抗体制剂
文献发布时间:2023-06-19 11:17:41
技术领域
本发明涉及一种抗血管生成素-2(ANG-2)-2和人血管内皮生长因子(VEGF,VEGF-A)的双特异性抗体(双特异性抗VEGF/ANG2抗体)的液体药物制剂以及一种制备和使用该制剂的方法。
背景技术
抗血管生成素-2(ANG-2)-2和人血管内皮生长因子(VEGF,VEGF-A)的双特异性抗体(双特异性抗VEGF/ANG2抗体)具有治疗意义,特别是作为治疗和预防性治疗血管疾病(眼部血管疾病)的药物。双特异性抗VEGF/ANG2抗体描述于例如WO2010040508、WO2011/117329或WO2014/009465。这些抗体抑制Vegf与VEGF受体的结合,同时使ANG-2与Tie2结合。
抗体分子作为蛋白质药物组的一部分,非常容易受到物理和化学降解的影响。化学降解包括涉及通过键形成或裂解来修饰蛋白质以得到新化学实体的任何过程。已知多种化学反应影响蛋白质。这些反应可能涉及水解(包括肽键的裂解)以及脱酰胺、异构化、氧化和分解。物理降解是指较高阶结构的变化,并且包括变性、表面吸附、聚集和沉淀。蛋白质稳定性受蛋白质本身的特征(例如氨基酸序列、糖基化模式)以及外部影响因素(诸如温度、溶剂pH、赋形剂、界面或剪切速率)的影响。因此,确定最佳制剂条件对于保护蛋白质在生产、储存和施用过程中免于发生降解反应很重要。(Manning,M.C.等人(1989),"Stability ofprotein pharmaceuticals",Pharm Res 6(11),903-918;Zheng,J.Y.,Janis,L.J.(2005),"Influence of pH,buffer species,and storage temperature onphysicochemical stability of a humanized monoclonal antibody LA298",Int.J.Pharmaceutics 308,46-51)。当制剂中应包含高浓度抗体时,特别难以获得稳定的治疗抗体液体制剂。
因此,本发明的目的是提供一种双特异性VEGF/ANG2抗体的液体(特别是高浓度)制剂,该制剂包含尽可能少的必需赋形剂,能够实现所需的给药,并且方便通过细针将双特异性抗体经玻璃体内施用施用于患者。
发明内容
本发明涉及一种双特异性抗VEGF/ANG2抗体的液体药物制剂以及一种制备和使用该制剂的方法。特别地,本发明的药物制剂用于经玻璃体内施用以治疗眼科疾病(如AMD和DME)。
在一个方面,本发明涉及一种液体药物制剂,该液体药物制剂包含:
-20mg/mL至150mg/mL双特异性抗VEGF/ANG2抗体,该抗体包含人IgG1亚类的重链恒定区
-15mM至35mM氯化钠
-15mM至25mM组氨酸醋酸盐缓冲剂
其pH值为5.5±0.5;其中
双特异性抗VEGF/ANG2抗体为二价的,并且包含与人VEGF特异性结合的第一抗原结合位点和与人ANG-2特异性结合的第二抗原结合位点,其中
i)与VEGF特异性结合的所述第一抗原结合位点在重链可变结构域中包含SEQ IDNO:1的CDR3H区、SEQ ID NO:2的CDR2H区和SEQ ID NO:3的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:4的CDR3L区、SEQ ID NO:5的CDR2L区和SEQ ID NO:6的CDR1L区;并且
ii)与ANG-2特异性结合的所述第二抗原结合位点在重链可变结构域中包含SEQID NO:9的CDR3H区、SEQ ID NO:10的CDR2H区和SEQ ID NO:11的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:12的CDR3L区、SEQ ID NO:13的CDR2L区和SEQ ID NO:14的CDR1L区,并且其中
iii)双特异性抗体包含人IgG1亚类的重链恒定区,该重链恒定区包含突变I253A、H310A和H435A以及突变L234A、L235A和P329G(根据Kabat EU索引编号)。
在一个实施例中,该双特异性抗VEGF/ANG2抗体为二价的,并且包含SEQ ID NO:17的氨基酸序列、SEQ ID NO:18的氨基酸序列、SEQ ID NO:19的氨基酸序列和SEQ ID NO:20的氨基酸序列。
在一个实施例中,该双特异性抗VEGF/ANG2抗体为法立昔单抗(faricimab)。
在一个实施例中,该制剂进一步包含
-1mM至20mM至少一种稳定剂。
在一个实施例中,该制剂进一步包含
-7.0mM±2.0mM甲硫氨酸。
在一个实施例中,该制剂进一步包含
-0.01%(w/v)至0.07%(w/v)表面活性剂。
在一个实施例中,该制剂进一步包含
-0.04%(w/v)±0.02%(w/v)聚山梨酯20。
在一个实施例中,该制剂进一步包含
-50mM至250mM张度剂。
在一个实施例中,该制剂进一步包含
-160mM±24mM蔗糖。
在一个实施例中,该制剂基本上不含可见颗粒。
在一个实施例中,该制剂为稳定制剂。
在一个实施例中,该制剂的渗透压为300mOsm/kg±100mOsm/kg。
在一个实施例中,该制剂用于玻璃体内施用。
在一个方面,该制剂用于治疗眼部血管疾病。
在一个实施例中,该眼部血管疾病选自由以下项组成的组:糖尿病性视网膜病变(DR)、糖尿病性黄斑水肿(DME)、视网膜静脉阻塞(RVO)、视网膜中央静脉阻塞(CRVO)、黄斑变性、湿性年龄相关性黄斑变性(湿性AMD)、早产儿视网膜病变(ROP)、新生血管性青光眼、色素性视网膜炎(RP)、视网膜血管瘤样增生、黄斑毛细血管扩张、缺血性视网膜病变、虹膜新生血管、眼内新生血管、角膜新生血管、视网膜新生血管、脉络膜新生血管和视网膜变性,特别地选自由以下项组成的组糖尿病性视网膜病变(DR)、糖尿病性黄斑水肿(DME)、视网膜静脉阻塞(RVO)、视网膜中央静脉阻塞(CRVO)、湿性年龄相关性黄斑变性(湿性AMD)。
本发明的一个方面提供了一种制备根据本发明所述的药物制剂的方法。
本发明的一个方面提供了一种包含根据本发明所述的药物制剂的小瓶。
本发明的一个方面提供了一种包含根据本发明所述的药物制剂的预充式注射器。
本发明的一个方面提供了一种根据本发明所述的冻干形式的液体药物制剂。
本发明提供了一种双特异性抗VEGF/ANG2抗体(包含IgG1恒定区)的液体药物制剂,其具有可用于眼科使用和玻璃体内应用的有价值的性质:该制剂具有低粘度和低浊度(即使在约120mg/L的高浓度下),该制剂稳定并且等渗。这尤其通过将20mg/mL至150mg/mL(特别是100mg/mL至140mg/mL)如本文所述的包含人IgG1亚类的重链恒定区的双特异性抗VEGF/ANG2抗体与15mM至35mM氯化钠和15mM至25mM组氨酸缓冲剂(PH值为5.5±0.5)组合来实现。
附图说明
图1由pH/缓冲剂筛选第I部分得到的制剂的浊度(图1A)和粘度(图1B)结果。图1比较了由pH/缓冲剂筛选I得到的制剂的浊度和粘度结果(条形下方:第一行:制剂样品编号;第二行:pH值;第三行:缓冲剂体系;第四行:离子强度)。
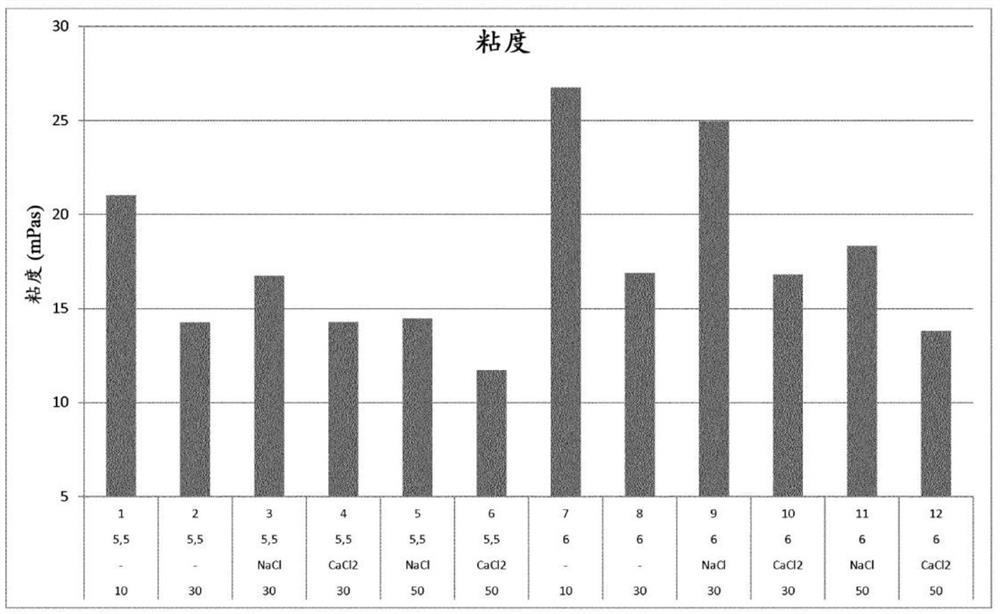
图2由pH/缓冲剂筛选第II部分得到的制剂的浊度(图2A)和粘度(图2B)结果(条形下方:第一行:制剂样品编号;第二行:pH值;第三行:不存在降粘剂-,存在NaCl或CaCl
图3由pH/缓冲剂筛选第II部分得到的制剂在初始状态以及在5℃和25℃储存8周后的高分子量物质(HMW)(条形下方:第一行:制剂样品编号;第二行:pH值;第三行:不存在降粘剂-,存在NaCl或CaCl
图4初始状态下和经过物理应激后的高分子量物质(HMW)的含量(条形下方:第一行:物理应激类型;第二行:%表面活性剂;第三行:制剂样品编号)。
图5由赋形剂筛选I得到的制剂的浊度(图5A)和粘度(图5B)(条形下方:第一行:不存在(-)作为降粘剂的NaCl或存在(+)甲硫氨酸作为稳定剂时的pH值;第二行:制剂样品编号)。
图6初始状态下和在5℃储存期间高分子量物质(HMW)的含量(条形下方:第一行:不存在(-)作为降粘剂的NaCl或存在(+)甲硫氨酸作为稳定剂时的pH值;第二行:制剂样品编号)。
图7初始状态下和在25℃储存期间高分子量物质(HMW)的含量(条形下方:第一行:不存在(-)作为降粘剂的NaCl或存在(+)甲硫氨酸作为稳定剂时的pH值;第二行:制剂样品编号)。
图8初始状态下和在5℃储存期间带电荷物质的含量(主峰(图8A)、酸性峰(图8B)和碱性峰(图8C))(条形下方:第一行:不存在(-)作为降粘剂的NaCl或存在(+)甲硫氨酸作为稳定剂时的pH值;第二行:制剂样品编号)。
图9初始状态下和在25℃储存期间带电荷物质的含量(主峰(图9A)、酸性峰(图9B)和碱性峰(图9C))(条形下方:第一行:不存在(-)作为降粘剂的NaCl或存在(+)甲硫氨酸作为稳定剂时的pH值;第二行:制剂样品编号)。
图10由赋形剂筛选II得到的蛋白质浓度为120mg/mL的优化制剂和参比制剂的浊度(图10A)和粘度(图10B)。
图11由赋形剂筛选II得到的蛋白质浓度为30mg/mL的优化制剂和参比制剂的浊度(图11A)和粘度(图11B)。
图12蛋白质浓度为120mg/mL的优化制剂和参比制剂在初始状态下(左侧条形)以及在5℃(中间条形)和25℃(右侧条形)储存13周后的高分子量物质(HMW)的含量。
图13蛋白质浓度为30mg/mL的优化制剂和参比制剂在初始状态下(左侧条形)以及在5℃(中间条形)和25℃(右侧条形)储存13周后的高分子量物质(HMW)的含量。
图14优化制剂和参比制剂在初始状态下(左侧条形)以及在5℃(中间条形)和25℃(右侧条形)储存13周后的带电荷物质的含量(主峰(图14A)、酸性峰(图14B)和碱性峰(图14C))。
具体实施方式
本发明涉及一种包含双特异性抗VEGF/ANG2的液体药物制剂,该双特异性抗VEGF/ANG2包含人IgG1亚类的重链恒定区。
术语“药物制剂物”是指处于允许活性成分的生物活性明确有效的形式,并且不含对于将被施用制剂的受试者有毒的另外组分的制备物。
如本文中与根据本发明所述的制剂结合使用的术语“液体”表示在大气压下和至少约2℃至约35℃(在一个实施例中,在约2℃与约25℃之间)的温度下呈液体的制剂。
药物制剂中包含的双特异性抗VEGF/ANG2抗体的浓度在约20mg/mL至约150mg/mL的范围内,特别地浓度为120mg/mL±18mg/mL,更特别地浓度为120mg/mL±12mg/mL。在另一个实施例中,浓度可为30mg/mL±4.5mg/mL。
如本文所用,“抗体”是指包含抗原结合位点的结合蛋白。如本文所用,术语“结合位点”或“抗原结合位点”表示配体实际结合的抗体分子的一个或多个区域。术语“抗原结合位点”包含抗体重链可变结构域(VH)和抗体轻链可变结构域(VL)(VH/VL对)。
抗体具体是指抗体对抗原的特定表位的选择性识别。例如,天然抗体为单特异性的。
根据本发明所述的“双特异性抗体”为具有两种不同的抗原结合特异性的抗体。本发明的抗体对两种不同的抗原具有特异性,其中VEGF为第一抗原并且ANG-2为第二抗原。
如本文所用,术语“单特异性”抗体表示具有一个或多个结合位点的抗体,每个结合位点与相同抗原的相同表位结合。
如在本申请中所用的术语“价”表示抗体分子中存在指定数目的结合位点。因此,术语“二价”“四价”和“六价”分别表示抗体分子中存在两个结合位点、四个结合位点和六个结合位点。根据本发明所述的双特异性抗体优选为“二价”。
如本文所用的术语“与人血管内皮生长因子(VEGF)和人血管生成素-2(ANG-2)结合的双特异性抗体”、“双特异性抗VEGF/ANG2抗体”和“双特异性
双特异性抗VEGF/ANG2抗体描述于例如WO2010/040508、WO2011/117329、WO2012/131078、WO2015/083978、WO2017/197199和WO2014/009465中。WO2014/009465描述了专门设计用于治疗眼部血管疾病的双特异性抗VEGF/ANG2抗体。WO2014/009465(其全文以引用方式并入本文)的双特异性抗VEGF/ANG2抗体尤其适用于治疗如本文所述的眼部血管疾病的治疗及其治疗方案。特别地,如WO2014/009465中所述的抗VEGF/ANG2抗体CrossMAbVEGFang2-0016也称为法立昔单抗(在World Health Organization(2017)."International Nonproprietary Names for Pharmaceutical Substances(INN).Proposed INN:List 118"WHO Drug Information.31(4)中),其为本发明的优选双特异性抗VEGF/ANG2抗体。
在一个实施例中,与人血管内皮生长因子(VEGF)和人血管生成素-2(ANG-2)结合的双特异性抗体为包含与人VEGF特异性结合的第一抗原结合位点和与人ANG-2特异性结合的第二抗原结合位点的双特异性抗VEGF/ANG2抗体,其中
i)与VEGF特异性结合的所述第一抗原结合位点在重链可变结构域中包含SEQ IDNO:1的CDR3H区、SEQ ID NO:2的CDR2H区和SEQ ID NO:3的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:4的CDR3L区、SEQ ID NO:5的CDR2L区和SEQ ID NO:6的CDR1L区;并且
ii)与ANG-2特异性结合的所述第二抗原结合位点在重链可变结构域中包含SEQID NO:9的CDR3H区、SEQ ID NO:10的CDR2H区和SEQ ID NO:11的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:12的CDR3L区、SEQ ID NO:13的CDR2L区和SEQ ID NO:14的CDR1L区,并且其中
iii)双特异性抗体包含人IgG1亚类的重链恒定区,该重链恒定区包含突变I253A、H310A和H435A以及突变L234A、L235A和P329G(根据Kabat EU索引编号)。
在一个实施例中,此类双特异性抗VEGF/ANG2抗体为二价的。
在一个实施例中,此类双特异性二价抗VEGF/ANG2抗体的特征在于:
i)与VEGF特异性结合的所述第一抗原结合位点包含作为重链可变结构域VH的SEQID NO:7的氨基酸序列和作为轻链可变结构域VL的SEQ ID NO:8的氨基酸序列,并且
ii)与ANG-2特异性结合的所述第二抗原结合位点包含作为重链可变结构域VH的SEQ ID NO:15的氨基酸序列和作为轻链可变结构域VL的SEQ ID NO:16的氨基酸序列。
在本发明的一个方面,根据本发明所述的此类双特异性二价抗体的特征在于包含:
a)特异性结合VEGF的第一全长抗体的重链和轻链;
b)特异性结合ANG-2的第二全长抗体的经修饰的重链和经修饰的轻链,其中恒定结构域CL和CH1彼此替换。
与人血管内皮生长因子(VEGF)和人血管生成素-2(ANG-2)特异性结合的双特异性抗体的该双特异性二价抗体形式描述于WO 2009/080253中(包括经杵臼结构修饰的CH3结构域)。基于该双特异性二价抗体形式的抗体称为CrossMAb。
在一个实施例中,此类双特异性二价抗VEGF/ANG2抗体的特征在于包含:
a)第一全长抗体的重链,其具有SEQ ID NO:17的氨基酸序列,和第一全长抗体的轻链,其具有SEQ ID NO:18的氨基酸序列;以及
b)第二全长抗体的经修饰的重链,其具有SEQ ID NO:19的氨基酸序列,和第二全长抗体的经修饰的轻链,其具有SEQ ID NO:20的氨基酸序列。
在一个实施例中,此类双特异性二价抗VEGF/ANG2抗体的特征在于包含SEQ IDNO:17的氨基酸序列、SEQ ID NO:18的氨基酸序列、SEQ ID NO:19的氨基酸序列和SEQ IDNO:20的氨基酸序列。
因此,本发明的一个实施例为一种双特异性二价抗体,其包含与人VEGF特异性结合的第一抗原结合位点以及与人ANG-2特异性结合的第二抗原结合位点,该双特异性二价抗体的特征在于包含SEQ ID NO:17的氨基酸序列、SEQ ID NO:18的氨基酸序列、SEQ IDNO:19的氨基酸序列和SEQ ID NO:20的氨基酸序列。
在一个实施例中,根据本发明所述的双特异性二价抗体的CH3结构域通过“杵臼结构(knob-into-holes)”技术改变,该技术在例如WO 96/027011、Ridgway,J.B.等人,Protein Eng 9(1996)617-621和Merchant,A.M.等人,Nat Biotechnol.16(1998)677-681中以若干实例详细描述。在该方法中,改变两个CH3结构域的相互作用表面,以增加包含这两个CH3结构域的两个重链的异源二聚化。(两个重链的)两个CH3结构域中的一个可为“杵(knob)”而另一个为“臼(hole)”。二硫键的引入使异二聚体稳定(Merchant,A.M等人,Nature Biotech 16(1998)677-681;Atwell,S.等人,J.Mol.Biol.270(1997)26-35)并且提高了产率。
在本发明的一个优选方面,根据本发明所述的双特异性抗VEGF/ANG2抗体的特征在于
一个重链的CH3结构域与另一个重链的CH3结构域各自在包含抗体CH3结构域之间初始界面的界面处相遇;
其中所述界面被改变以促进双特异性抗体的形成,其中该改变的特征在于:
a)一个重链的CH3结构域被改变,
使得在双特异性抗体内的一个重链的CH3结构域与另一个重链的CH3结构域的初始界面相遇的初始界面内,
氨基酸残基被具有较大侧链体积的氨基酸残基取代,从而在一个重链的CH3结构域的界面内产生突起,该突起可定位于另一个重链的CH3结构域的界面内的空腔中;
和
b)另一个重链的CH3结构域被改变,
使得在双特异性抗体内与第一CH3结构域的初始界面相遇的第二CH3结构域的初始界面内,
氨基酸残基被具有较小侧链体积的氨基酸残基取代,从而在第二CH3结构域的界面内产生空腔,该空腔可定位于第一CH3结构域的界面内的突起中。
因此,如本文所述的双特异性抗VEGF/ANG2抗体优选地特征在于
a)的全长抗体的重链的CH3结构域和b)的全长抗体的重链的CH3结构域各自在包含抗体CH3结构域之间的初始界面的改变的界面处相遇;
其中i)在一个重链的CH3结构域中
氨基酸残基被具有较大侧链体积的氨基酸残基取代,从而在一个重链的CH3结构域的界面内产生突起,该突起可定位于另一个重链的CH3结构域的界面内的空腔中;并且其中
ii)在另一个重链的CH3结构域中
氨基酸残基被具有较小侧链体积的氨基酸残基取代,从而在第二CH3结构域的界面内产生空腔,该空腔可定位于第一CH3结构域的界面内的突起中。
优选地,所述具有较大侧链体积的氨基酸残基选自由精氨酸(R)、苯丙氨酸(F)、酪氨酸(Y)、色氨酸(W)组成的组。
优选地,所述具有较小侧链体积的氨基酸残基选自由丙氨酸(A)、丝氨酸(S)、苏氨酸(T)、缬氨酸(V)组成的组。
在本发明的一个方面中,两个CH3结构域通过在每个CH3结构域的相应位置引入半胱氨酸(C)作为氨基酸来进一步改变,使得可以在两个CH3结构域之间形成二硫键。
在一个实施例中,双特异性抗体包含“突起链”的CH3结构域中的T366W突变以及“孔链”的CH3结构域中的T366S、L368A、Y407V突变。还可使用在CH3结构域之间的链间二硫键(Merchant,A.M等人,Nature Biotech 16(1998)677-681),例如通过将S354C突变引入一个CH3结构域并且将Y349C突变引入另一个CH3结构域来实现。
在另一个优选实施例中,双特异性抗体包含两个CH3结构域中的一个中的S354C和T366W突变以及两个CH3结构域的另一个中的Y349C、T366S、L368A、Y407V突变。在另一个优选实施例中,双特异性抗体包含两个CH3结构域中的一个中的Y349C、T366W突变以及两个CH3结构域的另一个中的S354C、T366S、L368A、Y407V突变(一个CH3结构域中的附加Y349C或S354C突变以及另一个CH3结构域中的附加S354C或Y349C突变形成链间二硫键),其中所述突变根据Kabat EU索引编号(Kabat,E.A.等人,Sequences of Proteins ofImmunological Interest,第5版,Public Health Service,National Institutes ofHealth,Bethesda,MD(1991))。
用于实施异源二聚化的其他CH3修饰技术被设想为本发明的替代方案,并且描述于例如WO 96/27011、WO 98/050431、EP 1870459、WO 2007/110205、WO 2007/147901、WO2009/089004、WO 2010/129304、WO 2011/90754、WO 2011/143545、WO 2012/058768、WO2013/157954和WO 2013/096291中。
在一个实施例中,替代性地使用EP 1 870 459A1中描述的异源二聚化方法。该方法基于在两个重链之间的CH3/CH3结构域界面的特定氨基酸位置引入带相反电荷的带电荷氨基酸的取代/突变。所述多特异性抗体的一个优选实施例是该多特异性抗体的一个重链的CH3结构域中的氨基酸R409D和K370E突变以及另一个重链的CH3结构域中的氨基酸D399K和E357K突变(根据Kabat EU索引编号)。
在另一个实施例中,所述多特异性抗体包含“突起链”的CH3结构域中的氨基酸T366W突变以及“孔链”的CH3结构域中的氨基酸T366S、L368A和Y407V突变;并且另外包含“突起链”的CH3结构域中的氨基酸R409D和K370E突变以及“孔链”的CH3结构域中的氨基酸D399K和E357K突变。
在一个实施例中,替代地使用WO2013/157953中描述的异源二聚化方法。在一个实施例中,一个重链的CH3结构域包含氨基酸T366K突变,并且另一个重链的CH3结构域包含氨基酸L351D突变。在又一实施例中,一个重链的CH3结构域进一步包含氨基酸L351K突变。在又一实施例中,另一个重链的CH3结构域进一步包含选自Y349E、Y349D和L368E(在一个实施例中,为L368E)的氨基酸突变。
在一个实施例中,替代地使用WO2012/058768中描述的异源二聚化方法。在一个实施例中,一个重链的CH3结构域包含氨基酸L351Y和Y407A突变,并且另一个重链的CH3结构域包含氨基酸T366A和K409F突变。在又一实施例中,另一个重链的CH3结构域进一步包含T411、D399、S400、F405、N390或K392位置处的氨基酸突变。在一个实施例中,所述氨基酸突变选自由以下项组成的组:
a)T411N、T411R、T411Q、T411K、T411D、T411E和T411W,
b)D399R、D399W、D399Y和D399K,
c)S400E、S400D、S400R和S400K,
d)F405I、F405M、F405T、F405S、F405V和F405W,
e)N390R、N390K和N390D,
f)K392V、K392M、K392R、K392L、K392F和K392E。
在又一实施例中,一个重链的CH3结构域包含氨基酸L351Y和Y407A突变,并且另一个重链的CH3结构域包含氨基酸T366V和K409F突变。在又一实施例中,一个重链的CH3结构域包含氨基酸Y407A突变,并且另一个重链的CH3结构域包含氨基酸T366A和K409F突变。在又一实施例中,另一个重链的CH3结构域进一步包含氨基酸K392E、T411E、D399R和S400R突变。
在一个实施例中,替代地使用WO2011/143545中描述的异源二聚化方法。在一个实施例中,将根据WO2011/143545所述的氨基酸修饰引入重链的CH3结构域中的以下位置,该位置选自由368和409组成的组。
在一个实施例中,替代性地使用WO2011/090762中描述的异源二聚化方法,该异源二聚化方法也使用上述杵臼结构技术。在一个实施例中,一个重链的CH3结构域包含氨基酸T366W突变,并且另一个重链的CH3结构域包含氨基酸Y407A突变。在一个实施例中,一个重链的CH3结构域包含氨基酸T366Y突变,并且另一个重链的CH3结构域包含氨基酸Y407T突变。
在一个实施例中,多特异性抗体为IgG2同种型,并且替代性地使用WO2010/129304中描述的异源二聚化方法。
在一个实施例中,替代地使用WO2009/089004中描述的异源二聚化方法。在一个实施例中,一个重链的CH3结构域包含带负电荷的氨基酸对K392或N392的氨基酸置换(在一个实施例中,包含谷氨酸(E)或天冬氨酸(D)置换;在又一实施例中,包含K392D或N392D突变),并且另一个重链的CH3结构域包含带正电荷的氨基酸对D399、E356、D356或E357的氨基酸置换(在一个实施例中,包含赖氨酸(K)或精氨酸(R)置换;在又一实施例中,包含D399K、E356K、D356K或E357K置换;并且在又一个实施例中,包含D399K或E356K突变)。在又一实施例中,一个重链的CH3结构域进一步包含带负电荷的氨基酸对K409或R409的氨基酸置换(在一个实施例中,包含谷氨酸(E)或天冬氨酸(D)置换;在又一实施例中,包含K409D或R409D突变)。在又一实施例中,一个重链的CH3结构域进一步或替代性地包含带负电荷的氨基酸对K439和/或K370的氨基酸置换(在一个实施例中,谷氨酸(E)或天冬氨酸(D)置换)。
在一个实施例中,替代地使用WO2007/147901中描述的异源二聚化方法。在一个实施例中,一个重链的CH3结构域包含氨基酸K253E、D282K和K322D突变,并且另一个重链的CH3结构域包含氨基酸D239K、E240K和K292D突变。
在一个实施例中,替代地使用WO2007/110205中描述的异源二聚化方法。
在一个优选实施例中,此类双特异性抗VEGF/ANG2抗体为二价的。
在一个实施例中,与人血管内皮生长因子(VEGF)和人血管生成素-2(ANG-2)结合的双特异性二价抗体为包含与人VEGF特异性结合的第一抗原结合位点以及与人ANG-2特异性结合的第二抗原结合位点的双特异性抗VEGF/ANG2抗体,其中
i)与VEGF特异性结合的所述第一抗原结合位点在重链可变结构域中包含SEQ IDNO:1的CDR3H区、SEQ ID NO:2的CDR2H区和SEQ ID NO:3的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:4的CDR3L区、SEQ ID NO:5的CDR2L区和SEQ ID NO:6的CDR1L区;并且
ii)与ANG-2特异性结合的所述第二抗原结合位点在重链可变结构域中包含SEQID NO:9的CDR3H区、SEQ ID NO:10的CDR2H区和SEQ ID NO:11的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:12的CDR3L区、SEQ ID NO:13的CDR2L区和SEQ ID NO:14的CDR1L区,并且其中
iii)双特异性抗体包含人IgG1亚类的重链恒定区,该重链恒定区包含突变I253A、H310A和H435A以及突变L234A、L235A和P329G(根据Kabat EU索引编号);并且其中
iv)在重链恒定区中,T366W突变包含在一个CH3结构域中,并且T366S、L368A、Y407V突变包含在另一个CH3结构域中(根据Kabat EU索引编号)。
在一个实施例中,与人血管内皮生长因子(VEGF)和人血管生成素-2(ANG-2)结合的双特异性二价抗体为包含与人VEGF特异性结合的第一抗原结合位点以及与人ANG-2特异性结合的第二抗原结合位点的双特异性抗VEGF/ANG2抗体,其中
i)与VEGF特异性结合的所述第一抗原结合位点在重链可变结构域中包含SEQ IDNO:1的CDR3H区、SEQ ID NO:2的CDR2H区和SEQ ID NO:3的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:4的CDR3L区、SEQ ID NO:5的CDR2L区和SEQ ID NO:6的CDR1L区;并且
ii)与ANG-2特异性结合的所述第二抗原结合位点在重链可变结构域中包含SEQID NO:9的CDR3H区、SEQ ID NO:10的CDR2H区和SEQ ID NO:11的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:12的CDR3L区、SEQ ID NO:13的CDR2L区和SEQ ID NO:14的CDR1L区,并且其中
iii)双特异性抗体包含人IgG1亚类的重链恒定区,该重链恒定区包含突变I253A、H310A和H435A以及突变L234A、L235A和P329G(根据Kabat EU索引编号);并且其中
iv)在重链恒定区中,S354C和T366W突变包含在一个CH3结构域中,并且Y349C、T366S、L368A和Y407V突变包含在另一个CH3结构域中(根据Kabat EU索引编号)。
在一个实施例中,此类双特异性二价抗VEGF/ANG2的特征在于包含SEQ ID NO:17的氨基酸序列、SEQ ID NO:18的氨基酸序列、SEQ ID NO:19的氨基酸序列和SEQ ID NO:20的氨基酸序列。
因此,本发明的一个实施例为一种双特异性二价抗体,其包含与人VEGF特异性结合的第一抗原结合位点以及与人ANG-2特异性结合的第二抗原结合位点,该双特异性二价抗体的特征在于包含SEQ ID NO:17的氨基酸序列、SEQ ID NO:18的氨基酸序列、SEQ IDNO:19的氨基酸序列和SEQ ID NO:20的氨基酸序列。
在一个优选实施例中,此类双特异性抗VEGF/ANG2抗体为法立昔单抗。
如本文所用的术语“VEGF”是指人血管内皮生长因子(VEGF/VEGF-A)、165-氨基酸人血管内皮细胞生长因子(人VEGF165的前体序列的氨基酸27-191:SEQ ID NO:25;氨基酸1-26代表信号肽)以及相关的121、189和206血管内皮细胞生长因子同种型,如以下文献所述:Leung,D.W.等人,Science 246(1989)1306-9;Houck等人,Mol.Endocrin.5(1991)1806-1814;Keck,P.J.等人,Science 246(1989)1309-12;和Connolly,D.T.等人,J.Biol.Chem.264(1989)20017-24;以及这些生长因子的天然存在的等位基因形式和加工过的形式。VEGF参与与肿瘤和眼内疾病相关的正常和异常血管生成和新生血管的调节(Ferrara,N.等人,Endocr.Rev.18(1997)4-25;Berkman,R.A.等人,J.Clin.Invest.91(1993)153-159;Brown,L.F.等人,Human Pathol.26(1995)86-91;Brown,L.F.等人,CancerRes.53(1993)4727-4735;Mattern,J.等人,Brit.J.Cancer.73(1996)931-934;和Dvorak,H.F.等人,Am.J.Pathol.146(1995)1029-1039)。VEGF是一种同二聚体糖蛋白,已从多种来源分离出来,并且包括多种同种型。VEGF对内皮细胞具有高度特异性的促有丝分裂活性。VEGF拮抗剂/抑制剂抑制VEGF与其受体VEGFR的结合。已知的VEGF拮抗剂/抑制剂包括如WO2014/009465所述的双特异性抗VEGF/ANG2抗体。
如本文所用的术语“ANG-2”是指人血管生成素-2(ANG-2)(替代性地缩写为ANGPT2或ANG2)(SEQ ID NO:24),其描述于例如以下文献中:Maisonpierre,P.C.等人,Science277(1997)55-60;和Cheung,A.H.等人,Genomics 48(1998)389-91。发现血管生成素-1和血管生成素-2为Ties的配体,Ties是在血管内皮内选择性表达的酪氨酸激酶家族(Yancopoulos,G.D.等人,Nature 407(2000)242-48)。现在,血管生成素家族包括四种确定的成员。血管生成素-3和血管生成素-4(Ang-3和Ang-4)可能在小鼠和人体内代表同一基因座的各种不同的对应物(Kim,I.等人,FEBS Let,443(1999)353-56;Kim,I.等人,J BiolChem 274(1999)26523-28)。ANG-1和ANG-2最初在组织培养实验中分别被鉴定为激动剂和拮抗剂(有关ANG-1,参见:Davis,S.等人,Cell 87(1996)1161-69;有关ANG-2,参见:Maisonpierre,P.C.等人,Science 277(1997)55-60)。所有已知的血管生成素都主要与其受体TIE2结合,并且Ang-1和Ang-2均以3nM(Kd)的亲和力与TIE2结合(Maisonpierre,P.C.等人,Science 277(1997)55-60)。ANG2拮抗剂/抑制剂抑制ANG2与其受体TIE2的结合。已知的ANG2拮抗剂/抑制剂包括如WO2014/009465所述的双特异性抗VEGF/ANG2抗体。
本发明的双特异性抗体的抗原结合位点包含六个互补决定区(CDR),这六个互补决定区在不同程度上有助于抗原结合位点的亲和力。有三个重链可变结构域CDR(CDRH1、CDRH2和CDRH3)和三个轻链可变结构域CDR(CDRL1、CDRL2和CDRL3)。CDR和框架区(FR)的范围通过与氨基酸序列的汇编数据库进行比较来确定,在该数据库中,已根据序列之间的变异性定义了那些区域。
本发明的抗体包含来源于人源性免疫球蛋白类别IgG1的免疫球蛋白恒定区。
如本文所用,术语“单克隆抗体”或“单克隆抗体组合物”是指具有单一氨基酸组成的抗体分子的制备物。
术语“嵌合抗体”是指包含来自一种来源或物种的可变区(即结合区)以及至少一部分来源于不同来源或物种的恒定区的抗体,其通常通过重组DNA技术进行制备。包含鼠可变区和人恒定区的嵌合抗体特别引人关注。本发明涵盖的其他形式的“嵌合抗体”是这样的抗体,相对于原始抗体,所述抗体中的恒定区已经经过了修饰或改变,以产生根据本发明所述的期望的性质,尤其是关于C1q结合和/或Fc受体(FcR)结合的性质。此类嵌合抗体也称为“类别转换抗体”。嵌合抗体是表达的免疫球蛋白基因的产物,其包含编码免疫球蛋白可变区的DNA片段以及编码免疫球蛋白恒定区的DNA片段。生产嵌合抗体的方法涉及常规的重组DNA,并且基因转染技术是本领域中所熟知的。参见例如以下文献:Morrison,S.L.等人,Proc.Natl.Acad.Sci.USA 81(1984)6851-6855;美国专利号5,202,238和5,204,244。
术语“人源化抗体”是指其中框架或“互补决定区”(CDR)经修饰以包含与亲本免疫球蛋白相比具有不同特异性的免疫球蛋白的CDR的抗体。在一个优选实施例中,将鼠CDR移植到人抗体的框架区以制备“人源化抗体”。参见例如以下文献:Riechmann,L.等人,Nature332(1988)323-327;和Neuberger,M.S.等人,Nature 314(1985)268-270。特别优选的CDR对应于那些代表识别上述嵌合抗体的抗原的序列。本发明涵盖的其他形式的“人源化抗体”是这样的抗体,相对于原始抗体,所述抗体中的恒定区已经经过了另外修饰或改变,以产生根据本发明的性质,特别是关于C1q结合和/或Fc受体(FcR)结合的性质。
如本文所用的术语“人抗体”旨在包括具有来源于人类种系免疫球蛋白序列的可变区和恒定区的抗体。人抗体是现有技术中所熟知的(van Dijk,M.A.和van de Winkel,J.G.,Curr.Opin.Chem.Biol.5(2001)368-374)。人抗体还可以在转基因动物(例如小鼠)中产生,这些动物在免疫后能够在不产生内源性免疫球蛋白的情况下产生人抗体的完整文库或特定部分。人类种系免疫球蛋白基因阵列在此类种系突变小鼠中的转染将导致在抗原攻击后产生人抗体(参见例如:Jakobovits,A.等人,Proc.Natl.Acad.Sci.USA 90(1993)2551-2555;Jakobovits,A.等人,Nature 362(1993)255-258;Bruggemann,M.等人,YearImmunol.7(1993)33-40)。人抗体也可以在噬菌体展示文库中产生(Hoogenboom,H.R.和Winter,G.,J.Mol.Biol.227(1992)381-388;Marks,J.D.等人,J.Mol.Biol.222(1991)581-597)。也可以使用Cole等人和Boerner等人的技术来制备人单克隆抗体(Cole等人,Monoclonal Antibodies and Cancer Therapy,Alan R.Liss,第77页(1985);和Boerner,P.等人,J.Immunol.147(1991)86-95)。如已经针对根据本发明所述的嵌合抗体和人源化抗体所提及的,如本文所用的术语“人抗体”还包括此类抗体,其在恒定区中经过修饰以产生根据本发明所述的性质,尤其是关于C1q结合和/或FcR结合的修饰,例如通过“类别转换”技术进行修饰,即Fc部分发生改变或突变(例如,从IgG1改变为IgG4和/或IgG1/IgG4突变)。
如本文所用,术语“重组人抗体”旨在包括通过重组方式制备、表达、产生或分离的所有人抗体,诸如从宿主细胞(诸如NS0或CHO细胞)或从转染人免疫球蛋白基因的动物(例如,小鼠)中分离的抗体,或者使用转染到宿主细胞中的重组表达载体所表达的抗体。此类重组人抗体具有重排形式的可变区和恒定区。根据本发明所述的重组人抗体经过体内体细胞超突变。因此,重组抗体的VH区和VL区的氨基酸序列是下述序列,尽管衍生自人类种系VH序列和VL序列并与之相关,但在天然条件下可以不存在于体内人类抗体种系品目中。
如本文所用的“可变区”(轻链(V
术语“表位”包括能够与抗体特异性结合的任何多肽决定簇。在某些实施例中,表位决定簇包括分子诸如氨基酸的化学活性表面基团、糖侧链、磷酰基或磺酰基,并且在某些实施例中,可具有特定的三维结构特征和/或特定的电荷特征。表位是与抗体结合的抗原区域。
术语“全长抗体”表示由两个“全长抗体重链”和两个“全长抗体轻链”组成的抗体。“全长抗体重链”是在N端至C端方向上由抗体重链可变结构域(VH)、抗体恒定重链结构域1(CH1)、抗体铰链区(HR)、抗体重链恒定结构域2(CH2)、抗体重链恒定结构域3(CH3)组成的多肽,缩写为VH-CH1-HR-CH2-CH3;如果是IgE亚型的抗体,则还可选地由抗体重链恒定结构域4(CH4)组成。优选地,“全长抗体重链”是在N末端至C末端方向上由VH、CH1、HR、CH2和CH3组成的多肽。“全长抗体轻链”是在N端至C端方向上由抗体轻链可变结构域(VL)和抗体轻链恒定结构域(CL)组成的多肽,缩写为VL-CL。抗体轻链恒定结构域(CL)可以是κ或λ。两条全长抗体链通过CL结构域和CH1结构域之间以及全长抗体重链的铰链区之间的多肽间二硫键链接在一起。典型的全长抗体的实例为天然抗体,例如IgG(例如IgG1和IgG2)、IgM、IgA、IgD和IgE。根据本发明所述的全长抗体可以来自单一物种,例如人,或者它们可以是嵌合抗体或人源化抗体。根据本发明所述的全长抗体通常包含两个抗原结合位点,每个抗原结合位点均由一对VH和VL形成,二者均特异性结合同一抗原。所述全长抗体的重链或轻链的C端表示所述重链或轻链的C端的最后一个氨基酸。所述全长抗体的重链或轻链的N端表示所述重链或轻链的N端的最后一个氨基酸。
如本申请内所用的术语“恒定区”或“恒定结构域”表示抗体中可变区以外的结构域的总和。恒定区不直接参与抗体与抗原的结合,但是表现出多种效应子功能。根据其重链的恒定区的氨基酸序列不同,可以将抗体分为以下类别:IgA、IgD、IgE、IgG和IgM,并且它们中的一些可以进一步分为亚类,例如IgG1、IgG2、IgG3和IgG4以及IgA1和IgA2。对应于不同类别的抗体的重链恒定区分别称为α、δ、ε、γ和μ。所有五个抗体类别中均存在的轻链恒定区称为κ和λ。
如本申请中所用的术语“来源于人源的恒定区”表示亚类IgG1、IgG2、IgG3或IgG4的人抗体的恒定重链区和/或恒定轻链κ或λ区。此类恒定区是现有技术中所熟知的,并且例如Kabat,E.A.所述(参见例如:Johnson,G.和Wu,T.T.,Nucleic Acids Res.28(2000)214-218;Kabat,E.A.等人,Proc.Natl.Acad.Sci.USA 72(1975)2785-2788)。
如本文所述用的术语“重链恒定结构域(或区)”定义包含重链恒定区的至少一部分的免疫球蛋白重链的C端区域。
该术语包括重链恒定结构域和变体重链恒定结构域的天然序列。变体重链恒定结构域包括例如恒定结构域中的突变,这些突变用于促进如上文针对杵臼结构技术所述的异源二聚化。还可以包括其他突变,例如L234A(Leu235Ala)、L235A(Leu234Ala)和P329G(Pro329Gly),因为具有此类突变的恒定结构域具有降低的FcR结合作用(尤其是它们不再与FcRγI、FcRγII和FcRγIII结合)。其尤其适用于减少潜在的副作用,例如血栓形成(Meyer,T.等人,J.Thromb.Haemost.7(2009)171-81)。此外,例如,恒定结构域中还可包含突变I253A、H310A和H435A(根据Kabat EU索引编号),因为具有此类突变的恒定结构域具有降低的FcRn结合作用(一个或多个突变)或消除FcRn结合作用(所有3个突变)。
在一个方面,人IgG重链恒定区从丙氨酸118(A118)(根据Kabat EU索引编号)延伸至重链的羧基端。然而,由宿主细胞产生的抗体可以经历对来自重链的C末端的一个或多个,特别是一个或两个氨基酸的翻译后切割。因此,由宿主细胞通过表达编码全长重链的特定核酸分子产生的抗体可以包括全长重链,或者所述抗体可以包括全长重链的切割变体。这可能是重链的最后两个C端氨基酸为甘氨酸(G446)和赖氨酸(K447,根据EU索引编码)的情况。因此,重链恒定结构域的C端赖氨酸(Lys447)或C端甘氨酸(Gly446)和赖氨酸(Lys447)可以存在或可以不存在。除非另外指明,否则包括重链恒定结构域的重链的氨基酸序列在本文中用C端甘氨酸-赖氨酸二肽表示。
“稳定”制剂是其中蛋白质(例如抗体)在储存时基本上保持其物理和化学稳定性及其生物活性的制剂;例如,当药物制剂在25℃储存8周后,该药物制剂中双特异性抗体的高分子量物质(HMW)含量低于10%(在一个实施例中,低于5%;在一个实施例中,低于2.5%)。在一个实施例中,当药物制剂在25℃储存52周后,该药物制剂中双特异性抗体的高分子量物质(HMW)含量低于10%(在一个实施例中,低于5%)。
在一个实施例中,“稳定液体药物制剂”是在冷藏温度(2-8℃)下储存至少12个月(特别是储存2年,更特别是储存3年)后未观察到明显变化的液体制剂。稳定性标准如下:通过体积排阻色谱法(SEC-HPLC)测得,不超过10%(特别是5%)的抗体单体发生降解。此外,通过目视分析,溶液呈无色或澄清至微乳白色。制剂的蛋白质浓度变化不超过+/-10%。形成不超过10%(特别是5%)的聚集体。稳定性通过本领域中的已知的方法诸如UV光谱、体积排阻色谱(SEC-HPLC)、离子交换色谱(IE-HPLC)、比浊法和目视检查来衡量。
浊度(单位为FTU(=福尔马肼浊度单位))
药物制剂的浊度可通过浊度计进行测定(例如,根据欧洲药典2.2.1“液体的澄清度和乳浊度”在Hach 2100AN浊度计上测定)。将样品体积约2mL的样品溶液转移至内径为11mm的玻璃比色杯中。将该玻璃比色杯放入浊度计中,并且根据参比悬浮液的校准曲线(1FTU、3FTU、10FTU、20FTU和100FTU测量浊度。
粘度(单位为mPa)
药物制剂的制剂样品的粘度可通过流变仪进行测定(例如,采用Anton PaarPhysica MCR 301旋转流变仪,锥度为25mm–0.5°,剪切速率为1000s
可见颗粒
在相应的检验设备(例如Seidenader检验设备V90-T上借助2倍放大镜)目视检查小瓶中的样品。将照明光源L1、L2和L3调整至设置5。在旋转运动过程中检查小瓶中的样品中是否存在颗粒。根据USP-NF<790>的要求(“基本上不含可见颗粒”),对于玻璃体内注射液,形成可见颗粒是不可接受的。USP-NF<790>规定,在检查肠胃外药物时,可以达到“基本上不含”的标准,并且观察到包含可见颗粒的单位数量不超过指定的数量。更具体地,对于接受100%检查的肠胃外药物,当批次达到0.65%或更低的可接受质量水平(AQL)时,即达到“基本上不含”的标准。另外,如果有必要评估已交付给客户的产品(例如,由于投诉或监管方面的原因),公司可以对20个单位进行取样和检查。如果样品中未观察到颗粒,则认为该批次“基本上不含”可见颗粒。
蛋白质浓度(单位为mg/mL)。
制剂样品的蛋白质浓度在Perkin Elmer的UV/Vis光度计λ35上通过紫外(UV)光吸收来测量。将制剂样品用20mM L-组氨酸-醋酸盐缓冲液(pH 5.5)稀释至蛋白质浓度约0.5mg/mL,然后装入1cm厚的比色皿中。测量比色皿在280nm和320nm的波长下的UV吸光度。
根据在280nm和320nm下测得的UV吸光度(分别为A280和A320)、1.70mL/(mg x cm)的消光系数(E)、1cm的厚度(d)以及对应于实际稀释液的稀释倍数(DF),通过以下公式计算蛋白质浓度:
pH
制剂样品的pH采用玻璃电极通过电位法测定。
离子强度
制剂的无量纲离子强度I根据公式1进行计算:
公式
在该表达式中,z是离子I的电荷数(阳离子为正,阴离子为负),b
带电荷的缓冲剂物质的重量摩尔浓度使用Henderson-Hasselbalch公式进行计算(Methods in Enzymology-Guide to Protein Purification,第182卷,M.P.Deutscher,Academic Press,Inc.,1990,第24ff页)。
渗透压
制剂样品的渗透压根据冰点降低原理在Gonotec的Osmomat 030 3P渗压计上进行测量。
表面活性剂
本发明的药物制剂包含表面活性剂以减少抗体的聚集和颗粒形成。如本文所用的术语“表面活性剂”表示用于保护蛋白质制剂免受机械应激(例如搅拌和剪切)影响的药用赋形剂。药用表面活性剂的实例包括聚氧乙烯脱水山梨醇脂肪酸酯(吐温)、聚氧乙烯烷基醚(例如以商品名Brij
优选地,表面活性剂为聚氧乙烯脱水山梨醇脂肪酸酯或泊洛沙姆。聚氧乙烯脱水山梨醇脂肪酸酯的实例为聚山梨酯20(以商品名吐温20
上述表面活性剂通常以0.01%(w/v)或更高的浓度(例如0.01%(w/v)至约0.09%(w/v))使用。本发明的药物组合物中的表面活性剂特别以约0.02%(w/v)至约0.06%(w/v)的范围使用,更特别地以约0.03%(w/v)至约0.05%(w/v)的范围使用,甚至更特别地以约0.04%(w/v)的浓度使用。
如本文所用的术语“泊洛沙姆”包括聚氧乙烯-聚氧丙烯三嵌段共聚物,其由聚氧丙烯的中心疏水链侧接两个聚氧乙烯亲水链组成,称为泊洛沙姆188,由组成,由BASF(Parsippany,N.J.)以商品名
缓冲剂
如本文所用的术语“缓冲剂”表示一种药用赋形剂,其用于稳定药物制剂的pH。合适的缓冲剂是本领域中熟知的,并且可参见文献。用于玻璃体内施用的典型药用缓冲剂包括但不限于组氨酸缓冲剂、柠檬酸盐缓冲剂、琥珀酸盐缓冲剂、醋酸盐缓冲剂、磷酸盐缓冲剂或其混合物。在此上下文中,特别关注的缓冲剂包括L-组氨酸(“组氨酸缓冲剂”)或L-组氨酸与L-组氨酸盐酸盐的混合物,其pH用本领域已知的酸或碱进行调节。特别关注的缓冲剂包括L-组氨酸(“组氨酸缓冲剂”),特别是pH通过乙酸(例如30%)或盐酸盐进行调节的L-组氨酸。上述缓冲剂通常以约2mM至约200mM或约5mM至约100mM的浓度使用,特别是以约10mM至约30mM或约15mM至约20mM的浓度使用,更特别地以约20mM的浓度使用。独立于所用的缓冲剂,可以用本领域中已知的酸或碱(例如乙酸、盐酸、磷酸、硫酸和柠檬酸、氢氧化钠和氢氧化钾,特别是乙酸)将pH调节为在4.5至7.0的范围内的值,特别是调节为在5.0至6.0的范围内的值,并且最特别地调节为pH 5.5±0.2。特别关注的缓冲剂是浓度为15-25mM(在一个实施例中为20mM±3mM,特别是20mM±2mM)、PH值为5.5±0.5(在一个实施例中,PH值为5.5±0.3;特别地,PH值为5.5±0.2)的组氨酸(L-组氨酸)。
稳定剂
术语“稳定剂”表示一种药用赋形剂,其用于保护活性药物成分和/或制剂免于在生产、储存和应用过程中发生化学和/或物理降解。蛋白质药物的化学和物理降解途径综述于以下文献中:Cleland等人(1993),Crit Rev Ther Drug Carrier Syst 10(4):307-77;Wang(1999)Int J Pharm 185(2):129-88;Wang(2000)Int J Pharm 203(1-2):1-60;和Chi等人(2003)Pharm Res 20(9):1325-36。稳定剂包括但不限于糖、氨基酸、多元醇、环糊精(例如羟丙基-β-环糊精、磺丁基乙基-β-环糊精、β-环糊精)、聚乙二醇(例如PEG 3000、PEG3350、PEG 4000、PEG 6000)、白蛋白、人血清白蛋白(HSA)、牛血清白蛋白(BSA)、盐(例如氯化钠、氯化镁、氯化钙)、螯合剂(例如,如下文所定义的EDTA)。在本发明中特别使用的稳定剂选自由糖、多元醇和氨基酸组成的组。更特别地,稳定剂选自由蔗糖、海藻糖、山梨糖醇和甲硫氨酸组成的组。
更优选地,稳定剂为甲硫氨酸。在本文所述的制剂中使用的甲硫氨酸首次用于眼部施用。临床前安全性试验表明,用于治疗眼病的甲硫氨酸在例如玻璃体内施用时表现出良好的安全性。
稳定剂在制剂中可以约2mM至约600mM的浓度存在,特别地,如果稳定剂为甲硫氨酸,其以约2mM至约15mM或5mM至12mM的浓度存在;更特别地,以约5mM至9mM或约7mM的浓度存在。
在一个优选实施例中,稳定剂为甲硫氨酸,并且甲硫氨酸浓度为7.0mM±2.0mM(在一个实施例中,甲硫氨酸浓度为7.0mM±1.0mM;在一个实施例中,甲硫氨酸浓度为7.0mM±0.7mM)。甲硫氨酸作为稳定剂特别有用,因为它还可以用作过氧化氢的清除剂,其中过氧化氢用于注射液或包装的预充式注射器的灭菌。
在一些实施例中,本发明的液体药物制剂包含作为第二稳定剂的抗氧化剂。“抗氧化剂”为防止活性药物成分氧化的药用赋形剂。抗氧化剂包括但不限于螯合剂,诸如EDTA、柠檬酸、抗坏血酸、丁基化羟基甲苯(BHT)、丁基化羟基茴香醚(BHA)、亚硫酸钠、对氨基苯甲酸、谷胱甘肽、没食子酸丙酯、半胱氨酸、甲硫氨酸、乙醇、苯甲醇和N-乙酰半胱氨酸。抗氧化剂可以约0.01mM至约100mM的浓度使用,特别是以约5mM至约50mM的浓度使用,并且更特别地以约5mM至约25mM的浓度使用。特别地,选择甲硫氨酸作为第二稳定剂,特别地,其浓度为约5mM至约25mM,更特别地,浓度为约10mM。
如本文所用的术语“糖”表示单糖或寡糖。单糖是不能被酸水解的单体碳水化合物,包括简单糖及其衍生物(例如氨基糖)。单糖的实例包括葡萄糖、果糖、半乳糖、甘露糖、山梨糖、核糖、脱氧核糖、神经氨酸。寡糖是由多于一个单体糖单元组成的碳水化合物,这些单体糖单元通过支化或链中的一个或多个糖苷键连接。寡糖内的单体糖单位可以相同或不同。根据单体糖单元的数量不同,寡糖为二糖、三糖、四糖、五糖等。与多糖相比,单糖和寡糖溶于水。寡糖的实例包括蔗糖、海藻糖、乳糖、麦芽糖和棉子糖。特别地,糖选自蔗糖和海藻糖,特别是蔗糖。
如本文所用的术语“氨基酸”一般表示具有位于羧基基团的α-位的氨基部分的药用有机分子。氨基酸的实例包括精氨酸、甘氨酸、鸟氨酸、赖氨酸、组氨酸、谷氨酸、天冬氨酸、异亮氨酸、亮氨酸、丙氨酸、苯丙氨酸、酪氨酸、色氨酸、甲硫氨酸、丝氨酸、脯氨酸,特别是甲硫氨酸。
如本文所用的术语“多元醇”表示指具有一个以上的羟基基团的药用醇。合适的多元醇包括但不限于甘露醇、山梨醇、甘油(丙三醇)、葡聚糖、阿糖醇、丙二醇、聚乙二醇以及它们的组合。多元醇可以10mM至约500mM的浓度使用,特别是以约10mM至约250mM的浓度使用,并且更特别地以约200mM至约250mM的浓度使用。
术语“稳定剂”还包括冻干保护剂。术语“冻干保护剂”表示用于保护不稳定的活性成分(例如蛋白质)在冻干过程以及随后的储存和复溶过程中免受不稳定条件的影响的药用赋形剂。冻干保护剂包括但不限于由糖、多元醇(诸如糖醇)和氨基酸组成的组。特别地,冻干保护剂可选自由以下项组成的组:糖,诸如蔗糖、海藻糖、乳糖、葡萄糖、甘露糖、麦芽糖、半乳糖、果糖、山梨糖、棉子糖、神经氨酸;氨基糖,诸如葡糖胺、半乳糖胺、N-甲基葡糖胺(“葡甲胺”);多元醇,诸如甘露醇和山梨醇;以及氨基酸,诸如甲硫氨酸或甘氨酸。冻干保护剂通常以约10mM至约600mM的浓度使用,特别是以约10mM至约250mM的浓度使用,并且更特别地以约100mM至约250mM的浓度使用。
张度剂
药物制剂还可包含张度剂。如本文所用的术语“张度剂”表示用于调节制剂的张度的药用张度剂。制剂可以是低渗、等渗或高渗制剂。等渗性通常涉及相对于溶液的渗透压,其通常相对于人血清的渗透压而言。根据本发明所述的制剂可以是低渗、等渗或高渗制剂,优选地,该药物制剂为等渗制剂。等渗制剂为液体或由固体形式(例如从冻干形式)复溶得到的液体并且表示与之相比的某种其他溶液诸如生理盐溶液和血清具有相似张度的溶液。合适的张度剂包括但不限于氯化钠、氯化钾、甘油以及选自由氨基酸、糖(特别是蔗糖)组成的组中的任意组分。在本发明的一个实施例中,优选张度剂为蔗糖。张度剂通常以约5mM至约1000mM、特别是约30mM至约500mM、更特别是约120mM至约200mM的浓度使用。用于本发明的等渗制剂中的张度剂通常以约50mM至约250mM、特别是约120mM至约200mM的浓度使用。更特别地,用于等渗制剂中的张度剂以130mM至190mM的浓度使用,并且甚至更特别地,在使用蔗糖作为张度剂的情况下,以约160mM±24mM的浓度使用。张度剂及其浓度选择为使得等渗制剂具有300mOsm/kg±100mOsm/kg的目标渗透压(特别是具有300mOsm/kg±50mOsm/kg的目标渗透压)。
在稳定剂和张度剂中,一组化合物可同时发挥两种作用,即它们同时用作稳定剂和张度剂。该化合物的实例可参见由糖类、氨基酸、多元醇、环糊精、聚乙二醇和盐组成的组。可同时用作稳定剂和张度剂的糖的一个实例为蔗糖和海藻糖,特别是蔗糖。
降粘剂
药物制剂可包含降粘剂。如本文所用的术语“降粘剂”表示用于降低制剂粘度的药用离子强度改性剂,其对于高浓度制剂以及在眼病的治疗中预计通过细针经玻璃体内施用的制剂非常(能够实现相对较快的施用,而无需使用高压注射)。典型降粘剂的实例为例如氯化钙或氯化钠。
佐剂
药物制剂还可包含佐剂,诸如防腐剂、润湿剂、乳化剂和分散剂。可通过灭菌程序并且通过加入各种抗菌和抗真菌剂(例如对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸等)以确保无微生物存在。防腐剂通常以约0.001%(w/v)至约2%(w/v)的浓度使用。防腐剂包括但不限于乙醇、苯甲醇、苯酚、间甲酚、对氯间甲酚、对羟基苯甲酸甲酯或对羟基苯甲酸丙酯、苯扎氯铵。
药物制剂还可包含少量的上述不同试剂,诸如缓冲剂、表面活性剂、稳定剂、离子强度改性剂,这些试剂的含量基本上不改变本发明药物制剂的技术特征,例如粘度为20mPas或更小(优选15mPas或更小)、浊度为30FTU或更小(优选25FTU或更小)、渗透压为300mOsm/kg±50mOsm/kg并且基本上不含可见颗粒。
使用
根据本发明所述的跟双特异性抗VEGF/ANG2抗体药物制剂可用于预防或治疗眼部血管疾病。为此,将用于玻璃体内施用的双特异性抗VEGF/ANG2抗体的药物制剂提供为液体等渗制剂,其粘度为20mPas或更小(优选15mPas或更小)、浊度为30FTU或更小(优选25FTU或更小)、渗透压为300mOsm/kg±50mOsm/kg,并且基本上不含可见颗粒。为此,双特异性抗VEGF/ANG2抗体的液体等渗药物制剂可提供于玻璃小瓶中,或呈预充式注射器的形式,特别是呈预充式玻璃注射器的形式。
对于经玻璃体内施用于眼睛的此类制剂,应避免使用如精氨酸等赋形剂,因为精氨酸作为组织纤溶酶原激活剂(t-PA)的载体对视网膜和视网膜色素上皮的毒性已见诸报道(参见例如:Benner J D,Morse LS,Toth CA等人,Arch Ophthalmol 19911091731-1736.1736;Johnson MW,Olsen KR,Hernandez E.等人,Arch Ophthalmool 1990108259-263.263;Irvine WD,Johnson MW,Heinandez E.等人,Arch Ophthalmol 1991109718-722.722;Johnson MW,Olsen KR,Heinandez E.,Retina,199111250-258.258)。因此,本发明的液体药物制剂基本上不含精氨酸(意指该制剂不含精氨酸或精氨酸的量低于可能有毒(通过作为组织纤溶酶原激活剂(t-PA)的载体而起作用)的浓度/含量)或不含精氨酸。低粘度对于实现商业规模的生产工艺(通过超滤提浓)并且确保轻松便捷的玻璃体内注射(在注射速度为50mm/min时,注射(滑行)力小于20N,特别是小于15N)至关重要。已证明,粘度小于15mPas的本发明的液体药物制剂能够通过30G注射针在5s注射时间内以小于5N的注射力注射。为此双特异性抗VEGF/ANG2抗体的药物制剂为液体等渗制剂,其粘度为15mPas或更小、浊度为25FTU或更小、渗透压为300mOsm/kg±50mOsm/kg并且基本上不含可见颗粒。为避免形成任何可见颗粒,本发明的液体药物制剂基本上不含氯化钙(意指该制剂不含氯化钙,或包含的氯化钙的量低于可能有助于形成可见颗粒的浓度/含量,使得该制剂仍然/基本上不含可见颗粒)或不含氯化钙。
术语“眼部血管疾病”和“血管性眼病”在本文可互换使用,包括但不限于眼内新生血管性综合征诸如糖尿病性视网膜病变、糖尿病性黄斑水肿、早产儿视网膜病变、新生血管性青光眼、视网膜静脉阻塞、视网膜中央静脉阻塞、黄斑变性、年龄相关性黄斑变性、色素性视网膜炎、视网膜血管瘤样增生、黄斑毛细血管扩张、缺血性视网膜病变、虹膜新生血管、眼内新生血管、角膜新生血管、视网膜新生血管、脉络膜新生血管和视网膜变性。(Garner,A.,Vascular diseases,In:Pathobiology of ocular disease,A dynamic approach,Garner,A.和Klintworth,G.K.主编,第2版,Marcel Dekker,New York(1994),第1625-1710页)。如本文所用,眼血管病症是指特征在于新血管的改变或不受调节的增殖和侵袭到眼组织诸如视网膜或角膜结构中的任何病理性病状。在一个实施例中,眼部血管疾病选自由以下项组成的组:湿性年龄相关性黄斑变性(湿性AMD)(也称为新生血管性年龄相关性黄斑变性(nAMD))、糖尿病性黄斑水肿(DME)、糖尿病性视网膜病变(DR)、非增生性糖尿病性视网膜病变(NPDR)、增生性糖尿病性视网膜病变(PDR)、黄斑囊样水肿(CME)、血管炎(例如视网膜中央静脉阻塞)、视网膜静脉阻塞(RVO)、视网膜中央静脉阻塞(CRVO)、视神经乳头水肿、视网膜炎、结膜炎、葡萄膜炎、脉络膜炎、多灶性脉络膜炎、眼部组织浆菌病、睑缘炎、干眼症(
与角膜新生血管相关联的其他疾病包括但不限于:流行性角膜结膜炎、维生素A缺乏症、角膜接触镜过度磨损、特应性角膜炎、上缘性角膜炎、翼状干燥性角膜炎、干燥综合征(sjogrens)、酒糟鼻、小水疱病(phlyctenulosis)、梅毒、分枝杆菌感染、化学烧伤、细菌性溃疡、真菌性溃疡、单纯疱疹感染、带状疱疹感染、原生动物感染、卡波西肉瘤、蚕食性角膜溃疡、特里昂边缘性角膜变性(Terrien's marginal degeneration)、边缘性角质层分离、类风湿性关节炎、系统性红斑狼疮、多发性动脉炎、创伤、韦格纳结节病(Wegenerssarcoidosis)、巩膜炎、史蒂芬约翰逊综合征(Steven's Johnson disease)、类天疱疮放射状角膜切开术和角膜移植排斥。
与视网膜/脉络膜新生血管相关联的疾病包括但不限于:糖尿病性视网膜病变、黄斑变性、镰形细胞贫血症、结节病、梅毒、弹性假黄瘤、佩吉特氏病、静脉阻塞、动脉阻塞、颈动脉阻塞性疾病、慢性葡萄膜炎/玻璃体炎、分枝杆菌感染、莱姆病、系统性红斑狼疮、早产儿视网膜病变、色素性视网膜炎、视网膜水肿(包括黄斑水肿)、伊尔斯病(Eales disease)、白塞氏病(Bechets disease)、引起视网膜炎或脉络膜炎的感染、假定眼组织胞浆菌病、贝斯特氏病(Bests disease)、近视、视窝(optic pits)、斯塔加特氏病(Stargartsdisease),睫状体扁平部炎、慢性视网膜脱离、高粘滞综合征、弓形体病、创伤和激光后并发症。其他疾病包括但不限于与红变(角部新生血管)相关联的疾病,以及由纤维血管或纤维组织异常增生引起的疾病(包括各种形式的增生性玻璃体视网膜病变)。
早产儿视网膜病变(ROP)是一种影响早产婴儿的眼病。据认为该疾病由视网膜血管生长紊乱引起,可能导致瘢痕形成和视网膜脱离。ROP可能是轻度的,并且可能自发消退,但是在严重情况下可能导致失明。因此,所有早产儿都存在发生ROP的危险,并且极低的出生体重是一个附加的风险因素。氧中毒和相对缺氧均可能导致ROP的发展。
黄斑变性是一种医学疾病,主要见于老年人群,其表现为眼内层中心(称为视网膜的黄斑区域)变薄、萎缩,并且在某些情况下出血。该疾病可能导致丧失中央视觉,从而导致无法看到细节、无法阅读或无法识别面部。根据美国眼科学会,该疾病是目前美国50岁以上人群中央视觉丧失(失明)的主要原因。尽管某些影响年轻人的黄斑营养不良有时被称为黄斑变性,但是该术语通常是指年龄相关性黄斑变性(AMD或ARMD)。
如本文所用的“年龄相关性黄斑变性(AMD)”是指视网膜的小中央部分(称为黄斑)退化时的严重眼病。湿性AMD(湿性AMD(wAMD),也称为新血管性AMD(nAMD))是一种晚期AMD,其特征在于黄斑下方脉络膜的血管生长异常。这称为脉络膜新生血管。这些血管导致血液和液体渗漏到视网膜中,导致视觉失真,使直线看起来呈波浪形,以及盲点和中央视觉丧失。这些异常血管最终形成疤痕,导致永久性中央视觉丧失。AMD的症状包括视觉中心存在黑暗的模糊区域;以及色觉减弱或改变。AMD可以在常规的眼科检查中检出。黄斑变性的最常见的早期症状之一是视网膜下存在玻璃疣状微小的黄色沉积物或色素结块。
色素性视网膜炎(RP)是一组遗传性眼病。在RP症状的发展过程中,夜盲症通常比隧道视觉先出现数年或甚至数十年。许多患有RP的人直到40多岁或50多岁才成为法定盲人,并且终生保留一定的视力。其他人则可能因为RP而完全失明,并且在某些病例中早在幼儿期就已完全失明。RP的进展因病例而异。RP是一种遗传性视网膜营养不良,属于一组遗传性疾病,其中视网膜的感光器(视杆细胞和视锥细胞)或视网膜色素上皮(RPE)异常导致进行性视力丧失。受影响的个体首先经历暗适应不良或夜盲症(夜盲),然后发生周围视野减小(称为隧道视觉),并且有时在病程晚期丧失中央视觉。
当液体和蛋白质沉积物积聚在眼黄斑(即视网膜的黄色中央区域)上方或下方时,导致其增厚和肿胀时,发生黄斑水肿。由于黄斑位于眼球后部视网膜中央附近,因此肿胀可能使人的中央视觉扭曲。该区域容纳紧密排列的视锥,提供明显、清晰的中央视觉,使人们能够看到实现范围内的形状、颜色和细节。囊性黄斑水肿是一种包括囊肿形成的黄斑水肿。
如本文所用,“糖尿病性黄斑水肿”(DME)是指一种影响糖尿病人(1型或2型)的严重眼病。当视网膜中的血管渗漏到黄斑中并且液体和蛋白质沉积物积聚在眼黄斑(即视网膜的黄色中央区域)上方或下方时并且导致增厚和肿胀时,发生黄斑水肿。由于黄斑位于眼球后部视网膜中央附近,因此肿胀可能使人的中央视觉扭曲。DME的主要症状包括但不限于视力模糊、漂浮、对比度损失、复视和最终视力丧失。DME的病理特征在于血液-视网膜屏障(在正常情况下防止水在视网膜中运动)的破坏,从而使液体在视网膜组织中积聚,并且存在视网膜增厚。DME目前在眼睛检查中得到诊断,该检查由视力测验(测定一个人可以在标准图表上阅读的最小字母)、扩张眼睛检查(用于检查疾病的症状)、影像学检查(诸如光学相干断层扫描(OCT)或荧光素血管造影(FA))和眼压计(一种测量眼内压力的仪器)组成。还实施以下研究以确定治疗方法:光学相干断层扫描(OCT)、荧光素血管造影和彩色立体眼底照相。DME可以大致分为两个主要类别:局灶性DME和弥漫性DME。局灶性DME的特征在于黄斑区存在分离且明显渗漏的特定区域,其中黄斑血流充足。弥漫性DME由黄斑周围的整个毛细血管床渗漏引起,而这种渗漏由眼睛内部的血液-视网膜屏障破坏引起。除局灶性DME和弥漫性DME以外,DME还根据临床检查结果分为有临床意义的黄斑水肿(CSME)、非CSME以及累及中央的CSME(CSME-CI,其涉及中央凹)。本发明包括治疗上述类别的DME的方法。
在本发明的一个实施例中,眼部血管疾病为选自由以下项组成的组:湿性年龄相关性黄斑变性(湿性AMD)、糖尿病性黄斑水肿(DME)、糖尿病性视网膜病变(DR)、非增生性糖尿病性视网膜病变(NPDR)、增生性糖尿病性视网膜病变(PDR)、血管炎(例如视网膜静脉阻塞(RVO)和视网膜中央静脉阻塞(CRVO))。
在一个实施例中,该眼部血管疾病选自由以下项组成的组:糖尿病性视网膜病变(DR)、糖尿病性黄斑水肿(DME)、视网膜静脉阻塞(RVO)、视网膜中央静脉阻塞(CRVO)、黄斑变性、湿性年龄相关性黄斑变性(湿性AMD)、早产儿视网膜病变(ROP)、新生血管性青光眼、色素性视网膜炎(RP)、视网膜血管瘤样增生、黄斑毛细血管扩张、缺血性视网膜病变、虹膜新生血管、眼内新生血管、角膜新生血管、视网膜新生血管、脉络膜新生血管和视网膜变性,特别地选自由以下项组成的组糖尿病性视网膜病变(DR)、糖尿病性黄斑水肿(DME)、视网膜静脉阻塞(RVO)、视网膜中央静脉阻塞(CRVO)、湿性年龄相关性黄斑变性(湿性AMD)。
在本发明的一个实施例中,眼部血管疾病为糖尿病性视网膜病变(DR)。
在本发明的一个实施例中,眼部血管疾病为糖尿病性黄斑水肿(DME)。
在本发明的一个实施例中,眼部血管疾病为视网膜静脉阻塞(RVO)。
在本发明的一个实施例中,眼部血管疾病为视网膜中央静脉阻塞(CRVO)。
在本发明的一个实施例中,眼部血管疾病为早产儿视网膜病变(ROP)。
在本发明的一个实施例中,眼部血管疾病为黄斑变性。
在本发明的一个实施例中,眼部血管疾病为年龄相关性黄斑变性(AMD)。
在本发明的一个实施例中,眼部血管疾病为湿性年龄相关性黄斑变性(wAMD)。
在本发明的一个实施例中,眼部血管疾病为脉络膜新生血管。
施用
根据本发明所述的液体药物制剂可通过玻璃体内(IVT)方式施用,例如制药领域中已知的那些方式(例如,采用适当的注射器)。对于玻璃体内注射,通常注射体积为50μL至100μL。玻璃体内注射通过使用一次性注射器和30G(25G至30G)注射针或配备适当的注射针的预充式注射器进行。可使用孔径为5μm的过滤器针将液体制剂从包含该制剂的小瓶中抽出。玻璃体内注射技术描述于例如以下文献中:D.Yorston,Community Eye Health.2014;27(87):47。
为此,双特异性抗VEGF/ANG2抗体的药物制剂为液体等渗制剂,其粘度为15mPas或更小,浊度为25FTU或更小,渗透压为300mOsm/kg±50mOsm/kg,并且基本上不含可见颗粒。
待用于体内施用的制剂必须为无菌的。这通过无菌滤膜过滤和无菌生产实践很容易实现。
用于制备制剂的方法
可通过本领域中处理已知的方法或工艺(例如超滤-渗滤、透析、添加与混合、冻干、复溶以及它们的组合)来制备根据本发明所述的药物制剂。根据本发明所述的制剂的制备例可参见下文。
在本发明的一个实施例中,药物制剂可通过以下生产方法或工艺进行生产,该生产方法或工艺包括以下步骤:
1.使用MWCO(截留分子量)在5kD与80kD之间(30kD至50kD)(30kD)的半透膜,通过超滤和渗滤,与渗滤缓冲剂(包含组氨酸-醋酸盐缓冲剂、或组氨酸-醋酸盐缓冲剂和氯化钠、或组氨酸-醋酸盐缓冲剂、氯化钠和甲硫氨酸、或组氨酸-醋酸盐缓冲剂、氯化钠、甲硫氨酸和蔗糖)进行缓冲剂交换。通常,渗滤缓冲剂与本体溶液的比率为5至20(5-10)。
2.替代1所述的操作,可使用透析缓冲剂(包含组氨酸-醋酸盐缓冲剂、或组氨酸-醋酸盐缓冲剂和氯化钠、或组氨酸-醋酸盐缓冲剂、氯化钠和甲硫氨酸、或组氨酸-醋酸盐缓冲剂、氯化钠、甲硫氨酸和蔗糖)以及MWCO在5kD与80kD之间(30kD至50kD)(30kD)的透析膜通过透析来实现缓冲剂交换。通常,透析缓冲剂与本体溶液的比率为5至20(5-10)。
3.使用MWCO(截留分子量)在5kD与80kD之间(30kD至50kD)(30kD)的渗滤膜,通过超滤将缓冲剂交换的本体溶液浓缩至蛋白质浓度超过120mg/mL(120mg/mL至160mg/mL或120mg/mL至200mg/mL)。
4.药物制剂的最终组成可通过添加相应赋形剂的储备溶液或通过适当的调节缓冲剂进行调节。通过混合使溶液均质化。
此外,生产方法或工艺可包括以下步骤:
5.最终配制的溶液可冷冻储存于-20℃以下(-40℃以下)。
6.在灌装到最终的主容器中之前,将溶液解冻
7.将多个容器或批次的药物制剂混合并且通过搅拌使其均质化
8.均质化药物制剂通过多个(至少两个)孔径为至少0.2μm或0.22μm的灭菌级过滤器过滤。
9.在无菌条件下,将无菌过滤后的溶液灌装到无菌小瓶或预充式注射器中,并且用弹性塞(分别是推杆塞和端盖)封闭。
10.检查灌装后的主容器是否存在缺陷和可见颗粒
11.将预充式注射器与相应的设备组件组装在一起,包装成无菌屏障系统,并且对外表面灭菌。
12.将小瓶和无菌注射器包装在最终的二级包装中
根据本发明所述的药物制剂也可提供为冻干形式或由冻干形式复溶的液体形式。“冻干形式”通过本领域中已知的冻干方法制得。冻干物通常具有约0.1%(w/w)至5%(w/w)的残留水分含量,并且以粉末或物理稳定的饼状物形式存在。“复溶形式”可通过在添加复溶介质后使冻干物快速溶解而获得。合适的复溶介质包括但不限于注射用水(WFI)、抑菌注射用水(BWFI)、氯化钠溶液(例如0.9%(w/v)NaCl)和葡萄糖溶液(例如5%(w/v)葡萄糖)。
抗体的生产
特别适用于本发明的抗VEGF/ANG2抗体通过重组手段进行生产。重组生产方法是现有技术中所熟知的,并且包括在原核和真核细胞中表达蛋白质,随后分离出抗体,并且通常将其纯化至药用纯度。为了在宿主细胞中表达上述抗体,利用标准方法将编码相应的经修饰的轻链和重链的核酸插入表达载体中。在适当的原核或真核宿主细胞(例如CHO细胞、NSO细胞、SP2/0细胞、HEK293细胞、COS细胞、PER.C6细胞、酵母或大肠杆菌细胞)中进行表达,并且从细胞(裂解后的上清液或细胞)中回收抗体。用于重组生产抗体的一般方法是现有技术中所熟知的,并且综述于以下论文中:Makrides,S.C.,Protein Expr.Purif.17(1999)183-202;Geisse,S.等人,Protein Expr.Purif.8(1996)271-282;Kaufman,R.J.,Mol.Biotechnol 16(2000)151-160;Werner,R.G.,Drug Res.48(1998)870-880。一种制备用于本发明的抗体的制备方法,包括以下步骤:a)用包含编码所述抗体的核酸分子的载体转化宿主细胞;b)在允许合成所述抗体分子的条件下培养宿主细胞;和c)从所述培养基中回收所述抗体分子。
抗体适合通过常规的免疫球蛋白纯化方法(诸如蛋白A-琼脂糖凝胶、羟磷灰石层析、凝胶电泳、透析或亲和层析)与培养基分离。编码单克隆抗体的DNA和RNA很容易通过常规方法进行分离和测序。杂交瘤细胞可用作此类DNA和RNA的来源。一旦分离,可以将DNA插入表达载体中,然后将该表达载体转染至至不另外产生免疫球蛋白的宿主细胞诸如HEK293细胞、CHO细胞或骨髓瘤细胞中,以在宿主细胞中获得重组单克隆抗体的合成。
双特异性抗体的氨基酸序列变体(或突变体)通过将适当的核苷酸改变引入抗体DNA中或通过核苷酸合成来制备。但是,此类修饰只能在非常有限的范围内进行。例如,修饰不改变上述抗体特性,诸如IgG同种型和抗原结合,但是可以改善重组生产的产率、蛋白质稳定性或促进纯化。
如本申请中所用的术语“宿主细胞”表示可以工程化以产生包含在本发明的制剂中的抗体的任何种类的细胞系统。在一个实施例中,将HEK293细胞和CHO细胞用作宿主细胞。
如本文所用,词语“细胞”、“细胞系”和“细胞培养物”可互换使用,并且所有此类名称都包括后代。因此,词语“转化体”和“转化的细胞”包括原代受试细胞和不考虑转移的数目从该细胞衍生的培养物。还应当理解,由于故意的或非故意的突变,所有后代可能在DNA含量上不是精确地相同的。包括如在原始转化细胞中筛选的具有相同功能或生物活性的变体子代。
NS0细胞中的表达描述于例如以下文献中:Barnes,L.M.等人,Cytotechnology 32(2000)109-123;Barnes,L.M.等人,Biotech.Bioeng.73(2001)261-270。瞬时表达描述于例如以下文献中:Durocher,Y.等人,Nucl.Acids.Res.30(2002)E9。可变结构域的克隆描述于例如以下文献中:Orlandi,R.等人,Proc.Natl.Acad.Sci.USA 86(1989)3833-3837;Carter,P.等人,Proc.Natl.Acad.Sci.USA 89(1992)4285-4289;和Norderhaug,L.等人,J.Immunol.Methods 204(1997)77-87。优选的瞬时表达系统(HEK 293)描述于例如以下文献中:Schlaeger,E.-J.和Christensen,K.,Cytotechnology 30(1999)71-83;和Schlaeger,E.-J.,J.Immunol.Methods 194(1996)191-199。
适用于原核生物的控制序列例如包括启动子,任选地操纵子序列,和核糖体结合位点。已知真核细胞利用启动子、增强子和多腺苷酸化信号。
当核酸与另一个核酸序列置于功能性关系中时,该核酸是“可操作连接的”。例如,将前序列或分泌前导序列的DNA与用于多肽的DNA(如果其表达为参与多肽分泌的前蛋白)可操作地连接;将启动子或增强子与影响序列的转录的编码序列可操作地连接;或者将核糖体结合位点与定位为便于翻译的编码序列可操作地连接。通常,“可操作地连接”意指所连接的DNA序列是连续的,并且对于分泌前导序列而言是连续的并且在读框中。但是,增强子不必是连续的。通过在方便的限制性酶切位点连接来完成连接。如果不存在此类位点,则根据常规做法使用合成的寡核苷酸衔接子或连接基。
进行抗体纯化以便通过标准技术消除细胞组分或其他污染物(例如其他细胞核酸或蛋白质),所述标准技术包括碱/SDS处理、CsCl显带、柱层析、琼脂糖凝胶电泳和本领域中熟知的其他技术。参见Ausubel,F.等人主编的Current Protocols in Molecular Biology(Greene Publishing and Wiley Interscience,New York,1987)。各种不同的蛋白质纯化方法已经非常成熟并且得到广泛应用,诸如微生物蛋白亲和层析(例如,蛋白A或蛋白G亲和层析)、离子交换色谱(例如,阳离子交换(羧甲基树脂)、阴离子交换(氨乙基树脂)和混合模式交换)、亲硫吸附(例如,用β-巯基乙醇和其他SH配体)、疏水相互作用或芳族吸附层析(例如,使用苯基-琼脂糖、亲杂氮芳基树脂或间氨基苯基硼酸)、金属螯合亲和层析(例如,使用Ni(II)-亲和材料和Cu(II)-亲和材料)、体积排阻色谱和电泳方法(诸如凝胶电泳、毛细管电泳)(Vijayalakshmi,M.A.,Appl.Biochem.Biotech.75(1998)93-102)。
本申请中公开的氨基酸序列:
SEQ ID NO:描述
SEQ ID NO:描述
下面列出本发明的实施例:
1.一种液体药物制剂,其包含:
-20mg/mL至150mg/mL双特异性抗VEGF/ANG2抗体,所述抗体包含人IgG1亚类的重链恒定区(在一个实施例中,包含30mg/mL±4.5mg/mL或120mg/mL±18mg/mL),
-15mM至35mM氯化钠(在一个实施例中,包含25mM±5mM;在一个实施例中,包含25mM±3.75mM氯化钠;特别地,包含25mM±2.5mM氯化钠),
-15mM至25mM组氨酸醋酸盐缓冲剂(在一个实施例中,包含20mM±3mM组氨酸醋酸盐缓冲剂;在一个实施例中,包含20mM±2mM组氨酸醋酸盐缓冲剂),
其PH值为5.5±0.5(在一个实施例中,PH值为5.5±0.3;特别地,PH值为5.5±0.2);
其中所述双特异性抗VEGF/ANG2抗体为二价的,并且包含与人VEGF特异性结合的第一抗原结合位点和与人ANG-2特异性结合的第二抗原结合位点,其中
i)与VEGF特异性结合的所述第一抗原结合位点在重链可变结构域中包含SEQ IDNO:1的CDR3H区、SEQ ID NO:2的CDR2H区和SEQ ID NO:3的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:4的CDR3L区、SEQ ID NO:5的CDR2L区和SEQ ID NO:6的CDR1L区;并且
ii)与ANG-2特异性结合的所述第二抗原结合位点在重链可变结构域中包含SEQID NO:9的CDR3H区、SEQ ID NO:10的CDR2H区和SEQ ID NO:11的CDR1H区,并且在轻链可变结构域中包含SEQ ID NO:12的CDR3L区、SEQ ID NO:13的CDR2L区和SEQ ID NO:14的CDR1L区,
并且其中
iii)所述双特异性抗体包含人IgG1亚类的重链恒定区,所述重链恒定区包含突变I253A、H310A和H435A以及突变L234A、L235A和P329G(根据Kabat EU索引编号)。
2.根据实施例1所述的药物制剂,其中双特异性抗VEGF/ANG2抗体为二价的,并且包含与人VEGF特异性结合的第一抗原结合位点以及与人ANG-2特异性结合的第二抗原结合位点,其中
i)与VEGF特异性结合的所述第一抗原结合位点包含作为重链可变结构域VH的SEQID NO:7的氨基酸序列和作为轻链可变结构域VL的SEQ ID NO:8的氨基酸序列,并且
ii)与ANG-2特异性结合的所述第二抗原结合位点包含作为重链可变结构域VH的SEQ ID NO:15的氨基酸序列和作为轻链可变结构域VL的SEQ ID NO:16的氨基酸序列。
3.根据实施例2所述的药物制剂,其中双特异性抗VEGF/ANG2抗体为二价的,并且包含与人VEGF特异性结合的第一抗原结合位点以及与人ANG-2特异性结合的第二抗原结合位点,其中
其中
iv)在所述重链恒定区中,S354C和T366W突变包含在一个CH3结构域中,并且Y349C、T366S、L368A和Y407V突变包含在另一个CH3结构域中(根据Kabat EU索引编号)。
4.根据实施例1至3中任一项所述的药物制剂,其中双特异性抗VEGF/ANG2抗体为二价的,并且包含SEQ ID NO:17的氨基酸序列、SEQ ID NO:18的氨基酸序列、SEQ ID NO:19的氨基酸序列和SEQ ID NO:20的氨基酸序列。
5.根据实施例1至3中任一项所述的药物制剂,其中双特异性抗VEGF/ANG2抗体为法立昔单抗。
6.根据实施例1所述的药物制剂,其中制剂包含
-120mg/mL±18mg/mL双特异性抗VEGF/ANG2抗体(特别是120mg/mL±12mg/mL双特异性抗VEGF/ANG2抗体)。
7.根据权利要求1至7中任一项所述的药物制剂,其用于玻璃体内施用。
8.根据实施例1至6中任一项所述的药物制剂,其中制剂基本上不含可见颗粒。
9.根据实施例1至8中任一项所述的药物制剂,其中制剂进一步包含
-1mM至20mM至少一种稳定剂(在一个实施例中,选自糖、多元醇和氨基酸)。
10.根据实施例1至8中任一项所述的药物制剂,其中制剂进一步包含
-7.0mM±2.0mM甲硫氨酸(在一个实施例中,包含7.0mM±1.0mM甲硫氨酸;在一个实施例中,包含7.0mM±0.7mM甲硫氨酸)。
11.根据实施例1至10中任一项所述的药物制剂,其中制剂进一步包含
-0.01%至0.07%表面活性剂(在一个实施例中,选自聚山梨酯20、聚山梨酯80或泊洛沙姆)。
12.根据实施例1至10中任一项所述的药物制剂,其中制剂进一步包含
-0.04%(w/v)±0.02(w/v)聚山梨酯20(在一个实施例中,包含0.03%(w/v)至0.07%(w/v);在一个实施例中,包含0.04%(w/v)±0.01(w/v);在一个实施例中,包含约0.04%(w/v))。
13.根据实施例1至12中任一项所述的药物制剂,其中制剂进一步包含
-50mM至250mM张度剂(在一个实施例中,张度剂选自蔗糖、海藻糖和山梨糖醇)。
14.根据实施例1至12中任一项所述的药物制剂,其中制剂进一步包含
-160mM±24mM蔗糖(在一个实施例中,包含160mM±16mM;在一个实施例中,包含约160mM)。
15.根据实施例1至14中任一项所述的药物制剂,其中制剂具有20mPas或更小(在一个实施例中,17mPas或更小;在一个实施例中,16mPas或更小;在一个实施例中,约15mPas或更小;在一个实施例中,15mPas或更小)的粘度。
16.根据实施例1至15中任一项所述的药物制剂,其中制剂具有浊度of 30FTU或更小(在一个实施例中,27FTU或更小;在一个实施例中,26FTU或更小;在一个实施例中,约25FTU或更小;在一个实施例中,25FTU或更小)的浊度。
17.根据实施例1至16中任一项所述的药物制剂,其中制剂具有在20与50之间的离子强度(在一个实施例中,具有在30与50之间的离子强度;在一个实施例中,具有在30与45之间的离子强度;在一个实施例中,具有30±10的离子强度)。
18.根据实施例1至17中任一项所述的药物制剂,其用于玻璃体内施用,其中制剂基本上不含氯化钙(或不含氯化钙)。
19.根据实施例1至17中任一项所述的药物制剂,其用于玻璃体内施用,其中制剂基本上不含精氨酸(或不含精氨酸)。
20.根据实施例1至17中任一项所述的药物制剂,其用于玻璃体内施用,其中制剂基本上不含精氨酸和氯化钙(或不含精氨酸和氯化钙)。
21.根据实施例1至5和实施例7至20中任一项所述的药物制剂,其中所述制剂包含(至少)以下组分或(至少)由以下组分组成:
-20mg/mL至150mg/mL双特异性抗VEGF/ANG2抗体(在一个实施例中,包含30mg/mL±4.5mg/mL或120mg/mL±18mg/mL,特别是120mg/mL±12mg/mL);
-15mM至35mM氯化钠(在一个实施例中,包含25mM±5mM;在一个实施例中,包含25mM±3.75mM氯化钠;特别是包含25mM±2.5mM氯化钠);
-15mM至25mM组氨酸醋酸盐缓冲剂(在一个实施例中,包含20mM±3mM组氨酸醋酸盐缓冲剂;在一个实施例中,包含20mM±2mM组氨酸醋酸盐缓冲剂);
-7.0mM±2.0mM甲硫氨酸(在一个实施例中,包含7.0mM±1.0mM甲硫氨酸;在一个实施例中,包含7.0mM±0.7mM甲硫氨酸);
-0.03%(w/v)至0.07%(w/v)聚山梨酯20(在一个实施例中,包含0.04%(w/v)±0.02(w/v);在一个实施例中,包含0.04%(w/v)±0.01(w/v);在一个实施例中,包含约0.04%(w/v));
-160mM±24mM蔗糖(在一个实施例中,包含160mM±16mM;在一个实施例中,包含约160mM);
-水(用于(眼科)注射液);
其PH值为5.5±0.5(在一个实施例中,PH值为5.5±0.3;特别地,PH值为5.5±0.2);
22.根据实施例1至21中任一项所述的药物制剂,其中制剂为稳定制剂。
23.根据实施例1至21中任一项所述的药物制剂,其中药物制剂在25℃储存8周后或在25℃储存52周后,药物制剂中双特异性抗体的高分子量物质(HMW)含量低于10%(在一个实施例中,低于5%)。
24.根据实施例1至21中任一项所述的药物制剂,其中药物制剂在2-8℃储存2年后(或在2-8℃储存3年后),药物制剂中双特异性抗体的主峰多于50%并且双特异性抗体的高分子量物质(HMW)低于10%(药物制剂在2-8℃储存2年后(或在2-8℃储存3年后),药物制剂中双特异性抗体的主峰多于55%并且双特异性抗体的高分子量物质(HMW)低于7%)。
25.根据实施例1至24中任一项所述的药物制剂,其中制剂的渗透压为300mOsm/kg±100mOsm/kg(在一个实施例中,为300mOsm/kg±50mOsm/kg)。
26.根据实施例1至25中任一项所述的药物制剂,其用于治疗眼部血管疾病。
27.根据实施例26的应用的药物制剂,其中眼部血管疾病选自由以下项组成的组:糖尿病性视网膜病变(DR)、糖尿病性黄斑水肿(DME)、视网膜静脉阻塞(RVO)、视网膜中央静脉阻塞(CRVO)、黄斑变性、湿性年龄相关性黄斑变性(湿性AMD)、早产儿视网膜病变(ROP)、新生血管性青光眼、色素性视网膜炎(RP)、视网膜血管瘤样增生、黄斑毛细血管扩张、缺血性视网膜病变、虹膜新生血管、眼内新生血管、角膜新生血管、视网膜新生血管、脉络膜新生血管和视网膜变性,特别地选自由以下项组成的组:糖尿病性视网膜病变(DR)、糖尿病性黄斑水肿(DME)、视网膜静脉阻塞(RVO)、视网膜中央静脉阻塞(CRVO)、湿性年龄相关性黄斑变性(湿性AMD)。
28.根据实施例26的应用的药物制剂,其中眼部血管疾病为糖尿病性视网膜病变。
29.根据实施例26的应用的药物制剂,其中眼部血管疾病为糖尿病性黄斑水肿(DME)。
30.根据实施例26的应用的药物制剂,其中眼部血管疾病为视网膜静脉阻塞(RVO)。
31.根据实施例26的应用的药物制剂,其中视网膜静脉阻塞为视网膜中央静脉阻塞(CRVO)。
32.根据实施例26的应用的药物制剂,其中眼部血管疾病为黄斑变性。
33.根据实施例26的应用的药物制剂,其中黄斑变性为年龄相关性黄斑变性(AMD)。
34.根据实施例26的应用的药物制剂,其中黄斑变性为湿性年龄相关性黄斑变性(wAMD)(也称为新生血管性年龄相关性黄斑变性(nAMD))。
35.根据实施例26的应用的药物制剂,其中眼部血管疾病为脉络膜新生血管。
36.一种用于制备根据实施例1至25中任一项所述的药物制剂的方法,
所述方法包括以下步骤:
-所述双特异性抗体本体溶液a)通过超滤和渗滤与渗滤缓冲剂进行缓冲剂交换,或b)使用透析缓冲剂通过透析进行缓冲剂交换,所述缓冲剂包含组氨酸-醋酸盐缓冲剂,或组氨酸-醋酸盐缓冲剂和氯化钠,或组氨酸-醋酸盐缓冲剂,氯化钠和甲硫氨酸,或组氨酸-醋酸盐缓冲剂,氯化钠,甲硫氨酸和蔗糖
-通过超滤浓缩所述缓冲剂交换的本体溶液
-通过添加相应赋形剂的储备溶液或通过适当的调节缓冲剂来调整所述药物制剂的最终组成,并且通过混合使所述液体药物制剂均质化。
37.一种小瓶,其包含根据实施例1至25中任一项所述的药物制剂。
38.一种预充式注射器,其包含根据实施例1至25中任一项所述的药物制剂。
39.根据实施例1至25中任一项所述的液体药物制剂的冻干形式。
实例
根据本发明所述的用于玻璃体内(IVT)施用的液体药品药物制剂的开发如下。
实例1:材料与方法
提供了按照WO2014/009465所述的方法制备和纯化的双特异性抗VEGF/ANG2抗体CrossMAb VEGFang2-0016(法立昔单抗),用于最初在20mM组氨酸-HCl缓冲剂(pH 5.5)中采用约130mg/mL至140mg/mL的浓度的进一步实验。
在制剂的制备过程中使用的材料(包括供应商)及其主要包装提供于表1和表2中。
表1用于制剂的化学品
表2主要包装
容器密封系统
2mL或6mL无色玻璃小瓶(1型玻璃)通过橡胶塞(D 777-1,13mm)和带翻转盖的铝质顶封装置封闭。
1.0mL带有鲁尔锥(luer cone)的无色预充式注射器(1型玻璃)通过GerresheimerBuende TELC端盖和West 4023/50推杆塞封闭。
0.5mL带有鲁尔锥的无色预充式注射器(1型玻璃)通过Vetter OVS端盖和West4023/50推杆塞封闭。Vetter OVS端盖由West 4023/50弹性体组成。
体积排阻色谱(SE-HPLC)
体积排阻色谱法(SEC)用于检测制剂中的可溶性高分子量物质(聚集体)和低分子量(LMW)水解产物。该方法采用TSK-Gel G3000SWXL,7.8×300mm,5μm(Tosoh Bioscience,目录号08541)或BioSuite 250,7.8×300mm,5μm(Waters,目录号186002165)进行。使用0.2M磷酸钾、0.25M KCl(pH 7.0)作为流动相,通过等度洗脱曲线分离完整单体、聚集体和片段,并且在280nm的波长下进行检测。
离子交换色谱(IE-HPLC))
离子交换色谱法(IEC)用于检测改变制剂中抗体的净电荷的化学降解产物。该方法采用YMC BioPro SP-F,100×4.6mm,5μm色谱柱(YMC,目录号SF00S05-1046WP)进行。使用20mM BES(N,N-双[2-羟乙基]-2-氨基乙烷磺酸)(pH 6.8)作为洗脱液A,并且使用20mMBES、488mM NaCl(pH 6.8)作为洗脱液B,其流速为0.8mL/min。在样品注入色谱柱之前,用洗脱液A将其稀释至3mg/mL。
梯度程序:
浊度(单位为FTU(=福尔马肼浊度单位))
根据欧洲药典2.2.1“液体的澄清度和乳浊度”,在Hach 2100AN浊度计上测量制剂样品的浊度。将样品体积约2mL的样品溶液转移至内径为11mm的玻璃比色杯中。将该玻璃比色杯放入浊度计中,并且根据参比悬浮液的校准曲线(1FTU、3FTU、10FTU、20FTU和100FTU测量浊度。
粘度(单位为mPa)
采用Anton Paar Physica MCR 301旋转流变仪测量制剂样品的粘度,所用锥度为25mm–0.5°,剪切速率为1000s
可见颗粒
在Seidenader检验设备V90-T上借助2倍放大镜,目视检查小瓶中的样品。将照明光源L1、L2和L3调整至设置5。在旋转运动过程中检查小瓶中的样品中是否存在颗粒。
蛋白质浓度(单位为mg/mL)。
制剂样品的蛋白质浓度在Perkin Elmer的UV/Vis光度计λ35上通过紫外(UV)光吸收来测量。将制剂样品用20mM L-组氨酸-醋酸盐缓冲液(pH 5.5)稀释至蛋白质浓度约0.5mg/mL,然后装入1cm厚的比色皿中。测量比色皿在280nm和320nm的波长下的UV吸光度。
根据在280nm和320nm下测得的UV吸光度(分别为A280和A320)、1.70mL/(mg x cm)的消光系数(E)、1cm的厚度(d)以及对应于实际稀释液的稀释倍数(DF),通过以下公式计算蛋白质浓度:
渗透压
制剂样品的渗透压根据冰点降低原理在Gonotec的Osmomat 030 3P渗压计上进行测量。
pH
制剂样品的pH采用玻璃电极通过电位法测定。
实例2:pH/缓冲剂筛选I
设置
pH/缓冲剂筛选的范围用于选择抗VEGF/ANG2抗体的商业制剂的最佳pH和缓冲剂,并且选择具有低粘度、低浊度和良好的稳定性行为(从而减少形成的可溶性聚集体和带电荷的变体)的制剂。
pH/缓冲剂筛选的第一部分包括三种缓冲系统:L-组氨酸/L-组氨酸盐酸盐(His/His-HCl)、L-组氨酸醋酸盐(His/醋酸盐)和醋酸钠(Na/醋酸盐),pH范围在5.3与6.5之间,缓冲剂强度范围在7mM与300mM之间,并且离子强度范围在5与86之间。活性制剂的设置如表3所示。
表3制剂代码:pH/缓冲剂筛选第I部分
材料与方法
在制剂的制备过程中使用的材料及其主要包装提供于表1和表2中。
使用截留分子量为10kD的Slide-A-Lyzer G2(Thermo Scientific),通过透析对原料药与表3所列的缓冲系统进行缓冲剂交换。从而将42mL原料药填充到透析设备中,并且用5L透析缓冲剂进行三次缓冲剂交换。
任选地,如果透析后蛋白质浓度低于120mg/mL,则使用Amicon Ultra 15、Ultracel 10K(Millipore)设备通过离心(20℃,4000rpm)对原料药进行浓缩。
然后,将透析后和任选浓缩后的原料药分别用透析缓冲剂稀释至目标蛋白质浓度120mg/mL,得到最终药品溶液。
每种药品溶液通过0.22μm Sterivex GV(Millipore)过滤器过滤,并且灌装到洁净并且无菌的6mL小瓶中,灌装体积为2.7mL。将小瓶加塞并卷压。
分析方法
实例1中描述了浊度和粘度的分析检测方法。
结果
图1比较了由pH/缓冲剂筛选I得到的制剂的浊度和粘度结果。
一般而言,具有高离子强度(例如,4、14、10、3、8和18)的制剂表现出高浊度(高于30FTU)和低粘度(约10mPas)。相反,离子强度低的制剂具有高粘度(约25mPas至40mPas)和低浊度(低于10FTU)。令人惊讶的是,可鉴定出采用缓冲系统组氨酸醋酸盐(13、6、4、14)的制剂具有约10mPas的低粘度和小于25FTU的浊度。
结论
测量由pH/缓冲剂筛选I得到的制剂的粘度和浊度。令人惊讶的是,采用组氨酸-醋酸盐缓冲系统并且具有45.5或更高的离子强度的制剂表现出低粘度和减小的浊度值。
实例3:pH/缓冲剂筛选II
设置
pH/缓冲剂筛选的范围用于选择抗VEGF/ANG2抗体CrossMAb VEGFang2-0016(法立昔单抗)抗VEGF/ANG2抗体的制剂的最佳pH和缓冲剂,并且选择具有低粘度、低浊度和良好的稳定性行为(从而减少形成的可溶性聚集体和带电荷的变体)的制剂。
pH/缓冲剂筛选的第二部分基于根据pH/缓冲剂筛选I的结果进行设计,并且包括缓冲剂L-组氨酸-醋酸盐(His/醋酸盐),pH范围在5.5与6.0之间,并且缓冲剂强度范围在14mM与59mM之间。通过提高缓冲剂强度或添加氯化钠(NaCl)或氯化钙(Ca Cl
表4制剂代码:pH/缓冲剂筛选第II部分
材料与方法
在制剂的制备过程中使用的材料及其主要包装提供于表1和表2中。
使用配备截留分子量为30kD的半透膜的Labscale TTF(Millipore),通过超滤-渗滤对原料药与表4中列出的缓冲系统进行缓冲剂交换。从而将120mL原料药填充到Labscale系统中,并且用1050mL渗滤缓冲剂进行缓冲剂交换。
在缓冲剂交换后,在Labscale系统中将原料药浓缩至蛋白质浓度为约150mg/mL。
然后,将浓缩的原料药用相应的缓冲剂和盐溶液的储备溶液稀释至目标蛋白质浓度为120mg/mL,得到根据表4所示的最终药品溶液。
每种药品溶液通过0.22μm Sterivex GV(Millipore)过滤器过滤,并且灌装到洁净并且无菌的6mL小瓶中,灌装体积为2.7mL。将小瓶加塞并卷压。
分析方法
浊度、粘度和SE-HPLC分析检测方法描述于实例I中。
稳定性研究计划
制剂于2-8℃和25℃可在8周内保持稳定。在稳定性研究开始时和储存8周后抽取样品并进行分析。
结果
图2比较了pH/缓冲剂筛选第二部分的浊度和粘度结果。一般而言,浊度随离子强度的增加而增加,并且与pH 5.5下的浊度相比,pH 6.0下的浊度有所增加。此外,氯化钠的存在也导致更高的浊度。与pH 6相比,在pH 5.5的条件下,粘度也有所降低。此外,至少30的离子强度有助于使粘度降至约15mPas的水平。比较不同的盐对粘度的影响发现,与氯化钠相比,氯化钙或更高的缓冲剂强度更有效地降低了粘度。
考虑到降低浊度和粘度这两个目标,具有至少30的离子强度并且PH值为5.5的制剂表现出约20FTU的降低的浊度水平以及约15mPas的粘度。
图3比较了pH/缓冲剂筛选II制剂在开始时(=0)的聚集体含量(HMWS)与在5℃和25℃储存8周后的含量。一般而言,HMW含量随离子强度的增加而增加。比较不同的盐的影响发现,高缓冲剂强度或氯化钠的存在导致聚集体的增加慢于存在氯化钠的情况下。较低的pH(5.5)对减少聚集体形成的影响较小。
结论
PH值为5.5并且离子强度为30的制剂提供了减小的浊度、低粘度和减少的聚集体形成的最佳平衡。可以用较高的缓冲剂强度或添加盐(氯化钠和氯化钙),对离子强度进行调节。一般而言,较高的缓冲剂强度或氯化钙的存在导致表现出更好的浊度和粘度行为。
实例4:表面活性剂筛选
设置
表面活性剂筛选的范围用于选择抗VEGF/ANG2抗体CrossMAb VEGFang2-0016(法立昔单抗)的表面活性剂类型和表面活性剂浓度。
制剂基质为120mg/mL VEGF/Ang-2抗体、20mM L-组氨酸-醋酸盐缓冲剂(pH 5.5)、25mM氯化钠和180mM蔗糖,其基于pH/缓冲剂筛选I和II的结果,并且能够使等渗制剂具有300mOsm/kg±50mOsm/kg的目标渗透压。
表面活性剂筛选测试了在0.01%与0.07%之间的不同表面活性剂浓度下的表面活性剂聚山梨酯20和泊洛沙姆188对Vegf-Ang2抗体的影响。此外,还对不含表面活性剂的制剂进行了测试。
表5汇总了表面活性剂筛选的测试制剂。
表5用于表面活性剂筛选的制剂代码
材料与方法
在制剂的制备过程中使用的材料及其主要包装提供于表1和表2中。
使用配备截留分子量为30kD的半透膜的Labscale TTF(Millipore),通过超滤-渗滤对原料药与20mM组氨酸-醋酸盐(pH 5.3)渗滤缓冲剂进行缓冲剂交换。从而将250mL原料药填充到Labscale系统中,并且用1700mL渗滤缓冲剂进行缓冲剂交换。
在缓冲剂交换后,在Labscale系统中将原料药浓缩至蛋白质浓度为约170mg/mL并且PH值为约5.5。
然后,将浓缩的原料药用相应的缓冲剂和盐溶液的储备溶液稀释至目标蛋白质浓度为120mg/mL,得到根据表5所示的最终药品溶液。
每种药品溶液通过0.22μm Sterivex GV(Millipore)过滤器过滤,并且灌装到洁净并且无菌的6mL小瓶中,灌装体积为2.7mL。将小瓶加塞并卷压。
分析方法
蛋白质浓度、pH、渗透压、浊度、粘度、可见颗粒和SE-HPLC分析检测方法描述于实例I中。
稳定性研究计划
在表面活性剂筛选中,应用的机械应激测试条件为:在2-8℃(200rpm)下水平振荡1周,在25℃(200rpm)下水平振荡1周,以及5次冻融循环(-40℃/5℃)。
结果
表6汇总了表面活性剂筛选样品的初始结果。所有制剂具有的蛋白质浓度为约120mg/mL并且PH值为5.5±0.1。实测渗透压在335mOsm/kg与350mOsm/kg之间,从而略高于目标值311mOsm/kg。所选制剂基质(120mg/mL Vegf-Ang2与pH 5.5的20mM组氨酸-醋酸盐缓冲剂以及25mM氯化钠和180mM蔗糖)可得到约15mPas的低粘度和减小的浊度(20FTU)。
将表面活性剂筛选制剂暴露于5℃和25℃的振荡应激以及冻融应激(五次冻融循环),并且分析可见颗粒和可溶性聚集体(HMWS)。
表7汇总了初始条件下和经过物理应激后的可见颗粒结果。所有制剂样品在初始条件下均不含颗粒。暴露于不同的物理应激条件后,不含表面活性剂的制剂GRM0071-01始终表现出许多颗粒。在暴露于三种物理应激方法的过程中,添加至少0.01%的聚山梨酯20可防止形成可见颗粒。
令人惊讶的是,添加泊洛沙姆无法防止在5℃振荡1周后形成可见颗粒,但它能够在25℃振荡和冻融应激条件下保护蛋白质。
图4显示了初始样品和经过应激的样品的可溶性聚集体含量(HMWS)。不含表面活性剂的制剂GRM0071-01(1)对振荡应激敏感,并且在5℃和25℃振荡1周后表现出的聚集体含量增加了多达10%。存在0.01%聚山梨酯20不足以完全阻止在25℃振荡1周后的可溶性聚集体的增加,因为聚集体含量从3%增加至3.8%。令人惊讶的是,为防止在25℃振荡1周后的可溶性聚集体增加,需要使用含量等于或高于0.03%的聚山梨酯20。
表6表面活性剂筛选样品在生产后得到的蛋白质浓度、pH、渗透压、浊度和粘度总结
表7初始条件下和经过物理应激的表面活性剂筛选样品的可见颗粒结果总结
结论
在振荡和冻融应激条件下,为完全稳定浓度为120mg/mL的抗VEGF/ANG2抗体,需要使用至少0.03%的聚山梨酯20。
在5℃的振荡应激条件下,表面活性剂泊洛沙姆无法保护浓度为120mg/mL的双特异性抗VEGF/ANG2抗体。
制剂基质(包含20mM组氨酸-醋酸盐缓冲剂(pH 5.5)、25mM氯化钠和180mM蔗糖)为120mg/mL抗VEGF/ANG2抗体制剂提供了可接受的浊度(约20FTU)和粘度结果(约15mPas)。
实例5:赋形剂筛选I
设置
赋形剂筛选的范围用于选择抗VEGF/ANG2抗体CrossMAb VEGFang2-0016(法立昔单抗)的商业制剂的最终组成。
基于之前的pH/缓冲剂筛选I和II以及表面活性剂筛选的结果,选择的制剂基质由120mg/mL Vegf-Ang2抗体、20mM组氨酸醋酸盐缓冲系统、160mM蔗糖和0.04%聚山梨酯20组成。在该制剂基质中,对pH(5.5与5.8)、盐(25mM氯化钠与8mM氯化钙)和甲硫氨酸(0mM与7mM)的影响进行测试。基于缓冲剂和盐浓度的贡献,将离子强度调节至40。
表8汇总了赋形剂筛选I的制剂。
表8赋形剂筛选第I部分的制剂代码
材料与方法
在制剂的制备过程中使用的材料及其主要包装提供于表1和表2中。
使用配备截留分子量为30kD的半透膜的Labscale TTF(Millipore),通过超滤-渗滤对原料药与20mM组氨酸-醋酸盐(pH 5.3)渗滤缓冲剂或20mM组氨酸-醋酸盐(pH 5.6)进行缓冲剂交换。从而将410mL原料药填充到Labscale系统中,并且用3000mL渗滤缓冲剂进行缓冲剂交换。
在缓冲剂交换后,在Labscale系统中将原料药浓缩至蛋白质浓度为约165mg/mL并且PH值为约5.5或pH 5.8。
然后,将浓缩的原料药用相应的缓冲剂和盐溶液的储备溶液稀释至目标蛋白质浓度为120mg/mL,得到根据表8所示的最终药品溶液。
每种药品溶液通过0.22μm Sterivex GV(Millipore)过滤器过滤,并且灌装到洁净并且无菌的6mL小瓶中,灌装体积为2.7mL。将小瓶加塞并卷压。
分析方法
蛋白质浓度、pH、渗透压、浊度、粘度、可见颗粒、SE-HPLC和IE-HPLC分析检测方法描述于实例I中。
稳定性研究计划
制剂存放于2-8℃可保持稳定长达20周,存放于25℃可保持稳定长达13周。此外,将样品在2-8℃(200rpm)下水平振荡1周,在25℃(200rpm)下水平振荡1周,并且经过5次冻融循环(-40℃/5℃)。
结果
表9汇总了赋形剂筛选I的初步结果。所有制剂的蛋白质浓度在125mg/mL与130mg/mL之间。目标PH值为5.5的制剂GRM0073-01至GRM0073-04的实测pH值为约5.6,而目标PH值为5.8的制剂GRM0073-05至GRM0073-08的实测pH值为约5.9。与包含8mM氯化钙的制剂(GRM0073-03、GRM0073-04、GRM0073-07和GRM0073-08)的渗透压(在273mOsm/kg与288mOsm/kg之间)相比,包含25mM氯化钠的制剂(GRM0073-01、GRM0073-02、GRM0073-05和GRM0073-06)具有更高的渗透压结果(在313mOsm/kg与322mOsm/kg之间)。
表10汇总了初始条件下和经过物理应激后以及在5℃和25℃储存13周后的可见颗粒结果。在生产后以及暴露于物理应激后(在5℃或25℃振荡1周,或经过五次冻融循环),所有制剂均不含颗粒。令人惊讶的是,包含8mM氯化钙的所有制剂在5℃和25℃储存13周后表现出可见颗粒,而包含25mM氯化钠的所有制剂均不含颗粒。
图5比较了包含25mM氯化钠的制剂的浊度和粘度结果。从而,PH值为5.5的制剂表现出较低的浊度(21FTU vs.25FTU)和较低的粘度(17mPas vs.21mPas)。
图6和图7分别显示了在5℃储存20周期间以及在25℃储存13周期间可溶性聚集体(HMWS)的增加。与PH值为5.5的制剂相比,PH值为5.8的制剂表现出的可溶性聚集体略有增加。令人惊讶的是,添加甲硫氨酸可减少可溶性聚集体的形成,并且可补偿较低的pH值对聚集的影响。
图8和图9分别比较了在5℃储存20周和在25℃储存13周后通过IEC测得的带电变体的变化。在5℃储存13周后,所有制剂表现出的主峰面积略微下降约1%。伴随酸性变体增加约0.2%,并且碱性峰面积增加约1%。不同的pH或存在的甲硫氨酸未引起任何明显的区别。尽管在25℃储存13周后主峰下降幅度大得多(约18%),但pH和甲硫氨酸未引起明显区别。在25℃储存期间,主峰的下降主要由酸性变体的增加所致。
表9赋形剂筛选I样品在生产后的蛋白质浓度、pH和渗透压结果总结
表10初始和经过应激的赋形剂筛选I样品的可见颗粒结果总结
结论
尽管氯化钙的存在导致粘度和浊度水平低于包含氯化钠的制剂(参见实例2和3),其出人意料地还引起可见颗粒的形成。根据USP-NF<790>的要求(“基本上不含可见颗粒”),对于玻璃体内注射液,形成可见颗粒是不可接受的。因此,与使用氯化钙相比,优选添加氯化钠作为离子强度调节剂来降低粘度。
此外,与PH值为5.8的制剂相比,PH值为5.5的制剂表现出更低的浊度和更低的粘度。但是,与PH值为5.5的制剂相比,PH值为5.8的制剂表现出略少的可溶性聚集体形成,但是添加7mM甲硫氨酸可以补偿这种效应。因此,添加甲硫氨酸可减少pH 5.5时可溶性聚集体的形成,同时仍可实现较低的粘度和浊度水平。
pH的差异(pH 5.5vs.5.8)或是否存在甲硫氨酸对带电变体的形成没有影响。
总之,包含25mM氯化钠(代替8mM氯化钙)以及20mM组氨酸-醋酸盐缓冲剂(pH 5.5)与7mM甲硫氨酸、160mM蔗糖和0.04%聚山梨酯20的制剂可提供低浊度和低粘度的无颗粒制剂,具有改善的稳定性。
实例6:赋形剂筛选II
设置
在赋形剂筛选的第二部分,稳定性的特征进一步在于预充式注射器和蛋白质浓度30mg/mL。
赋形剂筛选I得到一种优选制剂,该制剂由120mg/mL抗VEGF/ANG2抗体CrossMAbVEGFang2-0016(法立昔单抗)、20mM组氨酸-醋酸盐(pH 5.5)、25mM氯化钠、160mM蔗糖、7mM甲硫氨酸和0.04%聚山梨酯20组成,其灌装到玻璃小瓶中(对应于制剂GRM0073-02)。
也将该制剂灌装到预充式注射器(GRM0076-02)中。此外,在蛋白质浓度为30mg/mL的条件下,测试该制剂基质灌装到预充式注射器(GRM0077-02)或玻璃小瓶(GRM0077-06)中的稳定性。
为了比较,在30mg/mL(GRM0077-09)和120mg/mL(GRM0076-05)的条件下测试了灌装到玻璃小瓶中的参比制剂的稳定性。
表11汇总了赋形剂筛选II的制剂。
表11赋形剂筛选第II部分的制剂代码
材料与方法
在制剂的制备过程中使用的材料及其主要包装提供于表1和表2中。
使用配备截留分子量为30kD的半透膜的Labscale TTF(Millipore),通过超滤-渗滤对原料药进行缓冲剂交换。
为了在20mM组氨酸-醋酸盐缓冲剂(pH 5.5)中制备抗VEGF/ANG2抗体CrossMAbVEGFang2-0016(法立昔单抗),将约340mL原料药灌装到Labscale系统中,并且与2400mL的20mM组氨酸-醋酸盐(pH 5.2)渗滤缓冲剂进行缓冲剂交换。
使用约200mL原料药,在20mM组氨酸-盐酸盐缓冲剂(pH 6.0)中制备抗VEGF/ANG2抗体CrossMAb VEGFang2-0016(法立昔单抗)。将其灌装到Labscale系统中,并且与1400mL的20mM组氨酸-盐酸盐(pH 5.85)渗滤缓冲剂进行缓冲剂交换。
在缓冲剂交换后,在Labscale系统中将相应的原料药浓缩至蛋白质浓度为约165mg/mL并且PH值为约5.5或pH 6.0。
然后,将浓缩的原料药用相应的缓冲剂和盐溶液的储备溶液稀释至目标蛋白质浓度为30mg/mL至120mg/mL,得到根据表8所示的最终药品溶液。
每种药品溶液通过0.22μm Sterivex GV(Millipore)过滤器过滤,并且灌装到洁净并且无菌的6mL小瓶中(灌装体积为2.7mL),或灌装到洁净并且无菌的1mL预充式注射器中(灌装体积为1mL)。将小瓶加塞并卷压,而注射器而用推杆塞封闭。
分析方法
蛋白质浓度、pH、渗透压、浊度、粘度、可见颗粒和SE-HPLC分析检测方法描述于实例I中。
稳定性研究计划
制剂存放于2-8℃和24℃可保持稳定长达13周。此外,将样品在2-8℃(200rpm)下水平振荡1周,在25℃(200rpm)下水平振荡1周,并且经过5次冻融循环(-40℃/5℃)。
结果
表12汇总了赋形剂筛选II的初始结果。GRM0076-02(PFS中的优化制剂)和GRM0076-05(参比制剂)的目标蛋白质浓度120mg/mL分别与实测值119mg/mL或123mg/mL匹配。制备目标蛋白质浓度为30mg/mL的GRM0077-02(PFS中的优化制剂)、GRM0077-06(小瓶中的参比制剂)和GRM0076-05(参比制剂)。实际蛋白质浓度范围在30mg/mL与31mg/mL之间。
所有制剂的pH均接近目标pH,最大偏差仅为0.1个pH单位。
120mg/mL下的制剂的渗透压略高(在310mOsm/kg与320mOsm/kg之间),而30mg/mL制剂的渗透压在278mOsm/kg与394mOsm/kg之间。
表13汇总了初始条件下和经过物理应激后以及在5℃和25℃储存13周后的可见颗粒结果。在生产后以及暴露于物理应激后(在5℃或25℃振荡1周,或经过五次冻融循环),所有制剂均不含颗粒。令人惊讶的是,参比制剂在5℃和25℃储存13周后表现出许多颗粒。所有其他制剂均不含颗粒。
图10比较了蛋白质浓度为120mg/mL的优化制剂和参比制剂的浊度和粘度结果。优化制剂表现出明显更低的浊度(约23FTU),而临床使用的制剂的浊度则超过45FTU。有趣的是,两种制剂的粘度均低于14mPas。
图11显示了30mg/mL制剂的浊度和粘度。此处,制剂GRM0072-02与GRM0077-09之间的浊度差异较小,但是优化制剂仍然表现出较低的浊度。与120mg/mL制剂相比,两种30mg/mL制剂的粘度均非常低,并且低于2mPas。
图12和图13显示了在5℃和25℃储存13周后HMW物质含量的增加。120mg/mL和30mg/mL的优化制剂表现出的HMW物质含量的增加均小于参比制剂。在25℃储存13周后,稳定作用最为明显,其中优化制剂GRM0076-02中的HMW仅增加至2.4%,而制剂GRM0076-05则增加至高达2.9%。在30mg/mL制剂中也观察到相同的趋势,优化制剂的增加幅度较低(1.0%),而参比制剂增加至1.3%。
图14比较了在5℃和25℃储存13周后通过ICE测得的带电变体的变化。所有制剂在5℃储存13周后,其主峰面积均略有下降(约1%)。并且伴随有碱性峰面积发生相应的增加,而酸性峰面积则保持恒定。尽管在25℃储存13周后主峰下降幅度大得多(约10%),但是制剂之间未见明显差异。在25℃储存期间,主峰的下降主要由于酸性变体的增加(约8%)和碱性变体略有增加(1-2%)所致。在25℃储存13周后,参比制剂(配制成pH 6.0)中碱性变体的增加幅度略低(约1%)于优化制剂(增加约2%)。较低的蛋白质浓度和主容器对带电变体无影响。
表12赋形剂筛选II样品在生产后的蛋白质浓度、pH和渗透压结果总结
表13初始和经过应激的赋形剂筛选II样品的可见颗粒结果总结
结论
赋形剂筛选II的结果确认优化制剂优于参比制剂。120mg/mL的优化制剂的浊度从大于45FTU降至25FTU以下,同时保持粘度小于15mPas。低粘度对于实现商业规模的生产工艺(通过超滤提浓)并且确保轻松便捷的玻璃体内注射(注射力小于20N,特别是小于15N)至关重要。已经证明,粘度小于15mPas的优化制剂可通过30G注射针在5s的注射时间内以小于5N的注射力进行注射。
此外,30mg/mL的优化制剂仍然不含颗粒,而参比制剂在5℃和25℃储存13周后表现出可见颗粒。此外,优化制剂中HMW物质的增加幅度较低。
在30mg/mL和120mg/mL的蛋白质浓度下以及在小瓶和预充式注射器中,观察到优化制剂的稳定性得到改善。
总之,蛋白质浓度在30mg/mL与120mg/mL之间的优化制剂(包含PH值为5.5的组氨酸-醋酸盐缓冲剂、25mM氯化钠、7mM甲硫氨酸、160mM蔗糖和0.04%聚山梨酯20)可提供具有低浊度和低粘度的不含颗粒的制剂,并且在小瓶和预充式注射器中具有改善的稳定性。
实例7:甲硫氨酸在眼部适应症中的安全性(在用于玻璃体内施用的制剂中)
L-甲硫氨酸非临床毒性研究概述
在食蟹猴中进行毒性研究,其中甲硫氨酸(10mM)是媒介物和配制的供试品CrossMAb VEGFang2-0016(法立昔单抗)的组分。在本研究中,总共对12只动物(6只雄性/6只雌性)进行玻璃体内治疗,所用剂量为每只眼睛50μL,治疗两次并且两次之间间隔14天。左眼用媒介物(包含10mM甲硫氨酸)治疗,右眼用配制的供试品CrossMAb VEGFang2-0016(法立昔单抗)(也包含10mM甲硫氨酸)治疗。在本研究中,在食蟹猴中未见甲硫氨酸的眼部作用。
实施三项研究(在食蟹猴和新西兰白兔中),其中甲硫氨酸(5-25mM)是媒介物的组分,经玻璃体内施用(剂量也是每只眼睛50μL)最多六次,并且每次施用之间间隔14天。在这些研究中,在接受含甲硫氨酸-的媒介物治疗的任何动物中均未观察到甲硫氨酸的眼部作用。
非临床毒性研究概述提供于表14中。
表14包含甲硫氨酸的非临床毒性研究总结(经玻璃体内给药)
缩写:NAT=N-乙酰色氨酸;NZW=新西兰白色。
实例8:稳定性
一批药品(120mg/mL Vegf/Ang2抗体(法立昔单抗),在20mM L-组氨酸-醋酸盐(pH5.5)、160mM蔗糖、25mM氯化钠、7mM L-甲硫氨酸、0.04%聚山梨酯20中)通过0.22μm无菌过滤器过滤,并且灌装到洁净并且无菌的2mL小瓶中(灌装体积为0.24mL)。
生产得到的制剂的PH值为5.6、渗透压为320mOsm/kg并且蛋白质浓度为120mg/mL。
表15显示了药品批次GLI0219-01在5℃储存期间的稳定性数据。表16显示了在25℃储存期间的稳定性。
表15 Vegf-Ang2抗体(法立昔单抗)药品批次在5℃储存期间的稳定性数据
表16:Vegf-Ang2抗体(法立昔单抗)药品批次在25℃储存期间的稳定性数据
缩写
- 组合抗HLA‑DR抗体或抗TROP‑2抗体与微管抑制剂、PARP抑制剂、布鲁顿激酶抑制剂或磷酸肌醇3‑激酶抑制剂使癌症治疗结果显著改善
- 一种抗血小板抗体检测信号制剂的制备方法、抗血小板抗体检测信号制剂及应用