一种连续反应制备硫杂䓬类化合物的方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及药物领域,尤其涉及一种连续反应制备硫杂
背景技术
含有硫原子的杂环化合物因具有抗菌、除草、抗病毒、抗癌等生物活性而被广泛存在于医药、农药等领域中。例如,硫杂
在现有技术中,3,4-二氟-6,11-二氢二苯并[b,e]噻吩-11-醇(硫杂
在上述合成过程中,在第一步合成步骤中,使用正丁基锂(LDA),需要在无水无氧、-40℃低温下进行,工艺条件苛刻;第二步合成步骤中,使用的苯硫酚剧毒且有恶臭气味,安全风险较高;在终产物硫杂
发明内容
针对现有技术中存在的不足,本发明的目的在于提供一种连续反应制备硫杂
为实现上述发明目的,本发明采用的技术方案如下:
第一方面,提供了一种连续反应制备硫杂
制备化合物Ⅲ的有机相:
将具有式Ⅱ结构的化合物Ⅱ溶于有机溶剂中,制得化合物Ⅱ有机溶液,将溴酸钠的水溶液加入所述化合物Ⅱ有机溶液中,再缓慢加入亚硫酸氢钠的水溶液进行回流反应,反应完全后去除反应液中的水相,得到具有式Ⅲ结构的化合物Ⅲ的有机相;
制备化合物Ⅳ的有机相:
将所述具有式Ⅲ结构的化合物Ⅲ的有机相与碱的水溶液混合,再分批加入苯硫酚钠进行硫醚化反应,反应完全后,经冷却、酸化、去除反应液中的水相,得到具有式Ⅳ结构的化合物Ⅳ的有机相;
制备化合物Ⅰ:
所述具有式Ⅳ结构的化合物Ⅳ的有机相与酰氯试剂进行氯代反应,反应完全后,分批加入无水氯化铝进行傅克酰基化反应,得到具有式Ⅰ结构的化合物Ⅰ。
本发明提供的连续反应制备硫杂
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
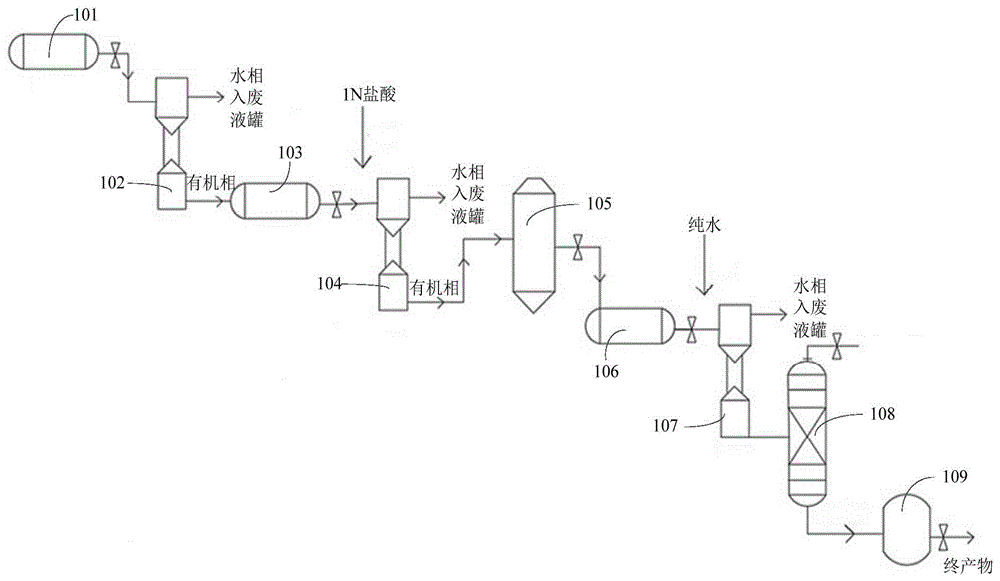
图1是本发明实施例的连续反应制备硫杂
图2是本发明实施例1制得的化合物Ⅲ的核磁共振氢谱谱图。
图3是本发明实施例1制得的化合物Ⅲ的核磁共振碳谱谱图。
图4是本发明实施例1制得的化合物Ⅳ的核磁共振氢谱谱图。
图5是本发明实施例1制得的化合物Ⅳ的核磁共振碳谱谱图。
图6是本发明实施例1制得的化合物Ⅰ的核磁共振氢谱谱图。
图7是本发明实施例1制得的化合物Ⅰ的核磁共振碳谱谱图。
具体实施方式
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明作出进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,但本发明的实施方式不限于此。
除另有定义外,以下实施例中所用的技术术语具有与本发明所属领域技术人员普遍理解的相同含义。以下实施例中所用的实验试剂,如无特殊说明,均为常规生化试剂;所述实验试剂用量,如无特殊说明,均为常规实验操作中试剂用量;所述实验方法,如无特殊说明,均为常规方法。
第一方面,本实施例提供了一种连续反应制备硫杂
制备化合物Ⅲ的有机相:
将具有式Ⅱ结构的化合物Ⅱ溶于有机溶剂中,制得化合物Ⅱ有机溶液,将溴酸钠的水溶液加入所述化合物Ⅱ有机溶液中,再缓慢加入亚硫酸氢钠的水溶液进行回流反应,反应完全后去除反应液中的水相,得到具有式Ⅲ结构的化合物Ⅲ的有机相;
制备化合物Ⅳ的有机相:
将所述具有式Ⅲ结构的化合物Ⅲ的有机相与碱的水溶液混合,再分批加入苯硫酚钠进行硫醚化反应,反应完全后,经冷却、酸化、去除反应液中的水相,得到具有式Ⅳ结构的化合物Ⅳ的有机相;
制备化合物Ⅰ:
所述具有式Ⅳ结构的化合物Ⅳ的有机相与酰氯试剂进行氯代反应,反应完全后,分批加入无水氯化铝进行傅克酰基化反应,得到具有式Ⅰ结构的化合物Ⅰ。
本发明提供的连续反应制备硫杂
在一些实施例中,在所述制备化合物Ⅲ的有机相的步骤中,所述有机溶剂为二氯甲烷、1,2-二氯乙烷、三氯甲烷或四氯化碳中的至少一种。
优选的,所述有机溶剂为1,2-二氯乙烷。
在一些实施例中,在所述制备化合物Ⅲ的有机相的步骤中,所述回流反应的温度为85℃~105℃(例如,可以是85℃、90℃、95℃、98℃、100℃、102℃或105℃等),反应时间为2h~6h(例如,可以是2h、3h、4h、5h或6h等)。
在一些实施例中,在所述制备化合物Ⅲ的有机相的步骤中,所述化合物Ⅱ、溴酸钠、亚硫酸氢钠之间的摩尔比为1:6:3~1:3:1.5。优选的,所述化合物Ⅱ、溴酸钠、亚硫酸氢钠之间的摩尔比为1:6:3。
在一些实施例中,在所述制备化合物Ⅳ的有机相的步骤中,所述硫醚化反应的温度为85℃~105℃(例如,可以是85℃、90℃、95℃、98℃、100℃、102℃或105℃等),反应时间为1.5h~3h(例如,可以是1.5h、2h或3h等);冷却温度为0℃~5℃(例如,可以是0℃、1℃、2℃、3℃、4℃或5℃等);酸化试剂为浓度为1mol/L的盐酸溶液。
在一些实施例中,所述制备化合物Ⅳ的有机相的步骤中,所述化合物Ⅲ、碳酸钾、苯硫酚钠之间的摩尔比为10:13:11。
在一些实施例中,在所述制备化合物Ⅰ的步骤中,所述酰氯试剂为草酰氯和/或氯化亚砜。
优选的,所述酰氯试剂为草酰氯。
在一些实施例中,在所述制备化合物Ⅰ的步骤中,所述氯代反应的温度为20℃~30℃(例如,可以是20℃、25℃或30℃等),反应时间为2h~4h(例如,可以是2h、3h或4h等);所述傅克酰基化反应的温度为80℃~90℃(例如,可以是80℃、85℃或90℃等),反应时间为2h~3h(例如,可以是2h、2.5h或3h等)。
在一些实施例中,在制备化合物Ⅰ的步骤中,所述化合物Ⅳ、酰氯试剂、无水氯化铝之间的摩尔比为10:12:15。
在一些实施例中,在分批加入无水氯化铝进行傅克酰基化反应之后,还包括:去除反应液中的水相,得到具有式Ⅰ结构的化合物Ⅰ的有机相;对所述化合物Ⅰ的有机相进行减压蒸馏,之后加入正庚烷打浆,再经过滤、结晶,得到所述具有式Ⅰ结构的化合物Ⅰ。
本发明实施例提供的连续反应制备硫杂
在一些实施例中,本发明实施例提供的方法还包括如下步骤:
具有式Ⅰ结构的化合物Ⅰ与四氢硼酸钠(NaBH
该化合物Ⅵ可以用作制备巴洛沙韦的关键中间体的原料。其中,巴罗沙韦的关键中间体的化学名称为(±)-12-(7,8-二氟-6,11-二氢二苯并[b,e]硫杂-11-基)-7-(羟基)-3,4,12,12a四氢-1H-[1,4]噁嗪基[3,4-c]吡啶基[2,1-f][1,2,4]三嗪-6,8-二酮,其结构式为
图1是本发明实施例提供的连续反应制备硫杂
结合图1,本发明实施例提供的连续反应制备硫杂
在一些实施例中,在制备化合物Ⅳ的有机相的步骤中,可以在将酸化反应后的反应液导入至第二萃取塔104,向第二萃取塔104加入体系体积的1%~5%的二甲基乙酰胺(DMAc)或者二甲基甲酰胺(DMF),搅拌均匀,以增加化合物Ⅳ在有机相的溶解度,提高萃取效果,然后再经液液分离,得到化合物Ⅳ的有机相。
在一些实施例中,在制备化合物Ⅰ的步骤中,可以在第三反应釜106与干燥塔105之间增加一个浓缩釜,从干燥塔105中流出的化合物Ⅳ的有机相可先经浓缩釜浓缩,去除其中的有机相,得到化合物Ⅳ浓缩产物,之后再将该化合物Ⅳ浓缩产物导入至第三反应釜106中,加入与在制备化合物Ⅲ的有机相的步骤中所使用的有机溶剂不同种类的有机溶剂溶解化合物Ⅳ浓缩产物,之后再加入酰氯试剂进行氯代反应。例如,在制备化合物Ⅲ的有机相的步骤中所使用的有机溶剂为1,2-二氯乙烷,那么可使用二氯甲烷、二氯乙烷或三氯甲烷来重新溶解化合物Ⅳ浓缩产物,之后再进行后续的氯代反应、傅克酰基化反应,得到最终产物。
本发明先后进行过多次试验,现举一部分试验结果作为参考对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
下述实施例中,核磁共振谱用Varian Mercury 400和600核磁共振仪记录,化学位移以δ(ppm)表示;质谱用Bruker ESQUIRE-3000型质谱仪记录,电喷雾电离(ESI);分离用硅胶未说明的均为200-300目。
实施例1
S1、制备化合物Ⅲ的有机相:
在第一反应釜101(溴代反应釜)中,将原料3,4-二氟-2-甲基苯甲酸(化合物Ⅱ)(5.16g,30mmol)溶于180mL 1,2-二氯乙烷中,制得化合物Ⅱ有机溶液,将溴酸钠(27.16g,180mmol)溶于180mL水,制得溴酸钠的水溶液,再将溴酸钠的水溶液加入上述化合物Ⅱ有机溶液中;另将亚硫酸氢钠(9.36g,90mmol)溶于50mL水后,得到亚硫酸钠的水溶液,再将亚硫酸钠的水溶液缓慢加入第一反应釜101中,升温至100℃回流反应约5h后降温,将反应液通入第一萃取塔102中,经液液分离除去水相,得到的化合物Ⅲ的有机相通入第二反应釜103(硫醚化反应釜)中进行下一步反应,此步的产率为85%。
对制得的化合物Ⅲ进行鉴定:
化合物Ⅲ的核磁共振氢谱谱图如图2所示,
1
化合物Ⅲ的核磁共振碳谱谱图如图3所示,
13
S2、制备化合物Ⅳ的有机相:
在第二反应釜103(硫醚化反应釜)中,取上述步骤S1制备的2-(溴甲基)-3,4-二氟苯甲酸(化合物Ⅲ)(2.51g,10mmol)的1,2-二氯乙烷溶液与碳酸钾(1.79g,13mmol)的水溶液30mL进行搅拌混合充分后,再分批加入苯硫酚钠(1.45g,11mmol),将反应液升温至100℃回流反应约2h后,将反应液冷却至0℃~5℃,经1N盐酸15mL(15mmol HCl)酸化后,酸化反应液进入第二萃取塔104,经液液分离除去水相,得到的具有式Ⅳ结构的化合物Ⅳ的有机相通入干燥塔105中,经干燥塔105干燥,之后再将干燥后的产物通入第三反应釜106(酰化反应釜)进行下一步反应,此步的产率为89%。
对制得的化合物Ⅳ进行鉴定:
化合物Ⅳ的核磁共振氢谱谱图如图4所示,
1
化合物Ⅳ的核磁共振碳谱谱图如图5所示,
13
S3、制备化合物Ⅰ:
在第三反应釜106(酰化反应釜)中,将上述制备的干燥3,4-二氟-2-((苯硫基)甲基)苯甲酸(化合物Ⅳ)(2.58g,10mmol)的1,2-二氯乙烷溶液与12mmol草酰氯在常温25℃下搅拌反应3h,待原料化合物Ⅳ消失后,在常温下向反应液中分批加入1.99g(15mmol)无水氯化铝,混合均匀后将反应液升温至85℃回流反应2h,待反应结束后加入50mL~100mL冷水淬灭反应,之后将反应混合液通入第三萃取塔107中,经液液分离除去水相,得到的具有式Ⅰ结构的化合物Ⅰ的有机相,再将该化合物Ⅰ的有机相通入溶剂回收塔108中进行减压蒸馏至残留溶剂约5mL后加入50mL正庚烷打浆,有浅黄色固体析出,即为产物化合物Ⅰ,过滤后进入结晶釜109结晶,最终得到产品化合物Ⅰ,纯度为99%,总收率57%。
对制得的化合物Ⅰ进行鉴定:
化合物Ⅰ的核磁共振氢谱谱图如图6所示,
1
化合物Ⅰ的核磁共振碳谱谱图如图7所示,
13
实施例2
S1、制备化合物Ⅲ的有机相:
化合物Ⅲ的合成路线和实施例1中的化合物Ⅲ的合成路线相同。
在第一反应釜101(溴代反应釜)中,将原料3,4-二氟-2-甲基苯甲酸(化合物Ⅱ)(5.16g,30mmol)溶于180mL二氯甲烷中,制得化合物Ⅱ有机溶液,将溴酸钠(27.16g,180mmol)溶于180mL水,制得溴酸钠的水溶液,再将溴酸钠的水溶液加入上述化合物Ⅱ有机溶液中;另将亚硫酸氢钠(9.36g,90mmol)溶于50mL水后,得到亚硫酸钠的水溶液,再将亚硫酸钠的水溶液缓慢加入第一反应釜101中,升温至100℃回流反应约5h后降温,将反应液通入第一萃取塔102中,经液液分离除去水相,得到的化合物Ⅲ的有机相通入第二反应釜103(硫醚化反应釜)中进行下一步反应,此步产率为75%。
S2、制备化合物Ⅳ的有机相:
化合物Ⅳ的合成路线与实施例1中的化合物Ⅳ的合成路线相同。
参照实施例1中的步骤S2的方法制备得到具有式Ⅳ结构的化合物Ⅳ的有机相,将该化合物Ⅳ的通入干燥塔105中,经干燥塔105干燥,之后再将干燥后的产物通入第三反应釜106(酰化反应釜)进行下一步反应,此步产率为80%。
S3、制备化合物Ⅰ:
化合物Ⅰ的合成路线与实施例1中的化合物Ⅰ的合成路线相同。
参照实施例1中的步骤S3的方法制备得到具有式Ⅰ结构的化合物Ⅰ,纯度为99%,总收率53%。
实施例3
S1、制备化合物Ⅲ的有机相:
化合物Ⅲ的合成路线和实施例1中的化合物Ⅲ的合成路线相同。
在第一反应釜101(溴代反应釜)中,将原料3,4-二氟-2-甲基苯甲酸(化合物Ⅱ)(5.16g,30mmol)溶于180mL三氯甲烷中,制得化合物Ⅱ有机溶液,将溴酸钠(27.16g,180mmol)溶于180mL水,制得溴酸钠的水溶液,再将溴酸钠的水溶液加入上述化合物Ⅱ有机溶液中;另将亚硫酸氢钠(9.36g,90mmol)溶于50mL水后,得到亚硫酸钠的水溶液,再将亚硫酸钠的水溶液缓慢加入第一反应釜101中,升温至100℃回流反应约5h后降温,将反应液通入第一萃取塔102中,经液液分离除去水相,得到的化合物Ⅲ的有机相通入第二反应釜103(硫醚化反应釜)中进行下一步反应,此步产率为82%。
S2、制备化合物Ⅳ的有机相:
化合物Ⅳ的合成路线与实施例1中的化合物Ⅳ的合成路线相同。
参照实施例1中的步骤S2的方法制备得到具有式Ⅳ结构的化合物Ⅳ的有机相,将该化合物Ⅳ的通入干燥塔105中,经干燥塔105干燥,之后再将干燥后的产物通入第三反应釜106(酰化反应釜)进行下一步反应,此步产率为83%。
S3、制备化合物Ⅰ:
化合物Ⅰ的合成路线与实施例1中的化合物Ⅰ的合成路线相同。
参照实施例1中的步骤S3的方法制备得到具有式Ⅰ结构的化合物Ⅰ,纯度为98%,总收率52%。
实施例4
S1、制备化合物Ⅲ的有机相:
化合物Ⅲ的合成路线和实施例1中的化合物Ⅲ的合成路线相同。
在第一反应釜101(溴代反应釜)中,将原料3,4-二氟-2-甲基苯甲酸(化合物Ⅱ)(5.16g,30mmol)溶于180mL四氯化碳中,制得化合物Ⅱ有机溶液,将溴酸钠(27.16g,180mmol)溶于180mL水,制得溴酸钠的水溶液,再将溴酸钠的水溶液加入上述化合物Ⅱ有机溶液中;另将亚硫酸氢钠(9.36g,90mmol)溶于50mL水后,得到亚硫酸钠的水溶液,再将亚硫酸钠的水溶液缓慢加入第一反应釜101中,升温至100℃回流反应约5h后降温,将反应液通入第一萃取塔102中,经液液分离除去水相,得到的化合物Ⅲ的有机相通入第二反应釜103(硫醚化反应釜)中进行下一步反应,此步产率为80%。
S2、制备化合物Ⅳ的有机相:
化合物Ⅳ的合成路线与实施例1中的化合物Ⅳ的合成路线相同。
参照实施例1中的步骤S2的方法制备得到具有式Ⅳ结构的化合物Ⅳ的有机相,将该化合物Ⅳ的通入干燥塔105中,经干燥塔105干燥,之后再将干燥后的产物通入第三反应釜106(酰化反应釜)进行下一步反应,此步产率为82%。
S3、制备化合物Ⅰ:
化合物Ⅰ的合成路线与实施例1中的化合物Ⅰ的合成路线相同。
参照实施例1中的步骤S3的方法制备得到具有式Ⅰ结构的化合物Ⅰ,纯度为98.5%,总收率53.8%。
实施例5
S1、制备化合物Ⅲ的有机相:
化合物Ⅲ的合成路线和实施例1中的化合物Ⅲ的合成路线相同。
参照实施例1中的步骤S1的方法制备得到的化合物Ⅲ的有机相通入第二反应釜103(硫醚化反应釜)中进行下一步反应,此步产率为89%。
S2、制备化合物Ⅳ的有机相:
化合物Ⅳ的合成路线与实施例1中的化合物Ⅳ的合成路线相同。
参照实施例1中的步骤S2的方法制备得到具有式Ⅳ结构的化合物Ⅳ的有机相,将该化合物Ⅳ的通入干燥塔105中,经干燥塔105干燥,之后再将干燥后的产物通入第三反应釜106(酰化反应釜)进行下一步反应,此步产率为90%。
S3、制备化合物Ⅰ:
化合物Ⅰ的合成路线与实施例1中的化合物Ⅰ的合成路线相同。
在第三反应釜106(酰化反应釜)中,将上述制备的干燥3,4-二氟-2-((苯硫基)甲基)苯甲酸(化合物Ⅳ)(2.58g,10mmol)的1,2-二氯乙烷溶液与12mmol氯化亚砜在常温25℃下搅拌反应3h,待原料化合物Ⅳ消失后,在常温下向反应液中分批加入1.99g(15mmol)无水氯化铝,混合均匀后将反应液升温至85℃回流反应2h,待反应结束后加入50mL~100mL冷水淬灭反应,之后将反应混合液通入第三萃取塔107中,经液液分离除去水相,得到的具有式Ⅰ结构的化合物Ⅰ的有机相,再将该化合物Ⅰ的有机相通入溶剂回收塔108中进行减压蒸馏至残留溶剂约5mL后加入50mL正庚烷打浆,有浅黄色固体析出,即为产物化合物Ⅰ,过滤后进入结晶釜109结晶,最终得到产品化合物Ⅰ,纯度为99%,总收率54.5%。
实施例6
S1、制备化合物Ⅲ的有机相:
化合物Ⅲ的合成路线和实施例1中的化合物Ⅲ的合成路线相同。
在第一反应釜101(溴代反应釜)中,将原料3,4-二氟-2-甲基苯甲酸(化合物Ⅱ)(5.16g,30mmol)溶于180mL三氯甲烷中,制得化合物Ⅱ有机溶液,将溴酸钠(27.16g,180mmol)溶于180mL水,制得溴酸钠的水溶液,再将溴酸钠的水溶液加入上述化合物Ⅱ有机溶液中;另将亚硫酸氢钠(4.68g,45mmol)溶于50mL水后,得到亚硫酸钠的水溶液,再将亚硫酸钠的水溶液缓慢加入第一反应釜101中,升温至100℃回流反应约5h后降温,将反应液通入第一萃取塔102中,经液液分离除去水相,得到的化合物Ⅲ的有机相通入第二反应釜103(硫醚化反应釜)中进行下一步反应,此步产率为81%。
S2、制备化合物Ⅳ的有机相:
化合物Ⅳ的合成路线与实施例1中的化合物Ⅳ的合成路线相同。
参照实施例1中的步骤S2的方法制备得到具有式Ⅳ结构的化合物Ⅳ的有机相,将该化合物Ⅳ的通入干燥塔105中,经干燥塔105干燥,之后再将干燥后的产物通入第三反应釜106(酰化反应釜)进行下一步反应,此步产率为92%。
S3、制备化合物Ⅰ:
化合物Ⅰ的合成路线与实施例1中的化合物Ⅰ的合成路线相同。
参照实施例1中的步骤S3的方法制备得到具有式Ⅰ结构的化合物Ⅰ,纯度为98.5%,总收率54.7%。
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围,均应包含在本发明的保护范围之内。
- 苄基氯类化合物和硫酚在无金属条件下一步反应制备亚砜化合物的方法
- 一种含有异羟肟酸的取代杂环类化合物及其制备方法和用途
- 一种苯并咪唑并手性杂环类化合物及其制备方法和应用
- 一种氮-烷基(氘代烷基)芳杂环和烷基(氘代烷基)芳基醚类化合物的制备方法
- 一种乙酰化二苯并-1,3-二氧杂环辛类化合物的制备方法与作为抗菌药物的应用
- 硫杂蒽酮类化合物及其制备方法和具有引发聚合反应功能的组合物以及制备共聚物的方法
- 硫杂蒽酮类化合物及其制备方法和具有引发聚合反应功能的组合物以及制备共聚物的方法