一种从铀及镧系元素混合物中分离制备铀及镧系元素的方法
文献发布时间:2024-04-18 20:02:18
技术领域
本发明涉镧系、锕系元素分离领域,特别涉及铀及镧系元素混合物分离并分别制备高纯铀及镧系元素的方法。
背景技术
铀及镧系元素的高效分离具有重要的经济和国防价值。例如乏燃料的回收涉及到将乏燃料中的铀和镧系核裂变产物完全分离并进行回收,这既可以去除乏燃料中具有中子毒性的镧系元素以实现核燃料的再生利用,也可以同时制备具有较高应用价值的镧系裂变产物。例如,乏燃料中的Pm147可被用作β粒子源,用于制造便携式X-射线仪,同时它还是一种放射性同位素热电发生器的燃料,可用来制造钷电池;Sr-90是乏燃料中含量较高的一种中寿命放射性裂变产物,半衰期为28.8年,可用于制造核燃料电池以及核仪器,此外还可以借助Sr
目前已经报道的用于分离铀和镧系元素分离的方法,包括液液萃取法、沉淀法和高效液相色谱法。在这些方法中,液液萃取法和沉淀法虽然生产成本较低且适合于大规模生产,但其分离效率较低,产物纯度普遍不高,不适合用于多种组分的高纯度同步分离,且对于微量低浓度放射性同位素无法实现快速有效分离。高效液相色谱法具有分离效率高且分离快速的优点,非常适合于复杂样品体系中多种组分的同步分离,同时通过使用制备级高效液相色谱装置可以有效改善色谱分离法样品处理量少的问题。因此,高效制备色谱法非常适合于乏燃料回收以及医用同位素分离这类样品组分相对复杂且对产物纯度要求较高的场景,尤其是对于同量异位素间的分离相较于物理分离手段具有很大优势。
由于镧系元素中相邻元素间的化学性质极为相似,因此对色谱分离材料的分离性能有很高的要求;另一方面,与镧系离子不同,锕系元素离子具有较强的疏水性,常规离子色谱柱通常无法同时实现对铀和镧系元素的有效保留。现在常用的一种镧系元素和锕系元素分离策略是将两根不同性质的色谱柱进行串联(如C18反相柱+阳离子交换柱的组合),然后通过第一根C18反相色谱柱先对锕系元素进行分离,然后在第二根离子交换色谱柱上对各个镧系元素进行分离。此方法需要两根色谱柱和两个六通阀联动才能完成分离过程,控制程序复杂,分离装置的制造成本较高,也增加了设备的维护和运行成本。
因此,针对制备高纯度铀及镧系元素的分离纯化方法所存在的问题,探索一种高效、简便且成本低的制备纯化新方法对于铀的回收以及高价值镧系同位素的生产具有重大意义。本发明在色谱法的基础上,采用含有疏水性基团且同时具有离子交换基团或特定离子螯合官能团的硅胶作为固定相制备单根色谱柱,使用有机酸或无机酸作为淋洗剂即可对铀及镧系元素混合溶液进行同步分离纯化,制备高纯铀及镧系元素;同时通过色谱柱后安装三通阀分流少部分流出液用于检测器检测,可实现实时成分监测。整个色谱分离过程操作简便,自动化程度高,监测准确度高,在铀及镧系元素的制备分离上有很好的应用前景。
发明内容
一种分离制备铀及镧系元素的方法,包括:
以含有疏水性碳链的离子交换基团或含有特定离子螯合基团的硅胶为固定相,以有机酸和/或无机酸溶液为流动相对所述含有铀及镧系元素离子的混合水溶液进行分离纯化;
在色谱柱后安装三通阀,进而将色谱柱流出液一分为二并分别通入检测器和组分收集器,少部分进入检测器实现对流出液组分信息的实时监控,绝大多数流出液进入组分收集器;
通过检测器实时检测各组分出峰时间,然后根据出峰时间及色谱峰宽确定收取各个组分的时间区段,以获得高纯度的铀及镧系元素。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述的离子交换型硅胶填料的键合相为含有烷基链的苯磺酸或2-羟基氧丙基磺酸基团,所述的螯合硅胶填料1的键合相为含有全氟代烷基链的β-二酮型螯合基团,所述的螯合硅胶填料2的键合相为氨基三乙酸基团。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述固定相填料的颗粒大小为2~100μm,孔径
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述的色谱柱规格为φ4.6~200mm,固定相填料的装填高度50~500mm。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述的色谱柱的载样量为十分之一到百万分之一。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述的离子交换型硅胶填料所用流动相为有机酸水溶液;螯合硅胶填料1所用流动相为有机酸和无机酸的甲醇/水溶液,即溶剂为甲醇和水的混合物,溶质为有机酸和无机酸中的一类或二类;螯合硅胶填料2所用流动相为无机酸的水溶液。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述有机酸为α-羟基异丁酸(α-HIBA)和2-羟基-2-甲基丁酸(HMBA)中的一种或二种,所述无机酸为硝酸、硫酸、盐酸和磷酸中的至少一种。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述离子色谱填料所用流动相中有机酸浓度为10~400mM,pH范围为2.2~6.5;所述螯合硅胶填料1所用流动相中有机酸浓度为10~200mM,无机酸浓度为0.1~200mM;所述螯合硅胶填料2所用流动相中无机酸浓度为10~200mM。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述流动相的流速为0.20~40mL/min。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述分离进行的温度为15~70℃。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述三通阀的分流比例可做到在1:1~1:1000(v/v)范围内按需变动。
进一步的,上述从铀及镧系元素混合物中分离制备铀及镧系元素的方法,其中,所述检测器为改造的电感耦合等离子体发射光谱仪(ICP-OES)和紫外-可见光检测器中的一种或二种。
技术优势:
1、本发明提出的分离纯化铀及镧系元素的方法不仅适用于一般性的铀及镧系元素的分离纯化,也适用于发射性铀及其裂变产物中的放射性镧系元素的分离纯化。
2、本发明提出的分离纯化铀及镧系元素的方法,仅使用单根色谱柱,通过一次或两次色谱分离过程即可获得高纯度的铀及镧系元素,纯度都可达到99.9%。
3、本发明提出的分离纯化铀及镧系元素的方法指标先进:分析速度快(<50min)、灵敏度高(ppb级别)、制备纯度高(>99.9%)、检出限低(0.03μg/L)、分离度大(≥2.2),且由于使用了实时监测流出液的方式,可准确收集各个已分离组分,保证了产品的纯度,同时避免了高价值组分的流失。
附图说明
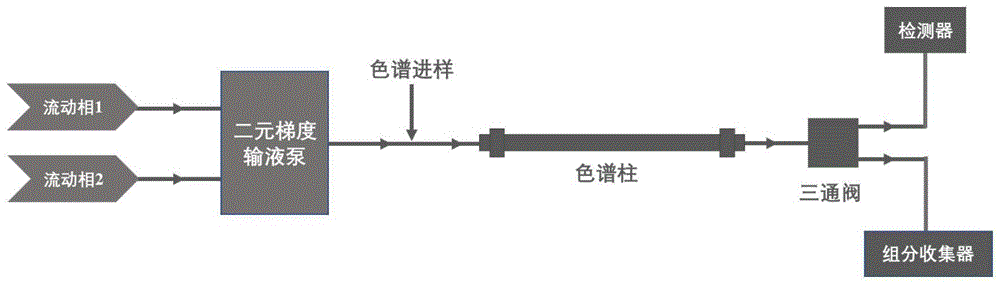
图1为本发明装置示意图;
图2为以离子色谱填料1为分离介质时铀及镧系元素混合液样品色谱流出曲线1;
图3为以离子色谱填料1为分离介质时铀及镧系元素混合液样品的色谱流出曲线2;
图4为以离子色谱填料1为分离介质时模拟乏燃料溶解液样品色谱流出曲线;
图5为以离子色谱填料2为分离介质时铀及镧系元素混合液样品的色谱流出曲线;
图6为以离子色谱填料2为分离介质时模拟乏燃料溶解液样品色谱流出曲线;
图7为以螯合离子色谱填料1为分离介质时镧系元素混合液样品的色谱流出曲线;
图8为以螯合离子色谱填料2为分离介质时镧系元素混合液样品的色谱流出曲线。
具体实施方式
为使本发明的目的、特征和优点能够更加明显易懂,下面结合实施例对本发明的具体实施方式做详细的说明。实施例中给出了本发明的若干实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容更加透彻全面。
本发明实施例基于现有铀及镧系元素分离纯化方法设备复杂,操作繁琐且制备产物纯度不高的问题,特别是对于乏燃料中铀的回收以及高价值镧系同位素的制备纯化,采用离子色谱法或螯合离子色谱法,通过简单快速、高效的分离方法,最终得到高纯铀和高纯镧系元素。
本发明实施例中所使用的制备色谱装置构成见附图1。
利用上述装置,开展分离制备铀及镧系元素的工艺流程如下:
样品溶液经六通阀进样,然后由二元梯度输液泵输送流动相进入六通阀带动样品溶液进入色谱柱进行分离,被分离的各组分按不同的时间流出色谱柱;色谱柱流出液经三通阀分流,其中绝大多数体积经三通阀进入组分收集器,其余小部分体积经分流后进入检测器,实现对流出液中各组分出峰时间的实时在线检测;随后根据出峰时间及色谱峰宽确定收取各个组分的时间区段,进而收集高纯度的铀及镧系元素流出液。对于一次分离流程无法完全分离的组分,需对其对应的流出液进行浓缩,然后优化洗脱条件重复上述色谱分离流程,以获得高纯度铀及镧系元素。
上述样品分离制备工艺阶段具有如下特征:
1、现有的铀及镧系元素的色谱分离一般通过两根色谱柱串联(如C18反相柱+阳离子交换柱)来实现,本发明使用单根色谱柱;
2、填料的特征在于:色谱填料为硅胶表面共价修饰填料,此填料所用硅胶裸球的颗粒大小为2~100μm,孔径
离子色谱填料的硅胶键合相中含有磺酸基和疏水性基团,其结构特征如式1所示。其中Silica Gel为硅胶,n=0-10,R是苯基(离子色谱填料1)或2-羟基氧丙基基团(离子色谱填料2)。
螯合离子色谱填料1的硅胶键合相中含有全氟代烷基链的β-二酮型螯合基团,其结构特征如式2所示。其中Silica Gel表示硅胶;n=0-10的整数。
螯合离子色谱填料2的硅胶键合相中含有氨基三乙酸基团,其结构特征如式3所示。其中Silica Gel表示硅胶;n=0-10的整数,m=0-10的整数,X为氧原子或碳原子。
3、流动相构成:
离子色谱填料1和离子色谱填料2所使用的流动相为有机酸的水溶液,其中的有机酸为α-羟基异丁酸(α-HIBA)和2-羟基-2-甲基丁酸(HMBA)中的一种或二种,有机酸的浓度范围为10~400mM,溶液的pH范围为2.2~6.5;
螯合离子色谱填料1所使用的流动相为有机酸和无机酸的甲醇/水溶液,即溶剂为甲醇和水的混合物,溶质为有机酸和无机酸中的一类或二类;所述有机酸为α-羟基异丁酸(α-HIBA)和2-羟基-2-甲基丁酸(HMBA)中的一种或二种,浓度范围为10~200mM;所述无机酸为硝酸、硫酸、盐酸和磷酸中的至少一种或二种以上,浓度范围为0.1~100mM;
螯合离子色谱填料2所使用的流动相为无机酸的水溶液,其中的无机酸为硝酸、硫酸、盐酸和磷酸中的至少一种或二种以上,浓度范围为10~200mM。
4、色谱柱规格为内径φ4.6~200mm,固定相填料的装填高度50~500mm,可根据分析和制备的需求更换。
5、色谱柱载样量为十分之一到百万分之一。
6、流动相的流速为0.2~40mL/min;柱温范围为15~70℃。
在本实施例的色谱分离过程中,铀及各镧系元素离子均可实现完全分离,且具有很好的分离度,因此,根据铀及各镧系元素离子的出峰时间收集从色谱柱流出的溶液,以获得铀及各镧系元素。如果一次色谱分离过程无法实现某些离子的高纯度分离,可通过调节洗脱条件进行二次色谱分离过程以获得高纯度产物,最终所得目标组分的纯度可达99.9%。
本发明实施例中的色谱分离工艺设备简单,可操作性强,在误差允许的范围内重复性很好,且运行成本较低。
下面将以具体的实施方式来说明本发明的内容及本发明所带来的积极效果。
实施例1
一种以离子色谱填料1为分离介质,从铀及镧系元素混合液中分离制备铀及镧系元素的方法,包括以下步骤
A、铀及镧系元素混合液的配置
称取适当质量的硝酸铀酰及14种三价镧系元素(镧、铈、镨、钕、钐、铕、钆、铽、镝、钬、铒、铥、镱、镥)硝酸盐,加入适量纯水溶解,然后倒入100mL容量瓶中定容,配制为含有0.1mg/L铀和浓度均为0.1mg/L的14种镧系元素离子的混合液。混合液通过0.22μm的滤膜过滤后,用于色谱分离过程。
B、淋洗液配制
称取20.82g的α-羟基异丁酸于烧杯中,加适量纯水溶解,待溶解完全后,用浓氨水调节溶液至pH=3.0,然后倒入500mL容量瓶里,用纯水稀释至刻度线,最终配制的淋洗液A中α-羟基异丁酸的浓度为400mM,pH为3.0;
同时,用上述方法再配制一种α-羟基异丁酸的浓度为400mM,pH为4.5的淋洗液。
C、离子色谱填料
离子色谱填料1的制备:将6g硅胶分散于25mL无水甲苯中,然后加入12mmol 3-苯基丙基三氯硅烷,100℃下反应24h,然后依次用甲苯、乙醇清洗过滤后干燥,制得3-苯基丙基三氯硅烷修饰后的硅胶。将25mmol氯磺酸溶于40mL1,2-二氯乙烷中,然后在冰水浴中加入5g 3-苯基丙基三氯硅烷修饰后的硅胶。随后将上述悬浮液在冰水浴中持续搅拌反应3h。反应结束后,所得固体材料依次用1,2-二氯乙烷、纯水和甲醇清洗后制得离子色谱填料1。所得离子色谱填料1粒径为5μm,孔径
D、填料装柱
称量所制得的离子色谱填料3.8g(颗粒大小为5μm,孔径大小为
安装色谱柱柱管与匀浆储液瓶相连,开始加压,泵的压力调节至40MPa。储液瓶中,选用甲基叔丁基醚作为顶替液,通过泵打入匀浆储液瓶中,替换原有的匀浆溶液。当排出液体积80ml时,停止加压,通过以上加工的色谱分离柱装到色谱仪器流路中,用于铀及镧系元素的分离;
E、离子交换色谱仪分离铀及镧系元素混合液
所用离子色谱分离条件如下所述,所得到的样品分离色谱图如附图2所示。
柱温:25℃;
流速:1mL/min;
进样量:10μL
流动相1:去离子水;
流动相2:400mM,pH=3.0的α-HIBA溶液;
流动相3:400mM,pH=4.5的α-HIBA溶液。
梯度洗脱条件:
根据附图2确定的收取各个组分的时间区段如下表所示:
对该实施例中收取的各个组分的元素分析结果证实,一次纯化各个组分的纯度均大于90%,收率大于80%,分别对上步收取的各个组分进行二次纯化后各组分的纯度可达到99.9%,最终收率大于60%。
根据铀和各镧系元素色谱峰的出锋时间不同分别收集从色谱柱流出的各类目标组分,以分别获得分离纯化的铀和各种单一镧系元素。
实施例2
过程和条件同实施例1,使用中实施例1制备的离子色谱填料1为分离介质,改变淋洗液组成,其他步骤均和实施例1相同。
淋洗液配制:称取10.41g的α-羟基异丁酸于烧杯中,加适量纯水溶解,待溶解完全后,用浓氨水调节溶液至pH=3.0,然后倒入500mL容量瓶里,用纯水稀释至刻度线,最终配制的淋洗液A中α-羟基异丁酸的浓度为200mM,pH为3.0;
同时,用上述方法再配制一种α-羟基异丁酸的浓度为400mM,pH为5.0的淋洗液。
所用离子色谱分离条件如下所述,所得到的样品分离色谱图如附图3所示。
柱温:25℃;
流速:1.0mL/min;
进样量:10μL
流动相1:去离子水;
流动相2:200mM,pH=3.0的α-HIBA溶液;
流动相3:400mM,pH=5.0的α-HIBA溶液。
梯度洗脱条件:
根据附图3确定的收取各个组分的时间区段如下表所示:
对该实施例中收取的各个组分的元素分析结果证实,一次纯化各个组分的纯度均大于90%,收率大于80%,分别对上步收取的各个组分进行二次纯化后各组分的纯度可达到99.9%,最终收率大于60%。
根据铀和各镧系元素色谱峰的出锋时间不同分别收集从色谱柱流出的各类目标组分,以分别获得分离纯化的铀和各种单一镧系元素。
实施例3
模拟乏燃料溶解液样品
过程和条件同实施例1,使用中实施例1制备的离子色谱填料1为分离介质,改变淋洗液组成、流动相流速、柱温等条件,其他步骤均和实施例1相同。
模拟乏燃料溶解液的配置:称取适当质量的硝酸铀酰及7种三价镧系元素(镧、铈、镨、钕、钐、铕、钆)硝酸盐,加入适量纯水溶解,然后倒入100mL容量瓶中定容,配制为含有5000ppm U和浓度均为250ppm的7种镧系元素离子的混合液。混合液通过0.22μm的滤膜过滤后,用于色谱分离过程。
淋洗液配制:称取5.20g的α-羟基异丁酸于烧杯中,加适量纯水溶解,待溶解完全后,用浓氨水调节溶液至pH=3.5,然后倒入500mL容量瓶里,用纯水稀释至刻度线,最终配制的淋洗液A中α-羟基异丁酸的浓度为100mM,pH为3.5;
同时,称取2.60g的α-羟基异丁酸于烧杯中,用上述方法再配制一种α-羟基异丁酸的浓度为50mM,pH为4.2的淋洗液。
所用离子色谱条件如下所述,所得到的样品的分析色谱流出曲线图如附图4所示。
柱温:30℃;
流速:1mL/min;
进样量:5μL
流动相1:去离子水;
流动相2:100mM,pH=3.5的α-HIBA溶液;
流动相3:50mM,pH=4.2的α-HIBA溶液。
梯度洗脱条件:
根据图4确定的收取各个组分的时间区段如下表所示:
对该实施例中收取的各个组分的元素分析结果证实,一次纯化各个组分的纯度均大于93%,收率大于85%,分别对上步收取的各个组分进行二次纯化后各组分的纯度可达到99.9%,最终收率大于70%。
根据铀和各镧系元素色谱峰的出锋时间不同分别收集从色谱柱流出的各类目标组分,以分别获得分离纯化的铀和各种单一镧系元素。
实施例4
一种以离子色谱填料2为分离介质,从铀及镧系元素混合液中分离制备铀及镧系元素的方法,包括以下步骤
模拟乏燃料溶解液的配置方法和实施例1相同
淋洗液配制:称取20.82g的α-羟基异丁酸于烧杯中,加适量纯水溶解,待溶解完全后,用浓氨水调节溶液至pH=3.0,然后倒入500mL容量瓶里,用纯水稀释至刻度线,最终配制的淋洗液A中α-羟基异丁酸的浓度为400mM,pH为3.0;
同时,用上述方法再配制一种α-羟基异丁酸的浓度为400mM,pH为4.2的淋洗液。
离子色谱填料2的制备:将6g硅胶分散于25mL无水甲苯中,然后加入12mmol 3-(2,3-环氧丙氧)丙基三甲氧基硅烷,100℃下反应24h,然后依次用甲苯、乙醇清洗过滤后干燥,制得3-苯基丙基三氯硅烷修饰后的硅胶。将25mmol亚硫酸氢钠溶于100mL水中,然后加入5g3-(2,3-环氧丙氧)丙基三甲氧基硅烷修饰后的硅胶。随后将上述悬浮液搅拌下加热至65℃反应24h。反应结束后,所得固体材料依次用1mol/L盐酸、纯水和甲醇清洗后制得离子色谱填料2。所得离子色谱填料2粒径为5μm,孔径
填料装柱方法和实施例1相同。
所用离子色谱分离条件如下所述,所得到的样品分离色谱图如图5所示。
柱温:22℃;
流速:1mL/min;
进样量:10μL
流动相1:去离子水;
流动相2:400mM,pH=3.0的α-HIBA溶液;
流动相3:400mM,pH=4.2的α-HIBA溶液。
梯度洗脱条件:
根据图5确定的收取各个组分的时间区段如下表所示:
/>
对该实施例中收取的各个组分的元素分析结果证实,除Dy和Y无法得到高纯度分离以外,其他各个组分一次纯化后的纯度均大于90%,收率大于80%,这些组分进行二次纯化后各组分的纯度可达到99.9%,最终收率大于60%。
根据铀和各镧系元素色谱峰的出锋时间不同分别收集从色谱柱流出的各类目标组分,以分别获得分离纯化的铀和各种单一镧系元素。
实施例5
模拟乏燃料溶解液样品
使用中实施例4制备的离子色谱填料2为分离介质,其他步骤均和实施例3相同。
所用离子色谱条件如下所述,所得到的样品的分析色谱流出曲线图如图6所示。
柱温:30℃;
流速:1mL/min;
进样量:5μL
流动相1:去离子水;
流动相2:100mM,pH=3.5的α-HIBA溶液;
流动相3:50mM,pH=4.2的α-HIBA溶液。
梯度洗脱条件:
/>
根据图6确定的收取各个组分的时间区段如下表所示:
对该实施例中收取的各个组分的元素分析结果证实,一次纯化各个组分的纯度均大于93%,收率大于85%,分别对上步收取的各个组分进行二次纯化后各组分的纯度可达到99.9%,最终收率大于70%。
根据铀和各镧系元素色谱峰的出锋时间不同分别收集从色谱柱流出的各类目标组分,以分别获得分离纯化的铀和各种单一镧系元素。
实施例6
一种以螯合离子色谱填料1为分离介质,从镧系元素混合液中分离制备各镧系元素的方法,包括以下步骤
镧系元素混合液的配置方法与实施例1相同。
淋洗液配制:称取10.41g的α-羟基异丁酸于烧杯中,加适量纯水溶解,待溶解完全后,然后倒入500mL容量瓶里,用纯水稀释至刻度线,最终配制的淋洗液A中α-羟基异丁酸的浓度为100mM;
同时,将1.38mL浓硝酸倒入500mL容量瓶里,用纯水稀释至刻度线,配置的淋洗液B中硝酸浓度为40mM。
螯合离子色谱填料1的制备:将10mmol的5-乙酰噻吩-2-羧酸溶于250mL甲醇中,然后加入3.3g甲醇钠,充分溶解后加入11mmol十三氟庚酸甲酯,80℃下反应40h。反应结束后反应液用1mol/L盐酸中和至中性,随后用乙醚萃取,旋蒸去除乙醚后用丙酮重结晶,得到2-噻吩甲酰十三氟辛酮-5-羧酸。将此产物溶于30mL甲醇中,并加入等当量N,N-碳酰二咪唑,充分混合后室温反应1h,随后加入5g氨丙基修饰的硅胶室温下反应24h,洗涤后制得螯合离子色谱填料1。所得螯合离子色谱填料2的粒径为5μm,孔径
填料装柱方法和实施例1相同。
具体的色谱条件如下所述,所得到的样品的分析色谱流出曲线图如图7所示。
柱温:40℃;
流速:0.8mL/min;
进样量:10μL
流动相1:甲醇;
流动相2:去离子水;
流动相3:200mM的α-HIBA溶液;
流动相4:40mM的HNO
梯度洗脱条件:
根据图7确定的收取各个组分的时间区段如下表所示:
对该实施例中收取的各个组分的元素分析结果证实,除Dy和Y无法得到高纯度分离以外,其他各个组分一次纯化后的纯度均大于70%,收率大于65%,这些组分进行二次纯化后各组分的纯度可达到99.9%,最终收率大于50%。
根据铀和各镧系元素色谱峰的出锋时间不同分别收集从色谱柱流出的各类目标组分,以分别获得分离纯化的铀和各种单一镧系元素。
实施例7
一种以螯合离子色谱填料2为分离介质,从铀及镧系元素混合液中分离制备铀及镧系元素的方法,包括以下步骤
铀及镧系元素混合液的配置方法与实施例1相同。
淋洗液配制:量取3.45mL浓硝酸倒入500mL容量瓶里,用纯水稀释至刻度线,配置的淋洗液中硝酸浓度为100mM。
螯合离子色谱填料2的制备:将6g硅胶分散于25mL无水甲苯中,然后加入12mmol3-(2,3-环氧丙氧)丙基三甲氧基硅烷,100℃下反应24h,然后依次用甲苯、乙醇清洗过滤后干燥,制得3-苯基丙基三氯硅烷修饰后的硅胶。将25mmol Nα,Nα-二(羧基甲基)-L-赖氨酸溶于100mL水中,然后加入5g 3-(2,3-环氧丙氧)丙基三甲氧基硅烷修饰后的硅胶。然后用氢氧化钠水溶液将反应液pH调节至pH=11,随后在50℃下反应24h。反应结束后,所得固体材料依次用1mol/L盐酸、纯水和甲醇清洗后制得螯合离子色谱填料2。所得螯合离子色谱填料2的粒径为5μm,孔径
填料装柱方法和实施例1相同。
具体的色谱条件如下所述,所得到的样品的分析色谱流出曲线图如图8所示。
柱温:55℃;
流速:1mL/min;
进样量:10μL
流动相1:去离子水;
流动相2:100mM的HNO
梯度洗脱条件:
根据图8确定的收取各个组分的时间区段如下表所示:
对该实施例中收取的各个组分的元素分析结果证实,除Eu和Gd无法得到高纯度分离以外,其他各个组分一次纯化后的纯度均大于70%,收率大于80%,这些组分进行二次纯化后各组分的纯度可达到99.9%,最终收率大于60%。
根据铀和各镧系元素色谱峰的出锋时间不同分别收集从色谱柱流出的各类目标组分,以分别获得分离纯化的铀和各种单一镧系元素。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。