一种氮稠杂环化合物的晶型及其制备方法
文献发布时间:2024-04-18 19:52:40
技术领域
本发明涉及化合物的晶型,更具体地,涉及一种由特定的X射线粉末衍射图定义的氮稠杂环化合物的新型晶型及其制备方法。
背景技术
化合物I,(S)-4- [(5,7-二氟苯并二氢吡喃-4-基)氧基]-N,N,2-三甲基-1 H-苯并 [d]咪唑-6-甲酰胺的结构式如下式所示:
化合物I
专利号CN101341149B的专利公开了化合物I及其用途,化合物I作为一种钾离子竞争性酸阻滞剂(P-CAB),用于治疗胃食管反流疾病和糜烂性食管炎的治疗。专利号CN107207478B的专利公开了衍射角为8.1°、10.0°、12.6°、14.9°、15.6°、16.5°、17.2°、19.6°、23.1°、24.2°、28.1°、30.2°和31.6°(2θ±0.2°)处具有峰的化合物I的晶型A。
然而由于晶型A在水中的溶解度较小,导致药品的生物利用度受到了极大限制。
发明内容
为了解决上述问题,本发明提供了一种氮稠杂环化合物的晶型,氮稠杂环化合物具有化合物I所示的结构:
化合物I
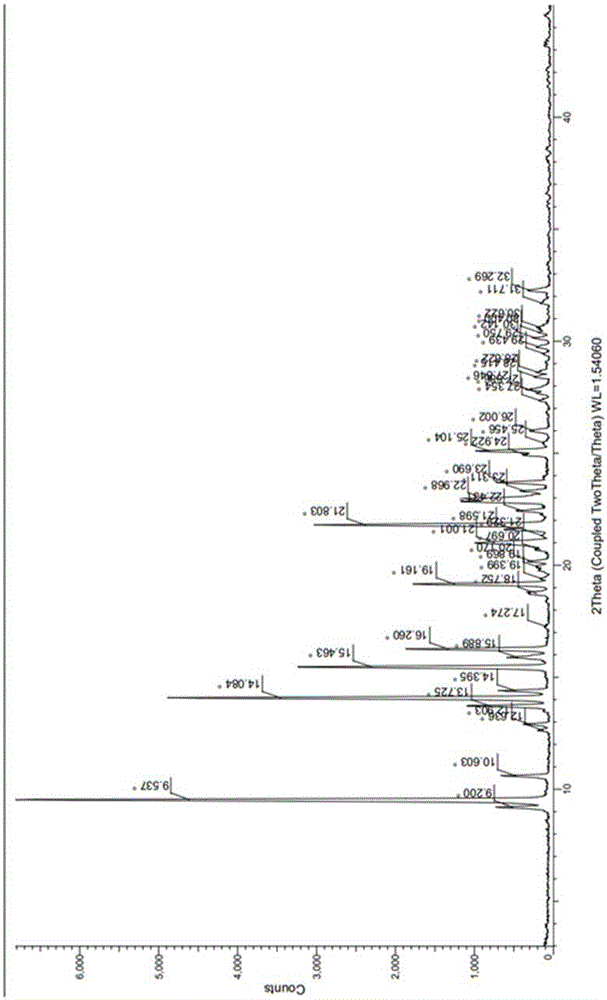
氮稠杂环化合物的晶型在使用Cu-Kα辐射时,以2θ值±0.2°表示的X射线粉末衍射图谱的特征衍射峰包括9.5、13.7、14.0、15.4、16.2、21.0、21.8、22.9、23.6和25.1。
化合物I以钾离子竞争性方式可逆性抑制H
作为一种优选的实施方式,在利用差示扫描量热法测量时,氮稠杂环化合物的晶型在150℃至165℃的温度下具有吸热峰。
本发明还提供了一种氮稠杂环化合物的其制备方法,制备方法如下:
S1.将化合物I加入第一溶剂中,得到混合物X;
混合物X中,化合物I和第一溶剂的比例为(8-12)g:(20-40)mL。
优选的,混合物X中,化合物I和第一溶剂的比例为(9-11)g:(20-40)mL。
进一步优选的,混合物X中,化合物I和第一溶剂的比例为10g:30mL。
第一溶剂为酯类溶剂。
酯类溶剂选自甲酸乙酯、乙酸乙酯、乙酸异丁酯、乙酸甲酯、乙酸丙酯的一种或多种。
优选的,酯类溶剂为乙酸乙酯。
S2.将混合物X升温至65-80℃,搅拌3-15min,以使化合物I溶清;降温至20-40℃;
优选的,步骤S2中将混合物X升温至70-75℃。
优选的,步骤S2中降温至25-35℃。
进一步优选的,步骤S2中降温至30℃。
S3.加入第二溶剂,降温至5-15℃后,保温搅拌50-70min;
第二溶剂加入的体积为第一溶剂的0.7-1.5倍。
优选的,第二溶剂加入的体积为第一溶剂的0.9-1.3倍,可列举的,第二溶剂加入的体积为第一溶剂的0.9、1.0、1.1、1.2或1.3倍。
第二溶剂选自甲基叔丁醚、环己烷、正庚烷、2,2,4-三甲基戊烷的一种或多种。
优选的,第二溶剂为甲基叔丁醚和/或正庚烷。
进一步优选的,第二溶剂为甲基叔丁醚或正庚烷。
优选的,步骤S3中加入第二溶剂,降温至10℃。
作为一种优选的实施方式,步骤S2和S3中,降温速率均为(8-12)℃/h。
优选的,步骤S2和S3中,降温速率均为(9-11)℃/h。
进一步优选的,步骤S2和S3中,降温速率均为10℃/h。
S4.过滤,取滤饼,干燥,即得。
有益效果:本发明通过简单的方法制备得到了一种溶解度高的氮稠杂环化合物的新晶型,与此同时,还意外地发现,通过控制本发明中的结晶方法中使用的有机溶剂,配合升降温度和保温过程,上述溶解度高的氮稠杂环化合物的新晶型还具有优异的稳定性和高的收率。尤其是在第一溶剂为酯类,第二溶剂为甲基叔丁醚和/或正庚烷时,且加入第二溶剂后降温至5-15℃再结合保温搅拌,所得的新的晶型的产率≥90%的同时,在水中的溶解度>0.05mg/mL,并且光稳定性和加速稳定性尤为显著。
附图说明
图1为实施例1所得的氮稠杂环化合物的晶型在使用Cu-Kα辐射时的X射线衍射图;
图2为实施例1所得的氮稠杂环化合物的晶型通过DSC(差示扫描量热法)而形成的温谱图。
实施方式
实施例
将100g (S)-4- [(5,7-二氟苯并二氢吡喃-4-基)氧基]-N,N,2-三甲基-1 H-苯并[d]咪唑-6-甲酰胺加入300ml乙酸乙酯中,升温至70℃,搅拌10min,使化合物1溶清,以10℃/h的降温速率降温至30℃,加入300ml甲基叔丁基醚,以10℃/h的降温速率降温继续降温至10℃,保温搅拌1小时,过滤,干燥,得到45g化合物I的晶型(产率:90%)。
实施例
将100g 化合物I加入300ml乙酸乙酯中,升温至75℃,搅拌10min,使化合物1溶清,以10℃/h的降温速率降温至30℃,加入300ml正庚烷,继续降温至10℃(降温速率10℃/h),保温搅拌1小时,过滤,干燥,得到46g化合物I的晶型(产率:92%)
对比例1
参照专利号CN 107207478 B的发明专利的说明书的实施例3,将50g 化合物I(化合物I即为(-)-4-[((4S)-5 ,7-二氟-3 ,4-二氢-2H-色烯-4-基)氧基]-N ,N ,2-三甲基-1H-苯并咪唑-6-甲酰胺)加入250mL纯净水中,加热混合溶液至其内部温度达到100℃。将混合溶液在相同温度下搅拌12h,然后缓慢冷却至室温后,继续搅拌36小时使晶体熟化,过滤,滤饼在40℃下真空干燥,即得化合物I的结晶形式A。
性能测试:
溶解度测试:取实施例1得到的化合物I的晶型和对比例1中得到的化合物I的结晶形式A各1g,分别加入到25ml容量瓶中,并在容量瓶分别加入水溶解定容后,转移至离心管,600-1500r/min离心1-2min后,取上清液置入液相色谱测定其中化合物I的含量,以得到化合物I在水中的溶解度。
按照上述方法,将水替换成pH=3.0的稀盐酸水溶液,测定化合物I在pH=3.0的稀盐酸水溶液中的溶解度。
结果如表1。
表1
光稳定性研究:参照中国药典9001原料药物与制剂稳定性试验指导原则规定试验。
将实施例1得到的化合物I的晶型和对比例1中得到的化合物I的结晶形式A各取2g开口放在光照箱内,在照度为4500lx±500lx的条件下,且光源总照度应不低于1.2 X10
表2
加速稳定性研究:参照中国药典9001原料药物与制剂稳定性试验指导原则规定试验。
将实施例1得到的化合物I的晶型和对比例1中得到的化合物I的结晶形式A放置在稳定性试验箱中,温度40℃±2℃、相对湿度75%±5%的条件下放置3个月,结果如表3。
表3
/>
- 一种氮掺杂碳材料负载钴催化剂及其制备方法和在N-杂环化合物催化氧化中的应用
- 一种多苯并五元芳杂环并香豆素稠杂环化合物的合成方法
- 含氮五元芳香杂环化合物及其制备方法和应用
- 含氮稠杂环化合物的盐、晶型及其制备方法、药物组合物和用途
- 含氮稠杂环化合物的盐的晶型及其制备方法和应用