糖原病Ia型治疗药
文献发布时间:2023-06-19 19:28:50
分案申请的相关信息
本案是分案申请。该分案的母案是申请日为2019年3月5日、申请号为201980006746.4、发明名称为“糖原病Ia型治疗药”的发明专利申请案。
[相关申请]
本申请享有以日本专利申请2018-043524号(申请日:2018年3月9日)、日本专利申请2018-128015号(申请日:2018年7月5日)、PCT专利申请PCT/JP2019/008713号(申请日:2019年3月5日)为基础申请的优先权。本申请是通过参照该基础申请而包含基础申请的全部内容。
技术领域
本发明涉及一种糖原病Ia型治疗药,更详细而言,本发明涉及一种将糖原病Ia型患者中具有c.648G>T变异的G6PC基因在mRNA水平修复而可使正常的G6PC蛋白质表达的寡核苷酸及含有其的医药。
背景技术
糖原病Ia型是以葡萄糖6磷酸脱磷酸化酶(G6PC)为原因基因的常染色体隐性遗传的代谢异常症,主要症状为低血糖、肝肿大、肾肿大。通过利用膳食疗法的血糖控制,生存预后大幅改善,但即便控制良好,其中超过一半也呈现肝腺瘤、白蛋白尿(非专利文献1)。报告了若干糖原病Ia型患者的好发变异,东亚地区引起剪接异常的G6PC c.648G>T变异作为好发变异而为人所知(非专利文献2)。
作为将寡核苷酸送达至肝脏的肝实质细胞的方法,业界报告了一种与作为可键结在去唾液酸醣蛋白受体(ASGPR)的配体而通过共价键上键结了N-乙酰基-D-半乳糖胺(GalNAc)等的核酸医药(反义或siRNA等)的复合体(非专利文献3及专利文献1~9)。在1个寡核苷酸键结了1至3个GalNAc。另外,作为键结了2个GalNAc的例,非专利文献4中有所记载。
现有技术文献
非专利文献
非专利文献1:Eur J Pediatr.2002年161卷Suppl 1:S20-34
非专利文献2:Hum Mutat.2008年,29卷,p.921-930.
非专利文献3:Methods in Enzymology,1999年,313卷,297~321页
非专利文献4:Bioorganic&Medicinal Chemistry Letters 26(2016)3690-3693
专利文献
专利文献1:国际公开第2009/073809
专利文献2:国际公开第2014/076196
专利文献3:国际公开第2014/179620
专利文献4:国际公开第2015/006740
专利文献5:国际公开第2015/105083
专利文献6:国际公开第2016/055601
专利文献7:国际公开第2017/023817
专利文献8:国际公开第2017/084987
专利文献9:国际公开第2017/131236
发明内容
[发明要解决的问题]
本发明的目的在于确立一种糖原病Ia型的分子治疗法。
[解决问题的技术手段]
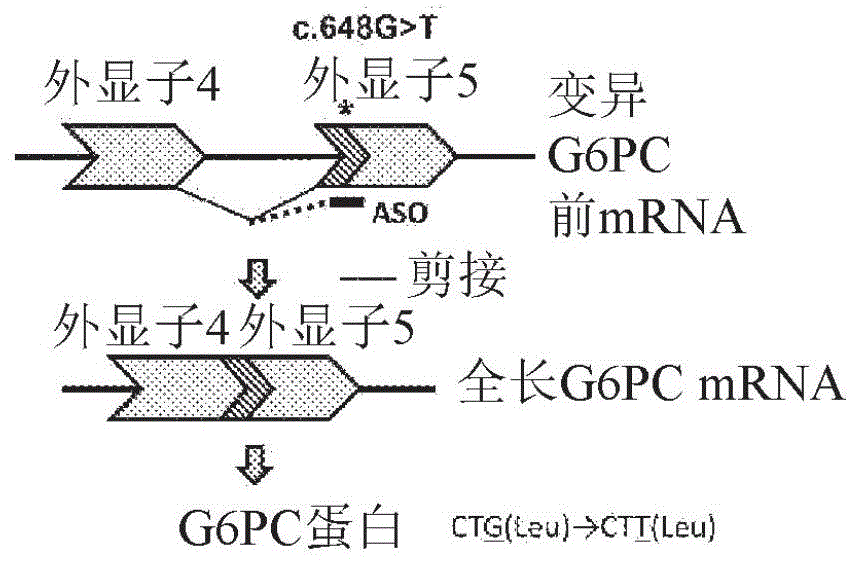
本发明人等确立了如下治疗法(图1),其通过对具有G6PC c.648G>T变异的糖原病Ia型患者投予反义寡核苷酸(ASO),而修复mRNA的异常剪接,诱导正常的G6PC蛋白质的产生。G6PC c.648G>T变异中产生mRNA的异常剪接,自内含子(intron)4与外显子(exon)5的接合处(junction)缺损外显子(exon)5部分的91个碱基,引起移码(frame shift)导致的酶的失活。当mRNA的异常剪接被修正时,可期待该翻译的氨基酸序列与正常型相同(CUG(Leu)→CUU(Leu)),因此在具有G6PC c.648G>T变异的患者中,通过该修复mRNA的异常剪接的治疗法,产生具有活性的正常的G6PC蛋白质,而可实现糖原病Ia型患者的低血糖发作的风险降低、器官肿大的改善、肝腺瘤风险的降低。本研究成为针对糖原病Ia型的世界首次的分子治疗的开发研究。
本发明的主旨如以下所述。
(1)一种寡核苷酸、其药理上容许的盐或溶剂合物,所述寡核苷酸含有与具有c.648G>T变异的G6PC基因的cDNA互补的核苷酸序列且碱基数为15~30,其含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第82号至第92号的任一部位的区域互补的序列。
(2)如(1)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸是碱基数为15~21的寡核苷酸,其含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第86号至第92号的任一部位的区域互补的序列。
(3)如(1)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸是碱基数为15~21的寡核苷酸,其含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第92号的部位的区域互补的序列。
(4)如(1)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸是碱基数为15~18的寡核苷酸,其含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第92号的部位的区域互补的序列。
(5)如(1)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸是碱基数为18的寡核苷酸,其含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第92号的部位的区域互补的序列。
(6)如(1)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸是碱基数为17的寡核苷酸,其含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第92号的部位的区域互补的序列。
(7)如(1)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸是碱基数为16的寡核苷酸,其含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第92号的部位的区域互补的序列。
(8)如(1)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸是碱基数为15的寡核苷酸,其含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第92号的部位的区域互补的序列。
(9)如(1)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸含有序列编号1~32、40~42、44~48的任一序列(其中,序列中的t也可为u,u也可为t)中连续的至少15个核苷酸的序列。
(10)如(1)至(9)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸进而在5'末端及/或3'末端附加了可在生物体内切断的寡核苷酸。
(11)如(1)至(10)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中构成寡核苷酸的糖及/或磷酸二酯键的至少1个被修饰。
(12)如(1)至(10)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中构成寡核苷酸的糖是D-呋喃核糖,糖的修饰是D-呋喃核糖的2'位的羟基的修饰。
(13)如(1)至(10)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中构成寡核苷酸的糖是D-呋喃核糖,糖的修饰是D-呋喃核糖的2'-O-烷基化及/或2'-,4'-交联化。
(14)如(1)至(10)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中构成寡核苷酸的糖是D-呋喃核糖,糖的修饰是D-呋喃核糖的2'-O-烷基化及/或2'-O,4'-C-烷撑化。
(15)如(1)至(14)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中磷酸二酯键的修饰是硫代磷酸酯。
(16)如(1)至(15)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸在5'末端及/或3'末端结合了GalNAc单元。
(17)如(1)至(15)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸在5'末端结合了GalNAc单元。
(18)如(16)或(17)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中GalNAc单元是式
[化1]
[式中,Ra表示式
[化2]
所表示的基团,Rb表示式
[化3]
所表示的基团或氢原子,XX表示式
[化4]
所表示的基团,G表示5-乙酰胺-2-羟基甲基-3,4-二羟基四氢吡喃-6-基(GalNAc),Z表示氧原子或硫原子,L
(19)如(16)或(17)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中GalNAc单元是式
[化5]
[式中,G、Z、L
(20)如(16)或(17)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中GalNAc单元是式
[化6]
[式中,G、Z、L
[化7]
所表示的基团]所表示的基团。
(21)如(16)或(17)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中GalNAc单元是式
[化8]
[式中,G、Z、L
[化9]
(式中,n'及m'表示与所述相同的含义)所表示的基团或氢原子]所表示的基团。
(22)如(16)或(17)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中GalNAc单元是或式
[化10]
[式中,G、Z、L
(23)如(16)或(17)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中GalNAc单元是式
[化11]
[式中,G、Z、L
(24)如(1)所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其中所述寡核苷酸是以式
[化12]
RO-Xg-Xf-Xe-Xd-Xc-Xb-Xa-T
[式中,R表示氢原子、XX基、或G基,T表示5'末端不具有羟基的寡核苷酸,Xg表示选自由X
[化13]
[化14]
[化15]
[化16]
[化17]
[化18]
[化19]
[化20]
[化21]
[化22]
[化23]
[化24]
[化25]
[化26]
[化27]
[化28]
[化29]
[化30]
[化31]
[化32]
[化33]
[化34]
(25)一种医药,其包含如(1)至(24)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物。
(26)一种糖原病Ia型治疗药,其包含如(1)至(24)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物。
(27)一种糖原病Ia型的治疗方法,其包括以医药上有效的量对受验者投予如(1)至(24)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物。
(28)如(1)至(24)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其用于糖原病Ia型的治疗方法。
(29)一种用于经口或非经口投予的调配物,其包含如(1)至(24)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物。
(30)如(1)至(24)的任一项所记载的寡核苷酸、其药理上容许的盐或溶剂合物,其用于作为医药使用。
[发明的效果]
根据本发明,在糖原病Ia型患者中,可将糖原病Ia型患者中具有c.648G>T变异的G6PC基因在mRNA水平修复,结果可表达G6PC蛋白质,实现糖原病Ia型患者的低血糖的正常化、肝肥大的正常化,抑制向肝癌的发展。
本说明书包含作为本案的优先权的基础的日本专利申请案、日本专利特愿2018-43524及日本专利特愿2018-128015的说明书及/或图式所记载的内容。
附图说明
图1(A)是对由G6PC基因生成G6PC蛋白质的过程进行说明的图。(B)是表示糖原病Ia型患者中具有c.648G>T变异的G6PC基因的mRNA的剪接模式的图。
图2表示对利用本发明的糖原病Ia型治疗药ASO进行治疗的原理进行说明的示意图。
图3是表示ASO(21e_001~21e_012)可键结在具有c.648G>T变异的G6PC基因的mRNA的外显子5的序列的图。
图4是表示ASO(21m_001~21m_012)可键结在具有c.648G>T变异的G6PC基因的mRNA的外显子5的序列的图。
图5是使用ASO,表现出利用qRT-PCR(quantitative real time polymerasechain reaction,定量实时聚合酶链反应)测得的修正G6PC mRNA的异常剪接的效果的图。(A)是使用ASO(21e_001~21e_012)的情形。(B)是使用ASO(21m_001~21m_012)的情形。RQ:相对定量(Relative Quantification)
图6是使用ASO修复G6PC mRNA的异常剪接,表现出利用LC-MS/MS(Liquidchromatography-mass spectrometry/Mass Spectrometry,液相色谱-质谱法/质谱法)测得的产生正常人G6PC特异性肽的效果的图。(A)是使用ASO(21e_001~21e_012)的情形。(B)是使用ASO(21m_001~21m_012)的情形。
图7是使用ASO修复G6PC mRNA的异常剪接而表现出G6PC酶活性的图。(A)是使用ASO(21e_001~21e_012)的情形。(B)是使用ASO(21m_001~21m_012)的情形。
图8是表示ASO(21e_001~21e_006及21e_013~21e_022)可键结在具有c.648G>T变异的G6PC基因的mRNA的外显子5上的序列的图。
图9是使用ASO(21e_001~21e_006及21e_013~21e_022),表现出利用qRT-PCR测得的修正G6PC mRNA的异常剪接的效果的图。RQ:相对定量(Relative Quantification)
图10是使用ASO(21e_001~21e_006及21e_013~21e_022)修复G6PC mRNA的异常剪接,表现出利用LC-MS/MS测得的产生正常人G6PC特异性肽的效果的图。
图11是表示ASO(18e_001~18e_017)可键结在具有c.648G>T变异的G6PC基因的mRNA的外显子5的序列的图。
图12是表示ASO(18m_001~18m_017)可键结在具有c.648G>T变异的G6PC基因的mRNA的外显子5的序列的图。
图13(A)是使用ASO(18e_001~18e_017),表现出利用qRT-PCR测得的修正G6PCmRNA的异常剪接的效果的图。(B)是使用ASO(18m_001~18m_017),表现出利用qRT-PCR测得的修正G6PC mRNA的异常剪接的效果的图。RQ:相对定量(Relative Quantification)
图14(A)是使用ASO(18e_001~18e_017)修复G6PC mRNA的异常剪接,表现出利用LC-MS/MS测得的产生正常人G6PC特异性肽的效果的图。(B)是使用ASO(18m_001~18m_017)修复G6PC mRNA的异常剪接,表现出利用LC-MS/MS测得的产生正常人G6PC特异性肽的效果的图。
图15是表示ASO(18e_018~18e_031)可键结在具有c.648G>T变异的G6PC基因的mRNA的外显子5的序列的图。
图16(A)是使用ASO(18e_018~18e_031),表现出利用qRT-PCR测得的修正G6PCmRNA的异常剪接的效果的图。(B)是使用ASO(18e_018~18e_031)修复G6PC mRNA的异常剪接,表现出利用LC-MS/MS测得的产生正常人G6PC特异性肽的效果的图。RQ:相对定量(Relative Quantification)
图17是表示ASO(21e_002、18e_005、21m_002、18e_022、18m_005、15e_001、15ed_001、18e_008、18e_025、18m_008、15e_002及15ed_002)可键结在具有c.648G>T变异的G6PC基因的mRNA的外显子5的序列的图。
图18是使用ASO(21e_002、18e_005、21m_002、18m_005、18e_022、15e_001、15ed_001、18e_008、18e_025、18m_008、15e_002、及15ed_002),表现出利用qRT-PCR测得的修正G6PC mRNA的异常剪接的效果的图。RQ:相对定量(Relative Quantification);Actn:β-肌动蛋白
图19是表示将实施例91至95的化合物向异型敲入小鼠投予时利用qRT-PCR测得的修正肝脏中的G6PC mRNA的异常剪接的效果的图。RQ:相对定量(RelativeQuantification);Actn:β-肌动蛋白;mpk:mg/kg
图20是表示将实施例91、及实施例96至103的化合物向异型敲入小鼠投予时利用qRT-PCR测得的修正肝脏中的G6PC mRNA的异常剪接的效果的图。RQ:相对定量(RelativeQuantification);Actn:β-肌动蛋白;mpk:mg/kg
图21是表示将实施例83、实施例87至91、及实施例104至107的化合物向异型敲入小鼠投予时利用qRT-PCR测得的修正肝脏中的G6PC mRNA的异常剪接的效果的图。RQ:相对定量(Relative Quantification);Actn:β-肌动蛋白;mpk:mg/kg
图22是表示将实施例104、及实施108至115的化合物向异型敲入小鼠投予时利用qRT-PCR测得的修正肝脏中的G6PC mRNA的异常剪接的效果的图。RQ:相对定量(RelativeQuantification);Actn:β-肌动蛋白;mpk:mg/kg
图23是表示将实施例105、113、及131至137的化合物向异型敲入小鼠投予时利用qRT-PCR测得的修正肝脏中的G6PC mRNA的异常剪接的效果的图。RQ:相对定量(RelativeQuantification);Actn:β-肌动蛋白;mpk:mg/kg
图24是表示利用实施例1的化合物所得的培养细胞中的异常剪接修复序列的片段解析的图。
图25是表示将实施例133、及143至149的化合物向异型敲入小鼠投予时利用qRT-PCR测得的修正肝脏中的G6PC mRNA的异常剪接的效果的图。RQ:相对定量(RelativeQuantification);mpk:mg/kg
图26是表示将实施例149、及152至160的化合物向异型敲入小鼠投予时利用qRT-PCR测得的修正肝脏中的G6PC mRNA的异常剪接的效果的图。RQ:相对定量(RelativeQuantification);mpk:mg/kg
具体实施方式
以下,对本发明的实施形态进一步详细说明。
本发明提供一种将糖原病Ia型患者中具有c.648G>T变异的G6PC基因在mRNA水平修复而可使正常的G6PC蛋白质表达的寡核苷酸、其药理上容许的盐或溶剂合物。本发明的寡核苷酸是含有与具有c.648G>T变异的G6PC基因的cDNA互补的核苷酸序列的碱基数为15~30的寡核苷酸,含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第82号至第92号的任一部位的区域互补的序列。
在本发明中,可以糖原病Ia型患者中进行c.648G>T变异的患者作为对象。不仅c.648G>T变异的同型接合的患者,而且也可以c.648G>T变异与其他变异的复合异型接合的患者作为对象。
G6PC基因的c.648G>T变异是智人葡萄糖-6-磷酸酶催化亚基(Homo sapiensglucose-6-phosphatase catalytic subunit)(G6PC),转录变体(transcript variant)1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G向T的变异。位于自外显子5的5'末端起第86号。本发明的寡核苷酸含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第86号至第92号的任一部位的区域互补的序列即可,优选含有与包含第92号的部位的区域互补的序列。
作为含有与具有c.648G>T变异的G6PC基因的cDNA互补的核苷酸序列且碱基数为15~30、并且含有与具有c.648G>T变异的G6PC基因的包含外显子5的5'末端起第82号至第92号的任一部位的区域互补的序列的寡核苷酸,可例示包含序列编号1~32、40~42、44~48的任一序列(其中,序列中的t或T也可为u或U,u或U也可为t或T)的全部或一部分的寡核苷酸。在本发明中,所谓“序列的一部分”通常为该序列整体的80%以上,适宜为85%,进而适宜为90%,最适宜为94%。本发明的寡核苷酸的碱基数为15~30较为适当,优选15~21,更优选15~18。
构成本发明的寡核苷酸(反义寡核苷酸)的核苷酸可为天然型DNA、天然型RNA、DNA/RNA的嵌合体、它们的修饰体的任一者,优选至少1个为修饰核苷酸。
作为本发明中的修饰核苷酸,可例示糖被修饰的核苷酸(例如,D-呋喃核糖被2'-O-烷基化的核苷酸、D-呋喃核糖被2'-O,4'-C-烷撑化的核苷酸、D-呋喃核糖被2'-,4'-交联的核苷酸)、磷酸二酯键被修饰(例如,硫代酯化)的核苷酸、碱基被修饰的核苷酸、将这些组合而成的核苷酸等。构成反义寡核苷酸的至少1个D-呋喃核糖被2'-O-烷基化而成的核苷酸或被2'-O,4'-C-烷撑化而成的核苷酸对RNA的结合力较高,对核酸酶的耐性较高,因此可期待高于天然型的核苷酸(即,寡DNA、寡RNA)的治疗效果。另外,构成寡核苷酸的至少1个磷酸二酯键被硫代酯化而成的核苷酸对核酸酶的耐性也较高,因此可期待高于天然型的核苷酸(即,寡DNA、寡RNA)的治疗效果。包含如上所述的被修饰的糖与被修饰的磷酸这两者的寡核苷酸对核酸酶的耐性更高,因此可期待更高的治疗效果。
关于本发明的寡核苷酸(反义寡核苷酸),作为糖的修饰的例,可列举:D-呋喃核糖的2'-O-烷基化(例如,2'-O-甲基化、2'-O-氨基乙基化、2'-O-丙基化、2'-O-烯丙基化、2'-O-甲氧基乙基化、2'-O-丁基化、2'-O-戊基化、2'-O-炔丙基化等)、D-呋喃核糖的2'-O,4'-C-烷撑化(例如,2'-O,4'-C-亚乙基化、2'-O,4'-C-亚甲基化、2'-O,4'-C-亚丙基化、2'-O,4'-C-四亚甲基化、2'-O,4'-C-五亚甲基化等)、D-呋喃核糖的2'-脱氧-2'-C,4'-C-亚甲氧基亚甲基化、S-cEt(2',4'-constrained ethyl,2',4'-限制性乙基)、AmNA(Amide-bridgednucleic acid,酰胺交联核酸)、3'-脱氧-3'-氨基-2'-脱氧-D-呋喃核糖、3'-脱氧-3'-氨基-2'-脱氧-2'-氟-D-呋喃核糖等。
关于本发明的寡核苷酸(反义寡核苷酸),作为糖的2'-,4'-交联修饰的例,可列举:D-呋喃核糖的2'-O,4'-C-烷撑化(例如,2'-O,4'-C-亚乙基化、2'-O,4'-C-亚甲基化、2'-O,4'-C-亚丙基化、2'-O,4'-C-四亚甲基化、2'-O,4'-C-五亚甲基化等)、D-呋喃核糖的2'-脱氧-2'-C,4'-C-亚甲氧基亚甲基化、S-cEt(2',4'-限制性乙基)、AmNA等。
关于本发明的寡核苷酸(反义寡核苷酸),作为磷酸二酯键的修饰的例,可列举:硫代磷酸酯键、甲基膦酸酯键、甲基硫代膦酸酯键、二硫代磷酸酯键、氨基磷酸酯(Phosphoroamidate)键等。
在本发明中,作为碱基的修饰的例,可列举:胞嘧啶的5-甲基化、5-氟化、5-溴化、5-碘化、N4-甲基化,胸腺嘧啶的5-去甲基化(尿嘧啶)、5-氟化、5-溴化、5-碘化,腺嘌呤的N6-甲基化、8-溴化,鸟嘌呤的N2-甲基化、8-溴化等。
本发明的寡核苷酸(反义寡核苷酸)可使用市售的合成仪(例如,PerkinElmer公司的利用亚磷酰胺(phosphoramidite)法获得的模型392)等,依照文献(Nucleic AcidsResearch,12,4539(1984))所记载的方法而合成。关于此时可使用的亚磷酰胺试剂,天然型的核苷及2'-O-甲基核苷(即,2'-O-甲基鸟苷、2'-O-甲基腺苷、2'-O-甲基胞苷、2'-O-甲基尿苷)可使用市售的试剂。关于烷基的碳数为2~6个的2'-O-烷基鸟苷、腺苷、胞苷及尿苷,如以下所述。
2'-O-氨基乙基鸟苷、腺苷、胞苷、尿苷可依照文献(Blommers etal.Biochemistry(1998),37,17714-17725.)而合成。
2'-O-丙基鸟苷、腺苷、胞苷、尿苷可依照文献(Lesnik,E.A.et al.Biochemistry(1993),32,7832-7838.)而合成。
2'-O-烯丙基鸟苷、腺苷、胞苷、尿苷可使用市售的试剂。
2'-O-甲氧基乙基鸟苷、腺苷、胞苷、尿苷可依照专利(US6261840)或文献(Martin,P.Helv.Chim.Acta.(1995)78,486-504.)而合成。
2'-O-丁基鸟苷、腺苷、胞苷、尿苷可依照文献(Lesnik,E.A.et al.Biochemistry(1993),32,7832-7838.)而合成。
2'-O-戊基鸟苷、腺苷、胞苷、尿苷可依照文献(Lesnik,E.A.et al.Biochemistry(1993),32,7832-7838.)而合成。
2'-O-炔丙基鸟苷、腺苷、胞苷、尿苷可使用市售的试剂。
关于2'-O,4'-C-亚甲基鸟苷、腺苷、胞苷、5-甲基胞苷及胸苷,可依照WO99/14226所记载的方法而制造,关于烷撑的碳数为2~5个的2'-O,4'-C-烷撑鸟苷、腺苷、胞苷、5-甲基胞苷及胸苷,可依照WO00/47599所记载的方法而制造。
D-呋喃核糖的被2'-脱氧-2'-C,4'-C-亚甲氧基亚甲基化的核苷可依照文献(Wang,G.etal.Tetrahedron(1999),55,7707-7724)而合成。
S-cEt(constrained ethyl,限制性乙基)可依照文献(Seth,P.P.etal.J.Org.Chem(2010),75,1569-1581.)而合成。
AmNA可依照文献(Yahara,A.et al.ChemBioChem(2012),13,2513-2516.)或WO2014/109384而合成。
在本发明中,核酸碱基序列可将腺嘌呤记载为(A)或(a),将鸟嘌呤记载为(G)或(g),将胞嘧啶记载为(C)或(c),将胸腺嘧啶记载为(T)或(t),及将尿嘧啶记载为(U)或(u)。可使用5-甲基胞嘧啶代替胞嘧啶。核酸碱基中尿嘧啶(U)或(u)与胸腺嘧啶(T)或(t)具有兼容性。尿嘧啶(U)或(u)与胸腺嘧啶(T)或(t)的任一者均可用于与互补链的腺嘌呤(A)或(a)形成碱基对。
使亚磷酰胺试剂偶合后,使硫、二硫化四乙基秋兰姆(TETD,Applied Biosystems公司)、Beaucage试剂(Glen Research公司)、或氢化黄原素(Xanthane Hydride)等试剂进行反应,由此可合成具有硫代磷酸酯键的反义寡核苷酸(Tetrahedron Letters,32,3005(1991),J.Am.Chem.Soc.112,1253(1990),PCT/WO98/54198)。
作为合成仪所使用的可控孔度玻璃(CPG),键结了2'-O-甲基核苷的可控孔度玻璃可利用市售者。另外,可依照文献(Oligonucleotide Synthesis,Edited by M.J.Gait,Oxford University Press,1984),将关于2'-O,4'-C-亚甲基鸟苷、腺苷、5-甲基胞苷及胸苷而依照WO99/14226所记载的方法所制造的核苷,关于烷撑的碳数为2~5个的2'-O,4'-C-烷撑鸟苷、腺苷、5-甲基胞苷及胸苷而依照WO00/47599所记载的方法所制造的核苷键结在CPG。通过使用被修饰的CPG(日本专利特开平7-87982的实施例12b所记载),可合成在3'末端结合了2-羟基乙基磷酸基的寡核苷酸。另外,如果使用3'-氨基-修饰基C3 CPG、3'-氨基-修饰基C7 CPG、甘油CPG(Glen Research)、3'-间隔基C3 SynBase CPG 1000、3'-间隔基C9SynBase CPG 1000(link technologies),则可合成在3'末端结合了羟基烷基磷酸基、或氨基烷基磷酸基的寡核苷酸。
本发明的寡核苷酸(反义寡核苷酸)可经由连接基及磷酸部而键结了GalNAc。
本发明中的所谓GalNAc单元是键结有键结了GalNAc的连接基的磷酸基,可进而具有1个键结键。GalNAc单元的磷酸基可与寡核苷酸的5'末端及/或3'末端键结,适宜为键结在寡核苷酸的5'末端。当GalNAc单元具有键结键时,该键结键可与羟基、GalNAc、键结了GalNAc的连接基、其他GalNAc单元的磷酸基、或寡核苷酸的3'末端的磷酸基键结。作为键结在1个GalNAc单元的GalNAc的个数,适宜为1至7个,更适宜为1至5个,尤其适宜为1至3个,最佳个数为2个。
在本发明中,1个GalNAc单元也可键结在寡核苷酸,多个GalNAc单元也可连续键结而键结在寡核苷酸。作为键结在1个寡核苷酸的GalNAc单元的个数,适宜为1至7个,更适宜为1至5个,尤其适宜为1至3个,最佳个数为1个。
在本发明中,可在与GalNAc单元键结的寡核苷酸(反义寡核苷酸)的5'末端及/或3'末端具有碱基序列与反义寡核苷酸不同、包含磷酸二酯键、且可在生物体内切断的寡核苷酸序列。作为可切断的寡核苷酸的链长,适宜为1至6个核苷酸,进而适宜为1至3个核苷酸。可切断的寡核苷酸只要可切断,则无特别限定,可列举全部包含DNA的天然型寡脱氧核苷酸、及全部包含RNA的天然型寡核苷酸等。作为碱基序列,只要为可切断的序列,则无特别限定,可列举5'-TCATCA-3'、5'-CATCA-3'、5'-ATCA-3'5'-TCA-3'、5'-CA-3'、5'-A-3'等。
作为本发明的GalNAc单元的例,例如为通式
[化35]
[式中,Ra表示式
[化36]
所表示的基团,Rb表示式
[化37]
所表示的基团或氢原子,XX表示式
[化38]
所表示的基团,G表示5-乙酰胺-2-羟基甲基-3,4-二羟基四氢吡喃-6-基(GalNAc),Z表示氧原子或硫原子,L
[化39]
[式中,G、Z、L
[化40]
[式中,G、Z、L
[化41]
所表示的基团]所表示的基团、式
[化42]
[式中,G、Z、L
[化43]
(式中,n'及m'表示与所述相同的含义)所表示的基团或氢原子]所表示的基团、式
[化44]
[式中,G、Z、L
[化45]
[式中,G、Z、L
在本发明中,GalNAc单元所键结的寡核苷酸例如为式
[化46]
RO-Xg-Xf-Xe-Xd-Xc-Xb-Xa-T
[式中,R表示氢原子、XX基、或G基,T表示5'末端不具有羟基的寡核苷酸,Xg表示具有键结键的GalNAc单元,Xa、Xb、Xc、Xd、Xe及Xf互相独立地表示具有键结键的GalNAc单元或单键]所表示的寡核苷酸。
作为Xa、Xb、Xc、Xd、Xe、Xf及Xg中的“具有键结键的GalNAc单元”,可列举如下基团。
[化47]
[化48]
[化49]
[化50]
/>
[化51]
[化52]
[化53]
[化54]
[化55]
[化56]
[化57]
[化58]
[化59]
[化60]
[化61]
另外,作为“具有键结键的GalNAc单元”的Xg、及R包含XX基或G基的RO-Xg-的“GalNAc单元”可列举如下基团。
[化62]
[化63]
[化64]
[化65]
[化66]
[化67]
[化68]
本发明的GalNAc单元所键结的寡核苷酸通过将含有GalNAc单元的酰胺与核苷的酰胺试剂同样地使用,可使用市售的合成仪(例如,PerkinElmer公司的利用亚磷酰胺法获得的模型392)等,依照文献(Nucleic Acids Research,12,4539(1984))所记载的方法而合成。当键结在寡核苷酸的5'末端时,可通过在结束寡核苷酸的部分的链伸长后,将含有GalNAc单元的酰胺进行偶合而合成。另外,当键结在寡核苷酸的3'末端时,可使用被修饰的CPG(日本专利特开平7-87982的实施例12b所记载)、3'-氨基-修饰基C3 CPG,3'-氨基-修饰基C7 CPG,甘油CPG,(Glen Research),3'-间隔基C3 SynBase CPG 1000,3'-间隔基C9SynBase CPG 1000(link technologies)等,将含有GalNAc单元的酰胺进行偶合,其后进行寡核苷酸的部分的链伸长,由此合成3'末端具有羟基烷基磷酸基、或氨基烷基磷酸基、进而键结了GalNAc单元的寡核苷酸。
含有GalNAc单元的酰胺的合成法
本发明的含有GalNAc单元的酰胺可依照下文所述的A法至I法而制造。在A法至I法中,各反应结束后,各反应的目标化合物是依照常规方法自反应混合物中采集。例如是通过如下方法获得:将反应混合物适当中和,另外,当存在不溶物时,通过过滤去除后,添加如水与乙酸乙酯的不混合的有机溶剂,将含有目标物的有机层分离,利用水等洗净后,利用无水硫酸钠等加以干燥后,将溶剂蒸馏去除。所获得的化合物可视需要通过常规方法、例如硅胶柱色谱法进行分离、精制。或可通过再沉淀、再结晶而精制。
A法
A法是用来制造化合物(8)的方法。如果使用与化合物(2)相反的立体构型的化合物,则化合物(8)中键结了-P(R
[化69]
式中R
步骤A-1是制造化合物(3)的步骤,于惰性溶剂中在酸的存在下,使化合物(1)与化合物(2)进行反应,而在环氧化物开环时生成醚键。
作为所述反应所使用的惰性溶剂,例如可列举:烃类、芳香族烃类、卤化烃类、醚类、乙腈等,优选二氯甲烷。
作为所述反应所使用的酸,例如可列举有机酸类、路易斯酸等。作为有机酸,例如可列举三氟甲磺酸等,作为路易斯酸,例如可列举三氟化硼-二乙醚络合物等。优选三氟甲磺酸。
反应温度根据化合物(1)及化合物(2)、酸、惰性溶剂等而不同,通常为-20℃至回流温度。优选30℃至45℃。
反应时间根据化合物(1)及化合物(2)、酸、惰性溶剂、反应温度等而不同,通常为15分钟至72小时,优选2小时至24小时。
步骤A-2是制造化合物(5)的步骤,于惰性溶剂中在酸的存在下,使化合物(3)及化合物(4)进行反应,而生成糖苷键。
作为所述反应所使用的惰性溶剂,例如可列举:烃类、芳香族烃类、卤化烃类、醚类、乙腈等,优选二氯甲烷。
作为所述反应所使用的酸,例如可列举有机酸类、路易斯酸等。作为有机酸,例如可列举三氟甲磺酸等,作为路易斯酸,例如可列举三氟化硼-二乙醚络合物等,优选三氟化硼-二乙醚络合物。
反应温度根据化合物(3)、酸、惰性溶剂等而不同,通常为-20℃至回流温度。优选30℃至45℃。
反应时间根据化合物(3)、酸、惰性溶剂、反应温度等而不同,通常为15分钟至72小时,优选2小时至24小时。
步骤A-3是制造化合物(6)的步骤,去除作为化合物(5)中的羟基的保护基的R
步骤A-4是制造化合物(7)的步骤,于惰性溶剂中在4,4'-二甲氧基三苯氯甲烷与碱的存在下,将R
作为所述反应所使用的惰性溶剂,例如可列举:烃类、芳香族烃类、卤化烃类、醚类、乙腈等,优选二氯甲烷或吡啶。
作为所述反应所使用的碱,例如可列举:三乙胺、N,N-二异丙基乙基胺、吡啶、4-二甲氨基吡啶等,优选N,N-二异丙基乙基胺或吡啶。
反应温度根据碱、惰性溶剂等而不同,通常为-20℃至回流温度。优选10℃至30℃。
反应时间根据碱、惰性溶剂、反应温度等而不同,通常为15分钟至24小时,优选1小时至4小时。
步骤A-5是制造化合物(8)的步骤,于惰性溶剂中在2-氰基乙基二异丙基氯亚磷酰胺与碱的存在下,将-P(R
作为所述反应所使用的惰性溶剂,例如可列举:烃类、芳香族烃类、卤化烃类、醚类、乙腈等,优选二氯甲烷。
作为所述反应所使用的碱,例如可列举:三乙胺、N,N-二异丙基乙基胺、吡啶、4-二甲氨基吡啶等,优选N,N-二异丙基乙基胺。
反应温度根据碱、惰性溶剂等而不同,通常为-20℃至回流温度。优选10℃至30℃。
反应时间根据碱、惰性溶剂、反应温度等而不同,通常为15分钟至24小时,优选1小时至4小时。
B法
B法是用来制造化合物(14)或分支型的化合物(19)的方法。如果使用与化合物(2)相反的立体构型的化合物,则化合物(14)或分支型的化合物(19)中键结了-P(R
[化70]
式中R
B-1步骤是制造化合物(10)的步骤,于惰性溶剂中在酸的存在下,使化合物(9)与化合物(2)进行反应,而在环氧化物开环时生成醚键。可与上文所述的步骤A-1同样地进行。
步骤B-2是制造化合物(11)的步骤,去除作为保护基的R
步骤B-3是制造化合物(13)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(11)及化合物(12)进行酰胺缩合。
所述反应所使用的惰性溶剂可根据所使用的缩合剂的种类而适当选择。例如可列举水、醇类、非质子性极性溶剂等,优选二氯甲烷或N,N-二甲基甲酰胺。
作为所述反应所使用的碱,例如可列举:三乙胺、N,N-二异丙基乙基胺、吡啶、4-二甲氨基吡啶等,优选N,N-二异丙基乙基胺。
作为所述反应所使用的缩合剂,例如可列举:碳二亚胺系缩合剂的WSC(1-[3-(二甲氨基)丙基]-3-乙基碳二亚胺)、DIC(N,N'-二异丙基碳二亚胺)、咪唑系缩合剂的CDI(N,N'-羰基二咪唑)、三嗪系缩合剂的DMT-MM(4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉鎓=氯化物n水合物)、脲阳离子系缩合剂的HATU(O-(7-氮杂苯并三唑-1-基)-N,N,N',N'-四甲基脲六氟磷酸盐)、TSTU(O-(N-丁二酰亚胺基)-N,N,N',N'-四甲基脲四氟硼酸盐)、鏻系缩合剂的PyBOP(1H-苯并三唑-1-基氧基三吡咯烷基鏻六氟磷酸盐)等,优选WSC或HATU。另外,也可视需要添加添加剂的HOAt(1-羟基-7-氮杂苯并三唑)、HOBt(1-羟基苯并三唑)、Oxyma(乙基(羟基亚氨基)氰基乙酸酯)。
反应温度根据缩合剂、碱、溶剂等而不同,通常为-20℃至回流温度。优选10℃至30℃。
反应时间根据缩合剂、碱、溶剂、反应温度等而不同,通常为15分钟至72小时,优选1小时至24小时。
化合物(13)向化合物(14)的转换可与上文所述的A法同样地进行。
步骤B-4是去除作为保护基的R
步骤B-5是将R
步骤B-6是将-P(R
步骤B-7是制造化合物(16)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(11)与化合物(15)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
步骤B-8是制造化合物(17)的步骤,去除作为保护基的R
步骤B-9是制造化合物(18)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(12)与化合物(17)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
化合物(18)向化合物(19)的转换可与上文所述的A法同样地进行。
步骤B-10是去除作为保护基的R
步骤B-11是将R
步骤B-12是将-P(R
C法
C法是用来合成制造化合物(26)或分支型的化合物(30)的方法。如果使用化合物(23)的二级羟基相反的立体构型的化合物,则化合物(26)或分支型的化合物(30)中键结了-P(R
[化71]
式中R
C-1步骤是制造化合物(21)的步骤,于惰性溶剂中在酸的存在下,使化合物(20)与化合物(4)进行反应,而生成糖苷键。可与上文所述的步骤A-2同样地进行。
步骤C-2是制造化合物(22)的步骤,去除作为化合物(21)的氨基的保护基的R
步骤C-3是制造化合物(24)的步骤,于惰性溶剂中在活化剂、碱的存在下,使化合物(23)转换为活性酯后,与化合物(22)进行反应,而进行氨基甲酸酯化。
作为所述反应所使用的惰性溶剂,例如可列举:烃类、芳香族烃类、卤化烃类、醚类等,优选二氯甲烷。
作为所述反应所使用的活化剂,只要为形成活化酯的活化剂,则无特别限定,例如可列举:碳酸双(五氟苯基)酯、碳酸双(4-硝基苯基)酯、氯甲酸4-硝基苯酯等。优选氯甲酸4-硝基苯酯。
作为所述反应所使用的碱,例如可列举:三乙胺、N,N-二异丙基乙基胺、吡啶、4-二甲氨基吡啶等,优选在制备活性酯时使用吡啶,继而在与胺进行反应时追加N,N-二异丙基乙基胺。
反应温度根据活化剂、碱、惰性溶剂等而不同,通常为-20℃至回流温度。优选10℃至30℃。
反应时间根据活化剂、碱、惰性溶剂、反应温度等而不同,通常为15分钟至24小时,优选1小时至4小时。
步骤C-4是制造化合物(25)的步骤,去除作为化合物(24)的羟基的保护基的R
化合物(25)向化合物(26)的转换可与上文所述的A法同样地进行。
步骤C-5是将R
步骤C-6是将-P(R
步骤C-7是制造化合物(28)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(22)与化合物(27)进行酰胺缩合。可与上文所述的B法步骤B-3同样地进行。
化合物(28)向化合物(29)的转换可与上文所述的方法同样地进行。
步骤C-8是去除作为化合物(28)的氨基的保护基的R
步骤C-9是进行氨基甲酸酯化的步骤,可与步骤C-3同样地进行。
步骤C-10是去除作为保护基的R
化合物(29)向化合物(30)的转换可与上文所述的A法同样地进行。
步骤C-11是将R
步骤C-12是将-P(R
D法
D法是用来制造化合物(35)的方法。可改变2次酰胺缩合的顺序,或根据各化合物的保护基的状态适当选择步骤。如果使用化合物(23)的二级羟基相反的立体构型的化合物,则化合物(35)中键结了-P(R
[化72]
式中R
步骤D-1是制造化合物(32)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(31)与化合物(12)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
步骤D-2是制造化合物(33)的步骤,去除作为化合物(32)的氨基的保护基的R
步骤D-3是制造化合物(34)的步骤,于惰性溶剂中在活化剂、碱的存在下,使化合物(23)转换为活性酯后,与化合物(33)进行反应,而进行氨基甲酸酯化。可与上文所述的步骤C-3同样地进行。
化合物(34)向化合物(35)的转换可与上文所述的C法同样地进行。
步骤D-4是去除作为保护基的R
步骤D-5是将R
步骤D-6是将-P(R
步骤D-7是制造化合物(36)的步骤,于惰性溶剂中在活化剂、碱的存在下,使化合物(23)转换为活性酯后,与化合物(31)进行反应,而进行氨基甲酸酯化。可与上文所述的步骤C-3同样地进行。
步骤D-8是仅去除作为化合物(36)的氨基的保护基的R
步骤D-9是制造化合物(34)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(37)与化合物(12)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
步骤D-10是制造化合物(39)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(38)与化合物(12)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
化合物(39)向化合物(35)的转换可通过步骤D-5及D-6进行。
E法
E法是用来利用D法所使用的化合物(31)或所合成的中间物(38)制造分支型的化合物(44)的方法。可改变2次酰胺缩合的顺序,或根据各化合物的保护基的状态适当选择步骤。如果使用化合物(23)的二级羟基相反的立体构型的化合物,则化合物(44)中键结了-P(R
[化73]
式中R
步骤E-1是制造化合物(40)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(31)与化合物(15)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
步骤E-2是仅去除作为化合物(40)的氨基的保护基的R
步骤E-3是制造化合物(42)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(41)与化合物(12)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
化合物(42)向化合物(43)的转换可与上文所述的D法同样地进行。
步骤E-4是去除作为化合物(42)的氨基的保护基的R
步骤E-5是进行氨基甲酸酯化的步骤。可与上文所述的D-3同样地进行。
化合物(43)向化合物(44)的转换可与上文所述的C法同样地进行。
步骤E-6是去除作为化合物(43)的羟基的保护基的R
步骤E-7是将R
步骤E-8是将-P(R
步骤E-9是制造化合物(45)的步骤,于惰性溶剂中在缩合剂、碱的存在下,将化合物(38)与化合物(15)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
化合物(45)向化合物(46)的转换可与上文所述的步骤E-2及E-3同样地进行。
步骤E-10是去除作为化合物(45)的氨基的保护基的R
步骤E-11是通过酰胺缩合导入化合物(15)的步骤。可与上文所述的步骤B-3同样地进行。
化合物(46)向化合物(44)的转换可通过步骤E-7及E-8进行。
F法
F法是用来利用A法所合成的中间物(3)制造分支型的化合物(49)的方法。如果使用化合物(3)的二级羟基相反的立体构型的化合物,则化合物(49)中键结了GalNAc的二级羟基的立体构型也变得相反。
[化74]
式中R
步骤F-1是制造化合物(47)的步骤,于惰性溶剂中在酸的存在下,使化合物(3)与化合物(4)进行反应,而生成2个糖苷键。可与上文所述的步骤A-2同样地进行。
步骤F-2是制造化合物(48)的步骤,去除作为化合物(47)的羟基的保护基的R
步骤F-3是制造化合物(49)的步骤,对羟基导入-P(R
G法
G法是用来利用C法所使用的中间物(22)而制造分支型的化合物(55)的方法。如果使用化合物(52)或化合物(56)的二级羟基相反的立体构型的化合物,则化合物(55)中键结了GalNAc的二级羟基的立体构型也变得相反。
[化75]
式中R
步骤G-1是于惰性溶剂中在缩合剂、碱的存在下,将化合物(22)与化合物(50)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
步骤G-2是去除作为保护基的R
步骤G-3是进行氨基甲酸酯化的步骤。可与上文所述的步骤C-3同样地进行。其中,当r=0时,将化合物(22)进行氨基甲酸酯化,当r=1时,将化合物(52)进行氨基甲酸酯化。
步骤G-4是去除作为保护基的R
步骤G-5是制造化合物(55)的步骤,对羟基导入-P(R
步骤G-6是进行氨基甲酸酯化的步骤。可与上文所述的步骤C-3同样地进行。其中,当r=0时,将化合物(22)进行氨基甲酸酯化,当r=1时,将化合物(52)进行氨基甲酸酯化。
步骤G-7是去除作为保护基的R
步骤G-8是进行氨基甲酸酯化的步骤。可与上文所述的C-3同样地进行。
H法
H法是用来利用C法所使用的中间物(22)或G法所使用的中间物(51)而制造分支型的化合物(61)的方法。化合物(52)或化合物(56)的立体构型并无限定。
[化76]
式中R
步骤H-1是进行氨基甲酸酯化的步骤。可与上文所述的步骤C-3同样地进行。其中,当r=0时,将化合物(22)进行氨基甲酸酯化,当r=1时,将化合物(51)进行氨基甲酸酯化。
步骤H-2是去除作为保护基的R
步骤H-3是制造化合物(61)的步骤,对羟基导入-P(R
步骤H-4是进行氨基甲酸酯化的步骤。可与上文所述的步骤C-3同样地进行。其中,当r=0时,将化合物(22)进行氨基甲酸酯化,当r=1时,将化合物(52)进行氨基甲酸酯化。
步骤H-5是去除作为保护基的R
步骤H-6是进行氨基甲酸酯化的步骤。可与上文所述的步骤C-3同样地进行。
I法
I法是用来制造分支型的化合物(73)、及化合物(74)的方法。例如,通过使用化合物(70),可适当选择GalNAc部分的6位羟基的保护基。当R
[化77]
式中R
步骤I-1是进行氨基甲酸酯化的步骤。可与上文所述的步骤C-3同样地进行。
步骤I-2是去除作为保护基的R
步骤I-3是于惰性溶剂中在缩合剂、碱的存在下,与化合物(22)进行酰胺缩合。可与上文所述的步骤B-3同样地进行。
步骤I-4是制造化合物(73)或化合物(74)的步骤,对羟基导入-P(R
本发明的寡核苷酸(反义寡核苷酸)可以医药的形式尤其用于糖原病Ia型的治疗。治疗包括预防及事后治疗的任一者。
本发明的寡核苷酸(反义寡核苷酸)也可以药理上容许的盐的形态使用。所谓“药理上容许的盐”是指寡核苷酸(反义寡核苷酸)的盐,作为此种盐,可列举:如钠盐、钾盐、锂盐的碱金属盐;如钙盐、镁盐的碱土金属盐;铝盐、铁盐、锌盐、铜盐、镍盐、钴盐等金属盐;如铵盐的无机盐;如叔辛基胺盐、二苄基胺盐、吗啉盐、葡糖胺盐、苯基甘氨酸烷基酯盐、乙二胺盐、N-甲基还原葡糖胺盐、胍盐、二乙胺盐、三乙胺盐、二环己基胺盐、N,N'-二苄基乙二胺盐、氯普鲁卡因盐、普鲁卡因盐、二乙醇胺盐、N-苄基-苯乙基胺盐、哌嗪盐、四甲基铵盐、三(羟甲基)氨基甲烷盐的有机盐等胺盐;如氢氟酸盐、盐酸盐、氢溴酸盐、氢碘酸盐的氢卤酸盐;硝酸盐、高氯酸盐、硫酸盐、磷酸盐等无机酸盐;如甲磺酸盐、三氟甲磺酸盐、乙磺酸盐的低级烷磺酸盐;如苯磺酸盐、对甲苯磺酸盐的芳基磺酸盐;乙酸盐、苹果酸盐、反丁烯二酸盐、丁二酸盐、柠檬酸盐、酒石酸盐、草酸盐、顺丁烯二酸盐等有机酸盐;如甘氨酸盐、赖氨酸盐、精氨酸盐、鸟氨酸盐、谷氨酸盐、天冬氨酸盐的氨基酸盐等,优选碱金属盐,更优选钠盐。这些盐可通过公知的方法制造。
另外,寡核苷酸(反义寡核苷酸)及其药理上容许的盐有时也以溶剂合物(例如,水合物)的形式存在,也可为此种溶剂合物。
当将本发明的寡核苷酸(反义寡核苷酸)、其药理上容许的盐或溶剂合物用于糖原病Ia的治疗时,可将其本身或与适宜的药理上容许的赋形剂、稀释剂等混合,通过锭剂、胶囊剂、颗粒剂、散剂或糖浆剂等经口投予,或者通过注射剂、栓剂、贴附剂或外用剂等非经口投予。
这些制剂是使用赋形剂(例如,如以下的有机系赋形剂:如乳糖、白糖、葡萄糖、甘露糖醇、山梨糖醇的糖衍生物;如玉米淀粉、马铃薯淀粉、α淀粉、糊精的淀粉衍生物;如结晶纤维素的纤维素衍生物;阿拉伯胶;葡聚糖;支链淀粉;如以下的无机系赋形剂:如轻质无水硅酸、合成硅酸铝、硅酸钙、硅酸铝镁的硅酸盐衍生物;如磷酸氢钙的磷酸盐;如碳酸钙的碳酸盐;如硫酸钙的硫酸盐等;等)、润滑剂(例如,硬脂酸;如硬脂酸钙、硬脂酸镁的硬脂酸金属盐;滑石;胶体二氧化硅;如蜂蜡、鲸蜡的蜡类;硼酸;己二酸;如硫酸钠的硫酸盐;二醇;反丁烯二酸;苯甲酸钠;DL亮氨酸;如月桂基硫酸钠、月桂基硫酸镁的月桂基硫酸盐:如无水硅酸、硅酸水合物的硅酸类;所述淀粉衍生物等)、结合剂(例如,羟丙基纤维素、羟丙基甲基纤维素、聚乙烯基吡咯烷酮、聚乙二醇、与所述赋形剂同样的化合物等)、崩解剂(例如,如低取代度羟丙基纤维素、羧甲基纤维素、羧甲基纤维素钙、内部交联羧甲基纤维素钠的纤维素衍生物;如羧甲基淀粉、羧甲基淀粉钠、交联聚乙烯基吡咯烷酮的被化学修饰的淀粉-纤维素类等)、乳化剂(例如,如膨润土、胶体硅酸镁铝的胶体性黏土;如氢氧化镁、氢氧化铝的金属氢氧化物;如月桂基硫酸钠、硬脂酸钙的阴离子表面活性剂;如氯化苄烷铵的阳离子表面活性剂;如聚氧乙烯烷基醚、聚氧乙烯山梨糖醇酐脂肪酸酯、蔗糖脂肪酸酯的非离子表面活性剂等)、稳定剂(如对羟基苯甲酸甲酯、对羟基苯甲酸丙酯的对羟基苯甲酸酯类;如氯丁醇、苄醇、苯基乙醇的醇类;氯化苄烷铵;如苯酚、甲酚的酚类;硫柳汞;去氢乙酸;山梨酸等)、矫味除臭剂(例如,通常使用的甜味料、酸味料、香料等)、稀释剂等添加剂,通过周知的方法而制造。
本发明的治疗药含有0.1~250μmoles/mL的寡核苷酸(反义寡核苷酸)即可,优选也可使其含有1~50μmoles/mL的寡核苷酸(反义寡核苷酸)、其药理上容许的盐或溶剂合物、0.02~10%w/v的碳水化合物或多元醇及0.01~0.4%w/v的药理上容许的表面活性剂。
作为所述碳水化合物,特别优选单糖类或二糖类。作为这些碳水化合物及多元醇的例,可列举:葡萄糖、半乳糖、甘露糖、乳糖、麦芽糖、甘露糖醇及山梨糖醇。这些可单独使用,也可并用。
另外,作为本发明中的表面活性剂的优选的例,可列举:聚氧乙烯山梨糖醇酐单~三-酯、烷基苯基聚氧乙烯、牛胆酸钠、胆酸钠、及多元醇酯。其中,特别优选聚氧乙烯山梨糖醇酐单~三-酯,其中,作为酯,特别优选油酸酯、月桂酸酯、硬脂酸酯及棕榈酸酯。这些可单独使用,也可并用。
另外,本发明的治疗药进而优选可含有0.03~0.09M的药理上容许的中性盐、例如氯化钠、氯化钾及/或氯化钙。
另外,本发明的治疗药进而优选可含有0.002~0.05M的药理上容许的缓冲剂。作为优选的缓冲剂的例,可列举:柠檬酸钠、甘氨酸钠、磷酸钠、三(羟甲基)氨基甲烷。这些缓冲剂可单独使用,也可并用。
进而,所述治疗药可以溶液状态供给。然而,由于存在必须保存一定时间的情形等,故而为了使寡核苷酸(反义寡核苷酸)稳定而防止治疗效果的降低,通常优选预先冷冻干燥,在这种情况下,在使用时以溶解液(注射用蒸馏水等)再构成(reconstruction),即制成投予的液体状态使用即可。因此,本发明的治疗药也包含用于以各成分成为特定的浓度范围的方式以溶解液再构成而使用的被冷冻干燥的状态的治疗药。为了促进冷冻干燥物的溶解性,可使其进而含有白蛋白、甘氨酸等氨基酸。
本发明的寡核苷酸(反义寡核苷酸)可使用国际公开第2015/005253所记载的脂质等将其封入,以如国际公开第2015/005253等所记载的核酸脂质纳米粒子或脂质体的形式投予。
当将本发明的寡核苷酸(反义寡核苷酸)、其药理上容许的盐或溶剂合物对人投予时,例如以成人每天约0.01~100mg/kg(体重)、优选0.1~20mg/kg(体重)的投予量,1次或分数次进行皮下注射、点滴静脉注射、或静脉注射即可,其投予量或投予次数可根据疾病的种类、症状、年龄、投予方法等适当变更。
本发明的寡核苷酸(反义寡核苷酸)、其药理上容许的盐或溶剂合物向糖原病Ia型患者的投予例如可以如下方式进行。即,通过业者所周知的方法制造寡核苷酸(反义寡核苷酸)、其药理上容许的盐或溶剂合物,通过常规方法对其进行杀菌处理,例如制备125mg/mL的注射用溶液。以寡核苷酸(反义寡核苷酸)的投予量相对于体重每1kg例如成为10mg的方式,例如以输液的形式将该溶液点滴投予至患者静脉内。投予例如是以1周的间隔进行,其后也一边通过血糖值、血中乳酸值、利用CT(Computed Tomography,计算机断层扫描技术)获得的肝肿大/肝糖值等确认治疗效果,一边适当重复该治疗。
实施例
以下,通过实施例具体说明本发明。再者,这些实施例是用来说明本发明的,并不限定本发明的范围。
(实施例1~11)
HO-A
使用核酸自动合成仪(“ABI 394DNA/RNA Synthesizer”Applied Biosystems制造),利用亚磷酰胺法(Nucleic Acids Research,12,4539(1984))进行合成。作为试剂,使用活化剂溶液-3(0.25mol/L的5-苄硫基-1H-四唑/乙腈溶液,和光纯药工业制造,产品编号013-20011)、CAP A for AKTA(1-甲基咪唑/乙腈溶液,Sigma-Aldrich制造,产品编号L040050)、Cap B1 for AKTA(乙酸酐/乙腈溶液,Sigma-Aldrich制造,产品编号L050050)、Cap B2 for AKTA(吡啶/乙腈溶液,Sigma-Aldrich制造,产品编号L050150)、DCA Deblock(二氯乙酸/甲苯溶液,Sigma-Aldrich制造,产品编号L023050)。作为用来形成硫代磷酸酯键的硫化试剂,以成为0.2M的方式使用乙腈(脱水,关东化学制造,产品编号01837-05)、吡啶(脱水,关东化学制造,产品编号11339-05)1:1(v/v)溶液溶解苯基乙酰基二硫醚(CARBOSYNTH制造,产品编号FP07495)使用。作为酰胺试剂,2'-O-Me核苷的亚磷酰胺(腺苷体产品编号ANP-5751,胞苷体产品编号ANP-5752,鸟苷体产品编号ANP-5753,尿苷体产品编号ANP-5754)是使用ChemGenes制造的亚磷酰胺。非天然型的亚磷酰胺是使用日本专利特开2000-297097的实施例14(5'-O-二甲氧基三苯甲基-2'-O,4'-C-亚乙基-6-N-苯甲酰基腺苷-3'-O-(2-氰基乙基N,N-二异丙基)亚磷酰胺)、实施例27(5'-O-二甲氧基三苯甲基-2'-O,4'-C-亚乙基-2-N-异丁酰基鸟苷-3'-O-(2-氰基乙基N,N-二异丙基)亚磷酰胺)、实施例22(5'-O-二甲氧基三苯甲基-2'-O,4'-C-亚乙基-4-N-苯甲酰基-5-甲基胞苷-3'-O-(2-氰基乙基N,N-二异丙基)亚磷酰胺)、实施例9(5'-O-二甲氧基三苯甲基-2'-O,4'-C-亚乙基-5-甲基尿苷-3'-O-(2-氰基乙基N,N-二异丙基)亚磷酰胺)的化合物。作为固相载体,使用Glen Unysupport 0.1μmol(GlenResearch制造),合成表述化合物。程序使用附属于核酸自动合成仪的0.2μmol尺度用,其中,酰胺体的缩合所需的时间设为600秒,硫化所需的时间设为150秒。
利用300μL的浓氨水处理具有目标序列的被保护的寡核苷酸相关物,由此自支持体切下低聚物,并且脱去磷原子上的保护基氰基乙基与核酸碱基上的保护基。使用ClarityQSP(Phenomenex制造),依照随附的操作说明进行精制。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第112号互补的序列。
实施例2至11的化合物也与实施例1同样地合成。将实施例1的数据、及实施例2至11记载于表1。
表1
表中的序列中大写字母表示2'-O,4'-C-亚乙基交联核酸,小写字母表示2'-OMe-RNA。2'-O,4'-C-亚乙基交联核酸的C是5-甲基胞嘧啶。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI(electrospray ionization,电喷雾电离)质量分析进行鉴定,将实测值示于表中。
(实施例12~22)
HO-A
使用核酸自动合成仪(BioAutomation制造的MerMade 192X),与实施例1同样地进行合成及精制,获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第112号互补的序列。
实施例13至22的化合物也与实施例12同样地合成。将实施例12的数据、及实施例13至22记载于表2。
表2
表中的序列中小写字母表示2'-OMe-RNA。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI质量分析进行鉴定,将实测值示于表中。
(实施例23~50)
HO-G
与实施例12同样地进行合成及精制,获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第111号互补的序列。
实施例24至50的化合物也与实施例23同样地合成。将实施例23的数据、及实施例24至50记载于表3。
表3
表中的序列中大写字母表示2'-O,4'-C-亚乙基交联核酸,小写字母表示2'-OMe-RNA。2'-O,4'-C-亚乙基交联核酸的C是5-甲基胞嘧啶。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI质量分析进行鉴定,将实测值示于表中。
(实施例51~69)
HO-U
与实施例12同样地进行合成。利用450μL的浓氨水处理具有目标序列的被保护的寡核苷酸相关物,由此自支持体切下低聚物,并且脱去磷原子上的保护基氰基乙基与核酸碱基上的保护基。将低聚物的混合溶液与Clarity QSP DNA上样缓冲液(Clarity QSP DNALoading Buffer)(Phenomenex制造)300μL混合并添加到Clarity SPE 96孔板(Phenomenex制造)上。依序添加Clarity QSP DNA上样缓冲液:水=1:1溶液1mL、水3mL、3%二氯乙酸(DCA)水溶液3mL、水6mL后,收集通过20mM Tris水溶液:乙腈=9:1溶液萃取的成分。将溶剂蒸馏去除后,获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第109号互补的序列。
实施例52至69的化合物也与实施例51同样地合成。将实施例51的数据、及实施例52至69记载于表4。
表4
表中的序列中大写字母表示2'-O,4'-C-亚乙基交联核酸,小写字母表示2'-OMe-RNA。2'-O,4'-C-亚乙基交联核酸的C是5-甲基胞嘧啶。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI质量分析进行鉴定,将实测值示于表中。
(实施例70~82)
HO-U
使用核酸自动合成仪(BioAutomation制造的MerMade 192X),与实施例12同样地进行合成及精制,而获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第109号互补的序列。
实施例71至82的化合物也与实施例70同样地合成。将实施例70的数据、及实施例71至82记载于表5。
表5
表中的序列中小写字母表示2'-OMe-RNA。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI质量分析进行鉴定,将实测值示于表中。
(参考例1)
HO-A
与实施例1同样地合成、精制,而获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第95号至第115号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7520.85)。
(参考例2~3)
HO-G
与实施例12同样地进行合成及精制,而获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第94号至第114号互补的序列。
参考例3的化合物也与参考例2同样地合成。将参考例2的数据、及参考例3记载于表6。
表6
/>
表中的序列中大写字母表示2'-O,4'-C-亚乙基交联核酸,小写字母表示2'-OMe-RNA。2'-O,4'-C-亚乙基交联核酸的C是5-甲基胞嘧啶。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI质量分析进行鉴定,将实测值示于表中。
(参考例4~15)
HO-C
与实施例51同样地进行合成及精制,而获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第96号至第113号互补的序列。
参考例5至15的化合物也与参考例4同样地合成。将参考例4的数据、及参考例5至15记载于表7。
表7
表中的序列中大写字母表示2'-O,4'-C-亚乙基交联核酸,小写字母表示2'-OMe-RNA。2'-O,4'-C-亚乙基交联核酸的C是5-甲基胞嘧啶。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA (NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI质量分析进行鉴定,将实测值示于表中。
(参考例16~20)
HO-C
与实施例70同样地进行合成及精制,而获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第96号至第113号互补的序列。
参考例17至20的化合物也与参考例16同样地合成。将参考例16的数据、及参考例17至20记载于表8。
表8
表中的序列中小写字母表示2'-OMe-RNA。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI质量分析进行鉴定,将实测值示于表中。
(实施例83~86)
HO-A
使用AKTA Oligopilot,使用Primer Support 5G Unylinker 350,100μmol(GEHealthcare制造)作为固相载体,与实施例1同样地进行合成。利用25mL的浓氨水处理具有目标序列的被保护的寡核苷酸相关物,由此自支持体切下低聚物,并且脱去磷原子上的保护基氰基乙基与核酸碱基上的保护基。使用phenomenex Clarity QSP 5g对低聚物的混合溶液进行脱保护、精制,将溶剂蒸馏去除后,获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。
实施例84至86的化合物也与实施例83同样地合成。将实施例83的数据、及实施例84至86记载于表9。
表9
表中的序列中大写字母表示2'-O,4'-C-亚乙基交联核酸,小写字母表示2'-OMe-RNA,标下划线的小写字母表示DNA。2'-O,4'-C-亚乙基交联核酸的C是5-甲基胞嘧啶。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI质量分析鉴定,将实测值示于表中。
(实施例87~90)
HO-A
与实施例1同样地进行合成。但使用Primer Support 5G Unylinker 350,10μmol(GE Healthcare制造)作为固相载体,且使用10μmol的程序。与实施例83同样地进行精制,而获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。
实施例88至90的化合物也与实施例87同样地合成。将实施例87的数据、及实施例88至90记载于表10。
表10
表中的序列中大写字母表示2'-O,4'-C-亚乙基交联核酸,小写字母表示2'-OMe-RNA。2'-O,4'-C-亚乙基交联核酸的C是5-甲基胞嘧啶。开始及结束表示智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起的编号。化合物是通过负离子ESI质量分析进行鉴定,将实测值示于表中。
(实施例91)
HO-X-X-X-A
与实施例1同样地进行合成。但使用Primer Support 5G Unylinker 350,10μmol(GE Healthcare制造)作为固相载体,且使用10μmol的程序。X部分使用文献(Bioorg.Med.Chem.(2016)24,26-32)所记载的GalNAc亚磷酰胺单元(phosphoramiditeunit)1缩合3次。与实施例83同样地进行精制,而获得目标化合物。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第107号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7131.24)。
(实施例92)
HO-X-X-X-A
与实施例91同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6818.18)。
(实施例93)
HO-X-X-X-T
与实施例91同样地进行合成。
本化合物的碱基序列具有与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第107号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7738.35)。
(实施例94)
HO-X-X-X-A
与实施例91同样地进行合成。
本化合物的碱基序列具有与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第107号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7141.22)。
(实施例95)
HO-X-X-X-A
与实施例91同样地进行合成。
本化合物的碱基序列具有与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第107号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7167.24)。
(参考例21)
(21A)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[5-(苄氧基羰基氨基)戊氧基]四氢吡喃-2-基]甲酯(化合物21A)的合成
[化78]
在文献已知化合物的N-苄氧基羰基-1-羟基戊基-5-胺(J.Am.Chem.Soc.,2006,128,4058-4073.)(8.05g,33.9mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三乙酰氧基-四氢吡喃-2-基]甲酯(WO2011053614)(12.0g,30.8mmol)的二氯甲烷(200mL)悬浮溶液中添加三氟甲磺酸(450μL,5.23mmol),在45℃下整夜搅拌。反应结束后,将溶剂减压蒸馏去除一半进行浓缩,添加到乙酸乙酯/饱和碳酸氢钠水溶液的混合溶液中。利用饱和碳酸氢钠水溶液、饱和食盐水/磷酸缓冲液(pH值7.0)洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物21A(17.0g,产率94%)。
1
C
(21B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-(5-氨基戊氧基)四氢吡喃-2-基]甲酯(化合物21B)的合成
[化79]
将步骤(21A)中合成的化合物21A(17.0g,30.0mmol)溶解于乙醇(200ml)中。添加10%钯/碳(湿基)(2g),于氢气环境下在室温下剧烈搅拌。反应结束后过滤,将所获得的通过液进行减压蒸馏去除而获得目标物21B(12.64g,产率97%)的粗产物。不再进一步进行精制,用于下一反应。
1
C
(21C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[5-[[(4S)-2,2-二甲基-1,3-二氧杂环戊烷-4-基]甲氧基羰基氨基]戊氧基]四氢吡喃-2-基]甲酯(化合物21C)的合成
[化80]
将(R)-(-)-2,2-二甲基-1,3-二氧杂环戊烷-4-甲醇(1.68g,12.8mmol)、三乙胺(3.22mL,23.1mmol)溶解于二氯甲烷(100mL)中。添加氯甲酸4-硝基苯酯(2.56g,12.7mmol),在室温下搅拌1小时。继而,添加三乙胺(5mL,36mmol)、步骤(21B)中合成的化合物21B(5.0g,11.56mmol)的二氯甲烷溶液(20mL),在45℃下搅拌1小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物21C(3.46g,产率51%)。
1
C
(21D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[5-[[(2S)-2,3-二羟基丙氧基]羰基氨基]戊氧基]四氢吡喃-2-基]甲酯(化合物21D)的合成
[化81]
在步骤(21C)中合成的化合物21C(4.38g,7.42mmol)中添加纯水(2mL)、三氟乙酸(6mL),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除。进而进行甲苯共沸,由此将水去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物21D(2.47g,产率61%)。
1
C
(21E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[5-[[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]羰基氨基]戊氧基]四氢吡喃-2-基]甲酯(化合物21E)的合成
[化82]
将步骤(21D)中合成的化合物21D(2.47g,4.52mmol)溶解于吡啶(15mL)中。添加4,4'-二甲氧基三苯氯甲烷(1.82g,5.38mmol),在室温下加以搅拌。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v,其后乙酸乙酯:甲醇=95:5,v/v)将其进行精制,而获得非晶状的目标物21E(1.84g,产率48%)。
1
C
(21F)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[5-[[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基丙氧基]羰基氨基]戊氧基]四氢吡喃-2-基]甲酯(化合物21F)的合成
[化83]
将步骤(21E)中合成的化合物21E(1.84g,2.16mmol)添加适量的吡啶并在减压下共沸后,溶解于二氯甲烷(21mL)中。添加N,N-二异丙基乙基胺(2.25mL,12.9mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.96mL,4.31mmol),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯共沸后获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物21F(1.87g,产率82%)。
1
(实施例96)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6830.15)。
(参考例22)
(22A)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[5-[[(4R)-2,2-二甲基-1,3-二氧杂环戊烷-4-基]甲氧基羰基氨基]戊氧基]四氢吡喃-2-基]甲酯(化合物22A)的合成
[化84]
将(S)-(+)-2,2-二甲基-1,3-二氧杂环戊烷-4-甲醇(1.68g,2.54mmol)、三乙胺(3.22mL,23.1mmol)溶解于二氯甲烷(100mL)中。添加氯甲酸4-硝基苯酯(2.56g,12.72mmol),在室温下搅拌1小时。继而,添加三乙胺(5mL,36mmol)、步骤(21B)中合成的化合物21B(5.2g,13mmol),在45℃下搅拌1小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物22A(3.82g,产率54%)。
1
C
(22B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[5-[[(2R)-2,3-二羟基丙氧基]羰基氨基]戊氧基]四氢吡喃-2-基]甲酯(化合物22B)的合成
[化85]
在步骤(22A)中合成的化合物22A(3.82g,6.47mmol)中添加纯水(2mL)、三氟乙酸(6mL),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除。进而进行甲苯共沸,由此将水去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物22B(2.49g,产率70%)。
1
C
(22C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[5-[[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]羰基氨基]戊氧基]四氢吡喃-2-基]甲酯(化合物22C)的合成
[化86]
将步骤(22B)中合成的化合物22B(2.49g,4.52mmol)溶解于吡啶(15mL)中。添加4,4'-二甲氧基三苯氯甲烷(1.84g,5.43mmol),在室温下加以搅拌。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v,其后乙酸乙酯:甲醇=95:5,v/v)将其进行精制,而获得非晶状的目标物22C(2.1g,产率54%)。
1
C
(22D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[5-[[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基丙氧基]羰基氨基]戊氧基]四氢吡喃-2-基]甲酯(化合物22D)的合成
[化87]
将步骤(22C)中合成的化合物22C(2.10g,2.50mmol)添加适量的吡啶并在减压下共沸后,溶解于二氯甲烷(20mL)中。添加N,N-二异丙基乙基胺(2.6mL,15mmol)、2-氰基乙基二异丙基氯亚磷酰胺(1.L1 ml,4.9mmol),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯共沸后获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物22D(1.63g,产率63%)。
1
(实施例97)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6830.14)。
(参考例23)
(23A)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[8-(苄氧基羰基氨基)辛氧基]四氢吡喃-2-基]甲酯(化合物23A)的合成
[化88]
在文献已知化合物的N-(8-羟基辛基)氨基甲酸苄酯(J.Med.Chem,1993,36,3721-3726.)(6.5g,23mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三氧基-四氢吡喃-2-基]甲酯(WO2011053614)(8.2g,21mmol)的二氯甲烷(150mL)悬浮溶液中添加三氟甲磺酸(310μL,3.6mmol),在45℃下整夜搅拌。反应结束后,将溶剂减压蒸馏去除一半进行浓缩,添加到乙酸乙酯/饱和碳酸氢钠水溶液的混合溶液中。利用饱和碳酸氢钠水溶液、饱和食盐水/磷酸缓冲液(pH值7.0)洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物23A(12.68g,产率99%)。
1
C
(23B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-(8-氨基辛氧基)四氢吡喃-2-基]甲酯(化合物23B)的合成
[化89]
将步骤(23A)中合成的化合物23A(12.68g,20.83mmol)溶解于乙醇(200mL)中。添加10%钯/碳(湿基)(2g),于氢气环境下在室温下剧烈搅拌。反应结束后过滤,将所获得的通过液进行减压蒸馏去除而获得目标物23B(10.1g)的粗产物。不再进一步进行精制,用于下一反应。
C
(23C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[8-[[(4S)-2,2-二甲基-1,3-二氧杂环戊烷-4-基]甲氧基羰基氨基]辛氧基]四氢吡喃-2-基]甲酯(化合物23C)的合成
[化90]
将(R)-(-)-2,2-二甲基-1,3-二氧杂环戊烷-4-甲醇(1.6g,12mmol)、三乙胺(3.1mL,22mmol)溶解于二氯乙烷(30mL)中。添加氯甲酸4-硝基苯酯(2.2g,12mmol),在室温下搅拌1.5小时。继而,添加三乙胺(4.5mL,32mmol)、步骤(23B)中合成的化合物23B(5.1g,11mmol),在45℃下搅拌4小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物23C(2.45g,产率36%)。
1
C
(23D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[8-[[(2S)-2,3-二羟基丙氧基]羰基氨基]辛氧基]四氢吡喃-2-基]甲酯(化合物23D)的合成
[化91]
在步骤(23C)中合成的化合物23C(2.45g,3.87mmol)中添加纯水(2mL)、三氟乙酸(6mL),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除。进而进行甲苯共沸,由此将水去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物23D(1.18g,产率51%)。
1
C
(23E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[8-[[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]羰基氨基]辛氧基]四氢吡喃-2-基]甲酯(化合物23E)的合成
[化92]
将步骤(23D)中合成的化合物23D(1.18g,1.99mmol)溶解于吡啶(20mL)中。添加4,4'-二甲氧基三苯氯甲烷(0.81g,2.39mmol),在室温下加以搅拌。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯而共沸后,获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v,其后乙酸乙酯:甲醇=95:5,v/v)将其进行精制,而获得非晶状的目标物23E(0.6g,产率34%)。
1
C
(23F)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[8-[[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基丙氧基]羰基氨基]辛氧基]四氢吡喃-2-基]甲酯(化合物23F)的合成
[化93]
将步骤(23E)中合成的化合物23E(0.60g,0.67mmol)添加适量的吡啶并在减压下共沸后,溶解于二氯乙烷(5mL)中。添加N,N-二异丙基乙基胺(0.70ml,4.0mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.30mL,1.3mmol),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯共沸后获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物23F(0.51g,产率69%)。
1
(实施例98)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6957.27)。
(参考例24)
(24A)
7-[(2S)-3-苄氧基-2-羟基丙氧基]庚烷-1-醇(化合物24A)的合成
[化94]
将苄基(S)-(+)-缩水甘油醚(5.0g,30mmol)溶解于二氯甲烷(150mL)中。添加1,7-庚二醇(6.0g,45mmol)、三氟化硼-二乙醚络合物(0.76mL,6.1mmol),在室温下搅拌2天。将反应溶液添加到乙酸乙酯/饱和碳酸氢钠水溶液的混合溶液中,利用乙酸乙酯进行萃取。利用饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50,v/v)将其进行精制,而获得无色油状的目标物24A(3.3g,产率37%)。
1
C
(24B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[7-[(2S)-3-苄氧基-2-羟基-丙氧基]庚氧基]四氢吡喃-2-基]甲酯(化合物24B-1)的合成
[化95]
及乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[7-[(2S)-2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基-3-苄氧基-丙氧基]庚氧基]-3,4-二乙酰氧基四氢吡喃-2-基]甲酯(化合物24B-2)的合成
[化96]
向步骤(24A)中合成的化合物15A(3.3g,11mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三乙酰氧基-四氢吡喃-2-基]甲酯(WO2011053614)(4.0g,10mmol)的二氯甲烷(200mL)悬浮溶液中添加三氟甲磺酸(0.15mL,1.7mmol),在45℃下搅拌3天。反应结束后,将溶剂减压蒸馏去除一半进行浓缩,添加到乙酸乙酯/饱和碳酸氢钠水溶液的混合溶液中,利用乙酸乙酯进行萃取。利用饱和碳酸氢钠水溶液、饱和食盐水/磷酸缓冲液(pH值7.0)洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物24B-1(4.5g,产率70%)及24B-2(1.9g,产率20%)。
(化合物24B-1)
1
C
(化合物24B-2)
1
C
(24C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[7-[(2R)-2,3-二羟基丙氧基]庚氧基]四氢吡喃-2-基]甲酯(化合物24C)的合成
[化97]
将步骤(24B)中合成的化合物24B-1(4.5g,7.2mmol)溶解于乙醇(100mL)中。添加10%钯/碳(湿基)(2.0g),于氢气环境下在室温下剧烈搅拌5小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除。添加适量的甲苯而共沸后,添加适量的吡啶进行共沸,而获得目标物24C的粗产物。不再进一步进行精制,用于下一反应。
1
C
(24D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[7-[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]庚氧基]四氢吡喃-2-基]甲酯(化合物24D)的合成
[化98]
将步骤(24C)中合成的化合物24C的粗产物溶解于吡啶(30mL)中。添加4,4'-二甲氧基三苯氯甲烷(2.7g,7.9mmol),在室温下搅拌13小时。反应结束后,将溶剂减压蒸馏去除。添加甲苯而共沸后,获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物24D(3.0g,产率2步49%)。
1
C
(24E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[7-[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙基-(二异丙氨基)膦基]氧基丙氧基]庚氧基]四氢吡喃-2-基]甲酯(化合物24E)的合成
[化99]
将步骤(24D)中合成的化合物24D(3.0g,3.6mmol)通过适量的吡啶共沸后,溶解于二氯甲烷(50mL)中。添加N,N-二异丙基乙基胺(2.5mL,14mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.88mL,3.9mmol),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物24E(3.0g,产率82%)。
1
(实施例99)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6785.22)
(参考例25)
(25A)
7-[(2R)-3-苄氧基-2-羟基丙氧基]庚烷-1-醇(化合物25A)的合成
[化100]
将苄基(R)-(-)-缩水甘油醚(5.0g,30mmol)溶解于二氯甲烷(150mL)中。添加1,7-庚二醇(6.0g,45mmol)、三氟化硼-二乙醚络合物(0.76mL,6.1mmol),在室温下搅拌2天。将反应溶液添加到乙酸乙酯/饱和碳酸氢钠水溶液的混合溶液中,利用乙酸乙酯进行萃取。利用饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50,v/v)将其进行精制,而获得无色油状的目标物25A(4.0g,产率44%)。
1
C
(25B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[7-[(2R)-3-苄氧基-2-羟基-丙氧基]庚氧基]四氢吡喃-2-基]甲酯(化合物25B-1)的合成
[化101]
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[7-[(2R)-2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基-3-苄氧基-丙氧基]庚氧基]-3,4-二乙酰氧基四氢吡喃-2-基]甲酯(化合物25B-2)的合成
[化102]
在步骤(25A)中合成的化合物25A(4.0g,14mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三乙酰氧基-四氢吡喃-2-基]甲酯(WO2011053614)(4.8g,12mmol)的二氯甲烷(200mL)悬浮溶液中添加三氟甲磺酸(0.18mL,2.1mmol),在45℃下搅拌3天。反应结束后,将溶剂减压蒸馏去除一半进行浓缩,添加到乙酸乙酯/饱和碳酸氢钠水溶液的混合溶液中,利用乙酸乙酯进行萃取。利用饱和碳酸氢钠水溶液、饱和食盐水/磷酸缓冲液(pH值7.0)洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物25B-1(4.8g,产率62%)及25B-2(1.9g,产率16%)。
(化合物25B-1)
1
C
(化合物25B-2)
1
C
(25C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[7-[(2S)-2,3-二羟基丙氧基]庚氧基]四氢吡喃-2-基]甲酯(化合物25C)的合成
[化103]
将步骤(25B)中合成的化合物25B-1(4.8g,7.7mmol)溶解于乙醇(100mL)中。添加10%钯/碳(湿基)(2.0g),于氢气环境下在室温下剧烈搅拌5小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除。添加适量的甲苯而共沸后,添加适量的吡啶进行共沸,而获得目标物25C的粗产物。不再进一步进行精制,用于下一反应。
1
C
(25D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[7-[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]庚氧基]四氢吡喃-2-基]甲酯(化合物25D)的合成
[化104]
将步骤(25C)中合成的化合物25C的粗产物溶解于吡啶(30mL)中。添加4,4'-二甲氧基三苯氯甲烷(2.7g,8.0mmol),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除。添加甲苯而共沸后,获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物25D(4.5g,产率2步70%)。
1
C
(25E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[7-[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙基-(二异丙氨基)膦基]氧基丙氧基]庚氧基]四氢吡喃-2-基]甲酯(化合物25E)的合成
[化105]
将步骤(25D)中合成的化合物25D(4.5g,5.4mmol)通过适量的吡啶共沸后,溶解于二氯甲烷(50mL)中。添加N,N-二异丙基乙基胺(3.7mL,21mmol)、2-氰基乙基二异丙基氯亚磷酰胺(1.3ml,5.9mmol),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯共沸后获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物25E(4.5g,产率81%)。
1
(实施例100)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6785.21)。
(参考例26)
(26A)
13-[(2S)-3-苄氧基-2-羟基丙氧基]十三烷-1-醇(化合物26A)的合成
[化106]
将苄基(S)-(+)-缩水甘油醚(4.0g,24mmol)溶解于二氯甲烷(150mL)中。添加十三烷-1,13-二醇(7.6g,35mmol)、三氟化硼-二乙醚络合物(0.61mL,4.9mmol),在室温下搅拌2天。将反应溶液添加到乙酸乙酯/饱和碳酸氢钠水溶液的混合溶液中,利用乙酸乙酯进行萃取。利用饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50,v/v)将其进行精制,而获得无色油状的目标物26A(3.8g,产率41%)。
1
C
(26B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[13-[(2S)-3-苄氧基-2-羟基-丙氧基]十三烷氧基]四氢吡喃-2-基]甲酯(化合物26B)的合成
[化107]
在步骤(26A)中合成的化合物26A(3.8g,9.9mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三乙酰氧基-四氢吡喃-2-基]甲酯(WO2011053614)(3.5g,9.0mmol)的二氯甲烷(60mL)悬浮溶液中添加三氟甲磺酸(0.13mL,1.5mmol),在45℃下搅拌17小时。反应结束后,将溶剂减压蒸馏去除一半进行浓缩,添加到乙酸乙酯/饱和碳酸氢钠水溶液的混合溶液中,利用乙酸乙酯进行萃取。利用饱和碳酸氢钠水溶液、饱和食盐水/磷酸缓冲液(pH值7.0)洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v)将其进行精制,而获得非晶状的目标物26B(3.7g,产率58%)。
1
C
(26C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[13-[(2R)-2,3-二羟基丙氧基]十三烷氧基]四氢吡喃-2-基]甲酯(化合物26C)的合成
[化108]
将步骤(26B)中合成的化合物26B(3.7g,5.2mmol)溶解于乙醇(100mL)中。添加10%钯/碳(湿基)(2.0g),于氢气环境下在室温下剧烈搅拌5小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:甲醇=100:0-90:10,v/v)将其进行精制,而获得无色油状的目标物26C(1.4g,产率43%)。
1
C
(26D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[13-[(2S)-3-[双(4-甲氧基苯基)-苯基甲氧基]-2-羟基-丙氧基]十三烷氧基]四氢吡喃-2-基]甲酯(化合物26D)的合成
[化109]
将步骤(26C)中合成的化合物26C(1.4g,2.3mmol)溶解于吡啶(10mL)中。添加4,4'-二甲氧基三苯氯甲烷(0.84g,2.5mmol),在室温下搅拌15小时。反应结束后,将溶剂减压蒸馏去除。添加甲苯而共沸后,获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50-20:80,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物26D(1.9g,产率91%)。
1
C
(26E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[13-[(2S)-3-[双(4-甲氧基苯基)-苯基甲氧基]-2-[2-氰基乙基-(二异丙氨基)膦基]氧基丙氧基]十三烷氧基]四氢吡喃-2-基]甲酯(化合物26E)的合成
[化110]
将步骤(26D)中合成的化合物26D(1.9g,2.1mmol)通过适量的吡啶共沸后,溶解于二氯甲烷(50mL)中。添加N,N-二异丙基乙基胺(1.4mL,8.0mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.49mL,2.2mmol),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物26E(1.7g,产率75%)。
1
(实施例101)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7037.48)。
(参考例27)
(27A)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[7-[(2R)-2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基-3-羟基-丙氧基]庚氧基]-3,4-二乙酰氧基四氢吡喃-2-基]甲酯(化合物27A)的合成
[化111]
将步骤(24B)中合成的化合物24B-2(1.9g,2.0mmol)溶解于乙醇(60mL)中。添加10%钯/碳(湿基)(2.0g),于氢气环境下在室温下剧烈搅拌8小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除。添加适量的甲苯而共沸后,添加适量的吡啶进行共沸,而获得目标物27A的粗产物(1.3g)。不再进一步进行精制,用于下一反应。
1
C
(27B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[7-[(2S)-2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基-3-[2-氰基乙氧基-(二异丙氨基)膦基]氧基丙氧基]庚氧基]-3,4-二乙酰氧基四氢吡喃-2-基]甲酯(化合物27B)的合成
[化112]
将步骤(27A)中合成的化合物27A的粗产物(1.3g)溶解于二氯甲烷(50mL)中。添加二异丙基乙基胺(1.0mL,5.8mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.36mL,1.6mmol),在室温下搅拌1小时。进而,添加2-氰基乙基二异丙基氯亚磷酰胺(0.36mL,1.6mmol),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯共沸后获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙腈=100:0-90:10,v/v)将其进行精制,而获得非晶状的目标物27B(0.56g,产率2步26%)。
1
(实施例102)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6517.17)。
(参考例28)
(28A)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[7-[(2S)-2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基-3-羟基-丙氧基]庚氧基]-3,4-二乙酰氧基四氢吡喃-2-基]甲酯(化合物28A)的合成
[化113]
将步骤(25B)中合成的化合物25B-2(1.9g,2.0mmol)溶解于乙醇(60mL)中。添加10%钯/碳(湿基)(2.0g),于氢气环境下在室温下剧烈搅拌5小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除。添加适量的甲苯而共沸后,添加适量的吡啶进行共沸,而获得目标物28A的粗产物(1.8g)。不再进一步进行精制,用于下一反应。
1
C
(28B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[7-[(2R)-2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基-3-[2-氰基乙氧基-(二异丙氨基)膦基]氧基丙氧基]庚氧基]-3,4-二乙酰氧基四氢吡喃-2-基]甲酯(化合物28B)的合成
[化114]
将步骤(28A)中合成的化合物28A的粗产物(1.8g)溶解于二氯甲烷(50mL)中。添加二异丙基乙基胺(1.5mL,8.4mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.51mL,2.3mmol),在室温下搅拌5.5小时。进而,添加2-氰基乙基二异丙基氯亚磷酰胺(0.51mL,2.3mmol),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯共沸后获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙腈=100:0-90:10,v/v)将其进行精制,而获得非晶状的目标物28B(1.5g,产率2步71%)。
1
(实施例103)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6517.10)。
(参考例29)
(29A)
(2R)-1-苄氧基-3-[2-[2-(2-羟基乙氧基)乙氧基]乙氧基]丙烷-2-醇(化合物29A)的合成
[化115]
将三乙二醇(6.9g,46mmol)、苄基(S)-(+)-缩水甘油醚(5.0g,30mmol)溶解于二氯甲烷(75mL)中,添加三氟化硼-二乙醚络合物(0.77mL,6.1mmol),在室温下搅拌16小时。反应结束后,添加饱和碳酸氢钠水溶液,并利用二氯甲烷进行萃取。利用饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(high performance liquid chromatography,高效液相色谱法)(GL Science,InertsilODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。将目标物溶解于二氯甲烷中,利用饱和食盐水、饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得油状的目标物29A(5.6g,产率58%)。
1
C
(29B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2R)-3-苄氧基-2-羟基-丙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物29B)的合成
[化116]
在步骤(29A)中合成的化合物29A(5.6g,18mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三乙酰氧基-四氢吡喃-2-基]甲酯(WO2011053614)(6.3g,16mmol)的二氯甲烷(100mL)悬浮溶液中添加三氟甲磺酸(0.24mL,2.8mmol),在45℃下搅拌16小时。反应结束后,添加饱和碳酸氢钠水溶液,并利用二氯甲烷进行萃取。利用饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GL Science,Inertsil ODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。将目标物溶解于乙酸乙酯中,利用饱和食盐水、饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得非晶状的目标物29B(4.9g,产率47%)。
1
C
(29C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2R)-2,3-二羟基丙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物29C)的合成
[化117]
将步骤(29B)中合成的化合物29B(4.9g,7.6mmol)溶解于乙醇(100mL)中。添加10%钯/碳(湿基)(4.0g),于氢气环境下在室温下剧烈搅拌8小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除。添加适量的甲苯进行共沸,而获得油状的目标物29C(4.0g,产率95%)。
1
C
(29D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基甲氧基]-2-羟基-丙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物29D)的合成
[化118]
在步骤(29C)中合成的化合物29C(4.0g,7.2mmol)中添加适量的吡啶,在减压下进行共沸。溶解于吡啶(30mL)中,添加4,4'-二甲氧基三苯氯甲烷(2.7g,8.0mmol),在室温下搅拌3小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯而共沸后,获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物29D(3.5g,产率57%)。
1
C
(29E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基丙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物29E)的合成
[化119]
将步骤(29D)中合成的化合物29D(3.5g,4.1mmol)通过适量的吡啶共沸后,溶解于二氯甲烷(40mL)中。添加N,N-二异丙基乙基胺(2.8mL,16mmol)、2-氰基乙基二异丙基氯亚磷酰胺(1.1mL,4.9mmol),在室温下搅拌3小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯共沸后获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物29E(2.2g,产率51%)。
1
(实施例104)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6839.16)。
(参考例30)
(30A)
(2R)-1-苄氧基-3-[2-[2-[2-(2-羟基乙氧基)乙氧基]乙氧基]乙氧基]丙烷-2-醇(化合物30A的合成)
[化120]
将四乙二醇(8.87g,45.7mmol)、苄基(S)-(+)-缩水甘油醚(4.66mL,30.5mmol)溶解于二氯甲烷(75mL)中,添加三氟化硼-二乙醚络合物(0.76mL,6.1mmol),在室温下整夜搅拌。反应结束后,添加饱和碳酸氢钠水溶液。利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GL Science,InertsilODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。将目标物溶解于二氯甲烷中,利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得油状的目标物30A(5.3g,产率49%)。
1
C
(30B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[2-[(2R)-3-苄氧基-2-羟基-丙氧基]乙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物30B的合成)
[化121]
在步骤30A中合成的化合物(5.3g,15mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三乙酰氧基-四氢吡喃-2-基]甲酯(WO2011053614)(5.2g,13mmol)的二氯甲烷(100mL)悬浮溶液中添加三氟甲磺酸(0.2mL,2.3mmol),在45℃下搅拌22小时。反应结束后,将溶剂减压蒸馏去除并进行浓缩,添加乙酸乙酯。利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GLScience,Inertsil ODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。将目标物溶解于二氯甲烷中,利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得非晶状的目标物30B(4.5g,产率49%)。
1
C
(30C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[2-[(2R)-2,3-二羟基丙氧基]乙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(30C的合成)
[化122]
将步骤(30B)中合成的化合物30B(4.5g,6.5mmol)溶解于乙醇(80mL)中。添加10%钯/碳(湿基)(4g),于氢气环境下在室温下剧烈搅拌8小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除而获得目标物30C(3.9g,产率定量)的粗产物。不再进一步进行精制,用于下一反应。
1
C
(30D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]乙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物30D的合成)
[化123]
在步骤(30C)中合成的化合物30C(3.9g,6.5mmol)添加适量的吡啶,在减压下进行共沸。溶解于吡啶(22mL)中,添加4,4'-二甲氧基三苯氯甲烷(4,4'-DimethoxytritylChloride)(2.4g,7.2mmol),在室温下搅拌2小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯而共沸后,获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物30D(3.12g,产率53%)。
1
C
(30E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]乙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物30E的合成)
[化124]
在步骤(30D)中合成的化合物30D(3.12g,3.47mmol)中添加适量的吡啶,在减压下共沸后溶解于二氯甲烷(35mL)中。添加N,N-二异丙基乙基胺(2.42mL,13.9mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.93mL,4.16mmol),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯,在减压下共沸后获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物30E(2.98g,产率78%)。
1
(实施例105)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6971.21)。
(参考例31)
(31A)
(2R)-1-苄氧基-3-[2-(2-羟基乙氧基)乙氧基]丙烷-2-醇(化合物31A的合成)
[化125]
将二乙二醇(5.0g,30.5mmol)、苄基(S)-(+)-缩水甘油醚(6.5,60.9mmol)溶解于二氯甲烷(75mL)中,添加三氟化硼-二乙醚络合物(0.76mL,6.1mmol),在室温下搅拌2小时。反应结束后,添加饱和碳酸氢钠水溶液。利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GL Science,InertsilODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。将目标物溶解于二氯甲烷中,利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得油状的目标物31A(5.3g,产率64%)。
1
C
(31B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[(2R)-3-苄氧基-2-羟基-丙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物31B的合成)
[化126]
在步骤31A中合成的化合物(4.0g,15mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三乙酰氧基-四氢吡喃-2-基]甲酯(WO2011053614)(5.2g,13mmol)的二氯甲烷(100mL)悬浮溶液中添加三氟甲磺酸(0.2mL,2.3mmol),在45℃下搅拌22小时。反应结束后,将溶剂减压蒸馏去除并进行浓缩,添加乙酸乙酯。利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GLScience,Inertsil ODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。将目标物溶解于二氯甲烷中,利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得非晶状的目标物31B(4.15g,产率52%)。
1
C
(31C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[(2R)-2,3-二羟基丙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物31C的合成)
[化127]
将步骤(31B)中合成的化合物31B(4.15g,6.9mmol)溶解于乙醇(80mL)中。添加10%钯/碳(湿基)(4g),于氢气环境下在室温下剧烈搅拌8小时。进而添加10%钯/碳(湿基)(1g),于氢气环境下在50℃下剧烈搅拌4小时。进而,添加10%钯/碳(湿基)(1g),于氢气环境下在50℃下剧烈搅拌8小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除而获得目标物31C(3.0g,产率86%)的粗产物。不再进一步进行精制,用于下一反应。
1
C
(31D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基甲氧基]-2-羟基-丙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物31D)的合成
[化128]
在步骤(31C)中合成的化合物31C(3.0g,5.9mmol)中添加适量的吡啶,在减压下进行共沸。溶解于吡啶(25mL)中,添加4,4'-二甲氧基三苯氯甲烷(2.2g,6.5mmol),在室温下搅拌2小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯而共沸后,获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物31D(1.7g,产率36%)。
1
C
(31E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基丙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物31E)的合成
[化129]
将步骤(31D)中合成的化合物31D(1.7g,2.1mmol)通过适量的吡啶共沸后,溶解于二氯甲烷(20mL)中。添加N,N-二异丙基乙基胺(1.5mL,8.4mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.56mL,2.5mmol),在室温下搅拌3小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯共沸后获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物31E(1.5g,产率71%)。
1
(实施例106)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6708.09)。
(参考例32)
(32A)
(2S)-1-苄氧基-3-[2-[2-(2-羟基乙氧基)乙氧基]乙氧基]丙烷-2-醇(化合物32A)的合成
[化130]
将三乙二醇(9.2g,61mmol)、苄基(R)-(-)-缩水甘油醚(5.0g,30mmol)溶解于二氯甲烷(75mL)中,添加三氟化硼-二乙醚络合物(0.77mL,6.1mmol),在室温下搅拌4小时。反应结束后,添加饱和碳酸氢钠水溶液,并利用二氯甲烷进行萃取。利用饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GLScience,Inertsil ODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。将目标物溶解于二氯甲烷中,利用饱和食盐水、饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得油状的目标物32A(6.4g,产率67%)。
1
C
(32B)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2S)-3-苄氧基-2-羟基-丙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物32B)的合成
[化131]
在步骤(32A)中合成的化合物32A(6.4g,20mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三乙酰氧基-四氢吡喃-2-基]甲酯(WO2011053614)(7.2g,18mmol)的二氯甲烷(200mL)悬浮溶液中添加三氟甲磺酸(0.28mL,3.1mmol),在45℃下搅拌20小时。反应结束后,添加饱和碳酸氢钠水溶液,并利用二氯甲烷进行萃取。利用饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GL Science,Inertsil ODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。将目标物溶解于乙酸乙酯中,利用饱和食盐水、饱和食盐水/饱和碳酸氢钠水溶液洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得非晶状的目标物32B(5.6g,产率47%)。
1
C
(32C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2S)-2,3-二羟基丙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物32C)的合成
[化132]
将步骤(32B)中合成的化合物32B(5.6g,8.7mmol)溶解于乙醇(100mL)中。添加20%氢氧化钯/碳(湿基)(1.5g),于氢气环境下在50℃下剧烈搅拌8小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除。添加适量的甲苯进行共沸,而获得油状的目标物32C(4.1g,产率85%)。
1
C
(32D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2R)-3-[双(4-甲氧基苯基)-苯基甲氧基]-2-羟基-丙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物32D)的合成
[化133]
在步骤(32C)中合成的化合物32C(4.1g,7.4mmol)中添加适量的吡啶,在减压下进行共沸。溶解于吡啶(30mL)中,添加4,4'-二甲氧基三苯氯甲烷(2.8g,8.2mmol),在室温下搅拌3小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯而共沸后,获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物32D(1.9g,产率30%)。
1
C
(32E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2R)-3-[双(4-甲氧基苯基)-苯基甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基丙氧基]乙氧基]乙氧基]乙氧基]四氢吡喃-2-基]甲酯(化合物32E)的合成
[化134]
将步骤(32D)中合成的化合物32D(1.9g,2.2mmol)通过适量的甲苯共沸后,溶解于二氯甲烷(22mL)中。添加N,N-二异丙基乙基胺(1.6mL,9.0mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.59mL,2.7mmol),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=50:50-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物32E(1.5g,产率64%)。
1
(实施例107)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6839.16)。
(实施例108)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7817.33)。
(实施例109)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:8796.73)。
(参考例33)
(33A)
N-[2-[2-[(2R)-3-苄氧基-2-羟基-丙氧基]乙氧基]乙基]氨基甲酸9H-芴-9-基甲酯(化合物33A)的合成
[化135]
将文献已知化合物的N-[2-(2-羟基乙氧基)乙基]氨基甲酸9H-芴-9-基甲酯(Bioconjugate Chemistry,2000,11,755-761)(3.11g,10mmol)与苄基(S)-(+)-缩水甘油醚(0.8g,5mmol)溶解于二氯甲烷(30mL)中,添加三氟化硼-二乙醚络合物(0.1mL,1mmol),在室温下搅拌16小时。反应结束后,添加饱和碳酸氢钠水溶液。利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=80:20-0:100,v/v)进行精制,而获得油状的目标物33A(0.74g,产率30%)。
1
C
(33B)
(2R)-1-[2-(2-氨基乙氧基)乙氧基]-3-苄氧基-丙烷-2-醇、三氟乙酸盐(化合物33B)的合成
[化136]
将步骤(33A)中合成的化合物33A(13.8g,28.1mmol)溶解于乙醇/四氢呋喃(50mL/50mL)中,添加1N氢氧化钠水溶液(100mL),在室温下搅拌2小时。反应结束后,将有机溶剂减压蒸馏去除,添加1N盐酸(100mL)。通过过滤将产生的固形物去除。利用乙酸乙酯对作为通过液的水层所含的副产物进行萃取,减压蒸馏去除后在所获得的残渣中添加乙腈并过滤。将通过液进行减压蒸馏去除而获得粗产物。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GL Science,Inertsil ODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除,而获得油状的目标物33B(7.1g,产率66%)。
1
(33C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2R)-3-苄氧基-2-羟基-丙氧基]乙氧基]乙基氨基]-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯(化合物33C)的合成
[化137]
将文献已知化合物的2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酸(WO2012037254)(6g,14.8mmol)、1-(-3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(3.4g,17.8mmol)、3H-1,2,3-三唑[4,5-b]吡啶-3-醇(2.0g,14.8mmol)、N,N-二异丙基乙基胺(9.0mL,51.8mmol)、步骤(33B)中合成的化合物33B(6.7g,17.5mmol)溶解于二氯甲烷(130mL)中,在室温下搅拌4小时。反应结束后,利用1N盐酸、饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:甲醇=100:0-90:10,v/v)进行精制,而获得非晶状的目标物33C(7.9g,产率81%)。
1
C
(33D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2R)-2,3-二羟基丙氧基]乙氧基]乙基氨基]-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯乙酸酯(化合物33D)的合成
[化138]
将步骤(33C)中合成的化合物33C(7.9g,12mmol)溶解于乙醇(80mL)中。添加20%氢氧化钯/碳(湿基)(2.0g),于氢气环境下在50℃下剧烈搅拌8小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除。获得非晶状的目标物33D(6.7g,产率98%)。
1
C
(33E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]乙氧基]乙基氨基]-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯(化合物33E)的合成
[化139]
在步骤(33D)中合成的化合物33D(6.8g,12mmol)中添加适量的吡啶,在减压下进行共沸。溶解于吡啶(40mL)中,添加4,4'-二甲氧基三苯氯甲烷(4.5g,13mmol),在室温下搅拌2小时。反应结束后,将溶剂减压蒸馏去除。添加适量的甲苯而共沸后,获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物33E(6.9g,产率66%)。
1
C
(33F)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]乙氧基]乙基氨基]-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯(化合物33F)的合成
[化140]
将步骤(33E)中合成的化合物33E(6.9g,7.9mmol)通过适量的甲苯共沸后,溶解于二氯甲烷(80mL)中。添加N,N-二异丙基乙基胺(5.5mL,32mmol)、2-氰基乙基二异丙基氯亚磷酰胺(2.1mL,9.5mmol),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物33F(6.6g,产率78%)。
1
(实施例110)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6878.11)。
(实施例111)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7882.43)。
(实施例112)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是在以下的HPLC条件下进行分析。分析管柱使用Clarity Oligo-MS C18(Phenomenex制造)(2.6μm,50×2.1mm),流动相A使用100mM六氟异丙醇、8mM三乙胺水溶液,流动相B使用甲醇,在流动相B于4分钟内自10%变化为25%的梯度、流速0.5mL/min、60℃下进行分析。化合物的HPLC保持时间为3.0分钟。
(参考例34)
(34A)
N-[(5S)-6-[2-[2-[(2R)-3-苄氧基-2-羟基-丙氧基]乙氧基]乙基氨基]-5-(叔丁氧基羰基氨基)-6-侧氧基-己基]氨基甲酸叔丁酯(化合物34A)的合成
[化141]
将(2S)-2,6-双(叔丁氧基羰基氨基)己酸(1.7g,4.9mmol)、1-(-3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(1.3g,6.9mmol)、3H-1,2,3-三唑[4,5-b]吡啶-3-醇(0.67g,4.9mmol)、N,N-二异丙基乙基胺(3.0mL,17mmol)、步骤(33B)中合成的化合物33B(2.1g,5.4mmol)溶解于二氯甲烷(40mL)中,在室温下搅拌4小时。进而,添加1-(-3-二甲氨基丙基)-3-乙基碳二酰胺盐酸盐(1.3g,6.9mmol)、N,N-二异丙基乙基胺(1.0ml,5.7mmol)并整夜搅拌。反应结束后,利用1N盐酸、饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得含有化合物34A的粗产物(3.1g)。不再进一步进行精制,将全部量用于下一反应中。
C
(34B)
(2S)-2,6-二氨基-N-[2-[2-[(2R)-3-苄氧基-2-羟基-丙氧基]乙氧基]乙基]己酰胺TFA盐(trifluoroacetate,三氟乙酸盐)(化合物34B)的合成
[化142]
将步骤(34A)中合成的含有化合物34A的粗产物(3.1g)溶解于二氯甲烷(24mL)/三氟乙酸(6mL)中,在室温下搅拌90分钟。反应结束后,将溶剂减压蒸馏去除,添加适量的二氯甲烷,再次将溶剂减压蒸馏去除。继而,添加适量的乙腈/蒸馏水(4/1),再次将溶剂减压蒸馏去除。重复所述操作直至可去除过量的三氟乙酸为止。获得含有化合物34B的粗产物(3.8g)。不再进一步进行精制,将全部量用于下一反应中。
C
(34C)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[2-[[(5S)-5-[[2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酰基]氨基]-6-[2-[2-[(2R)-3-苄氧基-2-羟基-丙氧基]乙氧基]乙基氨基]-6-侧氧基-己基]氨基]-2-侧氧基-乙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物34C)的合成
[化143]
将文献已知化合物的2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酸(WO2012037254)(3.5g,8.6mmol)、1-(-3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(2.3g,12mmol)、3H-1,2,3-三唑[4,5-b]吡啶-3-醇(1.3g,9.6mmol)、N,N-二异丙基乙基胺(10mL,59mmol)、步骤(34B)中合成的含有化合物34B的粗产物溶解于二氯甲烷(80mL)中,在室温下整夜搅拌。反应结束后,利用1N盐酸、饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:甲醇=100:0-70:30,v/v)进行精制,而获得非晶状的目标物34C(3.5g,产率62%(3步))。
1
C
(34D)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[2-[[(5S)-5-[[2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酰基]氨基]-6-[2-[2-[(2R)-2,3-二羟基丙氧基]乙氧基]乙基氨基]-6-侧氧基-己基]氨基]-2-侧氧基-乙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物34D)的合成
[化144]
将步骤(34C)中合成的化合物34C(3.5g,3.0mmol)溶解于乙醇(40mL)中。添加20%氢氧化钯/碳(湿基)(1.2g),于氢气环境下在50℃下剧烈搅拌8小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除。获得非晶状的目标物34D(3.14g,产率97%)。
1
C
(34E)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[2-[[(5S)-5-[[2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酰基]氨基]-6-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]乙氧基]乙基氨基]-6-侧氧基-己基]氨基]-2-侧氧基-乙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物34E)的合成
[化145]
在步骤(34D)中合成的化合物34D(3.14g,2.9mmol)中添加适量的吡啶,在减压下进行共沸。溶解于吡啶(10ml)中,添加4,4'-二甲氧基三苯氯甲烷(1.1g,3.2mmol),在室温下搅拌2小时。反应结束后,添加甲苯(20mL)、乙醇(1mL)、N,N-二异丙基乙基胺(1mL),将溶剂进行减压蒸馏去除后,获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物34E(2.5g,产率62%)。
1
C
(34F)
乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[2-[[(5S)-5-[[2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酰基]氨基]-6-[2-[2-[(2S)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]乙氧基]乙基氨基]-6-侧氧基-己基]氨基]-2-侧氧基-乙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物34F)的合成
[化146]
将步骤(34E)中合成的化合物34E(2.5g,1.8mmol)通过适量的甲苯/二氯甲烷(3/1)共沸后,溶解于二氯甲烷(18mL)中。添加N,N-二异丙基乙基胺(1.3mL,7.2mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.48mL,2.2mmol),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物34F(2.1g,产率73%)。
1
(实施例113)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6263.04)。
(实施例114)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7154.36)。
(实施例115)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:8046.64)。
(实施例116~127)封入了寡核苷酸的核酸脂质粒子的制备
将二硬脂酰基磷脂酰胆碱(1,2-二硬脂酰基-sn-甘油-3-磷酸胆碱(1,2-Distearoyl-sn-glycero-3-phosphocholine):以下表述为DSPC,NOF CORPORATION)、胆固醇(Cholesterol:以下表述为Chol,Sigma-Aldrich,Inc.)、国际公开第2015/005253所记载的实施例8的化合物(设为LP)、及N-[甲氧基聚(乙二醇)2000]胺甲酰基]-1,2-二肉豆蔻氧基丙基-3-胺(以下表述为PEG-C-DMA,NOF CORPORATION)以DSPC:Chol:LP:PEG-C-DMA=10:48:40:2的摩尔比而以在乙醇中总脂质浓度成为5mM的方式制备脂质溶液。
将寡核苷酸(21e_002、18e_005、21m_002、18e_005、18m_022、15e_001、15ed_001、18e_008、18e_025、18m_008、15e_002、及15ed_002)分别溶解于柠檬酸缓冲液(20mM,柠檬酸盐缓冲液(Citrate Buffer),pH值4.0)中,来制备寡核苷酸溶液。
将所述脂质溶液与寡核苷酸溶液以来自LP的氮原子(N)与来自寡核苷酸的磷原子(P)的比率(N/P)成为3的方式,使用NanoAssemblr Benchtop(注册商标。PrecisionNanosystems),在附属的微流路筒内加以混合(流速:12mL/min,混合体积比率:脂质溶液:寡核苷酸溶液=1:3,温度:室温),而获得核酸脂质粒子的分散液。继而,利用约2L的磷酸缓冲液(pH值7.4)将核酸脂质粒子的分散液透析12-18小时(Float-A-Lyzer G2,MWCO:1000kD,Spectra/Por),由此去除乙醇及进行中和,而获得封入了寡核苷酸的核酸脂质粒子被精制过的分散液。为了调整浓度,可适当通过超过滤(Amicon-Ultra,MWCO:100kD,MILLIPORE)将核酸脂质粒子的分散液浓缩。
对实施例116~127的封入寡核苷酸的核酸脂质粒子的特性进行评价。以下对各特性评价的方法进行说明。
(1)寡核苷酸的封入率
寡核苷酸的封入率是使用Quant-iT RiboGreen RNA分析套组(Invitrogen),依照随附文件进行测定。即,在0.015%Triton X-100表面活性剂存在下及非存在下,对核酸脂质粒子的分散液中的寡核苷酸进行定量,通过下式算出封入率。
{[表面活性剂存在下的寡核苷酸量]-[表面活性剂非存在下的寡核苷酸量]}/[表面活性剂存在下的寡核苷酸量]×100(%)
(2)寡核苷酸与脂质的比率
使用磷脂质C-Test Wako(和光纯药工业股份有限公司),依照随附文件测定核酸脂质粒子的分散液中的磷脂质量。即,在1%Triton X-100表面活性剂存在下,对试样中的磷脂质进行定量。
通过反相色谱法测定核酸脂质粒子的分散液中的胆固醇及LP量(系统:Agilent1100系列,管柱:Chromolith Performance RP-18封端100-3整体化HPLC-管柱(Merck),缓冲液A:0.01%三氟乙酸,缓冲液B:0.01%三氟乙酸,甲醇,梯度(B%):82-97%(0-17min),流速:2mL/min,温度:50℃,检测:205nm)。
根据磷脂质量、胆固醇量、及LP量与构成脂质体的脂质成分的组成比算出总脂质量,根据所述(1)的“表面活性剂存在下的寡核苷酸量”,通过下式算出寡核苷酸与脂质的比率。
[表面活性剂存在下的寡核苷酸浓度]/[总脂质浓度](wt/wt)
(3)平均粒径
脂质体的粒径是通过ζ电位/粒子尺寸NICOMPTM 380ZLS(PARTICLE SIZINGSYSTEMS)进行测定。表中的平均粒径表示体积平均粒径,±以下表示偏差。
将结果示于表11。
(表11)
*ASO/脂质(wt/wt):寡核苷酸与脂质的重量比。
根据以上的结果可明确,寡核苷酸被封入脂质粒子内,这些核酸脂质粒子具有约50nm至约70nm的平均粒径。
(实施例128)
HO-X
使用所述序列即15e_005代替实施例113所使用的序列,与实施例113同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6210.94)。
(实施例129)
HO-X
使用所述序列即15e_006代替实施例113所使用的序列,与实施例113同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第90号至第104号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6201.95)。
(实施例130)
HO-X
使用所述序列即15e_002代替实施例113所使用的序列,与实施例113同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第89号至第103号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6268.98)。
(参考例35)
(35A)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[3-(叔丁氧基羰基氨基)丙基氨基]-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯(化合物35A)的合成
[化147]
在文献已知化合物的2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酸(WO2012037254)(10g,24.7mmol)、N-(3-氨基丙基)氨基甲酸叔丁酯(4.7ml,27.1mmol)的N,N-二甲基甲酰胺(70ml)溶液中添加1-[双(二甲氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶3-氧化物六氟磷酸盐(11.3g,29.6mmol)与N,N-二异丙基乙基胺(8.6mL,49.3mmol),在室温下搅拌40分钟。反应结束后,添加乙酸乙酯,利用15%食盐水、0.5N盐酸、饱和食盐水、饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。将溶剂减压蒸馏去除,获得含有表述目标物的粗产物约14g。不再进一步进行精制,用于下一反应。
(35B)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-(3-氨基丙基氨基)-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯TFA盐(化合物35B)的合成
[化148]
将步骤(35A)中合成的粗产物(14g)溶解于二氯甲烷(40ml)中,至反应结束为止,添加三氟乙酸共计16ml。反应结束后,将溶剂减压蒸馏去除而获得粗产物。添加适量的二乙醚,进行倾析。添加适量的乙腈,进行减压蒸馏去除。进而,添加适量的二氯甲烷,进行减压蒸馏去除。获得含有表述目标物的粗产物约15.9g。不再进一步进行精制,用于下一反应。
C
(35C)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[3-[[(4R)-2,2-二甲基-1,3-二氧杂环戊烷-4-基]甲氧基羰基氨基]丙基氨基]-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯(化合物35C)的合成
[化149]
将(S)-(+)-1,2-亚异丙基甘油(6.53g,49.4mmol)、氯甲酸4-硝基苯酯(9.96g,49.4mmol)溶解于二氯甲烷(200ml)中,添加吡啶(7.95ml,98.8mmol),在室温下搅拌30分钟。在该反应液中添加步骤(35B)中合成的粗产物(15.9g)的二氯甲烷(100ml)溶液、三乙胺(20.5ml,148mmol),在室温下整夜搅拌。反应结束后,利用0.5N盐酸、饱和食盐水、饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:甲醇=100:0-65:35,v/v)进行精制,而以固体的形式获得目标物35C(9.0g,产率59%(3步))。
1
C
(35D)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[3-[[(2R)-2,3-二羟基丙氧基]羰基氨基]丙基氨基]-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯(化合物35D)的合成
[化150]
在步骤(35C)中合成的化合物(7.87g,12.7mmol)的甲醇溶液(160ml)中添加对甲苯磺酸(0.242g,1.27mmol),进行整夜搅拌。反应结束后,将溶剂减压蒸馏去除,而获得含有表述目标物的粗产物7.36g。不再进一步进行精制,用于下一反应。
C
(35E)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[3-[[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]羰基氨基]丙基氨基]-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯(化合物35E)的合成
[化151]
在步骤(35D)中合成的粗产物(7.36g)中添加二氯甲烷(150ml)、吡啶(10ml),将溶剂减压蒸馏去除。其后,溶解于吡啶(60ml)中,添加4,4'-二甲氧基三苯氯甲烷(5.59g,16.5mmol),在室温下搅拌30分钟。反应结束后,添加甲醇(1.03ml,25.4mmol)、N,N-二异丙基乙基胺(4.42mL,25.4mmol),将溶剂减压蒸馏去除。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-0:100,v/v,含有1%三乙胺)将其进行精制,而以固体的形式获得目标物35E(4.51g,产率40%)。
1
C
(35F)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-[2-[3-[[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]羰基氨基]丙基氨基]-2-侧氧基-乙氧基]四氢吡喃-2-基]甲酯(化合物35F)的合成
[化152]
将步骤(35E)中合成的化合物35E(4.51g,5.11mmol)通过适量的乙酸乙酯/甲苯(1/4)共沸后,溶解于二氯甲烷(90ml)中。添加N,N-二异丙基乙基胺(3.56ml,20.5mmol)、2-氰基乙基二异丙基氯亚磷酰胺(1.57ml,5.63mmol),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-50:50,v/v,含有1%三乙胺)将其进行精制,而以固体的形式获得目标物35F(3.97g,产率72%)。
1
(参考例36)
(36A)N-[2-(叔丁氧基羰基氨基)乙基]氨基甲酸[(4R)-2,2-二甲基-1,3-二氧杂环戊烷-4-基]甲酯(化合物36A)的合成
[化153]
将(S)-(+)-1,2-亚异丙基甘油(5.0g,37.5mmol)、氯甲酸4-硝基苯酯(7.55g,37.5mmol)溶解于二氯甲烷(150ml)中,添加吡啶(5.0ml,61.8mmol),在室温下搅拌1小时。在该反应液中添加N-(2-氨基乙基)氨基甲酸叔丁酯(6g,37.5mmol)的二氯甲烷溶液(40ml)、三乙胺(8.8ml,63.7mmol),在室温下搅拌1小时。反应结束后,反应结束后,利用0.5N盐酸、饱和食盐水、饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而以固体的形式获得含有目标物的粗产物(10.6g)。不再进一步进行精制,用于下一反应。
(36B)N-(2-氨基乙基)氨基甲酸[(2R)-2,3-二羟基丙基]酯TFA盐(化合物36B)的合成
[化154]
在步骤(36A)中合成的粗产物(10.6g)中添加三氟乙酸(16.6ml)、纯水(5.55ml),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除。继而,添加乙腈,将溶剂减压蒸馏去除。进而,添加乙腈/纯水(4/1),将溶剂减压蒸馏去除后,在减压下加以干燥。获得含有目标物的油状的粗产物(14.3g)。不再进一步进行精制,用于下一反应。
(36C)N-[(5S)-5-(苄氧基羰基氨基)-6-[2-[[(2R)-2,3-二羟基丙氧基]羰基氨基]乙基氨基]-6-侧氧基-己基]氨基甲酸苄酯(化合物36C)的合成
[化155]
在N,N'-二-Cbz-L-赖氨酸(13.7g)的N,N-二甲基甲酰胺(100ml)溶液中添加1-[双(二甲氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶3-氧化物六氟磷酸盐(15.0g,40mmol)与N,N-二异丙基乙基胺(12mL,66mmol)。进而,添加步骤(36B)中合成的粗产物(9.7g)与N,N-二异丙基乙基胺(12mL,66mmol)的N,N-二甲基甲酰胺(20ml)溶液,在室温下搅拌40分钟。反应结束后,添加乙酸乙酯,利用15%食盐水、0.5N盐酸、饱和食盐水、饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得固形物。利用1N盐酸、纯水将该固形物洗净后,利用二氯甲烷、二乙醚洗净,其后在减压下加以干燥,而获得含有目标物的粗产物(10.5g)。
C
(36D)N-[2-[[(2S)-2,6-二氨基己酰基]氨基]乙基]氨基甲酸[(2R)-2,3-二羟基丙基]酯盐酸盐(化合物36D)的合成
[化156]
将步骤(36C)中合成的粗产物(9.5g)溶解于四氢呋喃/1当量浓度的盐酸(36ml)中,添加10%钯碳(2.0g),于氢气环境下在室温下搅拌2.5小时。反应结束后进行过滤。将溶剂进行减压蒸馏去除后,利用高速浓缩装置V-10(Biotage)在减压下加以干燥。获得表述目标化合物的粗产物(6.4g)。不再进一步进行精制,用于下一反应。
(36E)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[2-[[(5S)-5-[[2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酰基]氨基]-6-[2-[[(2R)-2,3-二羟基丙氧基]羰基氨基]乙基氨基]-6-侧氧基-己基]氨基]-2-侧氧基-乙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物36E)的合成
[化157]
在文献已知化合物的2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酸(WO2012037254)(2.64g,6.5mmol)、步骤(36D)中合成的粗产物(1.37g,3.6mmol)的N,N-二甲基甲酰胺(30ml)溶液中添加1-[双(二甲氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶3-氧化物六氟磷酸盐(3.0g,8.0mmol)与N,N-二异丙基乙基胺(3.8mL,21.7mmol),在室温下搅拌40分钟。反应结束后,添加二乙醚(500ml),获得沉淀的残渣。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GL Science,Inertsil ODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。获得非晶状的目标物(2.23g,产率57%)。
1
C
(36F)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[2-[[(5S)-5-[[2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酰基]氨基]-6-[2-[[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]羰基氨基]乙基氨基]-6-侧氧基-己基]氨基]-2-侧氧基-乙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物36F)的合成
[化158]
在步骤(36E)中合成的化合物(7.36g,2.1mmol)中添加吡啶(10ml),将溶剂减压蒸馏去除。将该操作重复3次。其后,溶解于吡啶(10ml)中,添加4,4'-二甲氧基三苯氯甲烷(0.77g,2.3mmol),在室温下搅拌30分钟。反应结束后,添加乙醇(1.0ml)、N,N-二异丙基乙基胺(1.0mL),将溶剂减压蒸馏去除。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物(1.17g,产率41%)。
1
C
(36G)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[2-[[(5S)-5-[[2-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基乙酰基]氨基]-6-[2-[[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙基-(二异丙氨基)膦基]氧基-丙氧基]羰基氨基]乙基氨基]-6-侧氧基-己基]氨基]-2-侧氧基-乙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物36G)的合成
[化159]
将步骤(36F)中合成的化合物36F(1.17g,0.85mmol)通过适量的二氯甲烷/甲苯(1/4)共沸后,溶解于二氯甲烷(8ml)中。添加N,N-二异丙基乙基胺(0.59ml,3.4mmol)、2-氰基乙基二异丙基氯亚磷酰胺(0.23ml,1.0mmol),在室温下搅拌1小时。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:乙酸乙酯/甲醇(5/1)=100:0-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物36G(0.81g,产率61%)。
1
(参考例37)
(37A)乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4-二乙酰氧基-6-[3-(苄氧基羰基氨基)丙氧基]四氢吡喃-2-基]甲酯(化合物37A)的合成
[化160]
在N-(3-羟基丙基)氨基甲酸苄酯(6.4g,30.8mmol)与文献已知化合物的乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4,6-三乙酰氧基-四氢吡喃-2-基]甲酯(WO2011053614)(10g,25.7mmol)的二氯甲烷(200ml)悬浮溶液中添加三氟甲磺酸(450μl,5.1mmol),在45度下搅拌4小时。反应结束后,将溶剂减压蒸馏去除一半进行浓缩。利用饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除而获得粗产物。添加异丙醚(100m)并搅拌3小时。将所获得的固形物进行过滤,而获得目标物37A(11.9g)。不再进一步进行精制,用于下一反应。
1
C
(37B)乙酸[(2R,3R,4R,5R)-5-乙酰胺-3,4-二乙酰氧基-6-(3-氨基丙氧基)四氢吡喃-2-基]甲酯盐酸盐(化合物37B)的合成
[化161]
将步骤(37A)中合成的化合物37A(10g,18.6mmol)溶解于四氢呋喃(200ml)/1N盐酸(20ml)中。添加10%钯/碳(湿基)(1g),于氢气环境下在室温下剧烈搅拌1小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除而获得含有目标物37B的粗产物(8.97g)。不再进一步进行精制,用于下一反应。
C
(37C)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[(3S)-4-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基丙基酰胺]-3-(9H-芴-9-基甲氧基羰基氨基)-4-侧氧基-丁酰基]氨基]丙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物37C)的合成
[化162]
在N-[(9H-芴-9-基甲氧基)羰基]-L-天冬氨酸(1.7g,4.78mmol)、步骤(37B)中合成的化合物(5.48g,12.4mmol)的N,N-二甲基甲酰胺(15ml)溶液中添加1-[双(二甲氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶3-氧化物六氟磷酸盐(4.7g,12.4mmol)与N,N-二异丙基乙基胺(5.8mL,33.5mmol),在室温下搅拌1小时。添加乙酸乙酯,利用15%食盐水、0.5N盐酸、饱和食盐水、饱和碳酸氢钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得固形物。利用异丙醚将该固形物洗净后,在减压下加以干燥,而获得含有目标物37C的粗产物(5.4g)。不再进一步进行精制,用于下一反应。
C
(37D)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[(3S)-4-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基丙基氨基]-3-氨基-4-侧氧基-丁酰基]氨基]丙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯TFA盐(化合物37D)的合成
[化163]
在步骤(37C)中合成的化合物(5.4g,4.8mmol)的N,N-二甲基甲酰胺(25ml)溶液中添加哌啶(0.57ml,5.7mmol),在室温下搅拌1.5小时。反应结束后,添加三氟乙酸(0.31ml)。将反应液添加到适量的二乙醚中,获得沉淀的残渣。通过以0.1%三氟乙酸水溶液与0.1%三氟乙酸乙腈溶液作为洗脱液的反相HPLC(GL Science,Inertsil ODS-3)进行分离精制,将含有目标物的组分汇集,将溶剂减压蒸馏去除。获得非晶状的目标物(2.7g,产率55%)。
1
C
(37E)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[(3S)-4-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基丙基氨基]-3-[[(4R)-2,2-二甲基-1,3-二氧杂环戊烷-4-基]甲氧基羰基氨基]-4-侧氧基-丁酰基]氨基]丙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物37E)的合成
[化164]
使用步骤(37D)中合成的化合物(2.7g,2.6mmol),以与步骤(35C)同样的方式,获得非晶状的目标物(0.77g,产率27%)。
1
C
(37F)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[(3S)-4-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基丙基氨基]-3-[[(2R)-2,3-二羟基丙氧基]羰基氨基]-4-侧氧基-丁酰基]氨基]丙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物37F)的合成
[化165]
/>
使用步骤(37E)中合成的化合物(0.77g,0.72mmol),以与步骤(35D)同样的方式,获得含有目标物37F的粗产物(0.74g)。不再进一步进行精制,用于下一反应。
C
(37G)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[(3S)-4-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基丙基氨基]-3-[[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-羟基-丙氧基]羰基氨基]-4-侧氧基-丁酰基]氨基]丙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物37G)的合成
[化166]
使用步骤(37F)中合成的化合物(0.74g),以与步骤(35E)同样的方式,获得非晶状的目标物(0.63g,产率66%(2步))。
1
C
(37H)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[(3S)-4-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基丙基氨基]-3-[[(2R)-3-[双(4-甲氧基苯基)-苯基-甲氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]羰基氨基]-4-侧氧基-丁酰基]氨基]丙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物37H)的合成
[化167]
使用步骤(37G)中合成的化合物(0.63g,0.48mmol),以与步骤(35F)同样的方式,获得非晶状的目标物(0.52g,产率71%)。
1
(参考例38)
(38A)N-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二羟基-6-(羟甲基)四氢吡喃-2-基]氧基丙基]氨基甲酸苄酯(化合物38A)的合成
[化168]
在步骤(37A)中合成的化合物(12.4g,23mmol)的乙醇(200ml)悬浮溶液中添加28%甲醇钠甲醇溶液(0.85ml),在室温下加以搅拌。伴随反应的进行,一度成为透明的溶液。放置60分钟后获得沉淀物。将沉淀物过滤,利用乙醇、二异丙醚洗净后,在减压下加以干燥。以固体的形式获得含有目标物38A的粗产物(10g)。不再进一步进行精制,用于下一反应。
1
C
(38B)N-[(2R,3R,4R,5R,6R)-2-(3-氨基丙氧基)-4,5-二羟基-6-(羟甲基)四氢吡喃-3-基]乙酰胺盐酸盐(化合物38B)的合成
[化169]
将步骤(38A)中合成的化合物(22.8g,55.3mmol)溶解于四氢呋喃(280ml)/1N盐酸(61ml)中。添加10%钯/碳(湿基)(5.56g),于氢气环境下在室温下剧烈搅拌3.5小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除。添加适量的乙醇,将所产生的固形物过滤,利用乙醇、二乙醚洗净后,在减压下加以干燥。获得含有目标物38B的粗产物(17g)。不再进一步进行精制,用于下一反应。
1
(38C)N-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二羟基-6-(羟甲基)四氢吡喃-2-基]氧基丙基]氨基甲酸9H-芴-9-基甲酯(化合物38C)的合成
[化170]
将步骤(38B)中合成的化合物(16.4g,52.1mmol)溶解于四氢呋喃(200ml)/纯水(50ml)中,添加N,N-二异丙基乙基胺(12.7mL,72.9mmol)。在冰浴冷却下添加N-[(9H-芴-9-基甲氧基)羰氧基]丁二酰亚胺(21.1g,62.5mmol),在室温下搅拌1.5小时。反应结束后,将溶剂减压蒸馏去除。添加二乙醚(100ml),并搅拌1小时。利用乙醇、乙酸乙酯、二乙醚依序洗净固形物后,在减压下加以干燥,而获得含有目标物38C的粗产物(22.9g)。
1
C
(38D)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-4-乙酰氧基-2-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]-6-[3-(9H-芴-9-基甲氧基羰基氨基)丙氧基]四氢吡喃-3-基]酯(化合物38D)的合成
[化171]
在步骤(38C)中合成的化合物(1.56g,3.1mmol)的吡啶(20ml)悬浮液中添加4,4'-二甲氧基三苯氯甲烷(1.27g,3.7mmol),在室温下搅拌30分钟。继而,添加乙酸酐(1.18ml,12.5mmol),在室温下搅拌20小时。反应结束后,添加N,N-二异丙基乙基胺(1.1mL,6.2mmol)、乙醇(2ml),将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=75:25-0:100,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物38D(2.8g,定量)。
1
C
(38E)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-4-乙酰氧基-6-(3-氨基丙氧基)-2-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-3-基]酯(化合物38D)的合成
[化172]
在步骤(38D)中合成的化合物(2.49g,2.8mmol)的二氯甲烷(8ml)溶液中添加哌啶(0.45ml,4.5mmol),在室温下加以搅拌。如果反应不完全,就适当追加哌啶。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过NH-硅胶柱色谱法(二氯甲烷:甲醇=100:0-85:15,v/v)将其进行精制,而获得非晶状的目标物38E(1.48g,产率79%)。
1
C
(参考例39)
(39A)2-[[2-苄氧基-3-[(2-苄氧基-2-侧氧基-乙基)胺甲酰氧基]丙氧基]羰基氨基]乙酸苄酯(化合物39A)的合成
[化173]
将2-苄氧基-1,3-丙二醇(2.5g,14mmol)、氯甲酸4-硝基苯酯(5.8g,29mmol)溶解于四氢呋喃(100ml)中,添加吡啶(5.0ml,61.8mmol),在室温下搅拌20分钟。将所析出的固体过滤后,添加四氢呋喃(70ml)。继而,添加甘氨酸苄酯TFA盐(9.2g,33mmol)与N,N-二异丙基乙基胺(9.6mL,55mmol),在室温下整夜搅拌。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(己烷:乙酸乙酯=100:0-40:60,v/v)将其进行精制,而获得橡胶状的目标物39A(6.5g,产率84%)。
1
C
(39B)2-[[3-(羧甲基胺甲酰氧基)-2-羟基-丙氧基]羰基氨基]乙酸(化合物39B)的合成
[化174]
将步骤(39A)中合成的化合物39A(7.3g,2.8mmol)溶解于四氢呋喃(200ml)/纯水(50ml)中。添加20%氢氧化钯-活性碳(约50%含水)(3g),于氢气环境下在室温下剧烈搅拌5小时。反应结束后过滤,将所获得的通过液进行减压蒸馏去除而获得含有目标物39B的粗产物(3.2g)。不再进一步进行精制,用于下一反应。
1
(39C)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[2-[[3-[[2-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-[[双(4-甲氧基苯基)-苯基l-甲氧基]甲基]四氢吡喃-2-基]氧基丙基氨基]-2-侧氧基-乙基]胺甲酰氧基]-2-羟基-丙氧基]羰基氨基]乙酰基]氨基]丙氧基]-4-乙酰氧基-2-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-3-基]酯(化合物39C)的合成
[化175]
在步骤(39B)中合成的化合物39B(0.31g,1.05mmol)、步骤(38E)中合成的化合物38E(1.47g,2.21mmol)、N,N-二异丙基乙基胺(0.91mL,5.27mmol)的N,N-二甲基甲酰胺(9ml)溶液中添加1-[双(二甲氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶3-氧化物六氟磷酸盐(11.3g,29.6mmol),在室温下搅拌3小时。反应结束后,将反应液滴加至二乙醚(共计185ml)中。将上清液的溶剂去除后,通过硅胶柱色谱法(二氯甲烷:甲醇=100:0-15:85,v/v,含有1%三乙胺)将所获得的凝胶状的粗产物进行精制,而以固体的形式获得目标物39C(1.05g,产率63%)。
1
C
(39D)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[2-[[3-[[2-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-2-基]氧基丙基氨基]-2-侧氧基-乙基]胺甲酰氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]羰基氨基]乙酰基]氨基]丙氧基]-4-乙酰氧基-2-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-3-基]酯(化合物39D)的合成
[化176]
将步骤(39C)中合成的化合物39C(0.47g,0.30mmol)通过适量的乙酸乙酯/甲苯(1/1)共沸后,溶解于二氯甲烷(3ml)中。添加N,N-二异丙基乙基胺(210μl,1.18mmol)、2-氰基乙基二异丙基氯亚磷酰胺(79μl,0.36mmol),在室温下搅拌1小时。进而,添加2-氰基乙基二异丙基氯亚磷酰胺(20μl,0.09mmol),在室温下搅拌15分钟。反应结束后,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:甲醇=100:0-85:15,v/v,含有1%三乙胺)将其进行精制,而获得非晶状的目标物39D(0.34g,产率64%)。
1
(40A)2-[[3-苄氧基-2-[(2-苄氧基-2-侧氧基-乙基)胺甲酰氧基]丙氧基]羰基氨基]乙酸苄酯(化合物40A)的合成
[化177]
使用(+/-)-3-苄氧基-1,2-丙二醇(1.0g,5.5mmol)代替2-苄氧基-1,3-丙二醇,以与步骤(39A)同样的方式,获得目标物40A(2.8g,产率90%)。
1
C
(40B)2-[[2-(羧甲基胺甲酰氧基)-3-羟基-丙氧基]羰基氨基]乙酸(化合物40B)的合成
[化178]
使用化合物40A(2.8g,5.0mmol)代替化合物39A,以与步骤(39B)同样的方式,获得含有目标物40B的粗产物(1.2g)。
1
(40C)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[2-[[2-[[2-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-2-基]氧基丙基氨基]-2-侧氧基-乙基]胺甲酰氧基]-3-羟基-丙氧基]羰基氨基]乙酰基]氨基]丙氧基]-4-乙酰氧基-2-[[双(4-甲氧基苯基)-苯基l-甲氧基]甲基]四氢吡喃-3-基]酯(化合物40C)的合成
[化179]
/>
使用化合物40B(0.31g,1.05mmol)代替化合物39B,以与步骤(39C)同样的方式,获得目标物40C(0.74g,产率44%)。
1
C
(40D)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[[2-[[2-[[2-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-2-基]氧基丙基氨基]-2-侧氧基-乙基]胺甲酰氧基]-3-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]羰基氨基]乙酰基]氨基]丙氧基]-4-乙酰氧基-2-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-3-基]酯(化合物40D)的合成
[化180]
使用化合物40C(0.31g,1.05mmol)代替化合物39C,以与步骤(39D)同样的方式,获得目标物40D(0.60g,产率72%)。
1
(实施例131)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6261.97)
(实施例132)
HO-X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6917.09)
(实施例133)
X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6247.96)
(实施例134)
HO-X
使用所述序列即15e_005.1代替实施例132所使用的序列,与实施例132同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6919.09)
(实施例135)
HO-X
使用所述序列即15e_006.1代替实施例132所使用的序列,与实施例132同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第90号至第104号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6884.07)
(实施例136)
HO-X
使用所述序列即16e_001代替实施例132所使用的序列,与实施例132同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第107号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7276.10)
(实施例137)
HO-X
使用所述序列即17e_001代替实施例132所使用的序列,与实施例132同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第108号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:7636.19)
(实施例138)
X
使用所述序列即15e_005.1代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6250.00)
(实施例139)
X
使用所述序列即15e_006.1代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第90号至第104号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6214.96)
(实施例140)
X
使用所述序列即16e_001代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第107号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6607.02)
(实施例141)
X
使用所述序列即16e_002代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6609.03)
(实施例142)
X
使用所述序列即16e_003代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第90号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6586.00)
(实施例143)
X
使用所述序列即15e_001.5代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6233.97)
(实施例144)
X
使用所述序列即15e_005.5代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6235.97)
(实施例145)
X
使用所述序列即15e_006.5代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第90号至第104号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6200.94)
(实施例146)
X
使用所述序列即16e_001.5代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第107号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6593.02)
(实施例147)
X
使用所述序列即16e_002.5代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6595.01)
(实施例148)
X
使用所述序列即16e_003.5代替实施例133所使用的序列,与实施例133同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第90号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6571.98)
(实施例149)
HO-X
使用所述序列即15e_001.5代替实施例131所使用的序列,与实施例131同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6247.99)
(实施例150)
X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6247.99)
(实施例151)
X
将X的部分置换为X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6204.97)
(参考例41)
(41A)3-[[2-苄氧基-3-[(3-苄氧基-3-侧氧基-丙基)胺甲酰氧基]丙氧基]羰基氨基]丙酸苄酯(化合物41A)的合成
[化181]
使用3-氨基丙酸苄酯TFA盐(2.4g,8.17mmol)代替步骤(39A)所使用的甘氨酸苄酯TFA盐,以与步骤(39A)同样的方式,获得目标物41A(1.62g,产率80%)。
1
C
(41B)3-[[3-(2-羧基乙基胺甲酰氧基)-2-羟基-丙氧基]羰基氨基]丙酸(化合物41B)的合成
[化182]
使用化合物41A(1.62g,2.73mmol)代替步骤(39B)所使用的化合物39A,以与步骤(39B)同样的方式,获得目标物41B(0.89g,定量)。
1
(41C)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[3-[[3-[[3-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-2-基]氧基丙基氨基]-3-侧氧基-丙基]胺甲酰氧基]-2-羟基-丙氧基]羰基氨基]丙酰基氨基]丙氧基]-4-乙酰氧基-2-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-3-基]酯(化合物41C)的合成
[化183]
使用化合物41B(300mg,0.93mmol)代替步骤(39C)所使用的化合物39B,以与步骤(39C)同样的方式,获得目标物41C(950mg,产率63%)。
1
C
(41D)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[3-[[3-[[3-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-2-基]氧基丙基氨基]-3-侧氧基-丙基]胺甲酰氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基l]氧基-丙氧基]羰基氨基]丙酰基氨基]丙氧基]-4-乙酰氧基-2-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-3-基]酯(化合物41D)的合成
[化184]
使用化合物41C(945mg,0.58mmol)代替步骤(39D)所使用的化合物39C,以与步骤(39D)同样的方式,获得目标物41D(638mg,产率60%)。
1
(参考例42)
(42A)4-[[2-苄氧基-3-[(4-苄氧基-4-侧氧基-丁基)胺甲酰氧基]丙氧基]羰基氨基]丁酸苄酯(化合物42A)的合成
[化185]
使用4-氨基丁酸苄酯TFA盐(1.30g,4.21mmol)代替步骤(39A)所使用的甘氨酸苄酯TFA盐,以与步骤(39A)同样的方式,获得目标物42A(0.85g,产率78%)。
1
C
(42B)4-[[3-(3-羧基丙基胺甲酰氧基)-2-羟基-丙氧基]羰基氨基]丁酸(化合物42B)的合成
[化186]
/>
使用化合物42A(0.85g,1.4mmol)代替步骤(39B)所使用的化合物39A,以与步骤(39B)同样的方式,获得目标物42B(0.49g,定量)。
1
(42C)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[4-[[3-[[4-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-2-基]氧基丙基氨基]-4-侧氧基-丁基]胺甲酰氧基]-2-羟基-丙氧基]羰基氨基]丁酰基氨基]丙氧基]-4-乙酰氧基-2-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-3-基]酯(化合物42C)的合成
[化187]
使用化合物42B(300mg,0.86mmol)代替步骤(39C)所使用的化合物39B,以与步骤(39C)同样的方式,获得目标物42C(1.16g,产率82%)。
1
C
(42D)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[3-[[3-[[3-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-2-基]氧基丙基氨基]-4-侧氧基-丁基]胺甲酰氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]羰基氨基]丁酰基氨基]丙氧基]-4-乙酰氧基-2-[[双(4-甲氧基苯基)-苯基-甲氧基]甲基]四氢吡喃-3-基]酯(化合物42D)的合成
[化188]
使用化合物42C(1.16g,0.71mmol)代替步骤(39D)所使用的化合物39C,以与步骤(39D)同样的方式,获得目标物42D(795mg,产率61%)。
1
(参考例43)
(43A)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-3,4-二乙酰氧基-6-(5-氨基戊氧基)四氢吡喃-2-基]甲酯盐酸盐(化合物43A)的合成
[化189]
将步骤(21A)中合成的化合物21A(3.87g,6.83mmol)溶解于四氢吡喃(32ml)/1N盐酸(8ml)中。添加10%钯/碳(湿基)(2g),于氢气环境下在室温下剧烈搅拌。反应结束后过滤,将所获得的通过液进行减压蒸馏去除而获得目标物43A(3.1g,产率97%)的粗产物。不再进一步进行精制,用于下一反应。
C
(43B)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[5-[[3-[5-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基戊基胺甲酰氧基]-2-苄氧基-丙氧基]胺甲酰基氨基]戊氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物43B)的合成
[化190]
将2-苄氧基-1,3-丙二醇(600mg,3.29mmol)、氯甲酸4-硝基苯酯(1.39g,6.91mmol)溶解于四氢呋喃(22ml)中,添加吡啶(1.1ml,13.2mmol),在室温下搅拌15分钟。将所析出的固体过滤后,添加四氢呋喃(16ml)。继而,添加步骤(43A)中合成的化合物(43A)(3.09g,6.59mmol)的四氢呋喃(15ml)/二氯甲烷(5ml)溶液与N,N-二异丙基乙基胺(3.4mL,19.8mmol),于40℃下搅拌2小时。反应结束后,将溶剂减压蒸馏去除。添加二氯甲烷,利用0.5N盐酸、饱和食盐水、0.2N氢氧化钠水溶液、饱和食盐水洗净有机层后,利用无水硫酸钠进行干燥、过滤,将溶剂减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(二氯甲烷:甲醇=100:0-90:10,v/v)将其进行精制,而获得主要含有目标物43B的混合物(2.1g)。
C
(43C)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[5-[[3-[5-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基戊基胺甲酰氧基]-2-羟基-丙氧基]羰基氨基]戊氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物43C)的合成
[化191]
将步骤(43B)中获得的含有43B的混合物(2.1g)溶解于四氢吡喃(20ml)/纯水(1ml)中。添加1N盐酸(0.38ml)、10%钯/碳(湿基)(1g),于氢气环境下在室温下剧烈搅拌。反应结束后过滤,将所获得的通过液进行减压蒸馏去除,而获得粗产物。通过硅胶柱色谱法(乙酸乙酯:甲醇=100:0-90:15,v/v)将其进行精制,而以固体的形式获得目标物43C(1.38g,产率72%)。
1
C
(43D)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[5-[[3-[5-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基戊基胺甲酰氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]胺甲酰基氨基]戊氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物43D)的合成
[化192]
使用化合物43C(1.38g,1.37mmol)代替步骤(39D)所使用的化合物39C,以与步骤(39D)同样的方式,获得目标物43D(850mg,产率51%)。
1
(参考例44)
(44A)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[3-[[3-[[3-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基丙基氨基]-3-侧氧基-丙基]胺甲酰氧基]-2-羟基-丙氧基]羰基氨基]丙酰基氨基]丙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物44A)的合成
[化193]
在步骤(41B)中合成的化合物41B(200mg,0.62mmol)的N,N-二甲基甲酰胺(2ml)溶液中添加O-(N-丁二酰亚胺基)-N,N,N',N'-四甲基脲四氟硼酸盐(411mg,1.37mmol)、N,N-二异丙基乙基胺(0.32ml,1.86mmol),在室温下搅拌20分钟。将该反应液添加到步骤(37B)中合成的化合物37B(657mg,1.49mmol)的N,N-二甲基甲酰胺(2ml)溶液中后,添加N,N-二异丙基乙基胺(0.32ml,1.86mmol),在室温下搅拌60分钟。反应结束后,将反应液滴加至二乙醚(共计80ml)中。将上清液的溶剂去除后,通过硅胶柱色谱法(二氯甲烷:甲醇=100:0-75:25,v/v)将所获得的凝胶状的粗产物进行精制,而获得非晶状的目标物44A(408mg,产率60%)。
1
C
(44B)乙酸[(2R,3R,4R,5R,6R)-5-乙酰胺-6-[3-[3-[[3-[[3-[3-[(2R,3R,4R,5R,6R)-3-乙酰胺-4,5-二乙酰氧基-6-(乙酰氧基甲基)四氢吡喃-2-基]氧基丙基氨基]-3-侧氧基-丙基]胺甲酰氧基]-2-[2-氰基乙氧基-(二异丙氨基)膦基]氧基-丙氧基]羰基氨基]丙酰基氨基]丙氧基]-3,4-二乙酰氧基-四氢吡喃-2-基]甲酯(化合物44B)的合成
[化194]
使用化合物44A(1.53g,1.40mmol)代替步骤(39D)所使用的化合物39C,以与步骤(39D)同样的方式,获得目标物44B(510mg,产率28%)。
1
(实施例152)
X
将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6261.98)
(实施例153)
X
将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6264.00)
(实施例154)
X
将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第107号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6621.04)
(实施例155)
X
将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6623.05)
(实施例156)
X
将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6290.02)
(实施例157)
X
将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6175.96)
(实施例158)
X
将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6177.98)
(实施例159)
X
将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第107号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6535.03)
(实施例160)
X
将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6537.05)
(试验例1)使用培养细胞的利用实施例化合物的异常剪接的修复评价
293A细胞(人胚胎肾细胞)的培养
以如下方式进行293A细胞(R705-07 invitrogen)的培养。
将293A细胞于维持培养基(DMEM(Dulbecco's Modified Eagle Medium,达尔伯克改良伊格尔培养基)、10%FBS(Fetal Bovine Serum,胎牛血清))中维持培养。实验中以2×10
人G6PC全长质粒载体的制作
以如下方式制作人G6PC全长质粒载体(pcDNA hG6PC,pcDNA hG6PC(c.648G>T)+Int4)。
以人多组织(Human Multiple Tissue)cDNA板作为模板,使用下述G6PC cDNA扩增引物(Amplified primer)将G6PC cDNA扩增后,进而通过G6PC cDNA IF引物进行扩增。通过InFusion系统将经扩增的片段插入至pcDNA3.1的BamHI位点(pcDNA hG6PC)。
G6PC cDNA扩增引物:
正向引物5'-ATAGCAGAGCAATCACCACCAAGCC-3'(序列编号49)
反向引物5'-ATTCCACGACGGCAGAATGGATGGC-3'(序列编号50)
G6PC IF引物:
正向引物5'-TACCGAGCTCGGATCCACCACCAAGCCTGGAATAACTGC-3'(序列编号51)
反向引物5'-CTGGACTAGTGGATCCTGGCATGGTTGTTGACTTTAAAC-3'(序列编号52)
以人基因组DNA作为模板,使用下述G6PC Int4扩增引物将G6PC内含子4与包含一部分外显子5的区域扩增,进而通过G6PC Int4 IF引物将G6PC内含子4进行扩增。另外,使用下述hG6PC载体IF引物将STEP1-1制作的pcDNA hG6PC进行扩增。再者,对InFusion引物导入外显子5内的c.648G>T变异。通过InFusion系统将两片段链接,将G6PC内含子4序列插入至pcDNA hG6PC内(pcDNA hG6PC(c.648G>T)+Int4)。
G6PC Int4扩增引物:
正向引物5'-TCTGGGCTGTGCAGCTGAATGTCTG-3'(序列编号53)
反向引物5'-GTAGGGGATGACACTGACGGATGCC-3'(序列编号54)
G6PC Int4 IF引物:
正向引物5'-CTGGAGTCCTGTCAGGTATGGGC-3'(序列编号55)
反向引物5'-AGCTGAAAAGGAAGAAGGTAATGAG-3'(序列编号56)
hG6PC载体IF引物:
正向引物5'-TCTTCCTTTTCAGCTTCGCCATCGG-3'(序列编号57)
反向引物5'-CTGACAGGACTCCAGCAACAAC-3'(序列编号58)
实施例的化合物、及质粒载体的共转染
以如下方式将实施例的化合物、及质粒载体进行共转染。
制作下述A液与B液后加以混合。
当通过6孔板实施时,对于每孔,准备250μL的Opti-MEM培养基(gibco)、0.50μL质粒载体(1mg/mL)、4.0μL(最终:20nM)实施例中制造的化合物(12.5μM)作为A液,准备250μL的Opti-MEM培养基(gibco)、6.0μL Lipofectamine 2000(Invitrogen)作为B液,并将A液与B液加以混合。当通过12孔板实施时,对于每孔,准备125μL的Opti-MEM培养基(gibco)、0.25μL质粒载体(1mg/mL)、2.0μL(最终:20nM)实施例中制造的化合物(12.5μM)作为A液,准备125μL的Opti-MEM培养基(gibco)、3.0μL Lipofectamine2000(Invitrogen)作为B液,并将A液与B液加以混合。
将所述混合液在室温下培养20分钟后,添加到继代第二天的细胞中(共转染)。添加6小时后,更换为新鲜的维持培养基,自所述混合液添加起于CO
RNA萃取(体外)
以如下方式进行RNA的萃取。
通过冷PBS(Phosphate Buffered Saline,磷酸盐缓冲盐水)将共转染后培养24小时的细胞洗净1次。将RNeasy迷你套组或RNeasy 96套组(Qiagen)的细胞溶解液以每孔300μL的方式逐孔添加,在室温下培养5分钟后回收。对于回收液,依照包含DNase处理的套组的操作说明进行RNA精制。DNase处理是使用RNase-Free DNase set(Qiagen89254)。经精制、溶出的RNA是进行下文所述的反转录反应。
反转录反应
以如下方式进行反转录反应。
将萃取RNA调整为25~100mg/mL后,使用高容量RNA-to-cDNA套组(High CapacityRNA-to-cDNA kit)(Applied biosystems),以每份样品成为10μL缓冲液混合物(Buffermix)、1μL酶混合物(Enzyme mix)、9μL纯化水、+萃取RNA的方式混合,进行反转录反应(37℃下60min、95℃下5min、4℃下保持)。
相对于反转录反应产物20μL,通过纯化水80μL稀释成5倍,于-30℃下保存。
qRT-PCR(SYBR Green)
以下述方式设计qRT PCR引物(SYBR Green)。
修复hG6PC引物(SYBR):
正向引物5'-TTGTGGTTGGGATTCTGGGC-3'(序列编号59)
反向引物5'-ATGCTGTGGATGTGGCTGAA-3'(序列编号60)
hActin引物(SYBR):
正向引物5'-TGGCACCCAGCACAATGAA-3'(序列编号61)
反向引物5'-CTAAGTCATAGTCCGCCTAGAAGCA-3'(序列编号62)
另外,关于PCR反应液,每孔悬浮5μL的2×FAST SYBR Green Master Mix(AppliedBiosystems)、2μL纯化水、1μL的引物混合物(Primer mix)(10μM)、2μL的cDNA(经5倍稀释),通过viia7(Applied Biosystems)进行PCR反应(程序:SYBR Green Regents,FAST,包括熔解曲线)。
qRT-PCR(Taqman分析)
以下述方式设计修复hG6PC引物组、总hG6PC引物组,调整20×引物探针混合物(primer probe mix)(引物浓度:1000nM,探针浓度:250nM)。hActin引物组、mActin引物组直接使用原液。
修复hG6PC引物组(Taqman):
正向引物5'-GCTGCTCATTTTCCTCATCAAGTT-3'(序列编号63)
反向引物5'-TGGATGTGGCTGAAAGTTTCTGTA-3'(序列编号64)
探针5'-TCCTGTCAGGCATTGC-3'FAM(序列编号65)
hActin引物组(Taqman):ABI Hs01060665_g1 FAM
mActin引物组(Taqman):ABI Mm02619580_g1 FAM
18s引物组(Taqman):ABI Hs99999901_s1 FAM
另外,每孔悬浮5μL的2×Taqman Fast Advanced Master Mix(AppliedBiosystems)、2.5μL纯化水、0.5μL的20×引物探针混合物(10μM)、2μL的cDNA(经5倍稀释),调整PCR反应液(相对于每管),通过viia7或Quantstadio7(Applied Biosystems公司)进行PCR反应(程序:Taqman regents,FAST)。
利用LC-MS/MS的正常人G6PC特异性肽的定量
以如下方式进行蛋白质的萃取及正常人G6PC特异性肽的LC-MS/MS定量。
蛋白质溶解物的萃取、调整
通过冷PBS将共转染后培养24小时的细胞洗净1次。将RIPA(Radioimmunoprecipitation Assay,放射免疫沉淀分析)缓冲液(Nacalai Tesque)以每孔100μL的方式逐孔添加,在冰上培养后回收。对于回收液,在冰上培养20分钟后,将10000g于4℃下离心10分钟并将上清液回收。对上清液测定总蛋白质量,以成为0.4mg/mL的方式进行调整而制成蛋白质溶解物。
试剂的调整
DS264_100u:将稳定同位素标记肽GLGVD(L*)LWT(L*)EK(Scrum,L*:L-亮氨酸-
DS266_100u:将稳定同位素标记肽WCEQPEW(V*)HIDTTPFAS(L*)LK(L*:L-亮氨酸-
DS268_100u:将稳定同位素标记肽NLGTLFG(L*)GLA(L*)NSSMYR(Scrum,L*:L-亮氨酸-
IS溶液-1:悬浮50mL乙腈、0.5mL三氟乙酸(Nacalai)、20μL的DS264_100u、20μL的DS266_100u、20μL的DS268_100u。
IS溶液-2:悬浮50mL乙腈、50mL纯化水、0.5mL三氟乙酸(Nacalai)、20μL的DS264_100u、20μL的DS266_100u、20μL的DS268_100u。
0.1M Tris-HCl:将5mL的1M Tris-HCl缓冲溶液(pH值8.0)添加悬浮于45mL的纯化水中。
脲/EDTA(ethylenediamine tetraacetic acid,四乙酸乙二胺)溶液:将2.4g脲(Nacalai)、100μL的0.5M EDTA(sigma-aldorich)添加、悬浮于4.9mL的0.1M Tris-HCl中。
DTT溶液(20mg/mL):将20mg的DTT(dithiothreitol,二硫苏糖醇)(Wako)添加悬浮于1mL的纯化水中。
IAA溶液(50mg/mL):将50mg的IAA(iodoacetamide,碘乙酰胺)(sigma-aldorich)添加悬浮于1mL纯化水中。
胰蛋白酶/LysC Mix溶液(200μg/mL):将20μg胰蛋白酶/Lys-C Mix(1小瓶)(Promega)添加悬浮于100μL的再悬浮缓冲液(Promega)中。
消化处理
依照下述实施酶消化反应,调整LC-MS注射样品(相对于每份样品)。
悬浮20μL脲/EDTA溶液、10μL蛋白质溶解物、10μL的DTT溶液,在室温下静置60分钟后,添加悬浮2.5μL的IAA溶液并在室温下静置60分钟。其后,对于悬浮液,添加悬浮112.5μL的0.1M Tris-HCl与2.5μL胰蛋白酶/LysC Mix溶液,在37℃下培养一晚后,添加150μL的IS溶液-1、300μL的IS溶液-2制成LC-MS注射样品。
LC-MS分析、试样中浓度测定
使用Ultimate 3000(Thermo Fisher),Q Exactive plus(Thermo Fisher)实施LC-MS注射样品的LC-MS分析。使用内部标准法由质量范围(Mass range)820.0632-820.0782(m/z)算出下述肽(DS265)的试样中浓度。
DS265:WCEQPEWVHIDTTPFASLLK(序列编号66)
G6PC酶活性的测定
以下述方式实施所萃取的各检体的微粒体组分的G6PC的活性测定。
(1)试剂的制备
缓冲液A:100mM BIS-TRIS缓冲液,pH值6.5,37℃
于180mL纯化水中添加4.2g的BIS-TRIS(Sigma-Aldrich)。使用盐酸、纯化水调整为pH值6.5、37℃、200mL。
缓冲液B:HEPES 20mM,EDTA 1mM,蔗糖250mM(4℃保存)
在180mL纯化水中添加4.0mL的1.0M HEPES(Gibco)、0.4mL的0.5M EDTA(USB)、17g蔗糖(Wako)。将纯化水调整为200mL后,进行0.22μm过滤器的透过处理。
底物:200mM葡萄糖6-磷酸(4℃保存)
在88.65mL纯化水中添加5mg的D-葡萄糖6-磷酸钠(Sigma-Aldrich)。
TCA:20%三氯乙酸(室温、遮光保存)
在40mL纯化水中添加10mL三氯乙酸溶液(Sigma-Aldrich)。
标准液:磷标准液(Phosphorus Standard Solution),20μg/ml(Sigma-Aldrich)(4℃保存)
5M硫酸溶液(室温、遮光保存)
在67mL纯化水中添加25mL硫酸(Aldrich 258105)。
TSCR:Taussky-Shorr显色剂(用时制备)
在5M硫酸溶液20mL中添加2.4mg四水合钼酸铵(Ammonium MolybdateTetrahydrate)(Sigma-Aldrich),将溶解液添加于140mL的纯化水中。进而添加10g七水合硫酸亚铁(Ferrous Sulfate Heptahydrate)(Sigma-Aldrich),搅拌至溶解后,通过纯化水定容至200mL。
(2)微粒体组分的精制
依照下述程序对微粒体组分进行精制、调整。
通过冰冷PBS洗净实施例化合物、及已进行质粒载体的共转染的293A细胞(在6孔板中)后,将冰冷却缓冲液B添加到各孔中,并利用细胞刮勺进行回收。使用Dounce均质器在冰上(On ice)将所回收的细胞充分均质化后,将1000g在4℃下离心10min,将上清液回收。将该上清液13000g在4℃下离心60min,去除上清液,将颗粒于缓冲液B中悬浮。以悬浮液的蛋白浓度成为一定(0.3~1.0mg/mL)的方式通过缓冲液B进行调整。
(3)G6PC酶活性测定
通过下述方法测定蛋白浓度经调整的微粒体组分(样品)的G6PC酶活性。试剂的名称依照(1)。
对于各样品制作测试组与空白组。在测试组中悬浮150μL缓冲液A、50uL底物,在37℃下培养5分钟。在其中添加、悬浮5μL的样品,在37℃下准确培养5分钟后,添加45μL的TCA。充分悬浮后,在25℃下培养5分钟。在空白组中悬浮150μL缓冲液A、50uL底物,在37℃下培养5分钟。在其中添加悬浮5μL的样品、45μL的TCA并在25℃下培养者。将4000g的各样品的测试组及空白组在室温下离心10分钟,将该上清液分注于其他管中。对于各上清液100μL,添加100μL的TSCR,在室温下培养5分钟后,通过读板仪(Spectra Max M4 Molecular Devices)测定660nm的吸亮度。根据使用标准液的稀释体系算出的校准曲线、各样品的测试组与空白组的吸亮度、样品的蛋白浓度算出G6PC酶活性(U/mg:样品中的总蛋白质1mg在1分钟内分解的G6P量(μmol))。
使用人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)的利用实施例化合物
进行人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)与寡核苷酸(21e_001~012、及21m_001~012)的共转染,通过qRT-PCR(SYBR Green)评价是否修复G6PC(c.648G>T)引起的异常剪接。如图5A及5B所示,21e_002~012、及21m_002~012的化合物中观察到G6PC mRNA的异常剪接的正常化。另外,对利用LC-MS/MS的正常人G6PC特异性肽的产生进行研究,结果如图6A及6B所示,在21e_002~012、及21m_002~012的化合物中观察到正常人G6PC特异性肽的产生。进而,测定G6PC酶活性的结果为如图7A及7B所示,在21e_002~012、及21m_002~012的化合物中观察到G6PC酶活性。
使用人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)的利用实施例化合物
进行人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)与寡核苷酸(21e_001~006、及21e_013~022)的共转染,通过qRT-PCR(SYBR Green)评价是否修复G6PC(c.648G>T)引起的异常剪接。如图9所示,21e_002~006、及21e_015~022的化合物中观察到G6PCmRNA的异常剪接的正常化。另外,对利用LC-MS/MS的正常人G6PC特异性肽的产生进行研究,结果如图10所示,在21e_002~006、及21e_015~022的化合物中,观察到正常人G6PC特异性肽的产生。
使用人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)的利用实施例化合物
进行人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)与寡核苷酸(18e_001~017、及18m_001~017)的共转染,通过qRT-PCR(SYBR Green)评价是否修复G6PC(c.648G>T)引起的异常剪接。如图13A及13B所示,18e_005~017、及18m_005~017的化合物中观察到G6PC mRNA的异常剪接的正常化。另外,对利用LC-MS/MS的正常人G6PC特异性肽的产生进行研究,结果如图14A及14B所示,在18e_005~017、及18m_005~017的化合物中,观察到正常人G6PC特异性肽的产生。
使用人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)的利用实施例化合物
进行人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)与寡核苷酸(18e_018~031)的共转染,通过qRT-PCR(SYBR Green)评价是否修复G6PC(c.648G>T)引起的异常剪接。如图16A所示,18e_022~026、及18e_031的化合物中观察到G6PC mRNA的异常剪接的正常化。另外,对利用LC-MS/MS的正常人G6PC特异性肽的产生进行研究,结果如图16B所示,在18e_022~026、及18e_031的化合物中,观察到正常人G6PC特异性肽的产生。
使用人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)的利用实施例化合物
进行人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)与寡核苷酸(21e_002、18e_005、21m_002、18e_005、18m_022、15e_001、15ed_001、18e_008、18e_025、18m_008、15e_002、15ed_002、及作为对照的国际公开第2004/048570的实施例93的化合物)的共转染,通过qRT-PCR(SYBR Green)评价是否修复G6PC(c.648G>T)引起的异常剪接。如图18所示,21e_002、18e_005、21m_002、18e_005、18m_022、15e_001、15ed_001、18e_008、18e_025、18m_008、15e_002、及15ed_002的化合物中观察到G6PC mRNA的异常剪接的正常化。
(试验例2)使用模型小鼠的利用实施例化合物的异常剪接的修复评价
小鼠的制造
载体的制作
依照下述程序,制作G6PC KI载体。
以小鼠基因组DNA为模板,使用下述mG6PC 5'臂扩增引物,将G6PC 5'臂区域扩增。进而利用mG6PC 5'臂IF引物扩增后,插入pBluescriptII(+/-)的XhoI位点(G6PC 5'臂载体)。
mG6PC 5'臂扩增引物:
正向引物5'-GGGAAACATGCATGAAGCCCTGGGC-3'(序列编号67)
反向引物5'-TCCCTTGGTACCTCAGGAAGCTGCC-3'(序列编号68)
mG6PC 5'臂IF引物:
正向引物5'-CGGGCCCCCCCTCGAAAACTAGGCCTGAAGAGATGGC-3'(序列编号69)
反向引物5'-TACCGTCGACCTCGAGGGTTGGCCTTGATCCCTCTGCTA-3'(序列编号70)
继而,以小鼠基因组DNA为模板,使用下述mG6PC 3'臂扩增引物,将G6PC 3'臂区域扩增。进而,利用mG6PC 3'臂IF引物扩增后,插入G6PC 5'臂载体的NotI位点(G6PC5'+3'臂载体)。
mG6PC 3'臂扩增引物:
正向引物5'-GGTTGAGTTGATCTTCTACATCTTG-3'(序列编号71)
反向引物5'-GCAAGAGAGCCTTCAGGTAGATCCC-3'(序列编号72)
mG6PC 3'臂IF引物:
正向引物5'-AGTTCTAGAGCGGCCGCCCATGCAAAGGACTAGGAACAAC-3'(序列编号73)
反向引物5'-ACCGCGGTGGCGGCCAATGTTGCCTGTCTTCCTCAATC-3'(序列编号74)
以上文所述的pcDNA hG6PC(c.648G>T)+Int4作为模板,使用下述hG6PC+Int4 IF引物将hG6PC(c.648G>T)+内含子4进行扩增。另外,以G6PC 5'+3'臂载体作为模板,使用下述臂载体IF引物进行扩增。使用InFusion系统将两片段链接而制作G6PC KI载体。
hG6PC+Int4 IF引物:
正向引物5'-GGCCAACCCTGGAATAACTGCAAGGGCTCTG-3'(序列编号75)
反向引物5'-TTGCATGGTTGTTGACTTTAAACACCGAAGA-3'(序列编号76)
臂载体IF引物:
正向引物5'-TCAACAACCATGCAAAGGACTAGGAACAAC-3'(序列编号77)
反向引物5'-ATTCCAGGGTTGGCCTTGATCCCTCTGCTA-3'(序列编号78)
向pSPgRNA(addgene)的gRNA序列导入区域导入下述KI 5'gRNA、KI 3'gRNA序列,而制作pSPgRNA(KI 5')、pSPgRNA(KI 3')。
KI 5'gRNA:5'-GGGATCAAGGCCAACCGGCTGG-3'(序列编号79)
KI 3'gRNA:5'-TAAAGTCAACCGCCATGCAAAGG-3'(序列编号80)
显微注射
使用杀菌蒸馏水,以最终浓度成为G6PC KI载体10ng/μL、pSPgRNA(KI 5')5ng/μL、pSPgRNA(KI 3')5ng/μL、pSPCas9(addgene)5ng/μL的方式调整各种载体后,注射通过MILLEX-GV针筒过滤器(Millipore)的载体直至C57BL/6J小鼠受精卵的原核充分膨胀的程度为止(约2pL)。将该受精卵移植至受体C57BL/6J小鼠的输卵管,而获得F0小鼠。
基因型鉴定、F1系统化
通过以下的程序实施基因型鉴定(Genotyping)并进行F1系统化。
使用核酸自动提取装置(PI-200Kurabo)及专用套组自F0小鼠的尾组织提取基因组DNA。使用Amplitaq Gold Master mix(Thermo fisher)及使用下述KI筛选引物将提取DNA进行扩增(95℃下10min,将(95℃下30sec、60℃下30sec、72℃下30sec)循环35次,72℃下2min,4℃下保持)。
KI筛选引物:
正向引物5'-TACGTCCTCTTCCCCATCTG-3'(序列编号81)、
反向引物5'-CTGACAGGACTCCAGCAACA-3'(序列编号82)
对所述PCR产物实施凝胶电泳,以在433bp附近观察到频带的个体的基因组DNA作为模板,使用PrimeSTAR GXL(takara)及下述KI基因型鉴定引物进行扩增(98℃下2min,将(95℃下15sec、68℃下5min)循环38次,68℃下7min,15℃下保持)。
KI基因型鉴定引物(5'):
正向引物5'-TTCCTTCCAAAGCAGGGACTCTCTATGT-3'(序列编号83相同(1))
反向引物5'-CTTGCAGAAGGACAAGACGTAGAAGACC-3'(序列编号84相同(2))
KI基因型鉴定引物(3'):
正向引物5'-GAGTCTATATTGAGGGCAGGCTGGAGTC-3'(序列编号85)、
反向引物5'-TAGTCTGCCTGCTCACTCAACCTCTCCT-3'(序列编号86)
对所述PCR产物实施凝胶电泳。使用基因分析仪(Gentetic analyzer)(Lifetechnology)及下述KI定序引物对使用KI基因型鉴定引物(5')及KI基因型鉴定引物(3')均观察到扩增为所期待的序列长度(4705bp、4026bp)的个体的基因组DNA的KI基因型鉴定引物(5')的PCR产物进行直接定序。将可确认到所期待的序列的KI的个体设为KI阳性F0。
KI定序引物(5'):5'-GAGTCTATATTGAGGGCAGGCTGGAGTC-3'(序列编号87)
使KI阳性F0与C57BL/6J交配而获得F1。使用DNeasy 96血液及组织套组(NDeasy96Blood&Tissue Kit)(Qiagen)自F1的耳廓组织提取基因组DNA,使用PrimeSTARGXL(takara)及上文所述KI基因型鉴定引物(5')进行扩增(98℃下2min,将(95℃下15sec、68℃下5min)循环38次,68℃下7min,15℃下保持)。
对所述PCR产物实施凝胶电泳,将观察到扩增为所期待的序列长度(4705b)的个体设为KI阳性F1。使自KI阳性F1中筛选的1株进行繁殖,制成hG6PC(c.648G>T)+Int4 KI系统。
hG6PC(c.648G>T)+Int4系统的基因型鉴定
使用DNeasy 96血液及组织套组(Qiagen)自耳廓组织提取基因组DNA,使用KOD FX(TOYOBO)及下述KI基因型鉴定引物、mG6PC WT引物进行复合扩增(98℃下2min,将(95℃下15sec、68℃下5.5min)循环32次,68℃下5min,4℃下保持)。
KI基因型鉴定引物(5'):
正向引物5'-TTCCTTCCAAAGCAGGGACTCTCTATGT-3'(序列编号83相同(1))
反向引物5'-CTTGCAGAAGGACAAGACGTAGAAGACC-3'(序列编号84相同(2))
mG6PC WT引物:
正向引物5'-TAAATTTGACCAATGAGCACTGGAGGTC-3'(序列编号88)
反向引物5'-AAAATCATGTGTATGCGTGCCTTTCCTA-3'(序列编号89)'
对所述PCR产物实施凝胶电泳,以4705b附近的扩增作为KI等位基因,以2536bp附近的扩增作为mG6PC WT等位基因,判断新生小鼠的基因型鉴定(WT、Ht、Homo)。
小鼠的取材
以如下方式进行小鼠的取材。
在取材前一天傍晚设置用来避免食粪的地网,并且开始绝食。第二天,在麻醉导入下开腹,充分放血后,摘下肝脏、肾脏。各器官利用冰冷PBS洗净后,修整为合适的大小,保存到预先装入了均质化珠(NIKKATO)的管中。各器官通过液态氮瞬间冷却后在-80℃下保管。
RNA萃取(体内)
以如下方式进行RNA的萃取。
对于组织保存管,逐支添加600μL的RNeasy迷你套组或Qiacube系统(Qiagen)的细胞溶解液,通过tissue lyserII(Qiagen)以25kHz均质化2min。在冰浴冷却下培养10分钟后,以8000G在室温下离心10分钟并回收上清液。对于上清液,依照包含DNase处理的各套组的操作说明进行RNA精制。DNase处理使用RNase-Free DNase set(Qiagen)。经精制、溶出的RNA进行上文所述的反转录反应,依照上文所述的qRT-PCR(Taqman分析)进行修复G6PCmRNA的定量。
使用hG6PC(c.648G>T)+Int4Ht KI小鼠的利用实施例化合物的异常剪接的修复
对于hG6PC(c.648G>T)+Int4 Ht KI小鼠,可将实施例116至127的化合物溶解到PBS中,以成为3mg/kg的方式实施尾静脉投予。投予7天后,在绝食一晚的条件下采集小鼠的肝脏组织,可通过qRT-PCR(Taqman)评价是否修复G6PC(c.648G>T)引起的异常剪接。
使用hG6PC (c.648G>T)+Int4 Ht KI小鼠的利用实施例化合物的异常剪接的修
对于hG6PC(c.648G>T)+Int4 Ht KI小鼠,将实施例91至95的化合物溶解到大冢生食注中,以成为25mg/kg的方式实施皮下投予。投予7天后,在绝食一晚的条件下采集小鼠的肝脏组织,通过qRT-PCR(Taqman)评价是否修复G6PC(c.648G>T)引起的异常剪接,结果如图19所示,通过实施例91至95的化合物,在hG6PC(c.648G>T)+Int4 Ht KI小鼠的肝脏中可观察到mRNA的异常剪接的正常化。
使用hG6PC (c.648G>T)+Int4 Ht KI小鼠的利用实施例化合物的异常剪接的修
对于hG6PC(c.648G>T)+Int4 Ht KI小鼠,将实施例91、92、及96至103的化合物溶解到大冢生食注中,以成为25mg/kg的方式实施皮下投予。投予7天后,在绝食一晚的条件下采集小鼠的肝脏组织,通过qRT-PCR(Taqman)评价是否修复G6PC(c.648G>T)引起的异常剪接,结果如图20所示,通过实施例91、92、及96至103的化合物,在hG6PC(c.648G>T)+Int4Ht KI小鼠的肝脏中可观察到mRNA的异常剪接的正常化。
使用hG6PC(c.648G>T)+Int4HtKI小鼠的利用实施例化合物的异常剪接的修复评
对于hG6PC(c.648G>T)+Int4 Ht KI小鼠,将实施例83至91、及104至107的化合物溶解到大冢生食注中,以成为30mg/kg的方式实施皮下投予。投予7天后,在绝食一晚的条件下采集小鼠的肝脏组织,通过qRT-PCR(Taqman)评价是否修复G6PC(c.648G>T)引起的异常剪接,结果如图21所示,通过实施例83至91、及104至107的化合物,在hG6PC(c.648G>T)+Int4 Ht KI小鼠的肝脏中可观察到mRNA的异常剪接的正常化。
使用hG6PC(c.648G>T)+Int4HtKI小鼠的利用实施例化合物的异常剪接的修复评
对于hG6PC(c.648G>T)+Int4 Ht KI小鼠,将实施例104、及108至115的化合物溶解到大冢生食注中,以成为30mg/kg的方式实施皮下投予。投予7天后,在绝食一晚的条件下采集小鼠的肝脏组织,通过qRT-PCR(Taqman)评价是否修复G6PC(c.648G>T)引起的异常剪接,结果如图22所示,通过实施例83至91、及104至107的化合物,在hG6PC(c.648G>T)+Int4Ht KI小鼠的肝脏中可观察到mRNA的异常剪接的正常化。
使用hG6PC (c.648G>T)+Int4 Ht KI小鼠的利用实施例化合物的异常剪接的修
对于hG6PC(c.648G>T)+Int4 Ht KI小鼠,将实施例105、113、及131至137的化合物溶解到大冢生食注中,以成为30mg/kg的方式实施皮下投予。投予7天后,在绝食一晚的条件下采集小鼠的肝脏组织,通过qRT-PCR(Taqman)评价是否修复G6PC(c.648G>T)引起的异常剪接,结果如图23所示,通过实施例105、113、及131至137的化合物,在hG6PC(c.648G>T)+Int4 Ht KI小鼠的肝脏中可观察到mRNA的异常剪接的正常化。
(试验例3)异常剪接修复序列的片段解析
PCR反应与片段序列解析
以如下方式进行PCR反应与片段序列解析。
以下述方式设计hG6PC剪接验证引物。
hG6PC剪接验证引物:
正向引物5'-TTGTGGTTGGGATTCTGGGC-3'(序列编号90)
反向引物5'-TCCAGAGTCCACAGGAGGTC-3'(序列编号91)
以如下方式调整PCR反应液,进行PCR反应。
使23μL的Platinum
PCR产物是使用E-Gel(注册商标)琼脂糖凝胶电泳系统,通过E-gel Ex 2%琼脂糖(Invitrogen)进行电泳并解析。各片段通过NucleoSpin(注册商标)Gel and PCR Clean-up(MACHEREY-NAGEL)自凝胶中萃取,添加G6PC定序引物后,使用BigDye v3.1进行定序反应。利用Applied Biosystems 3730xl DNA Analyzer(Life technologies)确认碱基序列(图24)。
G6PC定序引物5'-GCTGTGCAGCTGAATGTCTG-3'(序列编号92)
利用实施例1的化合物(21e_002)的培养细胞中的异常剪接修复序列的片段解析
使用由仅将在使用人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)的利用实施例化合物的G6PC mRNA的异常剪接的修复评价(1)中所制作的人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)转染而成的检体、将人G6PC全长质粒载体(pcDNA hG6PC(c.648G>T)+Int4)与寡核苷酸(21e_002)共转染而成的检体所制作的cDNA作为模板,进行PCR反应与片段序列解析。如图24所示,通过实施例1的化合物(21e_002)可观察到缺损91个碱基的异常剪接的正常化。
(试验例4)使用模型小鼠的利用实施例化合物的异常剪接的修复评价
与试验例2同样地,使用模型小鼠对实施例化合物进行评价。
使用hG6PC(c.648G>T)+Int4HtKI小鼠的利用实施例化合物的异常剪接的修复评价(7)
对于hG6PC(c.648G>T)+Int4 Ht KI小鼠,将实施例133、及143至149的化合物溶解到大冢生食注中,以成为30mg/kg的方式实施皮下投予。投予7天后,在绝食一晚的条件下采集小鼠的肝脏组织,通过qRT-PCR(Taqman)评价是否修复G6PC(c.648G>T)引起的异常剪接,结果如图25所示,通过实施例133、及143至149的化合物,在hG6PC(c.648G>T)+Int4 HtKI小鼠的肝脏中可观察到mRNA的异常剪接的正常化。
使用hG6PC(c.648G>T)+Int4Ht KI小鼠的利用实施例化合物的异常剪接的修复
对于hG6PC(c.648G>T)+Int4 Ht KI小鼠,将实施例149、及152至160的化合物溶解到大冢生食注中,以成为30mg/kg的方式实施皮下投予。投予7天后,在绝食一晚的条件下采集小鼠的肝脏组织,通过qRT-PCR(Taqman)评价是否修复G6PC(c.648G>T)引起的异常剪接,结果如图26所示,通过实施例149、及152至160的化合物,在hG6PC(c.648G>T)+Int4 HtKI小鼠的肝脏中可观察到mRNA的异常剪接的正常化。
(实施例161)
X
使用所述序列即15e_001.6代替实施例133所使用的序列,将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6278.03)
(实施例162)
X
使用所述序列即15e_005.6代替实施例133所使用的序列,将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6280.03)
(实施例163)
X
使用所述序列即15e_001.7代替实施例133所使用的序列,将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第92号至第106号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6304.04)
(实施例164)
X
使用所述序列即15e_005.7代替实施例133所使用的序列,将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6306.03)
(实施例165)
X
使用所述序列即15e_005.8代替实施例133所使用的序列,将X
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6149.93)
(实施例166)
X
使用所述序列即15e_005.5.01代替实施例153所使用的序列,与实施例153同样地进行合成。但在核酸自动合成仪所使用的试剂中,作为表述序列合成所需的部分的氧化剂,以成为OXDIZER 0.05M(Sigma-Aldrich制造,产品编号L560250-04)、或0.02M的方式,使用四氢呋喃(脱水,关东化学制造,产品编号40993-05)、吡啶(脱水,关东化学制造,产品编号11339-05)、蒸馏水78:20:2(v/v/v)溶液溶解碘(关东化学制造,产品编号20035-00)来适当使用。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.00)
(实施例167)
X
使用所述序列即15e_005.5.02代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.00)
(实施例168)
X
使用所述序列即15e_005.5.03代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6247.99)
(实施例169)
X
使用所述序列即15e_005.5.04代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.00)
(实施例170)
X
使用所述序列即15e_005.5.05代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.02)
(实施例171)
X
使用所述序列即15e_005.5.06代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.02)
(实施例172)
X
使用所述序列即15e_005.5.07代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.02)
(实施例173)
X
使用所述序列即15e_005.5.08代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.02)
(实施例174)
X
使用所述序列即15e_005.5.09代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.01)
(实施例175)
X
使用所述序列即15e_005.5.10代替实施例166所使用的序列,与实施例166同样地进行合成。所使用的序列为所述序列15e_005.5.10。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.01)
(实施例176)
X
使用所述序列即15e_005.5.11代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6216.00)
(实施例177)
X
使用所述序列即15e_005.5.12代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6216.03)
(实施例178)
X
使用所述序列即15e_005.5.13代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6216.02)
(实施例179)
X
使用所述序列即15e_005.5.14代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6216.03)
(实施例180)
X
使用所述序列即15e_005.5.15代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6200.06)
(实施例181)
X
使用所述序列即15e_005.5.16代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6247.99)
(实施例182)
X
使用所述序列即15e_005.5.17代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.01)
(实施例183)
X
使用所述序列即15e_005.5.18代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.00)
(实施例184)
X
使用所述序列即15e_005.5.19代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.01)
(实施例185)
X
使用所述序列即15e_005.5.20代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.00)
(实施例186)
X
使用所述序列即15e_005.5.21代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.01)
(实施例187)
X
使用所述序列即15e_005.5.22代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.00)
(实施例188)
X
使用所述序列即15e_005.5.23代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.00)
(实施例189)
X
使用所述序列即15e_005.5.24代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.00)
(实施例190)
X
使用所述序列即15e_005.5.25代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6248.00)
(实施例191)
X
使用所述序列即15e_005.5.26代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.01)
(实施例192)
X
使用所述序列即15e_005.5.27代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6216.03)
(实施例193)
X
使用所述序列即15e_005.5.28代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6216.03)
(实施例194)
X
使用所述序列即15e_005.5.29代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6200.06)
(实施例195)
X
使用所述序列即15e_005.5.30代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6200.06)
(实施例196)
X
使用所述序列即15e_005.5.31代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6184.11)
(实施例197)
X
使用所述序列即15e_005.5.32代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6040.30)
(实施例198)
X
使用所述序列即15e_005.5.33代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.02)
(实施例199)
X
使用所述序列即15e_005.5.34代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.01)
(实施例200)
X
使用所述序列即15e_005.5.35代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.04)
(实施例201)
X
使用所述序列即15e_005.5.36代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.03)
(实施例202)
X
使用所述序列即15e_005.5.37代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.02)
(实施例203)
X
使用所述序列即15e_005.5.38代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.01)
(实施例204)
X
使用所述序列即15e_005.5.39代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.02)
(实施例205)
X
使用所述序列即15e_005.5.40代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.02)
(实施例206)
X
使用所述序列即15e_005.5.41代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6232.02)
(实施例207)
X
使用所述序列即15e_005.5.42代替实施例166所使用的序列,与实施例166同样地进行合成。
本化合物的碱基序列是与智人葡萄糖-6-磷酸酶催化亚基(G6PC),转录变体1,mRNA(NCBI-GenBank登录号NM_000151.3)的核苷酸编号728的G变异为T的c.648G>T变异G6PC基因的外显子5的5'末端起第91号至第105号互补的序列。化合物是通过负离子ESI质量分析进行鉴定(实测值:6216.00)
再者,在本说明书中,A
[化195]
[化196]
[化197]
[化198]
[化199]
[化200]
[化201]
[化202]
[化203]
/>
[化204]
[化205]
[化206]
将本说明书所引用的全部刊物、专利及专利申请案直接作为参考而并入本说明书中。
[产业上的可利用性]
本发明可用于糖原病Ia型的治疗。
[序列表非关键文字]
示出<序列编号1~48、93~95>反义寡核苷酸的序列。构成反义寡核苷酸的核苷酸可为天然型DNA、天然型RNA、DNA/RNA的嵌合体、它们的修饰体的任一者,优选至少1个为修饰核苷酸。
示出<序列编号49~65、67~92>引物的序列。
示出<序列编号66>肽的序列。