基于CRISPR-dCas9的基因靶向去甲基化方法及其应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于基因工程技术领域,涉及一种用于基因靶向去甲基化的系统及其应用。
背景技术
DNA甲基化在食管鳞癌的发生发展中发挥着重要作用,有许多异常甲基化基因被报道为食管鳞癌患者的诊断和预后标志物。由于DNA甲基化是可逆的,因此DNA甲基化在食管鳞癌的治疗中具有巨大的潜力。临床医生针对DNA甲基化异常的癌症患者主要使用DNA甲基转移酶抑制剂(DNA methyltransferase inhibitors,DNMTis)进行治疗。常用的临床药物包括阿扎胞苷和地西他滨,它们可以通过抑制DNA甲基化从而逆转抑癌基因的表达沉默,进而呈现出较好的肿瘤抑制效果,在血液癌症的治疗有一定的效果,但常见获得性耐药,且在实体瘤的治疗中效果并不理想。此类药物仅靶向作用于抑制DNA甲基化转移酶,缺乏基因靶向性,不能靶向于特定的基因,可能会扰乱其它基因原本正常的甲基化状态及转录水平,因此此类药物的疗效和安全性有待进一步考究。
CRISPR-Cas9是一种通用高效的针对基因组特定位点进行精确编辑的技术。核酸内切酶Cas9蛋白在细胞内sgRNA的引导下能够通过碱基互补配对原则识别靶标的基因组DNA,并由Cas9蛋白的活性结构域对NGG序列上游3-4碱基之间的DNA进行剪切,产生双链DNA损伤(double-strand break,DSB)。断裂的DNA可以通过非同源末端连接(non-homologousend joining)或者同源重组(homologous recombination)被修复,从而达到基因靶向敲除的目的。LentiCRISPR质粒是Broad Institute中心的张峰基于CRISPR-Cas9技术推出了一种可用于慢病毒包装的质粒,该质粒的优点为:一、可以通过慢病毒感染细胞的方式实现靶向基因的稳定性敲除;二、病毒感染成功的细胞可以通过药物进行筛选,方便易操作。dCas9(nuclease“dead”Cas9)蛋白是Cas9蛋白的突变体,即Cas9内切酶的RuvC1和HNH两个核酸酶活性区域同时发生突变,使其丧失DNA剪切作用,因此,dCas9蛋白的内切酶活性全部消失,只保留由gRNA引导进入基因组的能力。Tet1CD是可以进行DNA去甲基化的最小功能单位。通过选用LentiCRISPR质粒,在CRISPR/Cas9的基础上进一步将Cas9蛋白去掉核酸酶活性,改造为dCas9,并将其与Tet1CD进行连接,形成融合蛋白dCas9-Tet1CD,即可在细胞内sgRNA的引导下实现DNA的靶向去甲基化功能。
因此有必要提供一种精确的表观遗传编辑技术,逆转由于启动子高甲基化修饰导致的基因表达沉默,恢复基因的正常功能,调控表观遗传修饰的方式,为食管鳞癌的治疗提供更多可选选项。
发明内容
有鉴于此,本发明通过设计sgRNA以及可表达定位功能元件与去甲基化功能元件的载体,以慢病毒感染细胞的方式,配合药物筛选,在sgRNA引导下定位功能元件识别靶基因组DNA,再利用去甲基化功能元件对靶基因组DNA进行去甲基化,提供一种精确的表观遗传编辑技术,逆转由于启动子高甲基化修饰导致的基因表达沉默,恢复基因的正常功能,调控表观遗传修饰的方式,为因表观遗传修饰失常引起疾病的治疗提供更多可选选项。
本发明第一方面提供用于基因靶向去甲基化的sgRNA,包括sgZNF154-1、sgZNF154-2和/或
sgZNF154-3;其中sgZNF154-1包括如SEQ ID NO:13所示的正向序列及如SEQ IDNO:14所示的反向序列,sgZNF154-2包括如SEQ ID NO:15所示的正向序列及如SEQ ID NO:16所示的反向序列,sgZNF154-3包括如SEQ ID NO:17所示的正向序列及如SEQ ID NO:18所示的反向序列。
本发明第二方面提供用于基因靶向去甲基化的复合物,主要由如本发明第一方面所述的sgRNA和融合蛋白相互结合形成,所述的融合蛋白包括定位功能元件和去甲基化功能元件。
在本发明一实施例中,所述定位功能元件具有靶向和结合DNA的功能但无催化活性,包括Cas蛋白、锌指蛋白或TALENs蛋白,或其功能域,或其组合。
在本发明一优选实施例中,所述定位功能元件包括dCas9、dCpf1、dCas12、dCas13、dCms1、dMAD7,或其功能域,或其组合。
在本发明一优选实施例中,所述定位功能元件包括dCas9或其功能域。
在本发明一实施例中,所述去甲基化功能元件具有具有将甲基化胞嘧啶转换为非甲基化胞嘧啶的功能,包括ROS1、TET、DME、DML,或其组合。
在本发明一优选实施例中,所述定位功能元件包括Tet1、Tet2、Tet3或其功能域,或其组合。
在本发明一优选实施例中,所述定位功能元件包括Tet1或其功能域。
在本发明一实施例中,所述定位功能元件和去甲基化功能元件通过一个或多个下列组件连接:肽键、连接肽、核定位信号、表位标签,或其组合。
本发明第三方面提供用于基因靶向去甲基化的载体,包括如本发明第一方面所述的sgRNA和编码如本发明第二方面所述融合蛋白的核酸序列。
在本发明一实施例中,所述编码融合蛋白的核酸序列如SEQ ID NO:29所示。
本发明第四方面提供用于基因靶向去甲基化的宿主细胞,包括如本发明第一方面所述的sgRNA、如本发明第二方面所述的复合物和/或如本发明第三方面所述的载体。
本发明第五方面提供用于基因靶向的去甲基化方法,包括以下步骤:
1)靶向ZNF154启动子区域设计候选sgRNA;
2)将步骤1)所得sgRNA连接编码如本发明第二方面所述融合蛋白的核酸序列,构建靶向ZNF154启动子去甲基化的载体;
3)包装含步骤2)所得载体的宿主细胞;
4)将步骤3)所得宿主细胞加入食管癌细胞,药物筛选获取去甲基化成功的细胞;
5)筛选验证去甲基化效果好的载体或载体组合。
在本发明一实施例中,步骤2)编码所述融合蛋白的核酸序列如SEQ ID NO:29所示。
在本发明一实施例中,步骤3)所述宿主细胞为293T细胞。
在本发明一实施例中,步骤4)所述食管癌细胞为Kyse30细胞。
在本发明一实施例中,步骤4)所述药物为嘌呤霉素。
在本发明一实施例中,步骤5)所述筛选验证包括:a)数字PCR检测ZNF154启动子的甲基化率,b)qPCR检测ZNF154的mRNA水平,c)CCK8细胞增殖实验验证载体或载体组合对Kyse30细胞增殖抑制效果。
本发明第六方面提供如本发明第一方面所述的sgRNA、如本发明第二方面所述的复合物和/或如本发明第三方面所述的宿主细胞、如本发明第四方面所述的宿主细胞和/或如本发明第五方面所述的方法在对目标核酸进行去甲基化修饰中的应用。
本发明第七方面提供如本发明第一方面所述的sgRNA、如本发明第二方面所述的复合物和/或如本发明第三方面所述的宿主细胞、如本发明第四方面所述的宿主细胞和/或如本发明第五方面所述的方法在制备用于对目标核酸进行去甲基化修饰的试剂盒中的应用。
本发明有益效果如下:
1)本发明通过设计sgRNA以及可表达定位功能元件及去甲基化功能元件的载体,通过sgRNA引导下定位功能元件识别靶基因组DNA,再利用去甲基化功能元件对靶基因组DNA进行去甲基化,准确有效地实现了靶基因组DNA的稳定去甲基化;
2)本发明通过引入可用于慢病毒包装的LentiCRISPR质粒,通过慢病毒感染细胞的方式实现靶向基因的稳定性敲除,病毒感染成功的细胞可以通过药物进行筛选,方便易操作;
3)可精准地对ZNF154的启动子进行去甲基化,具备较好的去甲基化作用,且能有效抑制食管癌Kyse30细胞的增殖:本发明通过数字PCR检测ZNF154启动子的甲基化率、qPCR检测ZNF154的mRNA水平、CCK8细胞增殖实验等系统试验筛选验证得到去甲基化效果好的pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1载体以及pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1/2/3载体组合,其中采用sgZNF154-1/2/3组合对Kyse30细胞进行去甲基化处理后,ZNF154启动子的甲基化率相比对照组减少了8%,ZNF154的mRNA水平相比对照组上调了20倍。而小鼠皮下成瘤实验显示经其采用sgZNF154-1/2/3组合对Kyse30细胞进行去甲基化处理后的Kyse30细胞相比对照组生长速度明显减慢,其ZNF154启动子甲基化率相比对照组下调了大约10%-20%,ZNF154的mRNA水平相比对照组上调了200倍左右。
附图说明
图1为本发明实施例提供的质粒pLentiCRISPR-dCas9-Tet1CD的构建示意图;
图2为本发明实施例提供的质粒pLentiCRISPR-dCas9-Tet1CD的序列的结构图;
图3为本发明实施例提供的8条sgRNA在ZNF154启动子区的位置示意图;
图4为本发明实施例提供的8个质粒分别靶向ZNF154启动子去甲基化处理后数字PCR检测ZNF154启动子的甲基化率结果图;
图5为本发明实施例提供的8个质粒分别靶向ZNF154启动子去甲基化处理后qPCR检测ZNF154mRNA表达水平结果图;
图6为本发明实施例提供的质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1单独去甲基化处理Kyse30细胞后对其增殖影响结果图;
图7为本发明实施例提供的质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1/2/3组合去甲基化处理Kyse30细胞后对其增殖影响结果图;
图8为本发明实施例提供的质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1/2/3组合去甲基化处理Kyse30细胞后数字PCR检测ZNF154启动子的甲基化率结果图;
图9为本发明实施例提供的质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1/2/3组合去甲基化处理Kyse30细胞后qPCR检测ZNF154 mRNA表达水平结果图;
图10为本发明实施例提供的采用DNA甲基转移酶抑制剂对Kyse30细胞去甲基化处理后数字PCR检测ZNF154启动子的甲基化率结果图;
图11为本发明实施例提供的采用DNA甲基转移酶抑制剂对Kyse30细胞去甲基化处理后qPCR检测ZNF154 mRNA表达水平结果图;
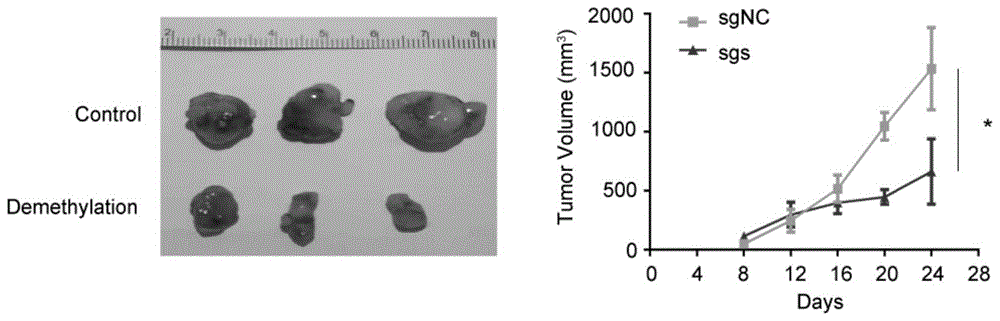
图12为本发明实施例提供的去甲基化细胞Kyse30注射小鼠皮下后皮下肿瘤拍照称量结果图;
图13为本发明实施例提供的去甲基化细胞Kyse30注射小鼠皮下后数字PCR检测皮下肿瘤ZNF154启动子的甲基化率结果图;
图14为本发明实施例提供的去甲基化细胞Kyse30注射小鼠皮下后qPCR检测皮下肿瘤ZNF154mRNA表达水平结果图。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,例如Sambrook等人,分子克隆:实验室手册(New York:Cold Spring HarborLaboratoryPress,1989)中所述的条件,或按照制造厂商所建议的条件。
本发明通过设计sgRNA以及可表达定位功能元件与去甲基化功能元件的载体,以慢病毒感染细胞的方式,配合药物筛选,在sgRNA引导下定位功能元件识别靶基因组DNA,再利用去甲基化功能元件对靶基因组DNA进行去甲基化,提供一种精确的表观遗传编辑技术,逆转由于启动子高甲基化修饰导致的基因表达沉默,恢复基因的正常功能,调控表观遗传修饰的方式,为因表观遗传修饰失常引起疾病的治疗提供更多可选选项。
本发明提供的基因靶向去甲基化系统中的sgRNA,包括sgZNF154-1、sgZNF154-2和/或
sgZNF154-3;其中sgZNF154-1包括如SEQ ID NO:13所示的正向序列及如SEQ IDNO:14所示的反向序列,sgZNF154-2包括如SEQ ID NO:15所示的正向序列及如SEQ ID NO:16所示的反向序列,sgZNF154-3包括如SEQ ID NO:17所示的正向序列及如SEQ ID NO:18所示的反向序列。
本发明提供的基因靶向去甲基化系统中的复合物,主要由靶向ZNF154的sgRNA和融合蛋白相互结合形成,所述的靶向ZNF154的sgRNA包括前述sgZNF154-1、sgZNF154-2和/或sgZNF154-3,所述的融合蛋白包括定位功能元件和去甲基化功能元件。
优选地,所述定位功能元件具有靶向和结合DNA的功能但无催化活性,包括Cas蛋白、锌指蛋白或TALENs蛋白,或其功能域,或其组合。
进一步优选地,所述定位功能元件包括dCas9、dCpf1、dCas12、dCas13、dCms1、dMAD7,或其功能域,或其组合。
进一步优选地,所述定位功能元件包括dCas9或其功能域。
优选地,所述去甲基化功能元件具有具有将甲基化胞嘧啶转换为非甲基化胞嘧啶的功能,包括ROS1、TET、DME、DML,或其组合。
进一步优选地,所述定位功能元件包括Tet1、Tet2、Tet3或其功能域,或其组合。
进一步优选地,所述定位功能元件包括Tet1或其功能域。
优选地,所述定位功能元件和去甲基化功能元件通过一个或多个下列组件连接:肽键、连接肽、核定位信号、表位标签,或其组合。
本发明提供的基因靶向去甲基化系统中的载体,包括靶向ZNF154的sgRNA和编码融合蛋白的核酸序列,所述的靶向ZNF154的sgRNA包括前述sgZNF154-1、sgZNF154-2和/或sgZNF154-3。
优选地,所述编码融合蛋白的核酸序列如SEQ ID NO:29所示。
本发明提供的基因靶向去甲基化系统中的宿主细胞,包括前述靶向ZNF154的sgRNA、复合物和/或载体。
本发明提供的基因靶向去甲基化系统的去甲基化方法,包括以下步骤:
1)靶向ZNF154启动子区域设计候选sgRNA;
2)将步骤1)所得sgRNA连接编码如本发明第二方面所述融合蛋白的核酸序列,构建靶向ZNF154启动子去甲基化的载体;
3)包装含步骤2)所得载体的宿主细胞;
4)将步骤3)所得宿主细胞加入食管癌细胞,药物筛选获取去甲基化成功的细胞;
5)筛选验证去甲基化效果好的载体或载体组合。
优选地,步骤2)编码所述融合蛋白的核酸序列如SEQ ID NO:29所示。
优选地,步骤3)所述宿主细胞为293T细胞。
优选地,步骤4)所述食管癌细胞为Kyse30细胞。
优选地,步骤4)所述药物为嘌呤霉素。
优选地,步骤5)所述筛选验证包括:a)数字PCR检测ZNF154启动子的甲基化率,b)qPCR检测ZNF154的mRNA水平,c)CCK8细胞增殖实验验证载体或载体组合对Kyse30细胞增殖抑制效果。
为更清楚展示本发明的技术方案,下面将结合具体实施例进一步阐述本发明。
实施例一、质粒LentiCRISPR-dCas9-Tet1CD的构建
1、针对LentiCRISPR质粒(Zhang Lab,pXPR_001)上不含Cas9的核酸序列以及pdCas9-Tet1-CD质粒(Addgene,plasmid#83340)上的dCas9-Tet1CD核酸序列设计PCR引物,并各自加上同源臂,按表1、表2、表3所示的PCR引物序列、反应体系和反应程序进行PCR扩增。
表1PCR引物序列(含同源臂)
表2PCR反应体系
表3PCR程序
2、用同源重组酶对步骤1所得PCR纯化产物进行体外同源重组(如图1所示),转化,体外同源重组的反应体系如表4所示。
表4体外同源重组的反应体系
注:X和Y的计算方法如下:
1)50–100ng载体对应2倍物质的量插入片段;
2)pmols=(weight in ng)x 1,000/(base pairs x 650daltons)
3)体积(μL)=质量/浓度
37℃孵育30min,进行细菌转化。挑取单克隆进行测序。经扩培后,用Tiangen质粒提取试剂盒提取质粒备用。命名为LentiCRISPR-dCas9-Tet1CD,其序列图谱如图2所示。
实施例二、靶向ZNF154启动子的去甲基化质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154的构建
1、针对靶基因序列设计8个靶向ZNF154启动子区域的候选序列,具体如下:AGGTTGGTGCAAAGGGTCCC(SEQ ID NO:5)、AGCTTCGCTTTTACTCCAAG(SEQ ID NO:6)、GACCCTTTGCACCAACCTCT(SEQ ID NO:7)、TGTAGTTTTCATAGATCCCG(SEQ ID NO:8)、TGGAGTAAAAGCGAAGCTCC(SEQ ID NO:9)、GGAGTAAAAGCGAAGCTCCA(SEQ ID NO:10)、TCGCTTTTACTCCAAGAGGT(SEQ ID NO:11)、AATGGCTTATCCAAGTCCTA(SEQ ID NO:12),在上述8个候选序列添加用于连接反应的粘性末端,合成Cas9 sgRNA序列,具体如表5所示。
表5靶向ZNF154启动子区域的Cas9 sgRNA序列
2、将单链sgRNA退火形成双链sgRNA
用去离子水分别对SEQ ID NO:13和SEQ ID NO:14、SEQ ID NO:15和SEQ ID NO:16、SEQ IDNO:17和SEQ ID NO:18、SEQ ID NO:19和SEQ ID NO:20、SEQ ID NO:21和SEQ IDNO:22、SEQ ID NO:23和SEQ ID NO:24、SEQ ID NO:25和SEQ ID NO:26、SEQ ID NO:27和SEQID NO:28所示的DNA序列进行溶解,使得各sgRNA序列的终浓度为10μM。PCR程序为:37℃30min,95℃5min,随后每分钟降低5℃,直至25℃为止,得到8条双链sgRNA,标记为sgZNF154-1、sgZNF154-2、sgZNF154-3、sgZNF154-4、sgZNF154-5、sgZNF154-6、sgZNF154-7、sgZNF154-8,8条sgRNA在在ZNF154启动子区的位置如图3所示。PCR反应体系如表6所示。
表6双链sgRNA合成体系
3、对LentiCRISPR-dCas9-Tet1CD进行酶切
用BsmBI对LentiCRISPR-dCas9-Tet1CD进行酶切,酶切体系如表7所示。
表7BsmBI酶切反应体系
反应条件为37℃,4-5h,得酶切产物,通过琼脂糖凝胶电泳,收取10000bp以上的条带,用TIANGEN DNA纯化试剂盒进行回收,并溶解于30μL去离子水中。标记为pLentiCRISPR-dCas9-Tet1CD,其核酸序列如SEQ ID NO:29所示。
4、构建质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154
将步骤2、步骤3的产物用快速连接酶试剂盒进行连接反应。反应体系如表8所示。
表8连接反应体系
室温反应15min。然后大肠杆菌转化,涂板,挑取单克隆,并进行测序。测序成功的质粒即为pLentiCRISPR-dCas9-Tet1CD-sgZNF154质粒。
实施例三、Kyse30细胞中对ZNF154启动子去甲基化
1、包装含有质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154的慢病毒
将构建的8条去甲基化质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154(1μg)分别与辅助质粒PXPAX2(2μg)、PMD.2G(1μg),转染试剂X-treme(20μL),opti-MEM(500μL)一起25℃孵育20min,加入到293T细胞中(10%胎牛血清的DMEM:5ml)。14h后换成5%胎牛血清的培养基DMEM,放置细胞培养箱中48h后,收集上清液,500g离心10min并用0.45μm的滤器过滤,获得病毒液。
2、获得去甲基化成功的细胞
向Kyse30细胞中(细胞汇合度50%-60%,6孔板,10%胎牛血清的1640:2ml)加入慢病毒液1ml,12h后换成正常培养基培养48h后,继续加入含有1μg/ml嘌呤霉素的培养基培养72h,即获得转染成功的细胞。接着将该细胞进行二次感染,并用正常培养基培养24h后,继续用含嘌呤霉素的培养基培养至15到20天。
在此其间用数字PCR检测ZNF154启动子的甲基化率(DNA浓度稀释至5-10ng/μL),结果如图4所示,sgZNF154-4、sgZNF154-5、sgZNF154-6、sgZNF154-7、sgZNF154-8等5条sgRNA处理后ZNF154启动子的甲基化率分别为93%、92%、91%、93%、97%,而对照组的甲基化率为93%,表明这5条sgRNA对ZNF154启动子几乎无去甲基化作用,而sgZNF154-1、sgZNF154-2、sgZNF154-3处理后ZNF154启动子的甲基化率分别为79%、88%、83%,表明sgZNF154-1、sgZNF154-2、sgZNF154-3可以在一定程度上对ZNF154的启动子进行去甲基化作用。
为进一步验证上述8条sgRNA的去甲基化效果,用qPCR检测ZNF154的mRNA水平,结果如图5所示,显示只有sgZNF154-1处理后ZNF154的mRNA水平大幅提高。
结论:质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154可以在一定程度上对ZNF154的启动子进行去甲基化作用。
实施例四、质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154对Kyse30细胞增殖影响实验
1、CCK8细胞增殖实验:
(1)取对数生长的细胞,消化成单个细胞,离心,重悬,计数。
(2)96孔板的每个孔接种1000个细胞,复孔5个,培养1周。
(3)培养期间换液一次。
(4)每孔加入10μL CCK8显色液,继续培养1h后,用酶标仪450nm测量吸光值。
结果,与对照组(sgNC)相比,sgZNF154-1(sg1)单独处理后对Kyse30细胞增殖抑制效果不明显
(如图6所示),而将sgZNF154-1、sgZNF154-2、sgZNF154-3组合处理后对Kyse30细胞增殖产生了明显的抑制效果(如图7所示)。
实施例五:sgZNF154-1/2/3组合对Kyse30细胞ZNF154启动子去甲基化
将实施例三步骤1、2操作步骤包装含有质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1/2/3的慢病毒,并获取去甲基化成功的细胞。
用数字PCR检测ZNF154启动子的甲基化率(DNA浓度稀释至5-10ng/μL),用qPCR检测ZNF154的mRNA水平。同时设置对照实验,利用DNA甲基转移酶抑制剂5-氮-2’-脱氧胞苷(5-aza-2’deoxycytidine,5-aza-dC)对Kyse30细胞去甲基化,具体步骤如下:1)将Kyse30按照2×105/孔的密度铺于6孔板中;2)每孔加入5-氮-2’-脱氧胞苷(5-aza-20-deoxycytidine,5-aza-dC)使其终浓度为2μM;3)2天换一次液,诱导4天,收集并裂解细胞用于甲基化和mRNA检测。
结果,采用sgZNF154-1/2/3组合对Kyse30细胞进行去甲基化处理后,数字PCR结果如图8所示,显示ZNF154启动子的甲基化率相比对照组减少了8%,,qPCR结果如图9所示,显示ZNF154的mRNA水平相比对照组上调了20倍。而采用药物对Kyse30细胞进行去甲基化处理后,数字PCR结果如图10所示,显示ZNF154启动子的甲基化率相比对照组减少了16%,qPCR结果如图11所示,显示ZNF154的mRNA水平相比对照组上调了8000倍左右。从上述结果可知使用DNA甲基转移酶抑制剂去甲基化效率更高,但由于缺乏基因靶向性,此类药物目前主要用于血液癌症的治疗,在实体瘤的治疗中效果并不理想,而本申请通过采用sgZNF154-1/2/3组合对Kyse30细胞进行去甲基化处理,利用sgRNA精确的表观遗传编辑技术,逆转由于启动子高甲基化修饰导致的基因表达沉默,恢复基因的正常功能,从而达到通过慢病毒感染细胞的方式实现靶向基因的稳定去甲基化,并通过药物筛选的方式获得去甲基化细胞的目的,从而提供一种方便易操作的基因靶向去甲基化技术。因此本申请提供的质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1/2/3组合可更精准地对ZNF154的启动子进行去甲基化且具备较好的甲基化作用。
实施例六:ZNF154启动子稳定去甲基化细胞Kyse30在小鼠功能实验中的应用
1、小鼠皮下成瘤实验
将去甲基化细胞Kyse30按4×10
结论:质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1/2/3组合在小鼠体内可以对Kyse30细胞的ZNF154启动子呈现出更好的去甲基化效果。
综上,本发明提供的通过采用质粒pLentiCRISPR-dCas9-Tet1CD-sgZNF154-1/2/3组合,利用sgRNA精确的表观遗传编辑技术,通过慢病毒感染细胞的方式,配合药物筛选,对Kyse30细胞进行去甲基化处理,从而实现靶向基因ZNF154的准确稳定去甲基化,进而调控ZNF154基因的表达,为食管癌的治疗提供一种新的思路与选择。
以上所述,仅为本发明的较佳实施例,并非对本发明作任何形式上和实质上的限制,凡熟悉本专业的技术人员,在不脱离本发明技术方案范围内,当可利用以上所揭示的技术内容,而作出的些许更动、修饰与演变的等同变化,均为本发明的等效实施例;同时,凡依据本发明的实质技术对以上实施例所作的任何等同变化的更动、修饰与演变,均仍属于本发明的技术方案的范围内。