌症样本),内部污染物尤其难以识别。因此,唯一被鉴定为明确交叉污染物并去除的内部污染物是分配给埃博拉病毒属的四个读数(两个读数来自布罗德研究的一个TCGA-LGG样本,两个读数来自HMS的一个TCGA-HNSC样本),几乎可以肯定来自TCGA研究收集期间(2006-2016)这些相同测序中心对2014年西非疫情的并行研究,以及分配给马尔堡病毒属的四个读数(来自布罗德研究的两个TCGA-OV样本),也可能具有相似的起源或假阳性(即埃博拉病毒和马尔堡病毒都是丝状病毒科)。这样做与先前发表的工作一致,该工作消除了与手头生物学无关的微生物分配。更不太可能的是,此类交叉污染物,特别是丰度极低的交叉污染物,会在许多中心和多年收集的癌症类型之间和内部产生一致的鉴别信号。对于其他可能的交叉污染物,使用生态预期群落的贝叶斯分析(如上所述)估计其贡献,而不是识别和消除它们。
首先,将五个伪污染物加标到原始数据集(图13a,右上)中,以通过净化、SNM和ML跟踪它们。这包括以下内容:(1)来自HMS所有样本的1,000次读取;(2)来自华盛顿大学医学院贝勒医学院HMS和加拿大迈克尔史密斯基因组科学中心的所有样本的1,000个读数;(3)来自所有测序中心的所有样本的1,000个读数;(4)从HMS中随机选择的100个样本中加标10
由于TCGA在样本处理过程中不包括任何阴性空白试剂管,因此本文描述的技术试图在属级上配对使用类似试剂和/或文库制备试剂盒的微生物黑名单。TCGA SOP主要使用QIAGEN产品(凯杰公司(Qiagen),瓦伦西亚,加利福尼亚州)提取组织中的DNA和RNA(DNA/RNA AllPrep试剂盒)和血液中的DNA(QiaAmp血液中型试剂盒)。Salter及其同事描述了宏基因组实验中DNA提取试剂盒的此种列表(n=94个属),包括来自QiaAmp试剂盒,这些试剂盒使用与TCGA血液提取中使用的基于二氧化硅膜的DNA纯化相同的DNA纯化,通过四年的“阴性空白”测序和三个高通量测序中心获得。基于来自污染物的序列通常具有与样本分析物浓度负相关的频率,鉴定了另外的推定外部污染。一个强大的统计框架最近验证了该原则16,提供了利用TCGA记录的样本DNA或RNA浓度作为识别推定污染物的手段的机会。该框架的两个主要假设是:(i)污染物在样本中以均匀的量添加;以及(ii)污染物DNA或RNA的量相对于真正的样本DNA或RNA(微生物或宿主)而言很小。然后使用相关的decontam R软件包(s://github.com/benjjneb/decontam)使用推荐的超参数阈值(P*=0.1)和更严格的方法(P*=0.5)进行过滤。注意,P*=0.5表示如果污染物模型或非污染物模型更好地拟合分布,则分类学被分类为“污染物”或“非污染”。由于发现测序中心对原始计数数据产生了实质性的变化,因此数据被相应地分批处理,从而在任何中心被鉴定为污染物的分类群随后在所有中心被丢弃(即batch.combine=decontam中的“最小值”)。污染物的推定列表(P*=0.1:n=283个属;P*=0.5:n=1,818个属)然后与微生物黑名单(n=94个属)合并/交叉,并从完整数据集中减去。对较小的组合污染物列表(n=377)进行人工文献检查,重新发现89个属是潜在的病原体或共生菌。这产生了三个新的数据集:“可能的污染物被去除”,“所有推定污染物被去除”和“最严格过滤”。作为进一步的保守措施,TCGA样本条形码(例如,采取TCGA-02-0001-01C-01D-0182-01;如NCI的文档s://docs.gdc.cancer.gov/Encyclopedia/pages/TCGA_Barcode/所示)并提取所有测序板-测序中心组合,如条形码的最后两组整数命名(即,本实例中为中心01的板0182或0182-01)。由于decontam计算批次中所有样本的分类群读取分数和分析物浓度之间的线性回归等效值,以确定给定的分类群是否被归类为污染物,因此每个板-中心组合需要10多个样本才能成为批次,总共有351个板-中心批次。使用P*=0.1(默认值),并且像以前一样,如果在351个批次中的任何一个批次中鉴定一个分类群为污染物(batch.combine=“最小值”),则将其从数据集中去除(n=421个分类群被去除)。与微生物黑名单交叉后,共去除497个属。这提供了第四个净化数据集,然后通过上述相同的SNM和ML管线处理所有这些数据集。
比较BWA、SHOGUN和Kraken数据之间的ML性能
BWA过滤发生在用于生成基于Kraken分配的同一数据库(n=59,974个微生物基因组(细菌、古细菌和病毒))上。然后,经由Voom-SNM以与Kraken数据相同的方式对过滤的BWA微生物计数数据进行批量校正,除了由于实验策略和减少的样本计数的测序中心之间的混淆,将DNA和RNA数据分别归一化。然后将来自原始Kraken来源数据的样本与BWA处理的样本进行匹配,并以与BWA数据相同的方式进行归一化。这导致了总共四个归一化数据集:DNABWA数据、RNA BWA数据、DNA Kraken子集数据和RNA Kraken子集数据。然后将所有四个归一化数据集输入用于ML,并相互比较它们的性能(图11a-h)。
用于SHOGUN分类分配的“生命网”数据库不包含病毒,SHOGUN处理了Kraken评估的所有TCGA样本的子集(13,517与17,625个样本)。因此,为了公平比较其下游ML性能,对原始Kraken计数数据进行了子集化,以去除所有已识别的病毒并匹配SHOGUN处理的相同样本。然后通过Voom(使用分位数归一化)和SNM算法(使用与上述主要TCGA分析相同的生物学和技术变量)对两个数据集进行相同的归一化,然后输入ML管线以鉴别癌症类型之间和类型内部。
补充诊断分析
当评估血液mbDNA对低级别癌症的适用性时,所有患有Ia-c期和IIa-c期分类肿瘤的患者被分组在一起,并丢弃所有其他的。为了与Guardant360和FoundationOne LiquidctDNA测定进行比较,过滤掉在其编码基因组上评估至少一种基因组改变的所有TCGA患者;这包括突变是否被认为是乘客或司机。其余患者用于如上所述的ML分析。
TCGA模拟,以估计验证研究所需的样本量
为了估计用于鉴别的前列腺癌、肺癌和皮肤癌(黑色素瘤)所需的样本数量,在两个不同的测序中心(布罗德,HMS)对TCGA血液样本进行了实证模拟,这些测序中心都在一种类型的平台上测序(Illumina HiSeq)。首先,使用Kraken来源的微生物计数数据,然后使用SHOGUN来源的微生物计数数据重复模拟。这最接近地模拟了验证研究的预期真实世界实验条件。
首先,从微生物计数的原始Kraken数据中子集化在Illumina HiSeq机器上测序的布罗德和HMS的所有TCGA PRAD、LUAD、LUSC和SKCM血液样本(布罗德:n=99;HMS:n=288)。使用的肺癌样本是混合来源的,因此LUAD和LUSC血液样本被组合成单一的非小细胞肺癌(NSCLC)伞状疾病类型;然而,这仅适用于布罗德样本,因为HMS的所有血液来源的肺癌样本都是LUAD起源的。这留下了以下样本细分:HMS:66LUAD,104PRAD,118SKCM;布罗德:42NSCLC(24LUAD、18LUSC)、17PRAD、40SKCM。然后,HMS和布罗德的每个原始计数数据集通过Voom(使用分位数归一化)和SNM算法独立归一化,使用疾病类型作为感兴趣的生物学变量,组织源位点作为技术变量,因为通过选择单个测序中心、数据类型和平台排除了所有其他技术因素。
对归一化数据集进行如下模拟:(1)随机分层采样从三个类别中选取相同数量的样本;(2)遗漏三类子样本中的一个样本;(3)在子样本中的所有剩余样本上建立ML模型,并应用于遗漏样本,以一定概率进行预测;(4)重复步骤2-3,直到所有样本都迭代完成;(5)使用观测类别列表和预测类别列表及其概率,估计多类别性能指标;(6)选择另一个具有相同样本量的分层随机样本,并将步骤2-5重复九次(总共十次),以估计多类别性能指标的标准误差;(7)对5-40的单类别样本量重复步骤1-6,步长为五个样本。如果分层采样大小大于一个类别中的样本的数量,则使用该类别中的所有样本。总的来说,这提供了良好执行多癌症鉴别所需的样本数量的估计(图16a)。经验性能估计(平均AUROC,平均AUPR)表明,每个癌症类别至少有15个样本就足够了。注意,由于TCGA不包括健康对照的理想样本量,因此无法估计健康对照的理想样本量。
临床队列选择和IRB协议编号
作为本研究的一部分,分析了来自加州大学圣地亚哥分校的169名患者的生物库、冷冻血浆样本。所有研究均由加州大学圣地亚哥分校的机构审查委员会(IRB)批准,并根据各自的IRB批准的协议,患者提供样本捐赠和研究的书面知情同意书。所有前列腺癌血浆样本(n=59)均符合IRB协议131550。所有肺癌和黑色素瘤血浆样本均符合IRB协议150348。所有无癌症和无HIV的健康对照受试者(n=69)均符合以下IRB协议编号:130296、091054、172092、151057和182064。
血浆来源的无细胞微生物DNA样本处理和测序
根据制造商的说明,使用QIAamp循环核酸试剂盒(QIAGEN)从每个样本的250μl血浆中提取总循环DNA,并用AMPure XP SPRI顺磁珠(贝克曼库尔特(Beckman Coulter))纯化。使用KAPA HyperPlus试剂盒(Kapa生物系统(Kapa Biosystems))和所述的标准Illumina索引适配器(IDT)由纯化的cfDNA制备测序文库。使用Agilent 4200TapeStation系统(高灵敏度DNA试剂盒)对样本库进行表征,并使用Illumina(新英格兰生物实验室(NewEngland Biolabs))NEBNext文库定量试剂盒进行qPCR定量。在NovaSeq 6000仪器(依诺米娜(Illumina))上进行配对端2×150bp测序(S4流动池),并在测序过程中将所有四个泳道的样本汇集。
血浆微生物组样本的生物信息处理
在所有样本上运行的单个NovaSeq 6000测序上共生成了21,600,141,264个读数。其中,19,046,611,360个读数被分配给人类样本(即去除阴性和阳性对照),总读数的2.186%被归类为非人类。使用Atropos对原始测序数据进行解复用和衔接子修剪。使用Trimmomatic通过以下设置完成另外的质量过滤-(ILLUMINACLIP:TruSeq3-PE-2.fa:2:30:7,MINLEN:50,TRAILING:20,AVGQUAL:20,SLIDINGWINDOW:20:20)。在标准TruSeq3衔接子中添加仅由G组成的另外的衔接子序列,以去除5'阅读端的尾随G序列。如果任一配偶使用具有快速局部参数集的Bowtie2映射到人类基因组(来自1000基因组计划的主要等位基因SNP参考),则丢弃读数对。然后使用FLASH合并配对端读数,参数如下-(最小重叠:20,最大重叠:150,失配比:0.01)。
然后,使用上面详述的相同工作流程和数据库(n=59,974个微生物基因组),或者使用本文详述的SHOGUN,通过Kraken处理过滤的、合并的读数。在单个血浆微生物组样本上处理样本(即,基于每个样本每个泳道,因为在运行过程中样本汇集到所有四个测序流动池中)。在Kraken或SHOGUN对每个样本每个泳道进行分类分配后,在分层聚类规程示出按样本ID而不是按流动池泳道进行一致分组后,对每个样本的跨泳道微生物计数进行汇总。对于SHOGUN来源数据,成功合并和未合并的读数都用作SHOGUN比对功能的输入,使用Bowtie2将读数映射到WoL数据库以生成分类谱,然后使用QIIME 2将其折叠到属级。过滤每个样本的分类谱,以去除相对丰度小于0.01%的所有分类群。
血浆微生物组技术验证和数据净化
为了评估测序运行和生物信息学微生物检测管线的性能,对照其他样本类型检查测序板上包括的加标孔与Aliivibrio fischeri(属:Aliivibrio)实验连续稀释液的差异丰度,并隔离了稀释液中丰度的对数倍数变化。这些技术阳性对照绘制在图16b-c中,用于Kraken和SHOGUN来源的分类分配。
在测序板上包括三种阴性空白对照:(1)DNA提取空白,其具有通过测序来自DNA提取阶段的试剂;(2)DNA文库制备空白,其具有通过测序来自文库制备阶段的试剂;(3)空对照孔,在文库制备过程中加入水,然后加入试剂,并含有飞溅和/或雾化的微生物核酸。与TCGA分析一样,decontam再次用于净化血浆微生物数据,除了它可以访问所有样本的阴性空白对照和DNA浓度(不包括后者的空对照孔)。作为保守措施,选择P*=0.5超参数值用于“流行”(即基于空白)和“频率”(即基于浓度)的净化模式的decontam;该超参数值相当于TCGA中最严格的净化,丢弃了总数据的>90%。对于流行模式,P*=0.5将把在阴性对照中比生物对照更流行的任何分类群标记为污染物;对于频率模式,P*=0.5将标记其模型(即回归模型)比使用读取分数和DNA浓度拟合污染物分布更好的任何分类群。对于Kraken计数数据,流行模式丢弃了21个分类群,频率模式丢弃了1,261个分类群(从1,753个原始分配中);对于SHOGUN计数数据,流行模式丢弃了57个分类群,频率模式丢弃了244个分类群(从1,181个原始分配中)。Kraken和SHOGUN的净化数据被输入下游归一化和ML管线。
血浆微生物组数据归一化、排列测试和ML
尝试使用GBM ML模型(与上述TCGA的架构相同)和留一(LOO)迭代ML(图16g)使用原始微生物计数数据预测年龄。
为了确认在该队列中对年龄和性别进行归一化的重要性,对每个因素进行了100次迭代,然后同时对两个因素进行了排列分析(图16h-j)。简而言之,执行以下四个步骤:(1)在所有样本中随机交换年龄和/或性别标签;(2)对原始数据运行Voom-SNM,使用疾病类型作为感兴趣的生物学变量,并将年龄和/或性别排列为技术因素;(3)使用70%-30%的训练-测试分割--使用固定的随机数种子和内部四重交叉验证进行ML分析,以鉴别分组的癌症样本和健康对照,以获得两类性能估计(AUROC,AUPR);(4)重复步骤1-3共100次,以创建零性能分布。接下来,使用正确的、固定的年龄和/或性别分配,在步骤3中随机选择随机数种子的同时,步骤2-3总共运行100次。最后,使用双侧曼-惠特尼U检验将该性能分布与其零分布的显著性直接进行比较。由于所有这些测试都非常显著(所有P≤1.5×10
对Voom-SNM归一化血浆微生物组样本的ML完全按照前面针对TCGA样本所描述的完成,除了采样模式之外,因为样本量小了几个数量级。首先,为了估计健康者与分组癌症鉴别的泛化,在500次迭代的训练期间,使用70%-30%的训练-测试分割进行“自举”,并进行四重交叉验证。允许使用替换进行采样,因为每个训练-测试分割(即每次迭代)都是唯一的;但是,在任何情况下,样本都不允许既是训练案例又是测试案例。所有500次迭代的结果性能指标的汇总统计数据估计了AUROC和AUPR分布以及置信区间(Cis)(图7b、图17a)。其次,使用LOO ML在健康对照和个体癌症类型之间进行成对和多类别鉴别。换句话说,一个样本被迭代地省略,一个模型在剩余的样本上迭代训练,并进行四重交叉验证以进行超参数调优,并且使用模型给出的概率对遗漏样本进行迭代预测。将所有样本的实际类别的最终列表与预测类别列表及其估计AUROC和AUPR指标的概率进行比较,如先前使用PRROC R软件包所述。通过取所有一个与所有其他比较的平均值来估计多类别性能,如Caret R软件包中的multiClassSummary()函数所报告的那样。
迭代二次抽样以评估较小样本量对黑色素瘤队列性能的贡献(图16k)如下:(1)对单一癌症类型和健康对照各16个样本(总共32个)进行随机分层采样;(2)执行LOO迭代ML并评估这32个样本的性能,以鉴别健康与癌症;(3)重复步骤1-2 100次,估计性能标准误差;(4)对三种癌症类型中的每一种重复步骤1-3。对PC和LC队列的迭代二次抽样也进行相同的过程,以研究样本量减少对其鉴别的影响。注意,在每个分层二次抽样期间使用整个黑色素瘤队列,因为目的是将其队列大小与其他样本量进行比较。
统计分析
所有统计分析均使用R版本3.4.3完成。ggpubr软件包(s://github.com/kassambara/ggpubr)在组间执行非参数统计检验,并在必要时考虑多个假设检验校正。注意,小于2.2×10
使用机器学习模型进行训练和推理
根据至少一个实施例,可以使用各种技术使用机器学习模型(如神经网络)进行训练和推理(例如,预测)。在至少一个实施例中,使用训练数据集训练未经训练的神经网络。未经训练的神经网络的初始权重参数可以设置为初始预定值、随机数等。在至少一个实施例中,训练框架用于使用训练数据集训练神经网络并更新神经网络的一个或多个权重。训练框架可以是任何合适的训练框架,如PyTorch框架、TensorFlow、Boost、Caffe、MicrosoftCognitive Toolkit/CNTK、MXNet、Chainer、Keras、Deeplearning4j或其他训练框架。在至少一个实施例中,训练框架训练未训练的神经网络,并使其能够使用本文描述的处理资源进行训练以生成经训练的神经网络。在至少一个实施例中,权重可以随机选择或者通过使用深度信念网络的预训练来选择。在至少一个实施例中,训练可以以监督、部分监督或无监督的方式进行。
在至少一个实施例中,使用监督学习训练未经训练的神经网络,其中训练数据集包括与用于输入的期望输出(例如,组织起源预测)配对的输入(例如,微生物谱),或者其中训练数据集包括具有已知输出的输入并且对神经网络的输出手动分级。在至少一个实施例中,未经训练的神经网络以监督方式训练并处理来自训练数据集的输入,并将所得输出与一组预期或期望的输出进行比较。在至少一个实施例中,错误通过未经训练的神经网络传播回去。在至少一个实施例中,训练框架调整在训练过程中控制未经训练的神经网络的权重。在至少一个实施例中,训练框架包括用于监测未经训练的神经网络向模型(如经训练的神经网络)收敛的程度的工具,这些工具适于基于输入数据(如新数据集)生成正确的答案(如在结果中)。在至少一个实施例中,训练框架反复训练未经训练的神经网络,同时使用损失函数和调整算法(如随机梯度下降)调整权重以细化未经训练的神经网络的输出。在至少一个实施例中,训练框架训练未经训练的神经网络,直到未经训练的神经网络达到所需的精度。在至少一个实施例中,经训练神经网络可以被部署以实现任意数量的机器学习操作。
在至少一个实施例中,未经训练的神经网络使用无监督学习进行训练,其中未经训练的神经网络试图使用未标记的数据来训练自身。在至少一个实施例中,无监督学习训练数据集将包括没有任何相关输出数据的输入数据或“基本事实”数据。在至少一个实施例中,未经训练的神经网络可以学习训练数据集内的分组,并且可以确定单个输入如何与未经训练的数据集相关。在至少一个实施例中,无监督训练可以用于在经训练的神经网络中生成自组织图,该神经网络能够执行有助于降低新数据集维数的操作。在至少一个实施例中,无监督训练还可以用于执行异常检测,其允许鉴定新数据集中偏离新数据集正常模式的数据点。
在至少一个实施例中,可以使用半监督学习,这是一种其中在训练数据集中包括标记数据和未标记数据的混合的技术。在至少一个实施例中,训练框架可以用于执行增量学习,如通过迁移学习技术。在至少一个实施例中,增量学习使经训练的神经网络能够适应新的数据集,而不会忘记在初始训练期间灌输在经训练神经网络中的知识。
图18是图示根据本公开的一个或多个示例性实施例可以执行一种或多种技术(例如,方法)的计算设备或计算机系统1800的实例的框图。
例如,图18的计算系统1800可以包括一个或多个处理器1802-1806。处理器1802-1806可以包括一个或多个内部级别的高速缓存(未示出)和总线控制器(例如,总线控制器1822)或总线接口(例如,I/O接口1820)单元,以直接与处理器总线1812交互。
处理器总线1812,也称为主机总线或前端总线,可以用于将处理器1802-1806与系统接口1824耦合。系统接口1824可以连接到处理器总线1812,以将系统1800的其他组件与处理器总线1812连接。例如,系统接口1824可以包括存储器控制器1818,用于将主存储器1816与处理器总线1812连接。主存储器1816通常包括一个或多个存储卡和控制电路(未示出)。系统接口1824还可以包括输入/输出(I/O)接口1820,以将一个或多个I/O桥1825或I/O设备1830与处理器总线1812连接。一个或多个I/O控制器和/或I/O设备可以与I/O总线1826连接,如I/O控制器1828和I/O设备1830,如图所示。
I/O设备1830还可以包括输入设备(未示出),如字母数字输入设备,包括用于向处理器1802-1806传达信息和/或命令选择的字母数字键和其他键。另一种类型的用户输入设备包括光标控制,如鼠标、轨迹球或光标方向键,用于向处理器1802-1806传送方向信息和命令选择以及用于控制显示设备上的光标移动。
系统1800可以包括动态存储设备,称为主存储器1816,或者随机存取存储器(RAM)或其他耦合到处理器总线1812的计算机可读设备,用于存储待由处理器1802-1806执行的信息和指令。主存储器1816还可以用于在处理器1802-1806执行指令期间存储临时变量或其他中间信息。系统1800可以包括只读存储器(ROM)和/或耦合到处理器总线1812的其他静态存储设备,用于存储处理器1802-1806的静态信息和指令。图18中概述的系统仅仅是可以使用或根据本公开的方面配置的计算机系统的一个可能的实例。
据一个实施例,响应于处理器1804执行包含在主存储器1816中的一个或多个指令的一个或多个序列,上述技术可以由计算机系统1800执行。这些指令可以从另一机器可读介质(如存储设备)读入主存储器1816。执行包含在主存储器1816中的指令序列可以导致处理器1802-1806执行本文描述的过程步骤。在替代实施例中,可使用电路系统来代替软件指令或与软件指令相结合。因此,本公开的实施例可包含硬件和软件组件两者。
根据一个实施例,处理器1802-1806可以包括张量处理单元(TPU)和/或其他人工智能加速器专用集成电路(ASIC),其可以实现神经联网和其他机器学习技术。在至少一个实施例中,机器学习模块1832是指执行本文描述的机器学习技术的软件和/或硬件,其可以包括训练和/或推理阶段。例如,机器学习模块1832可以经训练以鉴别转移性癌症的不同类型和/或阶段。
各种实施例可以在软件和/或固件中全部或部分实现。该软件和/或固件可以采用包含在非暂时性计算机可读存储介质中或上的指令形式。然后,这些指令可以由一个或多个处理器读取和执行,以使本文描述的操作的性能成为可能。这些指令可以是任何合适的形式,如但不限于,源代码、编译代码、解释代码、可执行代码、静态代码、动态代码等。此种计算机可读介质可以包括,用于以由一台或多台计算机可读的形式存储信息的任何有形的非暂时性介质,如但不限于只读存储器(ROM);随机存取存储器(RAM);磁盘存储介质;光存储介质;闪存等。
机器可读媒体包括用于以机器(例如计算机)可读的形式(例如软件、处理应用程序)存储或传输信息的任何机制。此类介质可采取但不限于非易失性介质和易失性介质的形式,且可包括可移除式数据存储介质、不可移除式数据存储介质和/或经由具有此类计算机程序产品的有线或无线网络架构可用的外部存储装置,包括一个或多个数据库管理产品、网络服务器产品、应用程序服务器产品和/或其他额外软件部件。可移动数据存储介质的实例包括压缩光盘只读存储器(CD-ROM)、数字多功能光盘只读存储器(DVD-ROM)、磁光盘、闪存驱动器等。不可移动数据存储介质的实例包括内部磁性硬盘、固态设备(SSD)等。一个或多个存储器设备(未示出)可以包括易失性存储器(例如,动态随机存取存储器(DRAM)、静态随机存取存储器(SRAM)等)和/或非易失性存储器(例如,只读存储器(ROM)、闪存等)。
包含用以根据当前描述的技术实现系统和方法的机制的计算机程序产品可驻存于主存储器1816中,其可被称为机器可读介质。应当理解的是,机器可读介质可以包含能够存储指令或对其进行编码以进行由机器执行的本公开的任何一个或多个操作的任何有形非暂时性介质,或能够存储由此类指令利用的或与此类指令相关联的数据结构和/或模块或对其进行编码的任何有形非暂时性介质。机器可读介质可包含存储一个或多个可执行指令或数据结构的单个介质或多个介质(例如,集中式或分布式数据库和/或相关联的高速缓存器和服务器)。
以下参考文献通过引用并入本文:
Bullman,S.等人,结直肠癌中梭杆菌属持久性和抗生素应答分析(Analysis ofFusobacterium persistence and antibiotic response in colorectal cancer)科学(Science)358,1443–1448(2017).
Dejea,C.M.等人,患有家族性腺瘤性息肉病的患者具有包含致瘤细菌的结肠生物膜(Patients with familial adenomatous polyposis harbor colonic biofilmscontaining tumorigenic bacteria)》科学359,592–597(2018).
Geller,L.T.等人,肿瘤内细菌在介导肿瘤对化疗药物吉西他滨的耐药性中的潜在作用(Potential role of intratumor bacteria in mediating tumor resistance tothe chemotherapeutic drug gemcitabine)》科学357,1156–1160(2017).
Gopalakrishnan,V.等人,肠道微生物组调节黑色素瘤患者对抗PD-1免疫疗法的应答(Gut microbiome modulates response to anti-PD-1immunotherapy in melanomapatients)科学359,97–103(2018).
Jin,C.等人,共生微生物群通过γδT细胞促进肺癌的发展(Commensalmicrobiota promote lung cancer development viaγδT cells)》细胞(Cell)176,998–1013.e16(2019).
Ma,C.等人,肠道微生物组介导的胆汁酸代谢经由NKT细胞调节肝癌(Gutmicrobiome-mediated bile acid metabolism regulates liver cancer via NKTcells)科学360,eaan5931(2018).
Matson,V.等人,共生微生物组与转移性黑色素瘤患者的抗PD-1疗效相关(Thecommensal microbiome is associated with anti-PD-1efficacy in metastaticmelanoma patients)科学359,104–108(2018).
Meisel,M.等人,微生物信号驱动Tet2缺陷宿主中的白血病前期骨髓增生(Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficienthost)》自然(Nature)557,580–584(2018).
Routy,B.等人,肠道微生物组影响基于PD-1的免疫疗法对上皮肿瘤的疗效(Gutmicrobiome influences efficacy of PD-1-based immunotherapy against epithelialtumors)科学359,91–97(2018).
Ye,H.等人,颠覆全身性糖代谢作为支持白血病细胞生长的机制(Subversion ofsystemic glucose metabolism as a mechanism to support the growth of leukemiacells)》癌细胞(Cancer Cell)34,659–673.e6(2018).
癌症基因组图谱研究网络等人,癌症基因组图谱泛癌症分析项目(The CancerGenome Atlas Pan-Cancer analysis project)》自然遗传(Nat.Genet.)45,1113-1120(2013).
Hanahan,D.&Weinberg,R.A.,癌症的标志(The hallmarks of cancer.)细胞100,57–70(2000).
Hanahan,D.&Weinberg,R.A.,癌症的标志:下一代(A.Hallmarks of cancer:thenext generation)》细胞144,646–674(2011).
Salter,S.J.等人,试剂和实验室污染会严重影响基于序列的微生物组分析(Reagent and laboratory contamination can critically impact sequence-basedmicrobiome analyses)BMC生物学(BMC Biol.)12,87(2014).
Glassing,A.,Dowd,S.E.,Galandiuk,S.,Davis,B.&Chiodini,R.J.,提取和测序试剂固有的细菌DNA污染可能会影响低细菌生物量样本中微生物群的翻译(Inherentbacterial DNA contamination of extraction and sequencing reagents may affectinterpretation of microbiota in low bacterial biomass samples)肠道病原体(GutPathog.)8,24(2016).
Davis,N.M.,Proctor,D.M.,Holmes,S.P.,Relman,D.A.&Callahan,B.J.,标记基因和宏基因组数据中污染物序列的简单统计识别与去除(Simple statisticalidentification and removal of contaminant sequences in marker-gene andmetagenomics data)微生物组学(Microbiome)6,226(2018).
Robinson,K.M.,Crabtree,J.,Mattick,J.S.A.,Anderson,K.E.&DunningHotopp,J.C.,在公共癌症基因组序列数据的二次数据分析中区分潜在的细菌-肿瘤关联与污染(Distinguishing potential bacteria-tumor associations from contaminationin a secondary data analysis of public cancer genome sequence data)微生物组学5,9(2017).
Eisenhofer,R.等人,低微生物生物量微生物组研究中的污染:问题和建议(Contamination in low microbial biomass microbiome studies:issues andrecommendations)微生物学进展(Trends Microbiol.)27,105–117(2019).
癌症基因组图谱研究网络,胃腺癌的综合分子表征(The Cancer Genome AtlasResearch Network.Comprehensive molecular characterization of gastricadenocarcinoma)》自然513,202–209(2014).
癌症基因组图谱研究网络,宫颈癌基因组和分子表征的整合(Integratedgenomic and molecular characterization of cervical cancer)》自然543,378–384(2017).
Tang,K.-W.,Alaei-Mahabadi,B.,Samuelsson,T.,Lindh,M.&Larsson,E.,人类癌症中病毒表达和宿主基因融合及适应的前景(The landscape of viral expression andhost gene fusion and adaptation in human cancer)》自然通讯(Nat.Commun.)4,2513(2013).
Minich,J.J.等人,KatharoSeq能够从低生物量样本中进行高通量微生物组分析(KatharoSeq enables high-throughput microbiome analysis from low biomasssamples)艾蒙系统(mSystems)3,e00218-17(2018).
Wood,D.E.&Salzberg,S.L.,Kraken:使用精确比对进行超快宏基因组序列分类(Kraken:ultrafast metagenomic sequence classification using exact alignments)基因组生物学(Genome Biol.)15,R46(2014).
Zhang,H.等人,人类高级别浆液性卵巢癌的综合蛋白质基因组学表征(Integrated proteogenomic characterization of human high-grade serous ovariancancer)》细胞166,755–765(2016).
Choi,J.-H.,Hong,S.-E.&Woo,H.G.,系统批次效应对体细胞序列变异的泛癌分析(Pan-cancer analysis of systematic batch effects on somatic sequencevariations)BMC生物信息学(BMC Bioinformatics)18,211(2017).
Lauss,M.等人,定量高通量数据集的技术变化的监测(Monitoring of technicalvariation in quantitative high-throughput datasets)癌症资讯(Cancer Inform.)12,193–201(2013).
Law,C.W.,Chen,Y.,Shi,W.&Smyth,G.K.,voom:精确砝码解锁RNA-seq读数计数的线性模型分析工具(voom:precision weights unlock linear model analysis toolsfor RNA-seq read counts)基因组生物学15,R29(2014).
Mecham,B.H.,Nelson,P.S.&Storey,J.D.,微阵列的监督归一化(Supervisednormalization of microarrays)生物信息学(Bioinformatics)26,1308–1315(2010).
Boedigheimer,M.J.等人,来自多个实验室的毒理基因组学研究对照动物的基线基因表达水平变异来源(Sources of variation in baseline gene expression levelsfrom toxicogenomics study control animals across multiple laboratories)BMC基因组学9,285(2008).
Scherer,A.,微阵列实验中的批处理效应和噪声:来源和解决方案(BatchEffects and Noise in Microarray Experiments:Sources and Solutions)(威利(Wiley),2009).
Hillmann,B.等人,评价浅层鸟枪宏基因组学信息含量(Evaluating theinformation content of shallow shotgun metagenomics)艾蒙系统3,e00069-18(2018).
Knights,D.等人,贝叶斯群落全培养非依赖性微生物源跟踪(Bayesiancommunity-wide culture-independent microbial source tracking)》自然方法(Nat.Methods)8,761–763(2011).
Integrative HMP(iHMP)研究网络联盟(Research Network Consortium),综合人类微生物组项目:在人类健康和疾病期间微生物组-宿主组学谱的动态分析(TheIntegrative Human Microbiome Project:dynamic analysis of microbiome-hostomics profiles during periods of human health and disease)细胞宿主和微生物(Cell Host Microbe)16,276–289(2014).
Yamamura,K.等人,食管癌组织中的人类微生物组梭杆菌核与预后有关(Humanmicrobiome Fusobacterium nucleatum in esophageal cancer tissue is associatedwith prognosis)临床癌症研究(Clin.Cancer Res.)22,5574–5581(2016).
Hsieh,Y.-Y.等人,台湾胃癌患者胃微生物群中梭状芽胞杆菌和梭杆菌丰度增加(Increased abundance of Clostridium and Fusobacterium in gastric microbiotaof patients with gastric cancer in Taiwan)科学报告(Sci.Rep.)8,158(2018).
Kostic,A.D.等人,PathSeq:通过人体组织深度测序来识别或发现微生物的软件(PathSeq:software to identify or discover microbes by deep sequencing ofhuman tissue)自然生物技术(Nat.Biotechnol.)29,393–396(2011).
Svircev,Z.等人,微囊藻毒素诱导肝毒性与肝癌发生的分子学方面(Molecularaspects of microcystin-induced hepatotoxicity and hepatocarcinogenesis)环境科学与健康杂志C-环境致癌作用与生态毒理学综述(J.Environ.Sci.Health C Environ.Carcinog.Ecotoxicol.Rev.)28,39–59(2010).
Jervis-Bardy,J.等人,通过对Illumina MiSeq数据的后测序处理,从细菌含量低的人类样本中获得准确的微生物群谱(Deriving accurate microbiota profiles fromhuman samples with low bacterial content through post-sequencing processingof Illumina MiSeq data)微生物组学3,19(2015).
Kwong,T.N.Y.等人,特定微生物菌血症与结直肠癌后续诊断的关系(Associationbetween bacteremia from specific microbes and subsequent diagnosis ofcolorectal cancer)肠胃病学(Gastroenterology)155,383–390.e8(2018).
Blauwkamp,T.A.等人,传染病微生物无细胞DNA测序测试的分析和临床验证(Analytical and clinical validation of a microbial cell-free DNA sequencingtest for infectious disease)自然微生物学4,663–674(2019).
Hong,D.K.等人,传染病的液体活检:对无细胞血浆进行测序以检测侵袭性真菌病患者的病原体DNA(Liquid biopsy for infectious diseases:sequencing of cell-freeplasma to detect pathogen DNA in patients with invasive fungal disease)诊断微生物学与传染病(Diagn.Microbiol.Infect.Dis.)92,210–213(2018).
Burnham,P.等人,尿无细胞DNA是一种用于监测尿路感染的多用途分析物(Urinary cell-free DNA is a versatile analyte for monitoring infections ofthe urinary tract)》自然通讯9,2412(2018).
De Vlaminck,I.等人,人类病毒组对免疫抑制和抗病毒治疗的时间应答(Temporal response of the human virome to immunosuppression and antiviraltherapy)细胞155,1178–1187(2013).
Huang,Y.-F.等人,分析早发型乳腺癌患者和健康女性血浆无细胞DNA中的微生物序列(Analysis of microbial sequences in plasma cell-free DNA for early-onsetbreast cancer patients and healthy females)BMC医学基因组学(BMC Med.Genomics)11(增刊1),16(2018).
Bettegowda,C.等人,检测早期和晚期人类恶性肿瘤中的循环肿瘤DNA(Detectionof circulating tumor DNA in early-and late-stage human malignancies)科学·转化医学(Sci.Transl.Med.)6,224ra24(2014).
Clark,T.A.等人,用于无细胞循环肿瘤DNA基因组分析的基于混合捕获的下一代测序临床测定的分析验证(Analytical validation of a hybrid capture-based next-generation sequencing clinical assay for genomic profiling of cell-freecirculating tumor DNA)分子诊断与治疗杂志(J.Mol.Diagn.)20,686–702(2018).
Sanders,J.G.等人,通过结合长短阅读优化排行榜宏基因组学的测序方案(Optimizing sequencing protocols for leaderboard metagenomics by combininglong and short reads)基因组生物学20,226(2019).
Huang S.等人,人体皮肤、口腔和肠道微生物组预测实际年龄(Human skin,oral,and gut microbiomes predict chronological age)艾蒙系统5,e00630-19(2020).
Zhu,Q.等人,10,575个基因组的系统发育组学揭示了细菌和古细菌结构域之间的进化接近性(Phylogenomics of 10,575genomes reveals evolutionary proximitybetween domains Bacteria and Archaea)自然通讯10,5477(2019).
Chiu,K.-P.&Yu,A.L.,无细胞DNA测序在血源性微生物表征中的应用和微生物-疾病相互作用研究方面的应用(Application of cell-free DNA sequencing incharacterization of bloodborne microbes and the study of microbe-diseaseinteractions)同行界(PeerJ)7,e7426(2019).
Lau,J.W.等人.,癌症基因组学云:协作、可重复和民主化-大规模计算研究的新范式(The Cancer Genomics Cloud:collaborative,reproducible,and democratized-anew paradigm in large-scale computational research)癌症研究77,e3–e6(2017).
Hoadley,K.A.等人,细胞起源模式主导来自33种癌症的10,000个肿瘤的分子分类(Cell-of-origin patterns dominate the molecular classification of 10,000tumors from 33types of cancer)细胞173,291–304.e6(2018).
Reynolds,S.M.等人,ISB癌症基因组云:一个灵活的基于云的癌症基因组研究平台(The ISB Cancer Genomics Cloud:a flexible cloud-based platform for cancergenomics research)癌症研究77,e7–e10(2017).
Ellrott,K.等人,使用多基因组管线进行肿瘤外显子组突变调用的可扩展开放科学方法(Scalable open science approach for mutation calling of tumor exomesusing multiple genomic pipelines)细胞体系(Cell Syst.)6,271–281.e7(2018).
癌症基因组图谱网络,人类乳腺肿瘤的全面分子画像(Comprehensive molecularportraits of human breast tumors)自然490,61–70(2012).
Cerami,E.等人,cBio癌症基因组学门户:探索多维癌症基因组学数据的开放平台(The cBio cancer genomics portal:an open platform for exploringmultidimensional cancer genomics data)癌症发现(Cancer Discov.)2,401–404(2012).
Gao,J.等人,使用cBioPortal对复杂的癌症基因组学和临床概况进行综合分析(Integrative analysis of complex cancer genomics and clinical profiles usingthe cBioPortal)科学信号(Sci.Signal.)6,pl1(2013).
Land,M.L.等人,32,000个基因组的质量分数(Quality scores for 32,000genomes)基因组科学标准(Stand.Genomic Sci.)9,20(2014).
Li,H.&Durbin,R.,利用伯罗斯-惠勒变换法的快速且准确的短读比对(Fast andaccurate short read alignment with Burrows-Wheeler transform)生物信息学25,1754–1760(2009).
Greathouse,K.L.等人,人类肺癌中微生物组与TP53之间的相互作用(Interaction between the microbiome and TP53 in human lung cancer)基因组生物学19,123(2018).
Shanmughapriya,S.等人,上皮性卵巢癌的病毒和细菌病因(Viral andbacterial aetiologies of epithelial ovarian cancer)欧洲临床微生物学与传染病杂志(Eur.J.Clin.Microbiol.Infect.Dis.)31,2311–2317(2012).
Banerjee,S.等人,卵巢癌肿瘤群(The ovarian cancer oncobiome)肿瘤标靶(Oncotarget)8,36225–36245(2017).
Langmead,B.&Salzberg,S.L.,用Bowtie 2进行快速间隙阅读比对(Fast gapped-read alignment with Bowtie 2)自然方法9,357-359(2012).
Bolyen,E.等人,使用QIIME 2进行可重复、交互式、可缩放和可扩展的微生物组数据科学(Reproducible,interactive,scalable and extensible microbiome datascience using QIIME 2)》自然生物技术37,852–857(2019).
Ritchie,M.E.等人,limma为RNA测序和微阵列研究的差异表达分析提供动力(limma powers differential expression analyses for RNA-sequencing andmicroarray studies)核酸研究43,e47(2015).
Robinson,M.D.,McCarthy,D.J.&Smyth,G.K.,edgeR:用于数字基因表达数据的差异表达分析的生物导体包(edgeR:a Bioconductor package for differentialexpression analysis of digital gene expression data)生物信息学26,139–140(2010).
McDonald,D.等人,生物观测矩阵(BIOM)格式或:我如何学会停止担忧并爱上ome-ome(The Biological Observation Matrix(BIOM)format or:how I learned to stopworrying and love the ome-ome)1,2047-217X-1-7(2012).
Friedman,J.H.,随机梯度提升(Stochastic gradient boosting)计算统计与数据分析(Comput.Stat.Data Anal.)38,367–378(2002).
Friedman,J.H.,贪婪函数近似:梯度提升机(Greedy function approximation:agradient boosting machine)统计年刊(Ann.Stat.)29,1189–1232(2001).
Kuhn,M.,使用caret软件包在R中构建预测模型(Building predictive modelsin Rusing the caret package)统计软件杂志(J.Stat.Softw.)28,1–26(2008).
Grau,J.,Grosse,I.&Keilwagen,J.,PRROC:在R中计算和可视化精度-召回和接收器工作特性曲线(PRROC:computing and visualizing precision-recall and receiveroperating characteristic curves in R)生物信息学31,2595–2597(2015).
Gire,S.K.等人,基因组监测阐明2014年疫情期间埃博拉病毒的起源和传播(Genomic surveillance elucidates Ebola virus origin and transmission duringthe 2014outbreak)科学345,1369–1372(2014).
Matranga,C.B.等人,从临床和生物样本中对拉沙和埃博拉RNA病毒进行无偏深入测序的增强方法(Enhanced methods for unbiased deep sequencing of Lassa andEbola RNA viruses from clinical and biological samples)基因组生物学15,519(2014).
Gonzalez,A.等人,在地铁里躲避疫情恐惧并征服鸭嘴兽(Avoiding pandemicfears in the subway and conquering the platypus)艾蒙系统1,e00050-16(2016).
Didion,J.P.,Martin,M.&Collins,F.S.,阿特罗波斯:测序读数的特异性、敏感性和快速微调(Atropos:specific,sensitive,and speedy trimming of sequencingreads)同行界5,e3720(2017).
Bolger,A.M.,Lohse,M.&Usadel,B.,Trimmomatic:Illumina序列数据的灵活调整器(Trimmomatic:A flexible trimmer for Illumina sequence data)生物信息学30,2114–2120(2014).
1000基因组项目联盟(The 1000Genomes Project Consortium),人类基因变异的全球参考(A global reference for human genetic variation.)自然526,68–74(2015).
T.&Salzberg,S.L.,FLASH:快速调整短序列长度以改善基因组组装(FLASH:fast length adjustment of short reads to improve genome assemblies)生物信息学27,2957–2963(2011).
Gonzalez,A.等人,Qiita:网络使能的快速微生物组荟萃分析(Qiita:rapid,web-enabled microbiome meta-analysis)自然方法15,796-798(2018).