一种达比加群酯重要中间体的合成方法
文献发布时间:2023-06-19 19:32:07
技术领域
本发明涉及一种达比加群酯重要中间体的合成方法,属于药物中间体制备领域。
背景技术
达比加群酯(dabigatran etexilate),化学名称为3-[[[2-[[[4-[[[(己氧基)羰基]氨基]亚氨甲基]苯基]氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基](吡啶-2-基)氨基]丙酸乙酯,由德国勃林格殷格翰公司研制的一种新型抗凝血药物,用于预防非瓣膜性房颤患者的卒中和全身性栓塞,达比加群酯是合成的直接凝血酶抑制剂,是达比加群(dabigatran)的前体药物,口服吸收后在体内准化为达比加群结合于凝血酶的纤维蛋白特异结合位点,阻止纤维蛋白原裂解为纤维蛋白,从而阻断了凝血瀑布网络的最后步骤及血栓形成。
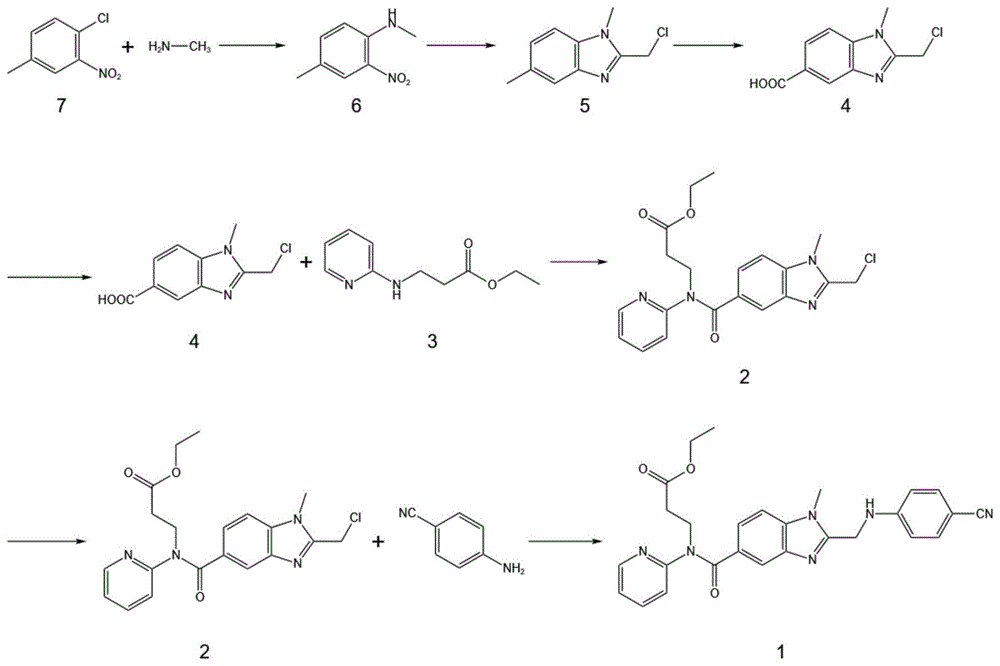
式1化合物3-[[[2-[[(4-氰基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯是制备达比加群酯的重要中间体,其化学结构式如下所示:
在专利W09837075中,首次公开了达比加群酯的制备方法,以4-氯-3-硝基苯甲酸为原料,在甲胺水溶液中进行取代反应生成4-甲胺基-3-硝基苯甲酸,与氯化亚砜反应生成4-甲胺基-3-硝基苯甲酰氯,在三乙胺催化下反应生成3-[(4-氨甲基-3-硝基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯,再使用钯炭催化得到3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯,在HOBT及EDCI作用下与N-(4-氰基苯基)氨基乙酸缩合得3-[[[2-[[[(4-氰基)苯基]氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基](吡啶-2-基)氨基]丙酸乙酯,在经过盐酸水解后与碳酸铵反应,最后经过还原反应制得达比加群酯,但该方案中3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯与N-(4-氰基苯基)氨基乙酸在EDCI和HOBT催化下缩合生成相应的苯并咪唑衍生物时,收率偏低,并且需要使用柱层析方式进行纯化,效率差,其合成路线如下所示:
在专利CN105481831B中,公开了以3-甲氧基-4-氨基苯甲酸甲酯为原料与碘甲烷在低温下反应制得3-甲氧基-4-(甲基氨基)苯甲酸甲酯,将3-甲氧基-4-(甲基氨基)苯甲酸甲酯与N-2吡啶-B-丙氨酸乙酯在有机溶剂和惰性气体中反应制得3-[4甲胺基-3-甲氧基-N-(2-吡啶基)-苯甲酰胺基]-丙烯酸乙酯,再与2-(4-氰基苯基氨基)乙酰胺在催化剂作用下制得成品将3-[[[2-[[(4-氰基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯,该方案中环合反应发生在后期,难以保证转化率,容易生成副产物,因此需要重结晶去除杂质后才能得到纯度较高的成品,其制备路线如下:
从以上文献中可以看出其共同存在的缺点是产物收率较低、产物纯度较低、生产流程多。
因此,目前亟需寻找一种产物收率高、产品纯度高、副产物生成少生产效率高的合成方法。
发明内容
本发明针对以上现有技术中存在的缺陷,提供一种达比加群酯重要中间体的合成方法,解决的问题是如何实现增加产物收率、提高产物纯度的制备方法。
本发明的目的是通过以下技术方案得以实现的,一种达比加群酯重要中间体的合成方法,该方法包括以下步骤:
S1:在有机溶剂中,使式7化合物在强碱的催化下与氨基甲烷发生缩合反应,生成式6化合物;
S2:在有机溶剂中,将S1步骤中得到的式6化合物在弱碱的催化下与卤代乙酸酐发生环合反应,生成式5化合物;
S3:将S2步骤中得到的式5化合物在高温环境下与NHPI和金属催化物发生氧化反应,生成式4化合物;
S4:在有机溶剂中,将S3步骤中得到的式4化合物与在EDCl的催化下与式3化合物发生缩合反应,生成式2化合物;
S5:将S4步骤中得到的式2化合物与对氨基苯腈在酸性条件催化下发生缩合反应生成式1化合物。
式1化合物合成路线为:
本发明通过3-硝基-4-氯甲苯与氨基甲烷反应生成N-甲基-(4-甲基-2-硝基)苯胺,再经过环合反应生成2-氯甲基-1,5-二甲基-1H-苯并咪唑,再经过氧化反应后生成2-氯甲基-1-甲基-1H-苯并咪唑-5-羧酸,与N-2吡啶-B-丙氨酸乙酯缩合生成式2化合物,式2化合物与对氨基苯腈在酸性条件催化下发生缩合反应生成式1化合物3-[[[2-[[(4-氰基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯,整体反应路线较为简单,环合反应发生在反应早期且在保护下进行,能够减少反应副产物,无需消耗大量的昂贵材料,产物收率高且产物纯度大。
在上述达比加群酯重要中间体的合成方法中,作为优选,所述S1步骤中有机溶剂可选用无水乙醇或乙醚其中一种。作为最优选,选用无水乙醇时反应稳定性最好。
在上述达比加群酯重要中间体的合成方法中,作为优选,所述S1步骤中强碱可选用氢氧化钠或氢氧化钾其中一种。作为最优选,选用氢氧化钠时反应效率最高,反应较为稳定。
在上述达比加群酯重要中间体的合成方法中,作为优选,所述S2步骤中有机溶剂可选用乙酸乙酯、四氢呋喃或乙酸丁酯其中一种。作为最优选,选用乙酸乙酯时产物纯度最高。
在上述达比加群酯重要中间体的合成方法中,作为优选,所述S2步骤中弱碱可选用碳酸钾、碳酸钠或碳酸氢钠其中一种。作为最优选,选用碳酸钾时反应速率最高。
在上述达比加群酯重要中间体的合成方法中,作为优选,所述S2步骤中卤代乙酸酐可选用氯代乙酸酐或溴代乙酸酐其中一种。作为最优选,选用氯代乙酸酐时生产成本较低。
在上述达比加群酯重要中间体的合成方法中,作为优选,所述S3步骤中氧化反应温度为90~110℃。作为最优选,反应温度为110℃时反应收率最大。
在上述达比加群酯重要中间体的合成方法中,作为优选,所述S3步骤中金属催化物可选用二氧化锰或钴盐其中一种。作为最优选,选用二氧化锰时生产成本较低。
在上述达比加群酯重要中间体的合成方法中,作为优选,所述S4步骤中有机溶剂可选用二氯甲烷、四氢呋喃或乙酸乙酯其中一种。作为最优选,选用二氯甲烷时反应稳定性最好,产物纯度较高。
在上述达比加群酯重要中间体的合成方法中,作为优选,所述S5步骤中酸性物质可选用乙酸、甲磺酸或对甲苯磺酸其中一种。作为最优选,选用乙酸时反应生成副产物较少,收率较高。
综上所述,本发明与现有技术相比,具有以下优点:
1.本发明中式6化合物N-甲基-(4-甲基-2-硝基)苯胺先进行环合反应后再与式3化合物进行缩合,避免先缩合后环合反应选择性较差生成其他副产物,能够增加收率,减少后处理难度;
2.本发明S3步骤中甲基氧化采用NHPI/金属催化,转化率高,选择性高,收率也更好。
附图说明
图1为本发明总合成路线。
具体实施方式
下面通过具体实施例,对本发明的技术方案作进一步具体的说明,但是本发明并不限于这些实施例。
实施例1
式6化合物的制备:
烧杯中放入85.5g式7化合物,加入500ml无水乙醇,加入20g氢氧化钠,加热至45℃,滴加15.53g氨基甲烷,滴毕,搅拌反应3h,溶液加入200ml乙酸乙酯萃取,脱溶液,合并有机相,使用饱和食盐水洗涤有机相,浓缩有机层,过滤,加入15g无水硫酸钠干燥,重结晶后烘干,得到产物式6化合物73.57g,产物收率为88.6%,产物纯度为98%。
实施例2
式5化合物的制备:
将500ml乙酸乙酯中加入83.03g式6化合物,控温18-22℃反应2h,添加77.28g氯代乙酸酐至溶液中,升温至65℃,搅拌反应2h,降温至40℃,添加34.55g碳酸钾,将搅拌反应50min。升温至45℃,加入44.07g甲基叔丁基醚反应30min,溶液中加入200ml乙酸乙酯萃取,脱去溶液,合并有机相,过滤,使用乙酸乙酯和饱和食盐水冲洗滤饼,加入15g无水硫酸镁干燥,重结晶后烘干,得到产物式5化合物83.93g,产物收率为86.5%,产物纯度为98.3%。
实施例3
式4化合物的制备:
在烧杯中加入97.03g式5化合物,加入4L四氢呋喃,将815.65g的NHPI与173.87g的二氧化锰混合后放入溶液中,通入0.3MPa的氧气,升温至110℃,搅拌反应3h,静置分层,水层加入200ml乙酸乙酯萃取,脱去溶液,合并有机相,使用二氯甲烷和饱和食盐水洗涤有机相,过滤,加入15g无水硫酸镁干燥,重结晶后烘干得到式4化合物104.07,产物收率为92.9%,产物纯度为98.6%。
实施例4
式2化合物的制备:
在氮气保护下将112.02g式4化合物加入到500mL二氯甲烷中,控制反应温度为0-5℃之间,缓慢加入97.06g式3化合物搅拌30min,升温至室温继续反应,TLC跟踪直到原料消耗完毕,溶液中加入200ml乙酸乙酯萃取,脱溶液,合并有机相,分别使用饱和碳酸氢钠、饱和食盐水洗涤有机相,过滤,加入15g无水硫酸钠干燥,重结晶后烘干,得到式2化合物粗产物,快速硅胶色谱纯化得到式2化合物176.26g,产物收率为88.1%,产物纯度为98.9%。
实施例5
式1化合物的制备:
烧杯中放入100.03g式2化合物,加入500ml无水乙醇,加入42.53g乙酸,升温至45℃,滴加29.53g对氨基苯腈,滴毕,搅拌反应2h,溶液中加入200ml乙酸乙酯萃取,脱溶液,合并有机相,使用饱和食盐水洗涤有机相3次,过滤,加入15g无水硫酸钠干燥,重结晶后烘干,得到最终产物式1化合物107.89g,产物收率为89.5%,产物纯度为99.4%。
实施例6
式6化合物的制备:
烧杯中放入85.5g式7化合物,加入500ml无水乙醇,加入28.05g氢氧化钾,加热至45℃,滴加15.53g氨基甲烷,滴毕,搅拌反应3h,溶液加入200ml乙酸乙酯萃取,脱溶液,合并有机相,使用饱和食盐水洗涤有机相,浓缩有机层,过滤,加入15g无水硫酸钠干燥,重结晶后烘干,得到产物式6化合物69.83g,产物收率为84.1%,产物纯度为97.3%。
实施例7
式5化合物的制备:
将500ml四氢呋喃中加入83.03g式6化合物,控温18-22℃反应2h,添加129.94g溴代乙酸酐至溶液中,升温至65℃,搅拌反应2h,降温至40℃,添加26.5g碳酸钠,将搅拌反应50min。升温至45℃,加入44.07g甲基叔丁基醚反应30min,溶液中加入200ml乙酸乙酯萃取,脱去溶液,合并有机相,过滤,使用乙酸乙酯和饱和食盐水冲洗滤饼,加入15g无水硫酸镁干燥,重结晶后烘干,得到产物式5化合物82.47g,产物收率为85%,产物纯度为97.1%。
实施例8
式5化合物的制备:
将500ml乙酸丁酯中加入83.03g式6化合物,控温18-22℃反应2h,添加77.28g氯代乙酸酐至溶液中,升温至65℃,搅拌反应2h,降温至40℃,添加21g碳酸氢钠,将搅拌反应50min。升温至45℃,加入44.07g甲基叔丁基醚反应30min,溶液中加入200ml乙酸乙酯萃取,脱去溶液,合并有机相,过滤,使用乙酸乙酯和饱和食盐水冲洗滤饼,加入15g无水硫酸镁干燥,重结晶后烘干,得到产物式5化合物82.18g,产物收率为84.7%,产物纯度为96.9%。
实施例9
式4化合物的制备
在烧杯中加入97.03g式5化合物,加入4L四氢呋喃,将815.65g的NHPI与173.87g的二氧化锰混合后放入溶液中,通入0.3MPa的氧气,升温至90℃,搅拌反应3h,静置分层,水层加入200ml乙酸乙酯萃取,脱去溶液,合并有机相,使用二氯甲烷和饱和食盐水洗涤有机相,过滤,加入15g无水硫酸镁干燥,重结晶后烘干得到式4化合物100.37,产物收率为89.6%,产物纯度为98.2%。
实施例10
式4化合物的制备:
在烧杯中加入97.03g式5化合物,加入4L四氢呋喃,将815.65g的NHPI与518.33g的乙酰丙酮钴混合后放入溶液中,通入0.3MPa的氧气,升温至50℃,搅拌反应3h,静置分层,水层加入200ml乙酸乙酯萃取,脱去溶液,合并有机相,使用二氯甲烷和饱和食盐水洗涤有机相,过滤,加入15g无水硫酸镁干燥,重结晶后烘干得到式4化合物97.9g,产物收率为87.4%,产物纯度为98.2%。
实施例11
式2化合物的制备:
在氮气保护下将112.02g式4化合物加入到500mL四氢呋喃中,控制反应温度为0-5℃之间,缓慢加入97.06g式3化合物搅拌30min,升温至室温继续反应,TLC跟踪直到原料消耗完毕,脱溶液,溶液中加入200ml乙酸乙酯萃取,合并有机相,过滤,分别使用饱和碳酸氢钠、饱和食盐水洗涤滤饼,重结晶后加入15g无水硫酸钠干燥,得到式2化合物粗产物,快速硅胶色谱纯化得到式2化合物175.65g,产物收率为87.8.%,产物纯度为98.5%。
实施例12
式2化合物的制备:
在氮气保护下将112.02g式4化合物加入到500mL乙酸乙酯中,控制反应温度为0-5℃之间,缓慢加入97.06g式3化合物搅拌30min,升温至室温继续反应,TLC跟踪直到原料消耗完毕,溶液中加入200ml乙酸乙酯萃取,脱溶液,合并有机相,分别使用饱和碳酸氢钠、饱和食盐水洗涤有机相,过滤,加入15g无水硫酸钠干燥,重结晶后烘干,得到式2化合物粗产物,快速硅胶色谱纯化得到式2化合物174.66g,产物收率为87.3%,产物纯度为98.1%。
实施例13
式1化合物的制备:
烧杯中放入100.03g式2化合物,加入500ml无水乙醇,加入53.63g甲磺酸,升温至45℃,滴加29.53g对氨基苯腈,滴毕,搅拌反应2h,溶液中加入200ml乙酸乙酯萃取,脱溶液,合并有机相,使用饱和食盐水洗涤有机相3次,过滤,加入15g无水硫酸钠干燥,重结晶后烘干,得到产物式1化合物105.36g,产物收率为87.4%,产物纯度为98.5%。
实施例14
式1化合物的制备:
烧杯中放入100.03g式2化合物,加入500ml无水乙醇,加入43.05g对甲苯磺酸,升温至45℃,滴加29.53g对氨基苯腈,滴毕,搅拌反应2h,溶液中加入200ml乙酸乙酯萃取,脱溶液,合并有机相,使用饱和食盐水洗涤有机相3次,过滤,加入15g无水硫酸钠干燥,重结晶后烘干,得到产物式1化合物104.76g,产物收率为86.9%,产物纯度为98.7%。
对比例
本实施例为已公开专利CN104418839B的实施例
将化合物F即3-({2-[(4-氰基-苯亚氨)-次甲基]-1-亚甲基-1H-苯并咪唑-5-羰基}-吡啶-2-亚胺)-丙酸乙酯)(9.6g,0.02mol)溶解于100mL无水乙醇中,用冰浴将体系的温度控制在0-5之间,慢慢的加入硼氢化钠(0.92g,0.025mol),继续反应30min,然后慢慢回至室温,TLC跟踪反应至完全。反应完毕后先减压除去50mL的无水乙醇在冰浴冷却的条件下慢慢加入0.05mol/L的AcOH淬灭反应,在用乙酸乙酯50mL×3萃取,合并有机相依次用饱和的食盐水洗涤、无水硫酸钠干燥,减压浓缩得到油状液体,用乙酸乙酯和正己烷的混合溶剂重结晶得到淡黄色的固体7.5g即化合物G(3-({2-[(4-氰基-苯亚氨)-亚甲基]-1-甲基-1H-苯并咪唑-5-羰基}-吡啶-2-亚胺)-丙酸乙酯),收率78%。
对比本发明中的制备方法,此实施例中收率较低,不利于大规模生产。
本发明的实施方式并不限于上述实施例所述,在不偏离本发明的精神和范围的情况下,本领域普通技术人员可以在形式和细节上对本发明做出各种改变和改进,而这些均被认为落入了本发明的保护范围。