一种聚乙二醇偶联药物、其制备方法及应用
文献发布时间:2023-06-19 11:21:00
技术领域
本发明属医药技术领域,涉及一种聚乙二醇偶联药物、其制备方法及应用。
背景技术
聚乙二醇化药物相对于原药有着巨大的优势,可以增加药物分子的水溶性(对紫杉醇、喜树碱或铂类这些溶解度极低的分子很重要);可以防止或减少药物产生结块、免疫原性和抗原性。大多数的小分子抗癌药在血液循环中只能保持数分钟,而高分子偶联抗癌药可以保持几十、几百小时甚至更长,这有利于因肿瘤毛细血管的渗漏而产生的“增强渗透和保留”效应,即EPR效应;由于流体力学体积增加而减弱了药物的肾消除,保护药物不被酶降解,延长药物在血浆内的半衰期,增加药物的生物利用度;通过EPR被动靶向或主动靶向使抗癌药高度富集在癌变的器官、组织或细胞,大幅度地降低因为小分子抗癌药充满全身而导致的毒副作用;将药物的细胞吸收局限于内吞路径,有利于向溶酶体方向的药物传输,从而避开p-糖蛋白泵出而导致的抗药性;刺激或恢复免疫功能,利于杀灭癌细胞。
美国NEKTAR公司和ENZON公司的聚乙二醇偶联药物技术取得了巨大的成功,迄今为止已经有15个药被美国FDA批准进入市场销售,此外还有36个临床新药在进行临床一期、二期、三期临床试验或进入NDA阶段。然而,以上所有的聚乙二醇化药物都是聚乙二醇化单药。
中国专利ZL201510996205.4公开了,将化疗药吉西他滨和Chk1抑制剂AZD7762同时接枝在一个四臂的聚乙二醇载体上,Chk1抑制剂本身没有抗癌疗效,但与化疗药吉西他滨联用可以增强化疗药的疗效。中国专利ZL201710761441.7和ZL201710761572.5公开了,将两种抗癌药同时接枝在聚乙二醇的一个接枝位点上,实现了不同的癌细胞生物信号通道和靶点抑制,以及不同的治疗方式之间的自由联用。
发明内容
本申请的发明人通过对聚乙二醇偶联药物的结构进行独特的设计,特别是选择合适的连接基团(包括尿素羰基、丁二酰基等扩展连接基团),可以非常方便地提高聚乙二醇偶联药物的载药量,并且可以非常方便地调节药物的位置以及双药或多药的种类和比例,由此提供了下述发明。
发明概述
在一个方面,本申请提供了一种如式(I)所示的聚乙二醇偶联药物或其药学上可接受的盐;
其中,PEG选自单臂或多臂的聚乙二醇链段,j为PEG1的臂数,X1、X2、Y、W1、W2具有如下文所述的定义。
在一个方面,本申请提供了一种药物组合物,其含有治疗和/或预防疾病有效量的本发明的聚乙二醇偶联药物或其药学上可接受的盐。
在一个方面,本申请提供了本发明的聚乙二醇偶联药物或其药学上可接受的盐在制备治疗和/或预防疾病(例如癌症)的药物中的用途,所述疾病为所述聚乙二醇偶联药物中的活性成分所治疗的疾病。
在一个方面,本申请提供了一种注射液,其包含本发明的聚乙二醇偶联药物或其药学上可接受的盐或药物组合物。
本申请还提供了制备本发明的聚乙二醇偶联药物或其药学上可接受的盐的方法以及中间体。
发明详述
在一个方面,本申请提供了一种如式(I)所示的聚乙二醇偶联药物或其药学上可接受的盐,
其中,PEG选自单臂或多臂的聚乙二醇链段;j为PEG1的臂数(例如为1、2、4或8);
X1选自:-NH-、
Y为Lys(赖氨酸残基)或Glu(谷氨酸残基);当j大于1(例如为2、4或8)时,会有多个(例如2个、4个或8个)Y同时存在,此时,各个Y之间可以相同或不同;
X2选自:
W1选自:N1-AC1、Q、
当j大于1(例如为2、4或8)时,会有多个(例如2个、4个或8个)W1同时存在,此时,各个W1之间可以相同或不同;
Q为
Z0、Z1、Z2各自独立地选自:
Z0、Z1、Z2之间可以相同或不同,并且,当同时存在多个Z0、多个Z1或多个Z2时,各个Z0之间、各个Z1之间或各个Z2之间可以相同或不同;
N1、N2、N3各自独立地为G(甘氨酸残基)或GFLG(甘氨酸-苯丙氨酸-亮氨酸-甘氨酸);N1、N2、N3之间可以相同或不同,并且,当同时存在多个N1、多个N2或多个N3时,各个N1之间、各个N2之间或各个N3之间可以相同或不同;
AC1、AC2、AC3为药物分子(例如具有抗肿瘤活性的药物分子),AC1、AC2、AC3之间可以相同或不同;并且当同时存在多个AC1、多个AC2或多个AC3时,各个AC1之间、各个AC2之间或各个AC3之间可以相同或不同;
W2选自:N1’-AC1’、Q’、
当j大于1(例如为2、4或8)时,会有多个(例如2个、4个或8个)W2同时存在,此时,各个W2之间可以相同或不同;
Q’为
Z0’、Z1’、Z2’各自独立地选自:
Z0’、Z1’、Z2’之间可以相同或不同,并且,当同时存在多个Z0’、多个Z1’或多个Z2’时,各个Z0’之间、各个Z1’之间或各个Z2’之间可以相同或不同;
N1’、N2’、N3’各自独立地为G或GFLG;N1’、N2’、N3’之间可以相同或不同,并且,当同时存在多个N1’、多个N2’或多个N3’时,各个N1’之间、各个N2’之间或各个N3’之间可以相同或不同;
AC1’、AC2’、AC3’为药物分子(例如具有抗肿瘤活性的药物分子),AC1’、AC2’、AC3’之间可以相同或不同;并且当同时存在多个AC1’、多个AC2’或多个AC3’时,各个AC1’之间、各个AC2’之间或各个AC3’之间可以相同或不同。
在某些实施方案中,本发明的聚乙二醇偶联药物或其药学上可接受的盐具有以下特征中的一个或多个:
(1)PEG选自单臂或四臂的聚乙二醇链段;
(2)PEG的数均分子量为5k-10k、10k-20k或20k-40k;
(3)AC1、AC2、AC3、AC1’、AC2’、AC3’各自独立地选自LPT、PCB、SB7、PKA、ABR、SN38;
(4)当Y为Lys时,X1与Lys上的α-氨基相连;
(5)当Y为Lys时,X1与Lys上的ε-氨基相连;
(6)当Y为Glu时,X1与Lys上的α-羧基相连;
(7)当Y为Glu时,X1与Lys上的γ-羧基相连;
(8)N1、N2均为GFLG;
(9)N1、N2、N3均为GFLG;
(10)N1、N2均为G;
(11)N1、N2、N3均为G;
(12)N1’、N2’均为GFLG;
(13)N1’、N2’、N3’均为GFLG;
(14)N1’、N2’均为G;
(15)N1’、N2’、N3’均为G。
在某些实施方案中,AC1和AC2选自以下组合:
(1)AC1为PCB,AC2为PCB;
(2)AC1为LPT,AC2为LPT;
(3)AC1为SN38,AC2为SN38。
在某些实施方案中,AC1’和AC2’选自以下组合:
(4)AC1’为LPT,AC2’为LPT;
(5)AC1’为SB7,AC2’为LPT;
(6)AC1’为PCB,AC2’为PCB;
(7)AC1’为ABR,AC2’为ABR。
在某些实施方案中,AC1为SN38,AC2为SN38,AC3为SN38。
在某些实施方案中,AC1’为SN38,AC2’为SN38,AC3’为SN38。
在某些实施方案中,本发明的聚乙二醇偶联药物或其药学上可接受的盐具有以下特征中的一个或多个:
(1)N1为GFLG,AC1为SB7;
(2)N1、N2均为GFLG,AC1、AC2均为LPT;
(3)N1、N2均为GFLG,AC1、AC2均为PCB;
(4)N1、N2、N3均为G,AC1、AC2、AC3均为SN38;
(5)N1为GFLG,AC1为PCB;
(6)N1’、N2’均为GFLG,AC1’、AC2’均为LPT;
(7)N1’、N2’均为GFLG,AC1’为SB7,AC2’为LPT;
(8)N1’、N2’均为GFLG,AC1’、AC2’均为PCB;
(9)N1’为GFLG,AC1’为PKA;
(10)N1’、N2’均为G,AC1’、AC2’均为ABR;
(11)N1’、N2’、N3’均为G,AC1’、AC2’、AC3’均为SN38;
(12)N1’、N2’均为GFLG,AC1’为SB7,AC2’为PCB;
(13)N1、N2均为G,AC1、AC2均为SN38;
(14)N1’为G,AC1’为SN38。
在某些实施方案中,本发明的聚乙二醇偶联药物或其药学上可接受的盐中,PEG为单臂聚乙二醇链段,X1连接在PEG的链端或链中。在某些实施方案中,当X1链接在PEG的链中时,X1为
在某些实施方案中,本发明的聚乙二醇偶联药物或其药学上可接受的盐中,Y为Lys。
在某些实施方案中,Y为Lys,X2为
X1选自:
W1选自:N1-AC1、
W2选自:Q’、
在某些实施方案中,PEG为数均分子量为10k-20k或20k-40k的单臂聚乙二醇链段。
在某些实施方案中,W1选自:
(1)
(2)
(3)N1-AC1,其中,N1为GFLG,在某些实施方案中,AC1为SB7。
在某些实施方案中,W2选自:
(1)
(2)Q’,其中,Z0’为
(3)
(4)
在某些实施方案中,所述聚乙二醇偶联药物具有选自以下的结构:
在某些实施方案中,Y为Lys,X2为
X1选自:
W1选自:N1-AC1、Q、
W2选自:N1’-AC1’、Q’、
在某些实施方案中,PEG为数均分子量为5k-20k或20k-40k的单臂或四臂聚乙二醇链段。
在某些实施方案中,W1选自:
(1)Q,其中,Z0为
(2)
(3)N1-AC1,其中,N1为GFLG,在某些实施方案中,AC1为PCB;
(4)
(5)
在某些实施方案中,W2选自:
(1)N1’-AC1’,其中,N1’为GFLG,在某些实施方案中,AC1’为PKA;
(2)
(3)N1’-AC1’,其中,N1’为GFLG,在某些实施方案中,AC1’为SB7;
(4)Q’,其中,Z0’为
在某些实施方案中,所述聚乙二醇偶联药物具有选自以下的结构:
在某些实施方案中,本发明的聚乙二醇偶联药物或其药学上可接受的中,Y为Glu。
在某些实施方案中,Y为Glu,X2为
在某些实施方案中,PEG为数均分子量为10k-20k的单臂聚乙二醇链段。
在某些实施方案中,N1为GFLG,AC1为SB7。
在某些实施方案中,Z2’为
在某些实施方案中,N1’、N2’均为GFLG,AC1’、AC2’均为LPT。
在某些实施方案中,所述聚乙二醇偶联药物具有以下结构:
在某些实施方案中,Y为Glu,X2为
在某些实施方案中,PEG为数均分子量为10k-20k的单臂聚乙二醇链段。
在某些实施方案中,N1为GFLG,AC1为SB7。
在某些实施方案中,Z2’、Z0’为
在某些实施方案中,N1’、N2’均为GFLG,AC1’、AC2’均为PCB。
在某些实施方案中,所述聚乙二醇偶联药物具有以下结构:
在某些实施方案中,Y为Glu,X2为
在某些实施方案中,PEG为数均分子量为20k-40k的四臂聚乙二醇链段。
在某些实施方案中,N1、N2均为G,AC1、AC2均为SN38。
在某些实施方案中,N1’为G,AC1’为SN38。
在某些实施方案中,所述聚乙二醇偶联药物具有以下结构:
本发明的聚乙二醇偶联药物包括但不限于:
在一个方面,本申请提供了一种药物组合物,其含有治疗和/或预防疾病有效量的本发明的聚乙二醇偶联药物或其药学上可接受的盐。在某些实施方案中,所述组合物还含有一种或多种药学上可接受的辅料。
本发明的药物组合物可以制成药学上可接受的任一剂型。例如,本发明的药物组合物可以配制为片剂、胶囊剂、丸剂、颗粒剂、溶液剂、混悬剂、糖浆剂、注射剂(包括注射液、注射用无菌粉末与注射用浓溶液)、栓剂、吸入剂或喷雾剂。
此外,本发明的药物组合物还可以以任何合适的给药方式,例如口服、肠胃外、直肠、经肺或局部给药等方式施用于需要这种治疗的患者或受试者。当用于口服给药时,所述药物组合物可制成口服制剂,例如口服固体制剂,如片剂、胶囊剂、丸剂、颗粒剂等;或,口服液体制剂,如口服溶液剂、口服混悬剂、糖浆剂等。当制成口服制剂时,所述药物组合物还可包含适宜的填充剂、粘合剂、崩解剂、润滑剂等。当用于肠胃外给药时,所述药物组合物可制成注射剂,包括注射液、注射用无菌粉末与注射用浓溶液。当制成注射剂时,所述药物组合物可采用现有制药领域中的常规方法来进行生产。当配制注射剂时,所述药物组合物中可以不加入附加剂,也可根据药物的性质加入适宜的附加剂。当用于直肠给药时,所述药物组合物可制成栓剂等。用于经肺给药时,所述药物组合物可制成吸入剂或喷雾剂等。
本发明所述的受试者为哺乳动物,例如牛科动物、马科动物、羊科动物、猪科动物、犬科动物、猫科动物、啮齿类动物、灵长类动物;其中,特别优选的受试者为人。
在某些实施方案中,所述药物组合物被制成注射剂的形式。
在一个方面,本申请提供了本发明的聚乙二醇偶联药物或其药学上可接受的盐在制备治疗和/或预防疾病(例如癌症)的药物中的用途,所述疾病为所述聚乙二醇偶联药物中的活性成分所治疗的疾病。
在某些实施方案中,所述癌症选自:结肠癌、白血病、淋巴瘤、膀胱癌、骨癌、脑瘤、髓母细胞瘤、胶质瘤、乳腺癌、腺瘤/类癌、肾上腺皮质癌、胰岛细胞癌、子宫颈癌、子宫内膜癌、卵巢癌、结肠直肠癌、皮肤癌、食管癌、眼癌、胆囊癌、胃癌、头颈癌、肝癌、黑色素瘤、卡波氏肉瘤、肾癌、口腔癌、肺癌、鼻咽癌、神经母细胞瘤、卵巢癌、胰腺癌、甲状腺癌、甲状旁腺阴茎癌、前列腺癌、尿道癌、阴道癌、外阴癌、肛门癌、肉瘤,以及所述癌症的转移。
在一个方面,本申请提供了一种注射液,其包含本发明的聚乙二醇偶联药物或其药学上可接受的盐、或本发明的药物组合物。在某些实施方案中,所述注射液以生理盐水作为载体。
在一个方面,本申请提供了一种制备本发明的聚乙二醇偶联药物或其药学上可接受的盐的方法(方法1),所述方法包括以下步骤:
步骤1:制备中间体W1-Y-X2-W2,其中,Y带有一个游离或活化的羧基;
步骤2:通过酰胺化反应,使带有氨基的PEG与中间体W1-Y-X2-W2上的Y相连,得到具有如式(I)所示结构的聚乙二醇偶联药物,所述氨基的个数为j,所述氨基为游离或活化的氨基;
其中,PEG、X2、Y、W1、W2、j如前文所定义。
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Glu,X1为-NH-,W1为N1-AC1,W2为
在某些实施方案中,方法1的中间体W1-Y-X2-W2通过包含以下步骤的方法制备:
步骤1:分别制备中间体W1-Y-X2和中间体W2,其中,X2上带有一个游离或活化的羧基,Y上带有一个被保护的羧基,W2上带有一个游离或活化的氨基;
步骤2:通过使X2上的游离或活化的羧基与W2上的游离或活化的氨基之间发生反应,使W1-Y-X2与W2相连;
步骤3:对Y上的被保护的羧基进行脱保护,任选地还进行活化,得到中间体W1-Y-X2-W2。
在一个方面,本申请提供了一种制备本发明的聚乙二醇偶联药物或其药学上可接受的盐的方法(方法2),所述方法包括以下步骤:
步骤1:制备中间体W1-Y-X2-W2,其中,Y带有一个游离或活化的氨基;
步骤2:通过酰胺化反应,使带有羧基的PEG与中间体W1-Y-X2-W2上的Y相连,得到具有如式(I)所示结构的聚乙二醇偶联药物,所述羧基的个数为j,所述羧基为游离或活化的羧基;
其中,PEG、X2、Y、W1、W2、j如前文所定义;
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Lys。
在某些实施方案中,方法2的中间体W1-Y-X2-W2通过包含以下步骤的方法制备:
步骤1:分别制备中间体W1-Y-X2和中间体W2,其中,Y上带有一个被保护的氨基,X2上带有一个游离或活化的羧基,W2上带有一个游离或活化的氨基;
步骤2:通过使X2上的游离或活化羧基与W2上的游离或活化的氨基反应,使W1-Y-X2与W2相连;
步骤3:对Y上的被保护的氨基进行脱保护,任选地还进行活化,得到中间体W1-Y-X2-W2。
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Lys,X1和X2均为
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Lys,X1为
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Lys,X1为
在某些实施方案中,方法2的中间体W1-Y-X2-W2通过包含以下步骤的方法制备:
步骤1:分别制备中间体W1-Y和中间体X2-W2,其中,Y上带有2个氨基,其中一个氨基是游离或活化的氨基,另一个氨基是被保护的氨基,X2上带有一个游离或活化的羧基;
步骤2:通过使Y上的游离或活化的氨基与X2上的游离或活化的羧基反应,使W1-Y和X2-W2相连;
步骤3:对Y上的被保护的氨基进行脱保护,任选地还进行活化,得到中间体W1-Y-X2-W2;
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Lys,X1为
在某些实施方案中,W1和W2上的各个N-AC都相同,方法2的中间体W1-Y-X2-W2通过包含以下步骤的方法制备:
步骤1:分别制备中间体N-AC、W1’-Y和中间体X2-W2’,其中,Y上带有2个氨基,其中一个氨基是游离或活化的氨基,另一个氨基是被保护的氨基,X2上带有一个游离或活化的羧基,W1’和W2’分别为W1和W2未连接N-AC的前体,W1’和W2’上带有相同的能够与N-AC反应的基团M(例如羟基),并且M被保护;
步骤2:通过使Y上的游离或活化的氨基与X2上的游离或活化的羧基反应,得到中间体W1’-Y-X2-W2’;
步骤3:在不脱除氨基保护的条件下,对M进行脱保护;
步骤4:将N-AC连接到W1’和W2’上;
步骤5:对Y上的被保护的氨基进行脱保护,任选地还进行活化,得到中间体W1-Y-X2-W2。
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Lys,X1为
在某些实施方案中,W1和W2上的各个N-AC都相同,方法2的中间体W1-Y-X2-W2通过包含以下步骤的方法制备:
步骤1:分别制备中间体N-AC、W1’-Y-X2和中间体W2’,其中,Y上带有一个被保护的氨基,X2上带有一个游离或活化的羧基,W2’上带有一个游离或活化的氨基,W1’和W2’分别为W1和W2未连接N-AC的前体,W1’和W2’上带有能够与N-AC反应的基团M(例如羟基),并且M被保护;
步骤2:通过使W2’上的游离或活化的氨基与X2上的游离或活化的羧基反应,得到中间体W1’-Y-X2-W2’;
步骤3:在不脱除氨基保护的条件下,对M进行脱保护;
步骤4:将N-AC连接到W1’和W2’上;
步骤5:对Y上的被保护的氨基进行脱保护,任选地还进行活化,得到中间体W1-Y-X2-W2。
本发明的聚乙二醇偶联药物或其药学上可接受的盐中,W1和W2上的各个N-AC可以都相同,同时X1可以为
步骤1:分别制备中间体N-AC、中间体W1’-Y-X2和中间体W2’,其中,X2上带有一个游离或活化的羧基,Y上带有一个被保护的氨基,W2’上带有一个游离或活化的氨基,W1’和W2’分别为W1和W2未连接N-AC的前体,W1’和W2’上带有能够与N-AC反应的基团M(例如羟基),并且M被保护;
步骤2:通过使W2’上的游离或活化的氨基与X2上的游离或活化的羧基反应,得到中间体W1’-Y-X2-W2’;
步骤3:在不脱除M的保护基的条件下,对Y上的氨基进行脱保护;
步骤4:使Y上的氨基与Boc-Gly-OH(氨基被保护的甘氨酸)上的羧基进行反应,得到中间体
步骤:5:在不脱除氨基保护的条件下,对M进行脱保护;
步骤6:将N-AC连接到W1’和W2’上;
步骤7:对X1上的被保护的氨基进行脱保护,任选地还进行活化,得到中间体
步骤8:通过酰胺化反应,使带有羧基的PEG与中间体
其中,PEG、X2、W1、W2、j如前文所定义。
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Lys。
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Lys,X1为
本发明的聚乙二醇偶联药物或其药学上可接受的盐中,W1和W2上的各个N-AC可以都相同。本申请提供了一种制备此类聚乙二醇偶联药物或其药学上可接受的盐的方法(方法4),所述方法包括以下步骤:
步骤1:分别制备中间体N-AC’、中间体W1’-Y-X2和中间体W2’,其中,X2上带有一个游离或活化的羧基,Y上带有一个被保护的羧基,W2’上带有一个游离或活化的氨基;W1’和W2’分别为W1和W2未连接N-AC’的前体,W1’和W2’上带有能够与N-AC’反应的基团M(例如羟基),并且M被保护;AC’是AC被保护基(例如TBDPS)保护的前体;
步骤2:使W2’上的游离或活化的氨基与X2上的游离或活化的羧基反应,得到中间体W1’-Y-X2-W2’;
步骤:3:在不脱除羧基保护的条件下,对M进行脱保护;
步骤4:将N-AC’连接到W1’和W2’上;
步骤5:对Y上的被保护的羧基进行脱保护,任选地还进行活化,得到中间体W1”-Y-X2-W2”,其中,Y带有一个游离或活化的羧基,W1”和W2”分别为W1和W2未脱除AC’上的保护基的前体;
步骤6:通过酰胺化反应,使带有氨基的PEG与中间体W1”-Y-X2-W2”相连,得到中间体
步骤7:脱除W1”和W2”上的AC’的保护基,得到具有如式(I)所示结构的聚乙二醇偶联药物;
其中,PEG、X1、X2、W1、W2、Y如前文所定义。
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Glu,X1为-NH-,X2为
在某些实施方案中,所述聚乙二醇偶联药物中,W1为Q,W2为N1’-AC1’。
在某些实施方案中,所述聚乙二醇偶联药物中,AC为SN38。
本发明的聚乙二醇偶联药物或其药学上可接受的盐中,W1和W2上的各个N-AC可以都相同,本申请提供了又一种制备此类聚乙二醇偶联药物或其药学上可接受的盐的方法(方法5),所述方法包括以下步骤:
步骤1:分别制备中间体N-AC’、中间体W1’-Y和中间体X2-W2’,其中,X2上带有一个游离或活化的羧基;Y上带有2个氨基,其中一个氨基是游离或活化的氨基,另一个氨基是被保护的氨基;W1’和W2’分别为W1和W2未连接N-AC’的前体,W1’和W2’上带有能够与N-AC’反应的基团M(例如羟基),并且M被保护;AC’是AC被保护基(例如TBDPS)保护的前体;
步骤2:通过使X2上的游离或活化的羧基与Y上的游离或活化的氨基反应,得到中间体W1’-Y-X2-W2’;
步骤3:在不脱除氨基保护的条件下,对M进行脱保护;
步骤4:将N-AC’连接到W1’和W2’上;
步骤5:对Y上的被保护的氨基进行脱保护,任选地还进行活化,得到中间体W1”-Y-X2-W2”,其中,Y带有一个游离或活化的氨基,W1”和W2”分别为W1和W2未脱除AC’上的保护基的前体;
步骤6:通过酰胺化反应,使带有羧基的PEG与中间体W1”-Y-X2-W2”上的Y相连,得到中间体
步骤7:脱除W1”和W2”上的AC’的保护基,得到具有如式(I)所示结构的聚乙二醇偶联药物;
其中,PEG、X1、X2、Y、W1、W2如前文所定义。
在某些实施方案中,所述聚乙二醇偶联药物中,Y为Lys,X1、X2均为
在某些实施方案中,所述聚乙二醇偶联药物中,W1为
在某些实施方案中,所述聚乙二醇偶联药物中,AC为SN38。
在一个方面,本申请提供了具有以下任一结构的化合物:
W1-Y-X2-W2、W1’-Y-X2-W2’、W1”-Y-X2-W2”、
其中,X1、X2、Y、W1、W2、W1’、W2’、W1’、W2’、PEG如前文所定义。
本申请还提供了以下化合物:
任选地,所述化合物中的游离氨基或游离羧基被保护或被活化。
在一个方面,本申请提供了上述化合物(作为中间体)在制备本发明的聚乙二醇偶联药物或其药学上可接受的盐中的用途。
术语定义
除非在下文中另有定义,本文中所用的所有技术术语和科学术语的含义意图与本领域技术人员通常所理解的相同。提及本文中使用的技术意图指在本领域中通常所理解的技术,包括那些对本领域技术人员显而易见的技术的变化或等效技术的替换。虽然相信以下术语对于本领域技术人员很好理解,但仍然阐述以下定义以更好地解释本发明。
如本文中所使用,“PEG”为聚乙二醇的缩写,是指重复单元为-CH
如本文中所使用,本发明的聚乙二醇偶联药物的“药学上可接受的盐”包括其酸加成盐及碱加成盐。例如盐酸盐、六氟磷酸盐、葡甲胺盐等。
如本文中所使用,结构式中的波浪线
如本文中所使用的,术语“有效量”指被给药后会在一定程度上缓解所治疗病症的一或多种症状的化合物的量。
如本文中所使用,术语“治疗”意指逆转、减轻、抑制这样的术语所应用的病症或病况或者这样的病症或病况的一或多种症状的进展,或预防这样的病症或病况或者这样的病症或病况的一或多种症状。
有益效果
本发明的聚乙二醇偶联药物可以具有较高的载药量,可以实现对药物的位置以及双药或多药的种类和比例的灵活调节。
下面将结合附图和实施例对本发明的实施方案进行详细描述,但是,本领域技术人员将理解,下列附图和实施例仅用于说明本发明,而不是对本发明的范围的限定。根据附图和优选实施方案的下列详细描述,本发明的各种目的和有利方面对于本领域技术人员来说将变得显然。
附图说明
图1为实验例一中,各组平均肿瘤体积增长趋势图。
图2为实验例一中,各组肿瘤重量抑制率示意图。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例中各缩写的含义如下:
部分原料的来源和结构如下:
M-NH
键凯,mPEG-CH
M-SCM-10K
键凯,
4ARM-SCM-40K/5K
键凯,
Y-SCM-40K
键凯,
实施例一化合物24-231的合成:
30-28
将原料Boc-GFLG-OBn(按文献合成,19.0g,32.6mmol)、10%Pd/C钯碳催化剂(300mg)加入到氢化反应装置中,然后加入DMF(50mL)使其溶解,并使溶剂没过搅拌子,封闭氢化反应装置,进行三抽三充(用真空水泵抽反应体系中的空气约3分钟—充氢气—抽氢气—充氢气---抽氢气---充氢气)后使氢化反应装置上的压力读数为18Psi,然后在常温下过夜反应。于第二日通过TLC点板发现反应完成后进行后处理,取出反应液均匀滴加到装有压实硅澡土的抽滤漏斗,用DMF(90mL)清洗反应装置,直至反应器被清洗干净不含产物为止,得到反应产物。
30-29
将30-28(17.9mmol)、LPT(8g,13.77mmol)、HBTU(7.83g,20.65mmol)和HOBT(2.79g,20.65mmol)置于500mL圆底烧瓶中然后加入DMF(100mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。然后缓慢滴加DIEA(10.24mL,61.96mmol),滴加完毕后低温下反应2小时,然后将反应装置放到室温条件下搅拌过夜反应。反应结束后,加入去离子水(1000mL)清洗DMF,析出淡黄色固体,干燥,得到产物14.53g。
14-128
将30-29(14.53g,13.77mmol)置于500mL的圆底烧瓶中,用二氯甲烷(150mL)溶解,然后加入TFA(15.34mL,206.55mmol)室温下搅拌反应过夜,反应停止,将反应液减压浓缩,加入饱和碳酸氢钠(200mL)中和TFA,用乙酸乙酯萃取水相中的产物,萃取3次(150mL×3),合并有机相再用无水硫酸钠干燥,抽滤,浓缩,干燥,干法上样,柱层析,用5%甲醇/0.5%氨水/二氯甲烷进行洗脱,收集浓缩,得到纯产品13.15g。
24-162
称取Boc-Glu-(OH)(0.8962g,3.6247mmol)、GFLG-LPT(7.1g,7.4307mmol,按14-128方法合成)、HBTU(4.1239g,10.8741mmol)、HOBT(1.4693g,10.8741mmol)投入500mL反应瓶中,加DMF(30mL)溶液,超声使反应物完全溶解,-5℃条件下搅拌30分钟后,缓慢滴加DIEA(5.3918mL,32.6223mmol),低温搅拌2小时后,放在室温下反应至结束。反应结束,将反应液移入2L分液漏斗,加乙酸乙酯(400mL)与饱和碳酸氢钠溶液(300mL),分离有机相,水相用乙酸乙酯萃取2次(100mL×2),水相中无产品,合并有机相,用饱和食盐水清洗1次(100mL),浓缩,蒸干得产品9.8g,超产2.2g,产率100%。
24-164
取24-162(7.69g,3.6247mmol),加入二氯甲烷(10mL),再加入TFA(8.0768mL,108.741mmol),超声使完全溶解,用磨口玻璃塞,常温搅拌反应。停止结束,将反应液蒸干,除去二氯甲烷,加正己烷(150mL),超声2分钟,静置10分钟,弃去上清液,下层加乙酸乙酯(10mL),超声均匀,加甲基叔丁基醚(100mL)与正己烷(100mL),抽滤,烘干得产品10g,超产3g,产率100%。
24-165
称取24-164(7.3g,3.6247mmol)、Boc-Gly-OH(0.6985g,3.9872mmol)、HBTU(2.0619g,5.4371mmol)、HOBT(0.7347g,5.4371mmol)投入500mL反应瓶中,加DMF(50mL)溶液,超声使反应物完全溶解,-5℃条件下搅拌30分钟后,缓慢滴加DIEA(2.9771mL,17.9424mmol),低温搅拌2小时后,放在室温下反应至结束。反应结束,在反应液中加甲基叔丁基醚(30mL),正己烷(300mL),超声2分钟,冰箱静置10分钟,上清液为杂质,丢弃,再重复以上操作2次,下层加乙酸乙酯(20mL),甲基叔丁基醚(50mL),正己烷(200mL),超声2分钟,抽滤,固体加20%甲醇/二氯甲烷(300mL),超声溶解,加硅胶粉(30g),用旋转蒸发仪蒸干,柱层析。用1%氨水+4%甲醇/二氯甲烷洗脱得产品5.8g,产率74%。
24-170
取24-165(5.8g,2.6614mmol),加入二氯甲烷(15mL),再加入TFA(4.9419mL,66.535mmol),超声使完全溶解,用磨口玻璃塞,常温搅拌反应。停止结束,将反应液蒸干,除去二氯甲烷,加甲基叔丁基醚(150mL),超声2分钟,加正己烷(50mL),抽滤,固体加20%甲醇/二氯甲烷(70mL),产品完全溶解,加硅胶粉(25g),用旋转蒸发仪蒸干,柱层析。用1%氨水+4%-5%甲醇/二氯甲烷梯度洗脱得产品4.8g,产率87%。
16-34
称取2-(-2氨基乙氧基)乙醇(18.8680g,190.2226mmol)倒入500mL圆底烧瓶中,加二氯甲烷(100mL)稀释,再加三乙胺(38.4972mL,380.4452mmol),搅拌条件下缓慢加入(Boc)
16-36
称取16-24(27.3g,132.8144mmol)加入500mL反应瓶中,通入氮气保护,加入叔丁醇钾的THF溶液,反应置于0℃条件下,加入溴乙酸乙酯(17.6265mL,159.3773mmol),搅拌三小时后,移至室温反应。反应结束,先将反应液蒸干,再加入去离子水与乙酸乙酯,分离有机相,水相用乙酸乙酯萃取至没有产品,合并有机相,用无水硫酸钠粉末干燥,抽滤,滤液干法上样,柱层析。用30%-100%乙酸乙酯/石油醚梯度洗脱得产品20g,产率52%。
24-36
称取16-36(17.9g,61.4402mmol)放入250mL反应瓶中,加1,4-二氧六环,搅拌条件下加入氢氧化锂(3.2386g,135.1685mmol),30分钟后加入去离子水至澄清。反应结束,用甲基叔丁基醚:正己烷=1:1萃取反应液3次(100mL×3)。水相用浓盐酸调至PH=1,用乙酸乙酯萃取三次(300mL×3),合并乙酸乙酯相,用饱和氯化钠溶解清洗3次(100mL×3),浓缩,干法上样,柱层析,用40%-100%乙酸乙酯/石油醚洗脱得产品10.1g,产率62%。
14-154
将化合物Boc-Glu-OH(10g,40.5mmol)、H-Glu-(OBn)
14-155
将化合物14-154(35.1g,40.5mmol)置于500mL圆底烧瓶中然后加入CH
14-163
将化合物14-155(31.06g,40.6mmol)、10-102(按照24-36方法合成,13.90g,52.8mmol)、HOBT(8.23g,60.9mmol)和HBTU(23.10g,60.9mmol)置于500mL原地烧瓶中然后加入DMF(150mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。然后缓慢滴加DIEA(30.20mL,182.7mmol),滴加完毕后,将反应装置于-5℃低温下搅拌反应2小时,再在室温下反应过夜。反应结束后,用饱和碳酸氢钠水洗,EA萃取三次,用饱和氯化钠水洗两次,取有机相蒸干、烘干。干法上柱,柱层析,用(0.2%-10%)甲醇/二氯甲烷进行洗脱,收集浓缩,得到产品22.63g,产率55.1%。
24-171
称取反应物Boc-LC-E[E(OBn)
24-172
称取24-170(5.5g,2.6453mmol)、HBTU(1.2540g,3.3066mmol)、HOBT(0.4468g,3.3066mmol)投入500mL反应瓶中,加24-171的DMF溶液中溶解,超声使反应物完全溶解,-5℃条件下搅拌30分钟,缓慢滴加DIEA(1.6396mL,9.9198mmol)低温搅拌2小时后,放在室温下反应至结束。反应结束,在反应液中加甲基叔丁基醚(150mL),超声5分钟,抽滤,加20%甲醇/二氯甲烷(20mL),加甲基叔丁基醚(100mL),正己烷(300mL)抽滤得产品5.5g,超产1.1g,产率100%。
24-173
取24-172(4.9g,0.5509mmol),加入二氯甲烷(15mL),再加入TFA(1.2275mL,16.5270mmol),超声使完全溶解,用磨口玻璃塞,常温搅拌反应。停止结束,将反应液蒸干,除去二氯甲烷,加乙酸乙酯(200mL),超声2分钟,抽滤,乙酸乙酯相为杂质,固体加20%甲醇/二氯甲烷(70mL),超声溶解,加硅胶粉,用旋转蒸发仪蒸干,柱层析。用1%氨水+5%-20%甲醇/二氯甲烷梯度洗脱得产品2.7g,产率56.25%。
MALDI-TOF MS:[M+Na
3-13
称取Boc-GFLG-OBn(按文献方法合成,2.0036g,3.4323mmoL)、pd/c(0.05502g)放入微型反应釜中,加入DMF(30mL),装好装置,打开进气与抽气阀门,将内环境抽成真空后关闭抽气阀门,通入H
3-14
称取SB7(SB-743921的简称)(1.4790g,2.8603mmoL)、HBTU(1.6270g,4.2905mmoL)、HOBT(0.5797g,4.2905mmoL)放入3-13的烧瓶中,将反应液放入低温恒温反应浴中(-5摄氏度),反应20分钟后,一滴一滴加入DIEA(2.2mL,12.8714mmoL),再反应2小时后取出置磁力搅拌器上,常温过夜。反应结束,将反应液移入2L分液漏斗中,加入饱和碳酸氢钠(300mL)与乙酸乙酯(400mL),充分震荡,分离有机相,将水相用乙酸乙酯萃取3次(150mL×3),合并有机相,用饱和氯化钠清洗三次(300mL),把有机相倒入2L的锥形瓶中,加入无水硫酸镁干燥,过滤,浓缩后,加入12g硅胶粉成固体溶液,干法上样,柱层析,用(2%甲醇:1%氨水:97%二氯甲烷、5%甲醇:1%氨水:94%二氯甲烷)依次洗脱得产品3.7g,产率100%。
MALDI-TOFMS:[M+H
3-16
取3-14(3.6g,3.6304mmoL)放入250mL烧瓶中,加二氯甲烷(15mL)溶解,加TFA(2.7mL,36.304mmoL)常温搅拌过夜。反应结束,将反应液蒸干除去溶剂,乙酸乙酯溶解后移入2L分液漏斗中,加入饱和碳酸氢钠(300mL水相变成碱性)与乙酸乙酯(400mL),充分震荡,分离有机相,将水相用乙酸乙酯萃取3次(100mL×3),合并有机相,用饱和碳酸氢钠清洗一次(100mL),饱和氯化钠清洗2次(100mL×2),把有机相倒入2L的锥形瓶中,加入无水硫酸钠干燥,过滤,浓缩后,加入10g硅胶粉蒸干成固体溶液,干法上样,柱层析,用(5%甲醇:1%氨水:94%二氯甲烷)洗脱,收集蒸干得产品1.7587g,产率54.33%。
24-197
称取3-16(0.5g,0.5608mmol)、Boc-Glu-OtBu(0.2505g,0.5888mmol)、HBTU(0.3190g,0.8412mmol)、HOBT(0.1137g,0.8412mmol)投入100mL反应瓶中,加DMF(20mL)溶液,超声使反应物完全溶解,-5℃条件下搅拌30分钟后,缓慢滴加DIEA(0.4170mL,2.5236mmol),低温反应至结束。反应结束,将反应液移入1L分液漏斗,加乙酸乙酯(200mL)与去离子水(150mL),分离有机相,水相用乙酸乙酯萃取2次(100mL×2),水相中无产品,合并有机相,用饱和食盐水清洗1次(100mL),浓缩,蒸干得产品1.2g,超产0.48g,产率100%。
24-203
将24-197(0.72g,0.5543mmol)置于250mL的圆底烧瓶中,用DMF(10mL)溶解,然后加入吗啉(1.4495mL,16.6288mmol)室温下搅拌反应120分钟。反应结束后,将反应液移入1L分液漏斗,加乙酸乙酯(200mL)与去离子水(150mL),分离有机相,水相用乙酸乙酯萃取2次(100mL×2),水相中无产品,合并有机相,用饱和食盐水清洗1次(100mL),浓缩,蒸干得产品1g,超产0.41g,产率100%。
24-204
将24-203(0.6g,0.5573mmol)置于250mL的圆底烧瓶中,用DMF(10mL)溶解,放入-5℃条件下低温恒温浴中,加入DIEA(0.4606mL,2.7865mmol)室温下搅拌反应30分钟,加入丁二酸酐(0.1673mL,1.6718mmol),反应结束后,将反应液移入1L分液漏斗,加乙酸乙酯(200mL)与去离子水(150mL),分离有机相,水相用乙酸乙酯萃取2次(100mL×2),水相中无产品,合并有机相,用饱和食盐水清洗1次(100mL),浓缩,蒸干得产品1g,超产0.35g,产率100%。
24-225
称取24-173(0.5g,0.0569mmol)、24-204(0.0803g,0.0682mmol)、HBTU(0.0324g,0.0854mmol)、HOBT(0.0115g,0.0854mmol)投入250mL反应瓶中,加DMF(20mL)溶液,超声使反应物完全溶解,-5℃条件下搅拌30分钟后,缓慢滴加DIEA(0.0424mL,0.2561mmol),低温反应至结束。反应结束,将反应液移入1L分液漏斗,加乙酸乙酯(20mL)与去离子水(150mL)超滤,固体用20%甲醇/二氯甲烷溶解,加硅胶粉,蒸干,柱层析。用1%氨水+5%-7%甲醇/二氯甲烷梯度洗脱得产品0.3g,产率60%。
24-227
取24-225(0.5g,0.0502mmol),加入二氯甲烷(10mL),再加入TFA(0.0933mL,1.2558mmol),超声使完全溶解,用磨口玻璃塞,常温搅拌反应。反应结束,在反应液中直接加甲基叔丁基醚(100mL)与正己烷(150mL),抽滤,固体加20%甲醇/二氯甲烷(50mL),超声溶解,加硅胶粉(3g),用旋转蒸发仪蒸干,柱层析。用1%氨水+6%-7%甲醇/二氯甲烷梯度洗脱得产品0.3g,产率60%。
24-231
称取反应物24-227(0.3g,0.0303mmoL)、M-NH
实施例二化合物31-188的合成
33-22
称取14-163(4g,3.95mmoL),加TFA(8.8mL,118.6mmoL)。二氯甲烷(10mL)。溶解。搅拌反应。反应结束,反应液浓缩,加乙酸乙酯(100mL)与饱和NaHCO
31-121
将14-155(7.0g,9.14mmol)加入到装有500mL烧瓶中,加入DMF(10mL)使其溶解,再缓慢滴加DIEA(6.04mL,36.56mmol),丁二酸苷(2.74g,27.42mmol)继续反应,置于室温下搅拌反应过夜。反应结束,加入饱和NaCl(400mL)洗涤,EA(300mL)萃取3次,将EA相旋蒸干,干法上样,柱层析,用2%甲醇/二氯甲烷-10%甲醇/二氯甲烷进行梯度洗脱,收集浓缩,蒸干后放入真空箱中干燥,得到纯产品31-121:5.1g,产率99%。
31-149
将31-146(按33-22方法合成,13.76g,15.1mmol)、Fmoc-Lys(Boc)-OH(7.78g,16.61mmol)、HOBT(3.06g,22.65mmol)和HBTU(8.59g,22.65mmol)置于500mL圆底烧瓶中,然后加入DMF(30mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。然后缓慢滴加DIEA(15mL,90.75mmol),滴加完毕后低温下反应2小时,然后将反应装置放到室温条件下搅拌过夜反应。反应结束后,将反应液转入2L分液漏斗中,加入饱和NaCl(400mL)洗涤,EA(300mL)萃取3次,将EA相旋蒸干,干法上样,柱层析,用1%甲醇/二氯甲烷-3%甲醇/二氯甲烷进行梯度洗脱,收集浓缩,蒸干后放入真空箱中干燥,得到纯产品7.7g,混产物13.9g。
31-155
将化合物31-149(5.1g,3.74mmol)置于250mL圆底烧瓶中然后加入DMF(15mL),使其溶解,将混合液常温条件下搅拌。然后滴加吗啉(9.79mL,112.37mmol),滴加完毕后常温下反应2小时。反应结束后,用饱和氯化钠(200mL)水洗,EA(200mL)萃取3次,将有机相旋蒸干,干法上样,柱层析,用1%甲醇/0.5%氨水/二氯甲烷-2%甲醇/0.5%氨水/二氯甲烷进行梯度洗脱,收集浓缩,蒸干后放入真空箱中干燥,得到产品2.31g,产率54.1%。
31-165
将31-121(1.84g,2.13mmol)、31-155(2.31g,2.03mmol)、HOBT(0.41g,3.04mmol)和HBTU(1.15g,3.04mmol)置于500mL圆底烧瓶中,然后加入DMF(25mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。然后缓慢滴加DIEA(1.51mL,9.12mmol),滴加完毕后低温下反应2小时,然后将反应装置放到室温条件下搅拌过夜反应。反应结束后,将反应液转入2L分液漏斗中,加入饱和NaHCO
31-172
将原料31-165(0.59g,0.30mmol)、10%Pd/C钯碳催化剂(100mg)加入到氢化反应装置中,然后加入DMF(40mL)使其溶解,并使溶剂没过搅拌子,封闭氢化反应装置,进行三抽三充(用真空水泵抽反应体系中的空气约3分钟—充氢气—抽氢气—充氢气---抽氢气---充氢气)后使氢化反应装置上的压力读数为30Psi,然后在常温下过夜反应。于第二日通过TLC点板发现反应完成后进行后处理,取出反应液均匀滴加到装有压实硅澡土的抽滤漏斗,用DMF(30mL)清洗反应装置,直至反应器被清洗干净不含产物为止,得到反应产物。
31-173
将31-172(0.30mmol)、14-128(2.5g,2.62mmol)、HOBT(0.49g,3.6mmol)和HBTU(1.36g,3.6mmol)置于500mL圆底烧瓶中,然后加入DMF(90mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。然后缓慢滴加DIEA(1.78mL,10.8mmol),滴加完毕后低温下反应2小时,然后将反应装置放到室温条件下搅拌过夜反应。反应结束后,加入正己烷(500mL)和甲基叔丁基醚(500mL)沉降,过滤得到固体产物,干燥,理论产物重2.63g。
31-179
将31-173(2.63g,0.30mmol)置于500mL的圆底烧瓶中,用二氯甲烷(20mL)溶解,然后加入TFA(0.33mL,4.5mmol)室温下搅拌反应2h,反应结束后,将反应液旋蒸干,加入甲基叔丁基醚(150mL)沉降,抽滤,得到固体产物,将得到的固体产物用二氯甲烷(100mL)和甲醇(10mL)溶解,干法上样,柱层析,用6%甲醇/1%氨水/二氯甲烷-9%甲醇/1%氨水/二氯甲烷进行梯度洗脱,收集浓缩,蒸干后放入真空箱中干燥,得到产品1.16g,产率44.6%。
31-188
将化合物31-179(0.39g,0.045mmol)、Y-NHS-40K(1.8g,0.041mmol)置于250mL圆底烧瓶中然后加入DMF(20mL),使其溶解,遮光缓慢搅拌反应一周。每天点TLC板,监测反应,反应结束后,用正己烷(500mL)、甲基叔丁基醚(200mL)沉降,抽滤,取固体烘干。干法上柱,柱层析,用5%甲醇/1%氨水/二氯甲烷-8%甲醇/1%氨水/二氯甲烷进行洗脱,收集浓缩,旋蒸干,烘干。将产物用无水乙醇(50mL)溶解,加入甲基叔丁基醚(100mL)、正己烷(50mL)沉降,抽滤,取固体烘干,得到产品1.58g,产率72.1%。
实施例三化合物24-243的合成
24-205
称取反应物15-91(按31-149路线合成,0.33g,0.1503mmol)投入微型反应釜中,加入10%Pd/C(30mg),加DMF(30mL)溶解,通入H
24-206
称取24-170(1.5g,0.721mmol)、HBTU(0.3420g,0.9018mmol)、HOBT(0.1219g,0.9018mmol)投入250mL反应瓶中,加24-205的DMF溶液中溶解,超声使反应物完全溶解,-5℃条件下搅拌30分钟,缓慢滴加DIEA(0.4472mL,2.7054mmol),低温搅拌2小时后,放在室温下反应至结束。反应结束,在反应液中加甲基叔丁基醚(100mL),正己烷(150mL),超声5分钟,放入冰箱静置20分钟,倾倒上清液,下层加乙酸乙酯(20mL),超声2分钟,加正己烷(100mL),抽滤,加20%甲醇/二氯甲烷(70mL)溶解,加入硅胶粉蒸干。柱层析。用1%氨水+5%-7%甲醇/二氯甲烷梯度洗脱得产品1g,产率76.9%。
24-223
将24-206(1g,0.1082mmol)置于250mL的圆底烧瓶中,用DMF(10mL)溶解,然后加入吗啉(0.2827mL,3.2448mmol)室温下搅拌反应。反应结束后,在反应液中加甲基叔丁基醚(150mL),正己烷(100mL),超声5分钟,抽滤,加20%甲醇/二氯甲烷(50mL)溶解,加入硅胶粉蒸干。柱层析。用1%氨水+7%甲醇/二氯甲烷洗脱得产品0.7g,产率70%。
19-107
将19-100(按24-197方法合成,4g,3.0794mmol)置于500mL的圆底烧瓶中,用二氯甲烷(50mL)溶解,然后加入TFA(6.8617mL,92.3823mmol)室温下搅拌反应过夜,反应停止,将反应液减压浓缩,加入饱和碳酸氢钠(200mL)中和TFA,用乙酸乙酯萃取水相中的产物,萃取3次(150mL×3),合并有机相再用无水硫酸钠干燥,抽滤,浓缩,干燥,得产品4g,产率100%。
15-98
取反应物19-107(1g,0.8033mmol)、14-128(0.7675g,0.8033mmol)、HBTU(0.4570g,1.2050mmol)、HOBT(0.1628g,1.2050mmol)加入到250mL反应瓶中,置于低温0℃条件下搅拌反应30分钟,再缓慢滴加DIEA(0.5975mL,3.6149mmol),于此条件下继续搅拌反应3小时后在常温搅拌反应。将反应液移入1L分液漏斗,加乙酸乙酯(200mL)与去离子水(150mL),分离有机相,水相用乙酸乙酯萃取2次(100mL×2),水相中无产品,合并有机相,用饱和食盐水清洗1次(100mL),浓缩有机相,加入硅胶粉(10g)蒸干,柱层析。用1%氨水+4%-9%甲醇/二氯甲烷梯度洗脱得产品:1.3g,产率:76.3%。
15-100
取15-98(0.3g,0.6249mmol),加DMF溶解,加入吗啉(1.6mL,18.7471mmol),置于常温条件下搅拌反应1小时。反应结束,加甲基叔丁基醚(30mL)与正己烷沉降(250mL),抽滤得粉末产品,干燥,得产品1.1g,产率90.2%。
15-102
取15-100(1.1g,0.5618mmol)、Boc-Gly-OH(0.0984g,0.5618mmol)、HBTU(0.3196g,0.8427mmol)、HOBT(0.1139g,0.8427mmol)依次加入到250mL反应瓶中,加DMF(30mL)溶解,于低温0℃条件下搅拌反应30分钟,再缓慢滴加DIEA(0.4mL,2.5280mmol),于此条件下继续搅拌反应3小时后常温搅拌反应。反应结束,加甲基叔丁基醚(30mL)与正己烷沉降(200mL),抽滤得粉末产品,干燥,得产品1.3g,产率100%。
15-103
取15-102(1.2g,0.5618mmol),量取二氯甲烷(10mL)加入到反应瓶中,再加入TFA(1.3mL,16.8540mmol)。常温条件下搅拌反应3小时,反应结束,加甲基叔丁基醚(30mL)与正己烷沉降(200mL),抽滤,干燥得产品1.1g,产率100%。
15-105
取15-103(1.2g,0.5618mmol),加DMF(20mL)溶解,再加入丁二酸酐(0.1687mL,1.6854mmol),置于低温条件下搅拌反应30分钟,缓慢滴加DIEA(0.5mL,2.8090mmol),30分钟后常温搅拌反应3小时。反应结束,在反应液中加甲基叔丁基醚(30mL)与正己烷沉降(200mL),抽滤得固体,用20%甲醇/二氯甲烷溶解加硅胶粉,蒸干,柱层析。用1%氨水+4%-9%甲醇/二氯甲烷梯度洗脱得产品0.9g,产率76.3%。
24-237
称取24-223(0.7g,0.0776mmol)、15-105(0.1969g,0.0931mmol)、HBTU(0.0441g,0.1164mmol)、HOBT(0.0157g,0.1164mmol)投入250mL反应瓶中,加DMF(40mL)溶液,超声使反应物完全溶解,-5℃条件下搅拌30分钟后,缓慢滴加DIEA(0.0577mL,0.3492mmol),低温反应至结束。反应结束,在反应液中加甲基叔丁基醚(150mL),正己烷(150mL),超声5分钟,倾倒上清液,下层加乙酸乙酯(10mL),超声2分钟,加甲基叔丁基醚(100mL),正己烷(50mL),抽滤,加20%甲醇/二氯甲烷(50mL)溶解,加入硅胶粉蒸干。柱层析。用1%氨水+6%-8%甲醇/二氯甲烷洗脱得产品0.5g,产率62%。
24-241
取24-237(0.5g,0.0450mmol),加入二氯甲烷(10mL),再加入TFA(0.0835mL,1.1240mmol),超声使完全溶解,用磨口玻璃塞,常温搅拌反应。反应结束,在反应液中直接加甲基叔丁基醚(150mL)与正己烷(100mL),抽滤,固体加20%甲醇/二氯甲烷(50mL),超声溶解,加硅胶粉(3g),用旋转蒸发仪蒸干,柱层析。用1%氨水+6%-8%甲醇/二氯甲烷梯度洗脱得产品0.2g,产率40%。
24-243
称取24-241(0.2g,0.0180mmol)加入DMF(20mL)溶解,再加M-SCM-40K(0.7419g,0.01798mmol),超声溶解。低速避光反应。反应结束,在反应液中加甲基叔丁基醚(150mL),正己烷(70mL)析出固体,抽滤,加20%甲醇/二氯甲烷溶解,加硅胶粉(3g),蒸干,柱层析。用1%氨水+6%-7%甲醇/二氯甲烷梯度洗脱,收集产品蒸干,加无水乙醇(3mL),超声均匀,加甲基叔丁基醚(150mL),加正己烷(50mL),抽滤,继续加溶解无水乙醇(3mL),甲基叔丁基醚与正己烷沉降,反复溶解沉降三次,抽滤,烘干,得产品0.66g,产率73%。
实施例四化合物33-82的合成
33-41
称取Boc-L-Lys(Fmoc)-OH(1.42g,1.49mmoL)、33-22(2.6g,2.9mmoL)、HOBT(0.589g,4.365mmoL)、HBTU(1.65g,4.36mmoL),置于250mL烧瓶中,DMF溶液(20mL)溶解,置于低温恒温浴中(-5℃),搅拌30分钟后,滴加DIEA(2.1mL,13.08mmoL),低温反应两小时后,常温搅拌反应。反应结束,反应液加乙酸乙酯(100mL)与饱和NaCl溶液(70mL)萃取,分离有机相,水相用乙酸乙酯萃取3次(50mL×3)。合并有机相,蒸干。柱层析,用50%-100%乙酸乙酯/石油醚梯度洗脱。重3.9克,产率100%。
33-44
称取33-41(2.9mmoL)置于250mL烧瓶中,加DMF溶液(25mL)溶解,加入吗啉(5.0mL,58mmoL),常温搅拌反应,3小时后,反应结束,反应液加乙酸乙酯(100mL)与去离子水溶液(70mL)萃取,分离有机相,水相用乙酸乙酯萃取3次(50mL×3)。合并有机相,蒸干投下一步。产物重1.7克,产率51%
33-49
称取反应物33-44(1.7g,1.49mmoL),用DMF溶液(30mL)溶解。滴加DIEA(0.985mL,5.96mmoL),30分钟后加入丁二酸酐(0.447g,4.47mmoL)。超声溶解,搅拌反应。反应结束,反应液加乙酸乙酯(100mL)与去离子水溶液(70mL)萃取,分离有机相,水相用乙酸乙酯萃取3次(50mL×3)。合并有机相,蒸干投下一步。产率100%
33-55
称取33-49(0.26g,1.49mmoL)、33-22(1.49mmoL)、HOBT(0.3g,2.235mmoL)、HBTU(0.84g,2.235mmoL),置于250mL烧瓶中,DMF溶液(20mL)溶解,置于低温恒温浴中(-5℃),搅拌30分钟后,滴加DIEA(1.18mL,6.705mmoL),低温反应两小时后,常温搅拌反应。反应结束,反应液加乙酸乙酯(100mL)与饱和NaCl溶液(70mL)萃取,分离有机相,水相用乙酸乙酯萃取3次(50mL×3)。合并有机相,蒸干投下一步。产率100%
33-58
反应物33-55(1.49mmoL),加TFA(2.7mL,37.25mmoL)。二氯甲烷(10mL)。溶解。搅拌反应。反应结束,反应液浓缩,加甲基叔丁基醚(50mL),正己烷(100mL)沉降。抽滤。蒸干,产率100%。
33-60
称取Boc-Gly-OH(0.26g,1.49mmoL)、33-58(1.49mmoL)、HOBT(0.3g,2.235mmoL)、HBTU(0.84g,2.235mmoL),置于250mL烧瓶中,DMF溶液(80mL)溶解,置于低温恒温浴中(-5℃),搅拌30分钟后,滴加DIEA(1.18mL,6.705mmoL),低温反应两小时后,常温搅拌反应。反应结束,加乙酸乙酯(100mL)与纯水(70mL)分离有机相,水相用乙酸乙酯萃取3次(50mL×3)。柱层析,干法上样,用2%甲醇/二氯甲烷--5%甲醇/二氯甲烷梯度洗脱。产品重1克。产率33%。
MALDI-TOF MS:[M+H
33-70
称取33-60(0.2448g,0.1118mmoL)、10%Pd/C(70mg)放入微型反应釜中,加入DMF(30mL),装好装置,通入H
33-73
称取24-170(2.0g,0.9619mmoL)、33-70(0.098g,0.0.1118mmoL)、HOBT(0.18g,1.34mmoL)、HBTU(0.508g,1.34mmoL),置于250mL烧瓶中,DMF溶液(80mL)溶解,置于低温恒温浴中(-5℃),搅拌30分钟后,滴加DIEA(0.069mL,0.383mmoL),低温反应两小时后,常温搅拌反应。反应结束,用甲基叔丁基醚(50mL),正己烷(100mL)沉降。柱层析,干法上样,用1%氨水:5%甲醇/二氯甲烷--1%氨水:10%甲醇/二氯甲烷梯度洗脱。产品重0.5克。
MALDI-TOF MS:[M+K
33-77
反应物33-73(0.5g,0.0278mmoL),加TFA(0.06mL,0.835mmoL)。二氯甲烷(10mL)。溶解。搅拌反应。反应结束,反应液浓缩,加甲基叔丁基醚(50mL),正己烷(100mL)沉降。抽滤。柱层析,干法上样,用1%氨水:4%甲醇/二氯甲烷--1%氨水:5%甲醇/二氯甲烷梯度洗脱。产品重0.2克。
33-82
反应物33-77(0.2g)加入M-SCM-40K(0.44g),加入DMF溶液(20mL)溶解,低速搅拌避光反应。反应结束,加甲基叔丁基醚(50mL),正己烷(100mL)沉降。抽滤。柱层析,干法上样,用二氯甲烷--1%氨水:6%甲醇/二氯甲烷梯度洗脱。产品蒸干,加无水乙醇(10mL),超声成均相,加入正己烷(50mL)沉降。反复沉降三次。真空干燥得产品0.3克。产率48%。
实施例五化合物15-139的合成
15-125
将24-203(0.5g,0.4644mmol)置于250mL的圆底烧瓶中,用二氯甲烷(10mL)超声溶解,加入TEA(0.26mL,1.8576mmol),0℃条件下搅拌反应30分钟。再缓慢滴加氯甲酸苯酯(0.1167mL,0.9288mmol)低温反应结束。2小时后反应结束,加甲基叔丁基醚与正己烷,抽滤得产品0.4g。
30-28
将原料Boc-GFLG-OBn(按文献合成,19.0g,32.6mmol)、10%Pd/C钯碳催化剂(300mg)加入到氢化反应装置中,然后加入DMF(50mL)使其溶解,并使溶剂没过搅拌子,封闭氢化反应装置,进行三抽三充(用真空水泵抽反应体系中的空气约3分钟—充氢气—抽氢气—充氢气---抽氢气---充氢气)后使氢化反应装置上的压力读数为18Psi,然后在常温下过夜反应。于第二日通过TLC点板发现反应完成后进行后处理,取出反应液均匀滴加到装有压实硅澡土的抽滤漏斗,用DMF(90mL)清洗反应装置,直至反应器被清洗干净不含产物为止,得到反应产物。
30-30
将30-28(13.97mmol)、PCB(5g,11.17mmol)、HBTU(6.35g,16.76mmol)和HOBT(2.26g,16.76mmol)置于500mL圆底烧瓶中然后加入DMF(100mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。然后缓慢滴加DIEA(8.31mL,50.28mmol),滴加完毕后低温下反应2小时,然后将反应装置放到室温条件下搅拌过夜反应。反应结束后,加入去离子水(1000mL)清洗DMF,析出黄色固体,干燥,得到产物10.3g。
30-33
将30-30(10.3g,11.17mmol)置于500mL的圆底烧瓶中,用二氯甲烷(50mL)溶解,然后加入TFA(12.45mL,167.55mmol)室温下搅拌反应过夜,反应停止,将反应液减压浓缩,加入正己烷(50mL)和甲基叔丁基醚(400mL)沉降,重复3次,过滤得到固体产物,将得到的固体产物用二氯甲烷和甲醇溶解,干法上样,柱层析,用5%甲醇/1%氨水/二氯甲烷进行洗脱,收集浓缩,干燥,得到产品8.6g,产率94%。
41-1
将Boc-Glu-OH(1.2g,4.8mmol),GFLF-PCB(按30-33方法合成,8.2g,9.7mmol),HBTU(5.5g,14.4mmol)和HOBT(1.9g,14.4mmol)置于500mL圆底烧瓶中然后加入DMF(50mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。再缓慢滴加DIEA(7.1mL,43.2mmol)继续反应,反应2h后在室温下搅拌过夜。反应结束后,加入去离子水(200mL)清洗DMF,析出淡黄色固体,烘干,得到产品22g(超重)。
41-2
将41-1(22g,11.8mmol)置于500mL圆底烧瓶中,加入二氯甲烷(10mL)使其溶解,然后滴加TFA(26.3mL,354mmol),室温下搅拌过夜。反应结束后,将反应液浓缩,加入正己烷(100mL)、甲基叔丁基醚(300mL)沉降,抽滤,取固体、烘干备用。干法上柱,柱层析,用1%氨水/3%甲醇/二氯甲烷—1%氨水/5%甲醇/二氯甲烷进行梯度洗脱,收集浓缩,得到产品5.9g,产率70.2%。
41-4
将41-2(5.9g,3.36mmol),Boc-LC-OH(按24-36方法合成,0.88g,3.36mmol),HBTU(1.9g,5.04mmol)和HOBT(0.68g,5.04mmol)置于250mL圆底烧瓶中然后加入DMF(20mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。再缓慢滴加DIEA(2.5mL,15.12mmol)继续反应,反应2h后在室温下搅拌过夜。反应结束后,加入正己烷(100mL)、甲基叔丁基醚(300mL)沉降,抽滤,取固体、烘干备用.投下一步反应。
41-6
将41-4(7.9g,3.9mmol)置于250mL圆底烧瓶中,加入二氯甲烷(10mL)使其溶解,然后滴加TFA(8.7mL,117mmol),室温下搅拌过夜。反应结束后,将反应液浓缩,加入正己烷(100mL)、甲基叔丁基醚(300mL)沉降,抽滤,取固体、烘干备用。干法上柱,柱层析,用1%氨水/3%甲醇/二氯甲烷—1%氨水/4%甲醇/二氯甲烷—1%氨水/5%甲醇/二氯甲烷进行梯度洗脱,收集浓缩,得到产品。
24-1
称取反应物14-163(0.2g,0.2104mmol)投入微型反应釜中,加入10%Pd/C(30mg),加DMF(30mL)溶解,通入H
15-131
称取41-6(2g,1.0525mmol)、HBTU(0.4170g,1.2630mmol)、HOBT(0.1707g,1.2630mmol)投入250mL反应瓶中,加24-1的DMF溶液中溶解,超声使反应物完全溶解,-5℃条件下搅拌30分钟,缓慢滴加DIEA(0.6mL,3.7891mmol)低温搅拌2小时后,放在室温下反应至结束。反应结束,在反应液中加甲基叔丁基醚(150mL),超声5分钟,抽滤,加20%甲醇/二氯甲烷(20mL)溶解,加硅胶粉,蒸干,柱层析。用1%氨水+6%-15%甲醇/二氯甲烷梯度洗脱得产品1.7g,产率98%。
15-133
取15-131(1.7g,0.2105mmol),加入二氯甲烷(10mL),再加入TFA(0.47mL,6.3300mmol),超声使完全溶解,用磨口玻璃塞,常温搅拌反应。停止结束,将反应液蒸干,除去二氯甲烷,加乙酸乙酯(20mL),超声2分钟,加甲基叔丁基醚(150mL),正己烷(70mL)抽滤,固体加20%甲醇/二氯甲烷,超声溶解,加硅胶粉,用旋转蒸发仪蒸干,柱层析。用1%氨水+4%-6%甲醇/二氯甲烷梯度洗脱得产品0.7g,产率43.8%。
15-135
称取15-133(0.6g,0.0743mmol)、15-125(0.1333g,0.1114mmol)投入250mL反应瓶中,加DMF(20mL)溶液,加DIEA(0.1mL),在100℃条件下搅拌反应至结束。反应结束,在反应液中加甲基叔丁基醚(150mL),超声5分钟,抽滤烘干得产品0.6g。
15-136
取15-135(0.6g,0.0649mmol),加入二氯甲烷(10mL),再加入TFA(0.1446mL,1.9463mmol),超声使完全溶解,用磨口玻璃塞,常温搅拌反应。反应结束,在反应液中直接加甲基叔丁基醚(100mL)与正己烷(150mL),抽滤,固体加20%甲醇/二氯甲烷(50mL),超声溶解,加硅胶粉(3g),用旋转蒸发仪蒸干,柱层析。用1%氨水+3%-8%甲醇/二氯甲烷梯度洗脱得产品0.2g,产率30%。
15-139
称取反应物15-136(0.2g,0.0222mmoL)、M-NH
实施例六化合物29-138的合成
24-58
称取PKA(18.3g,31.2028mmol),HBTU(17.75g,46.8042mmol)、HOBT(6.3241g,46.8042mmol)置于圆底烧瓶中,加入Boc-GFLG-OH的DMF溶液溶解,置于-5℃条件搅拌30分钟后缓慢滴加DIEA(23.2076mL,140.4126mmol),反应2小时后,置于常温下搅拌过夜反应。反应结束,在反应液中加入甲基叔丁基醚与正己烷,静置倾倒上清液,上清液中没有产品,油状产品中加少量乙酸乙酯,超声,再加甲基叔丁基醚与正己烷沉降,反复用乙酸乙酯与甲基叔丁基醚、正己烷沉降四次,蒸干得产品40g,超产7g,直接投下一步反应。
24-60
取24-58(33g,31.2028mmol),加入二氯甲烷(20mL),再加入TFA(69.5279mL,936.084mmol),超声使完全溶解,用磨口玻璃塞,常温搅拌反应。反应结束,现将反应液蒸干,除去二氯甲烷,再加入正己烷(200mL)超声10分钟,静置20分钟,倾倒上清液,加入甲基叔丁基醚至析出固体,抽滤,再加入甲基叔丁基醚超声,抽滤,反复用甲基叔丁基醚清洗四次,抽滤得产品35g,产率100%。
29-110
将30-33(4.0g,4.87mmol)、Boc-Asp-OH(0.5g,2.21mmol)、HBTU(0.90g,6.64mmol)、HOBT(2.52g,6.64mmol)加入到装有的500mL烧瓶中,加入适量DMF(50mL)使其溶解,置于-5℃下搅拌反应30分钟。再缓慢滴加DIEA(3.3mL,19.9mmol),经5分钟滴加完毕。反应继续在-5℃下搅拌反应1小时后,将其置于室温下搅拌反应过夜。反应结束后,用正己烷(100mL)震荡,倒掉上清液,重复上述操作三次,再加甲基叔丁基醚(80mL)和少量正己烷(10mL)震荡,倒掉上清液,重复三次,反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,真空干燥箱干燥,得到粗产品29-110。
29-112
将29-110(4.17g,2.21mmol)加入250mL烧瓶中,加入二氯甲烷(8mL)和TFA(2.5mL,33.15mmol)使之溶解,反应在室温下搅拌过夜。反应结束,用旋转蒸发仪将反应液蒸至油状,再加入甲基叔丁基醚(60mL),反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,真空烘箱干燥,得到粗产品29-112:3.84g,产率100%。
29-114
将29-112(3.84g,2.21mmol)、Fmoc-Lys(Boc)-OH(1.14g,2.43mmol)、HBTU(1.26g,3.32mmol)、HOBT(0.45g,3.32mmol)加入到装有的500mL烧瓶中,加入适量DMF(50mL)使其溶解,置于0℃下搅拌反应30分钟。再缓慢滴加DIEA(1.6mL,9.95mmol),经3分钟滴加完毕。反应继续在0℃下搅拌反应过夜。反应结束后,用正己烷(100mL)震荡,倒掉上清液,重复上述操作三次,再加甲基叔丁基醚(80mL)和少量正己烷(10mL)震荡,倒掉上清液,重复三次,反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,真空干燥箱干燥,得到粗产品29-112:4.8g,产率100%。
29-117
将29-114(4.8g,2.21mmol)加入到250mL的烧瓶中,加入DMF(10mL)使其溶解,再加入吗啉(5.8mL,66.3mmol)常温下搅拌反应1小时。反应结束,用正己烷(100mL)震荡,倒掉上清液,重复上述操作三次,再加甲基叔丁基醚(80mL)和少量正己烷(10mL)震荡,倒掉上清液,重复三次,反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,然后用甲醇/二氯甲烷(1:4)溶液(100mL)溶解,加入硅胶粉(20g),蒸干至粉末状,干法上样,柱层析,用5.5%-8%甲醇的二氯甲烷混合溶液梯度洗脱,收集浓缩,真空烘箱干燥,得到产品29-117:2.9g,产率67%。
26-124
将29-117投入500mL的烧瓶中,加入二氯甲烷超声溶解,加入DIEA(0.56mL,3.43mmol),反应置于0℃,搅拌20分钟,再缓慢滴加氯甲酸苯酯(0.2mL,1.65mmol),滴加完毕,再0℃下反应1小时。反应结束后,用正己烷(100mL)震荡,倒掉上清液,重复上述操作三次,再加甲基叔丁基醚(80mL)和少量正己烷(10mL)震荡,倒掉上清液,重复三次,反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,真空干燥箱干燥,得到粗产品29-124:2.7g,产率100%。
26-135
将29-124(1.8g,0.8mmol),24-60(1.3g,1.2mmol)投入到500mL烧瓶中,加入DMF(15mL)使之溶解,反应置于80℃下搅拌反应过夜。反应结束,用正己烷(100mL)震荡,倒掉上清液,重复上述操作三次,再加甲基叔丁基醚(80mL)和少量正己烷(10mL)震荡,倒掉上清液,重复三次,反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,然后用甲醇/二氯甲烷(1:4)溶液(100mL)溶解,加入硅胶粉(20g),蒸干至粉末状,干法上样,柱层析,用4%-8%甲醇的二氯甲烷混合溶液梯度洗脱,收集浓缩,真空烘箱干燥,得到产品29-135:1.2g,产率52%。
26-137
将29-135(1.2g,0.41mmol)加入250mL烧瓶中,加入二氯甲烷(8mL)和TFA(0.5mL)使之溶解,反应在室温下搅拌过夜。反应结束,用旋转蒸发仪将反应液蒸至油状,再加入甲基叔丁基醚(60mL),反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,然后用甲醇/二氯甲烷(1:4)溶液(100mL)溶解,加入硅胶粉(10g),蒸干至粉末状,干法上样,柱层析,用1%氨水和4%-7%甲醇的二氯甲烷混合溶液梯度洗脱,收集浓缩,真空烘箱干燥,得到产品29-137:0.8g,产率67%。
29-138
将29-137(0.4g,0.14mmol),M-SCM-5K(购于键凯)(0.77g,0.15mmol)投入到250mL烧瓶中,加入DMF(15mL)使之溶解,反应在室温下低速避光搅拌反应一周。反应结束,加入甲基叔丁基醚(30mL)振荡,有析出固体,抽滤,滤饼用甲基叔丁基醚(40mLⅹ3)清洗,滤饼再用甲醇/二氯甲烷(1:4)溶液(100mL)溶解,加入硅胶粉(10g),蒸干,呈粉末状固体,干法上样,柱层析,用1%氨水和5%-7%甲醇的二氯甲烷混合溶液梯度洗脱,收集浓缩,蒸干至固体,真空烘箱干燥,得到粗产品,粗产品用无水乙醇(15mL)和二氯甲烷(5mL)溶解,加入甲基叔丁基醚(100mLⅹ3),析出粉末状固体,抽滤,滤饼用甲基叔丁基醚(40mLⅹ3)清洗,滤饼用真空烘箱干燥,得到产品29-138:0.5g,产率45%。
实施例七化合物26-256的合成
26-217
将阿比特龙(简写为:ABR,8.0g,22.89mmol),Boc-Gly-OH(5.2g,29.76mmol),DMAP(0.28g,2.3mmol)投入1L的圆底烧瓶中,加入二氯甲烷(400mL),使之溶解,放入低温下反应0.5小时后,加入DCC(11.8g,57.2mmol),用布遮光在0℃下搅拌反应2小时后移至室温下搅拌反应过夜。反应结束后,抽滤,将滤液蒸干,真空箱干燥,得到粗产品26-217:11.6g,产率100%。
26-218
将26-217(11.6g,22.89mmol)加入250mL烧瓶中,加入二氯甲烷(8mL)和TFA(25.4980mL,343.35mmol)使之溶解,反应在室温下搅拌过夜。反应结束,用旋转蒸发仪将反应液蒸至油状,再加入乙酸乙酯(50mL),超声溶解,再加入石油醚(80mL),有固体析出,抽滤。重复上述操作5-6次,收集滤饼,真空箱干燥,得到粗产品26-218:9.3g,产率100%。
38-9
将Boc-Gly-OH(4.0g,15.1924mmol)、E(OBn)
26-221
将38-9(7.0276mmol)和10%Pd/c(0.03g)投入到氢化反应釜中,然后加入DMF(30mL)使其溶解,封闭氢化反应装置,进行三抽三充(用真空水泵抽反应体系中的空气约3分钟—充氢气—抽氢气—充氢气---抽氢气---充氢气)后使氢化反应装置上的压力读数为0.18MPa,然后在常温下过夜反应。反应结束,反应液用硅藻土过滤,滤饼用DMF(20mLⅹ3)清洗,得到产品26-221,产率100%。
26-222
将26-218(6.0g,14.7689mmol)、26-221(7.0276mmol)、HBTU(8.0g,21.0828mmol)、HOBT(2.85g,21.0828mmol)加入500mL烧瓶中,加入适量DMF(50mL)使其溶解,置于-5℃下搅拌反应30分钟。再缓慢滴加DIEA(10.45mL,63.2484mmol),经5分钟滴加完毕。反应继续在-5℃下搅拌反应1小时,然后移至室温下搅拌反应过夜。反应结束,用正己烷(100mL)震荡,倒掉上清液,重复上述操作三次,再加甲基叔丁基醚(80mL)和少量正己烷(10mL)震荡,倒掉上清液,重复三次,反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,真空箱干燥,得到粗产品26-222:产率100%。
26-227
将26-222(9.1g,7.0276mmol)加入250mL烧瓶中,加入二氯甲烷(8mL)和TFA(7.8mL,105.414mmol)使之溶解,反应在室温下搅拌过夜。反应结束,用旋转蒸发仪将反应液蒸至油状,再加入甲基叔丁基醚(60mL),反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,然后用甲醇/二氯甲烷(1:4)溶液(100mL)溶解,加入硅胶粉(20g),蒸干至粉末状,干法上样,柱层析,用3%-6%甲醇的二氯甲烷混合溶液梯度洗脱,收集浓缩,真空烘箱干燥,得到产品26-227:4.3g,产率52%。
26-229
将14-163(0.8961g,0.8862mmol)和10%Pd/c(0.03g)投入到氢化反应釜中,然后加入DMF(30mL)使其溶解,封闭氢化反应装置,进行三抽三充(用真空水泵抽反应体系中的空气约3分钟—充氢气—抽氢气—充氢气---抽氢气---充氢气)后使氢化反应装置上的压力读数为0.18MPa,然后在常温下过夜反应。反应结束,反应液用硅藻土过滤,滤饼用DMF(20mLⅹ3)清洗,得到产品26-229,产率100%。
26-230
将26-227(4.3g,3.6333mmol)、26-229(0.8862mmol)、HBTU(2.0165g,5.3171mmol)、HOBT(0.7185g,5.3171mmol)加入500mL烧瓶中,加入适量DMF(50mL)使其溶解,置于-5℃下搅拌反应30分钟。再缓慢滴加DIEA(10.45mL,63.2484mmol),经5分钟滴加完毕。反应继续在-5℃下搅拌反应1小时,然后移至室温下搅拌反应过夜。反应结束,用正己烷(100mL)震荡,倒掉上清液,重复上述操作三次,再加甲基叔丁基醚(80mL)和少量正己烷(10mL)震荡,倒掉上清液,重复三次,反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,真空箱干燥,得到粗产品26-230:产率100%。
26-240
将26-230(4.7g,0.8862mmol)加入250mL烧瓶中,加入二氯甲烷(8mL)和TFA(0.9872mL,13.293mmol)使之溶解,反应在室温下搅拌过夜。反应结束,用旋转蒸发仪将反应液蒸至油状,再加入甲基叔丁基醚(60mL),反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,然后用甲醇/二氯甲烷(1:4)溶液(100mL)溶解,加入硅胶粉(20g),蒸干至粉末状,干法上样,柱层析,用4%-8%甲醇的二氯甲烷混合溶液梯度洗脱,收集浓缩,真空烘箱干燥,得到产品26-240:3.3g,产率72%。
40-5
将3-16(2.0g,2.24mmol)、Fmoc-L-Lys(Boc)-OH(1.15g,2.46mmol)、HBTU(1.27g,3.36mmol)、HOBT(0.45g,3.36mmol)加入到装有的250mL烧瓶中,加入适量DMF(30mL)使其溶解,置于0℃下搅拌反应30分钟。再缓慢滴加DIEA(1.67mL,10.08mmol),经5分钟滴加完毕。反应继续在0℃下搅拌反应过夜。反应结束后,将反应液转移到1L的分液漏斗中,加入纯水(100mL)和乙酸乙酯(80mL)进行萃取,得到有机相,水相用乙酸乙酯(80mL ⅹ3)进行萃取,合并有机相,有机相用饱和氯化钠溶液(70mLⅹ3)进行清洗,浓缩蒸干,真空箱干燥,得到粗产品40-5,产率100%。
40-6
将40-5(3.01g,2.24mmol)加入到250mL的烧瓶中,加入DMF(10mL)使其溶解,再加入吗啉(3.9mL,44.8mmol)常温下搅拌反应1小时。反应结束后,将反应液转移到1L的分液漏斗中,加入纯水(100mL)和乙酸乙酯(80mL)进行萃取,得到有机相,水相用乙酸乙酯(80mLⅹ3)进行萃取,合并有机相,有机相用饱和氯化钠溶液(70mLⅹ3)进行清洗,浓缩蒸干,真空箱干燥,得到粗产品40-6,产率100%。
40-7
将40-6(2.51g,2.24mmol)加入到装有500mL烧瓶中,加入DMF(10mL)使其溶解,再缓慢滴加DIEA(1.48mL,8.96mmol),丁二酸苷(0.67g,6.72mmol)继续反应,置于室温下搅拌反应过夜。反应结束,用正己烷(100mL)震荡,倒掉上清液,重复上述操作三次,再加甲基叔丁基醚(80mL)和少量正己烷(10mL)震荡,倒掉上清液,重复三次,反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,然后用甲醇/二氯甲烷(1:4)溶液(100mL)溶解,加入硅胶粉(8g),蒸干至粉末状,干法上样,柱层析,用1%氨水和5%-7%甲醇的二氯甲烷混合溶液梯度洗脱,收集浓缩,真空烘箱干燥,得到产品40-7:1.2g,产率48%。
26-242
将40-7(0.6g,0.5276mmol)、26-240(2.5g,0.4796mmol)、HBTU(0.2728g,0.7194mmol)、HOBT(0.1g,0.7194mmol)加入250mL烧瓶中,加入适量DMF(50mL)使其溶解,置于-5℃下搅拌反应30分钟。再缓慢滴加DIEA(0.3573mL,2.1583mmol),经2分钟滴加完毕。反应继续在-5℃下搅拌反应1小时,然后移至室温下搅拌反应过夜。反应结束,用正己烷(100mL)震荡,倒掉上清液,重复上述操作三次,再加甲基叔丁基醚(80mL)和少量正己烷(10mL)震荡,倒掉上清液,重复三次,反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,真空箱干燥,得到粗产品26-242:产率100%。
26-248
将26-242(3.2g,0.5276mmol)加入250mL烧瓶中,加入二氯甲烷(8mL)和TFA(0.5877mL,7.914mmol)使之溶解,反应在室温下搅拌过夜。反应结束,用旋转蒸发仪将反应液蒸至油状,再加入甲基叔丁基醚(60mL),反应液中粉末状固体析出,抽滤,滤饼用甲基叔丁基醚(40mL x 3)清洗,收集滤饼,然后用甲醇/二氯甲烷(1:4)溶液(100mL)溶解,加入硅胶粉(20g),蒸干至粉末状,干法上样,柱层析,用3%-8%甲醇的二氯甲烷混合溶液梯度洗脱,收集浓缩,真空烘箱干燥,得到产品26-248:1.7g,产率55%。
26-256
将26-242(0.2110g,0.3571mmol),M-CM-40K(购于键凯,1.5g,0.3752mmol)HBTU(0.2031g,0.5357mmol)、HOBT(0.072g,0.5357mmol)加入250mL烧瓶中,加入适量DMF(40mL)使其溶解,置于0℃下搅拌反应30分钟。再缓慢滴加DIEA(0.5312mL,3.2139mmol),经30分钟滴加完毕。然后将反应移至室温下,反应在室温下低速避光搅拌反应一周。反应结束,加入甲基叔丁基醚(30mL)振荡,有析出固体,抽滤,滤饼用甲基叔丁基醚(40mLⅹ3)清洗,滤饼再用甲醇/二氯甲烷(1:4)溶液(100mL)溶解,加入硅胶粉(8g),蒸干,呈粉末状固体,干法上样,柱层析,用5%-9%甲醇的二氯甲烷混合溶液梯度洗脱,收集浓缩,蒸干至固体,真空烘箱干燥,得到粗产品,粗产品用无水乙醇(15mL)和二氯甲烷(5mL)溶解,加入甲基叔丁基醚(100mLⅹ3),析出粉末状固体,抽滤,滤饼用甲基叔丁基醚(40mLⅹ3)清洗,滤饼用真空烘箱干燥,得到产品26-242:1.2g,产率71%。
实施例八化合物28-229的合成
22-262
将25-254(按30-33方法合成,3.86g,4.6954mmol)、Fmoc-L-Lys(Boc)-OH(2g,4.2686mmol)、HBTU(2.43g,6.4029mmol)、HOBT(0.87g,6.4029mmol)加入到500mL烧瓶中,加入20mL DMF使其溶解,置于-5摄氏度下搅拌反应30min,再缓慢滴加DIEA(3.2mL,19.2086mmol),滴加完毕,继续反应1小时,然后置于室温下搅拌反应过夜。反应结束,加入正己烷(100mLx3)沉降,下层油状固体用少量二氯甲烷溶解,加入甲基叔丁基醚,析出固体,烘干,得产品22-262:9.1g,产率100%
22-263
将22-262(9.1g,4.2686mmol)倒入250mL烧瓶中,加入DMF(20mL)使之溶解,再加吗啉(3.72ml,42.686mmol),反应在常温下搅拌反应1小时。反应结束,加入正己烷(100mLx3)沉降,下层油状固体用少量二氯甲烷溶解,加入甲基叔丁基醚,析出固体,滤饼用甲醇/二氯甲烷(1:5)溶解,加入40mL硅胶粉,蒸干,干法上样,柱层析,洗脱剂用(0%-0.5%氨水:2%-4%甲醇:98%-95.5%二氯甲烷)进行洗脱,收集滤液,浓缩蒸干,得产品22-263:5.8g,产率100%
MALDI-TOF MS:[M+Na
22-267
将22-263(5.8g,4.2686mmol)倒入250mL烧瓶中,加入二氯甲烷20mL使之溶解,再加三乙胺(3ml,21.343mmol),将反应液置于0度搅拌20-30min,缓慢滴加氯甲酸苯酯(1.6ml,12.8058mmol),滴毕。在0度反应2-3h。反应结束,将反应液旋蒸为原体积的二分之一时,加入正己烷100mL和甲基叔丁基醚200mL,析出固体,滤饼用甲醇/二氯甲烷(1:5)溶解,加入60mL硅胶粉,蒸干,干法上样,柱层析,洗脱剂用(1%-4%甲醇:99%-96%二氯甲烷)进行洗脱,收集滤液,浓缩蒸干,得产品22-267:5.7g,产率100%
22-273
将22-267(4.99g,4.2686mmol)、22-272(按3-16方法合成,3.8g,4.2686mmol)加入到500mL烧瓶中,加入20mL DMF使其溶解,置于45-80摄氏度油浴下搅拌反应3-4h。反应结束,加入正己烷(100mLx3)沉降,下层油状固体用少量二氯甲烷溶解,加入甲基叔丁基醚,析出固体,滤饼用甲醇/二氯甲烷(1:5)溶解,加入40mL硅胶粉,蒸干,干法上样,柱层析,洗脱剂用(0.5%氨水:1%-5%甲醇:98.5%-94.5%二氯甲烷)进行洗脱,收集滤液,浓缩蒸干,得产品22-273:7.5g,产率89.3%
MALDI-TOF MS:[M+Na
22-277
将22-273(7.5g,3.8115mmol)加入到500mL的烧瓶中,加入二氯甲烷(10mL),再加入TFA(2.8mL,38.1148mmol),反应在室温下搅拌过夜。反应结束,将将反应液浓缩至10mL,加入甲基叔丁基醚(200mL),析出粉末,抽滤,滤饼用甲基叔丁基醚(50mL x3)清洗,再用(20%甲醇:80%二氯甲烷)溶液(200mL)溶解,加入硅胶粉(60mL),蒸干呈粉末固体,干法上样,柱层析,用1%氨水:4%-7%甲醇的二氯甲烷混合溶液进行洗脱,收集浓缩蒸干,得产品22-277:2.4g,产率34%。
MALDI-TOF MS:[M+H
28-211
将25-75(0.1839g,0.1819mmol,按14-163方法合成)投入氢化反应釜中,再加入10%Pd/C(0.0100g),再加入DMF(30mL)使之溶解,通入氢气,压力Pa=1.6MPa,反应在室温下搅拌过夜。反应结束后,将反应釜取出,将反应液通过硅藻土过滤,用DMF(20mL x 3)清洗。得到产品28-211:0.1183g,产率:100%。
28-212
将28-211(0.1183g,0.1819mmol),22-277(1.56g,0.8353mmol)、HBTU(0.4139g,1.0914mmol)、HOBT(0.1475g,1.0914mmol)投入250mL中,将反应置于-5℃条件下搅拌约10分钟,然后缓慢滴加DIEA(0.54mL,3.2742mmol),继续在-5℃反应1小时,然后将反应移到室温下搅拌过夜。反应结束,加入正己烷(150mL)和甲基叔丁基醚(40mL),进行沉降,倾倒上层液体,下层油状物继续加入正己烷(150mL)和甲基叔丁基醚(40mL),如此重复4次,呈粘稠状油状物,加入甲基叔丁基醚(100mL)析出固体,抽滤,滤饼用甲基叔丁基醚(50mLⅹ3)清洗,滤饼用甲醇(40mL)和二氯甲烷(160mL)混合溶液溶解,加入硅胶粉(15g),蒸干呈粉末状固体,干法上样,柱层析,用1%氨水:5%-7%甲醇的二氯甲烷混合溶液进行洗脱,收集浓缩蒸干,得产品28-212:1.1g,产率75%。
28-218
将28-212(1.1g,0.1367mmol)加入到250mL的烧瓶中,加入二氯甲烷(15mL),再加入TFA(2.0152mL,13.67mmol),反应在室温下搅拌过夜。反应结束,将反应液浓缩至少量,加入甲基叔丁基醚(150mL),析出粉末状固体,抽滤,滤饼用甲基叔丁基醚(50mL x 3)清洗,再用甲醇(30mL)和二氯甲烷(120mL)混合溶液溶解,加入硅胶粉(15g),蒸干呈粉末状固体,干法上样,柱层析,用1%氨水:6%-8%甲醇的二氯甲烷混合溶液进行洗脱,收集浓缩蒸干,得产品28-218:0.75g,产率69%。
28-229
将28-218(0.3771g,0.0474mmol)投入250mL中,加入DMF(25mL)使其溶解,加入Y-SCM-40K(1.8748g,0.0431mmol),超声溶解,反应于室温下避光低速搅拌7天。反应结束,加入正己烷(150mL)和甲基叔丁基醚(50mL),倾倒上层清液,下层液体加入正己烷(150mL)和甲基叔丁基醚(50mL),如此重复4次,呈粘稠油状物,加入甲醇(30mL)和二氯甲烷(120mL)溶液溶解,加入硅胶粉(25g),蒸干,呈粉末状固体,干法上样,柱层析,用1%氨水和6%-8%甲醇的二氯甲烷混合溶液洗脱,收集浓缩,蒸干至固体,真空烘箱干燥1小时,加入无水乙醇(10mL)和二氯甲烷(20mL)溶解,再加入甲基叔丁基醚(30mL)和正己烷(100mL),析出固体,过滤,滤饼用甲基叔丁基醚(50mLⅹ2)清洗,滤饼用真空烘箱干燥,得产品28-229:1.53g,产率:68%。
实施例九化合物43-8的合成
28-252
将Boc-Glu-(OH)(OBn)(5g,14.82mmol),H-Glu-(OBn)
28-253
将28-252(9.58g,14.82mmol)投入到500mL烧瓶中,加入二氯甲烷(30mL)使其溶解,在搅拌状态下加入三氟乙酸(13.68mL,148.2mmol),反应在室温下搅拌过夜。反应结束,将反应液浓缩,将反应液转移到1L分液漏斗中,加入饱和碳酸氢钠(300mL)和乙酸乙酯(200mL),振摇,萃取,得有机相,水相用乙酸乙酯(200mL x 2)清洗,合并有机相,用饱和食盐水(200mL x 2)清洗,浓缩蒸干,烘箱干燥,得产品28-253:8.1g,产率100%。
28-254
将Boc-LC-OH(3.55g,13.47mmol,按24-36方法合成),28-253(8.1g,14.82mmol),HBTU(7.61g,20.21mmol),HOBT(2.73g,20.21mmol)加入到500mL烧瓶中,用DMF(50mL)溶解后,在将反应置于-5℃条件下搅拌约30分钟,然后缓慢滴加DIEA(10.02mL,60.62mmol),滴加完毕后,反应继续在-5℃搅拌1小时,然后移至室温搅拌过夜。反应结束后,将反应液转移到1L分液漏斗中,加入饱和碳酸氢钠(250mL)和乙酸乙酯(200mL),振摇,萃取。水相用乙酸乙酯(150mL x 2)清洗,合并有机相,用饱和食盐水(200mL x 2)清洗,浓缩蒸干,真空烘箱中干燥,得到产品28-254:10.7g,产率100%。
28-256
将28-254(10.66g,13.47mmol)投入到500mL烧瓶中,加入二氯甲烷(30mL)使其溶解,在搅拌状态下加入三氟乙酸(15mL,202.05mmol),反应在室温下搅拌过夜。反应结束,将反应液浓缩,将反应液转移到1L分液漏斗中,加入饱和碳酸氢钠(300mL)和乙酸乙酯(200mL),振摇,萃取,得有机相,水相用乙酸乙酯(150mL x 2)清洗,合并有机相,用饱和食盐水(200mL x 2)清洗,浓缩蒸干,加入甲醇(30mL)和二氯甲烷(120mL)溶解,加入硅胶粉(25g),蒸干呈粉末状固体,干法上样,柱层析,用0.5%氨水和3%甲醇的二氯甲烷的混合溶液洗脱,得产品28-256:7.8g,产率84%。
28-263
将Fmoc-L-Lys(Boc)-OH(1.76g,3.76mmol),28-256(2.6g,3.76mmol),HBTU(2.14g,5.64mmol),HOBT(0.76g,5.64mmol)加入到500mL烧瓶中,用DMF(50mL)溶解后,在将反应置于-5℃条件下搅拌约30分钟,然后缓慢滴加DIEA(2.8mL,16.92mmol),滴加完毕后,反应继续在-5℃搅拌反应3小时。反应结束后,将反应液转移到1L分液漏斗中,加入去离子水(200mL)和乙酸乙酯(200mL),振摇,萃取。水相用乙酸乙酯(150mL x 2)清洗,合并有机相,用饱和食盐水(200mL x 2)清洗,浓缩蒸干,真空烘箱中干燥,得到产品28-263:4.2g,产率100%。
37-3
将28-263(5.7g,4.99mmol)投入250mL的烧瓶中,加入DMF(20mL),使其溶解,加入吗啉(8.69mL,99.8mmol),反应在室温下搅拌1小时,反应结束,将反应液转移到1L的分液漏斗中,加入去离子水(200mL)和乙酸乙酯(200mL),进行萃取,得有机相,水相用乙酸乙酯(200mL x 2)清洗,合并有机相,有机相用饱和食盐水(200mL x 2)清洗,浓缩蒸干,加入甲醇(30mL)和二氯甲烷(120mL)溶解,加入硅胶粉(20g),蒸干呈粉末状固体,干法上样,柱层析,用0.5%氨水和3%甲醇的二氯甲烷的混合溶液洗脱,得产品37-3:2.7g,产率58%。
28-262
将28-256(5.2g,7.5mmol)置于250mL的圆底烧瓶中,用二氯甲烷(15mL)超声溶解,加入TEA(2.1mL,15mmol),0℃条件下搅拌反应30分钟。再缓慢滴加氯甲酸苯酯(0.94mL,7.5mmol)低温反应结束。2小时后反应结束,用二氯甲烷与饱和氯化钠溶解萃取处理,蒸干有机相得产品产品5g,产率83%。
37-8
将37-3(2.7g,2.9mmol)、28-262(2.4g,2.9mmol)投入到250mL的烧瓶中,加入DMF(35mL)溶解,反应在80℃搅拌过夜,反应结束,将反应液转移到1L的分液漏斗中,加入乙酸乙酯(200mL)和饱和碳酸氢钠(200mL)萃取,得有机相,水相相用乙酸乙酯(150mL x 3)清洗,加入硅胶粉(20g),蒸干呈粉末状固体,干法上样,柱层析,用0.5%氨水和1%甲醇的二氯甲烷的混合溶液洗脱,得产品37-8:1.3g,产率28%。
43-1
将37-8(1.3g,0.79mmol)投入氢化反应釜中,再加入Pd/C(0.0300g),再加入DMF(30mL)使之溶解,通入氢气,压力Pa=1.8MPa,反应在室温下搅拌过夜。反应结束后,将反应釜取出,将反应液通过硅藻土过滤,用DMF(20mL x 3)清洗,作为下一步反应的原料。
43-2
将G-SN38-TBDPS(3.314g,4.819mmol)、43-1(0.86g,0.79mmol)、HBTU(2.7g,7.11mmol)、HOBT(0.96g,7.11mmol)加入到250mL烧瓶中,将反应置于0℃搅拌约30分钟,然后缓慢滴加DIEA(3.5mL,21.33mmol),滴加完毕,继续于0℃搅拌3小时。反应结束,将反应液转移到1L的分液漏斗中,加入饱和食盐水(200mL)和乙酸乙酯(250mL),进行萃取,得有机相,水相用乙酸乙酯(200mL x 1)清洗,合并有机相,有机相用饱和食盐水(200mL x 1)清洗,浓缩蒸干,加入甲醇(20mL)和二氯甲烷(80mL)溶解,加入硅胶粉(15g),蒸干呈粉末状固体,干法上样,柱层析,用4%-5%甲醇的二氯甲烷混合溶液洗脱,收集浓缩,蒸干,烘箱干燥,得产品43-2:2.1g,产率53%。
43-4
将43-2(2.1g,0.412mmol)投入到250mL烧瓶中,加入二氯甲烷(5mL)溶解,在搅拌状态下加入三氟乙酸(9.3mL,125.5545mmol),反应在室温下搅拌过夜。反应结束,先将反应液浓缩至少量,再向反应液中加入甲基叔丁基醚(150mL)沉降,呈粉末状固体,过滤,用甲基叔丁基醚(20mL x 3)洗涤滤饼。滤饼加入甲醇(20mL)和二氯甲烷(80mL)溶解,加入硅胶粉(10g),蒸干呈粉末状固体,干法上样,柱层析,用6%甲醇的二氯甲烷混合溶液洗脱,收集浓缩,蒸干,烘箱干燥,得产品43-4:1.3g,产率62%。
将43-4(1.3g,0.2592mmol)投入250mL烧瓶中,加入DMF(20mL)使其溶解,再加入4ARM-CM-40K(2.5512g,0.0642mmol,购于键凯),HBTU(0.2191g,0.5778mmol),HOBT(0.0781g,0.5778mmol),反应置于-5℃下搅拌20分钟,缓慢滴加DIEA(0.493mL,2.9853mmol),滴定完毕,反应半小时后取出,于室温下低速避光搅拌反应一周。反应结束后,加入正己烷(150mL)和甲基叔丁基醚(40mL)振荡,倒掉上清液,如此重复4次呈粘稠油状物,加入二氯甲烷(10mL)溶解,加入甲基叔丁基醚(150mL),析出固体,抽滤,滤饼用甲基叔丁基醚(40mLⅹ3)清洗,滤饼再用甲醇(20mL)和二氯甲烷(80mL)溶解,加入硅胶粉(10g),蒸干,呈粉末状固体,干法上样,柱层析,用6%-7%甲醇的二氯甲烷混合溶液进行洗脱,收集浓缩,蒸干至固体,真空烘箱干燥,得到产品43-6:2.8g,产率:75%。
43-8
将43-6(2.8g,0.047mmol)用四氢呋喃(25mL)溶解,加入四丁基氟化铵三水合物(,0.0418g,0.1325mmol,简称TBAF·3H
实施例十化合物22-278的合成
22-278
将22-277(0.5g,0.2677mmol)加入到500mL烧瓶,再加10mL DMF使之溶解,置于-5摄氏度下搅拌反应30min,再缓慢滴加DIEA(0.09mL,0.5354mmol),滴加完毕,再加入4ARM-SCM-40K(2.25g),将反应置于常温下低速搅拌一周。反应结束后,先加入正己烷(50mL x3),下层油状只有很少的量时,加入甲基叔丁基醚(20mL)沉降,析出固体,滤饼用甲醇/二氯甲烷(1:5)溶解,加入30mL硅胶粉,蒸干,干法上样,柱层析,洗脱剂用(0.5%-1%氨水:4%-6%甲醇:95.5%-93%二氯甲烷)进行洗脱,收集滤液,浓缩蒸干,得产品22-278:1.745g,产率67%。
实施例十一化合物22-284的合成
22-279
将4ARM-NH2.HCL-40K(2.97g,0.07585mmol)倒入250mL烧瓶中,加入二氯甲烷20mL使之溶解,再加三乙胺(0.26ml,1.8177mmol),将反应液置于0摄氏度下搅拌20-30min,缓慢滴加氯甲酸苯酯(0.05ml,0.3793mmol),滴毕。在0摄氏度下反应2-3h。反应结束,将反应液旋蒸为原体积的二分之一时,加入甲基叔丁基醚200mL,析出固体,再次用甲基叔丁基醚100mL洗涤固体2次,烘干,得产品22-279:2.9g,产率96.7%
22-284
22-279(1.925g,0.04868mmol)、22-277(0.4g,0.2142mmol)加入到500mL烧瓶,再加10mL DMF使之溶解,置于70摄氏度下遮光低速搅拌一周。反应结束后,先加入正己烷(50mL x 3),下层油状只有很少的量时,加入甲基叔丁基醚(20mL)沉降,析出固体,滤饼用甲醇/二氯甲烷(1:5)溶解,加入30mL硅胶粉,蒸干,干法上样,柱层析,洗脱剂用(1%氨水:5%-8%甲醇:94%-91%二氯甲烷)进行洗脱,收集滤液,浓缩蒸干,得产品22-284:1g,产率44%。
实施例十二化合物44-10的合成
25-229
将Boc-Asp-OH(6.0g,25.7268mmol),HBTU(29.2699g,77.1803mmol),HOBT(10.4286g,77.1803mmol)和H-Gly-OBn·TosOH(17.3599g,51.4536mmol)加入到500mL圆底烧瓶中,用DMF(100mL)溶解后,在将反应置于-5℃条件下搅拌约30分钟,然后缓慢滴加DIEA(38.3mL,231.5410mmol),滴加完毕后,反应继续在-5℃搅拌反应1小时后,转移至室温下搅拌反应2小时。反应结束后,将反应液转移到2L分液漏斗中,加入饱和碳酸氢钠溶液(400mL)和乙酸乙酯(300mL),振摇,萃取。有机相中继续加入饱和氯化钠(300mL),振摇,萃取。有机相蒸干,置于真空烘箱中干燥。得到产品25-229:13.5727g,产率:100%。
25-230
将25-229(13.5727g,25.7268mmol)投入到100mL圆底烧瓶中,加入二氯甲烷(10mL)溶解,在搅拌状态下加入TFA(19.1mL,257.268mmol),反应在室温下搅拌过夜。反应结束后,先将反应液浓缩蒸干除去二氯甲烷。然后将反应液转移到2L分液漏斗中,加入饱和碳酸氢钠溶液(400mL)和乙酸乙酯(300mL),振摇,萃取。有机相中继续加入饱和氯化钠(300mL),振摇,萃取。有机相蒸干,置于真空烘箱中干燥。得到产品25-230:9.3g,产率:84.57%。
25-231
将25-230(9.3g,21.7569mmol),HBTU(11.2515g,29.6685mmol),HOBT(4.0088g,29.6685mmol)和Fmoc-Glu(OtBu)-OH(8.4154g,19.7790mmol)加入到500mL圆底烧瓶中,用DMF(50mL)溶解后,在将反应置于-5℃条件下搅拌约30分钟,然后缓慢滴加DIEA(19.6mL,118.6740mmol),滴加完毕后,反应继续在-5℃搅拌反应3小时后。反应结束后,将反应液转移到2L分液漏斗中,加入饱和氯化钠溶液(400mL)和乙酸乙酯(300mL),振摇,萃取。有机相中继续加入饱和氯化钠(300mL),振摇,萃取。有机相蒸干,置于真空烘箱中干燥。得到产品25-231:16.5137g,产率:100%。
25-232
将25-231(16.5137g,19.779mmol)投入到500mL圆底烧瓶中,加入DMF(20mL)溶解,在搅拌状态下加入吗啉(25.8mL,296.6850mmol),置于室温下搅拌反应2小时。反应结束后,先将反应液转移到2L分液漏斗中,加入饱和氯化钠溶液(300mL)和乙酸乙酯(200mL),振摇,萃取。再向有机相中继续加入饱和氯化钠溶液(300mL),振摇,萃取。然后,向有机相中加入去离子水(300mL),振摇,萃取。最后,有机相浓缩蒸干,置于真空烘箱中干燥。得到产品25-232:12.1303g,产率:100%。
25-246
将25-232(12.1303g,19.779mmol)用DMF(50mL)溶解后,在将反应置于-5℃条件下搅拌约10分钟,然后缓慢滴加DIEA(19.6mL,118.6740mmol),滴加完毕后,反应继续在-5℃搅拌反应30分钟,取出后,向其中加入丁二酸酐(5.9379g,59.337mmol),并置于室温下反应3小时。反应结束后,将反应液转移到2L分液漏斗中,加入饱和氯化钠溶液(400mL)和乙酸乙酯(300mL),振摇,萃取。有机相中继续加入饱和氯化钠(300mL),振摇,萃取。有机相蒸干,置于真空烘箱中干燥。得到产品25-246:14.0973g,产率:100%。
25-249
将25-246(14.0973g,19.7790mmol),HBTU(11.2515g,29.6685mmol),HOBT(4.0088g,29.6685mmol)和H-Gly-OBn·TosOH(6.9402g,20.5702mmol)加入到500mL圆底烧瓶中,用DMF(100mL)溶解后,在将反应置于-5℃条件下搅拌约30分钟,然后缓慢滴加DIEA(19.6mL,118.674mmol),滴加完毕后,反应继续在-5℃搅拌反应1小时后,转移至室温下搅拌反应过夜。反应结束后,将反应液转移到2L分液漏斗中,加入饱和碳酸氢钠溶液(400mL)和乙酸乙酯(300mL),振摇,萃取。有机相中继续加入饱和氯化钠(300mL),振摇,萃取。有机相蒸干。将其用20%甲醇/二氯甲烷(100mL)混合溶剂溶解,加入80mL硅胶粉,蒸干,干法上样,柱层析。用洗脱剂(1%-3%甲醇:99%-97%乙酸乙酯)进行洗脱,收集液体浓缩蒸干。得到产品25-249:9.8g,产率:73.52%。
25-258
将25-249(4.0482g,5.0061mmol)投入氢化反应釜中,加入0.1mg钯炭,再加入DMF(40mL)使之溶解,最后通入氢气,压力P=1.6MPa,在室温下搅拌反应过夜。反应结束后,将反应釜取出,将反应液通过硅藻土过滤,用DMF(20mL*3)清洗反应釜。然后,向滤液中加入正己烷(150mL)和甲基叔丁基醚(30mL)沉降3次,下层油状物蒸干,置于真空烘箱中干燥。得到产品25-258:2.9513g,产率:100%。
25-259
将25-258(0.9921g,1.6828mmol),G-SN38-TBDPS(3.5g,5.5534mmol)和4-二甲氨基吡啶(DMAP,0.1234g,1.0097mmol)加入到500mL圆底烧瓶中,用二氯甲烷(100mL)溶解后,将反应置于0℃条件下搅拌约30分钟,然后加入二环己基碳二亚胺(DCC,2.0833g,10.0968mmol),反应置于0℃搅拌反应过夜。反应结束后,首先将反应液过滤除去DCC,并用二氯甲烷(20mL)洗涤滤饼3次。向其中加入硅胶粉(50mL),蒸干,干法上样,柱层析。用洗脱剂(3%-7%甲醇:93%-93%二氯甲烷)进行洗脱,收集浓缩蒸干,得到产品25-259:1.5191g,产率:37.18%。
MALDI-TOF MS:[M+Na
25-280
将25-259(1.51g,0.6219mmol)投入到100mL圆底烧瓶中,加入二氯甲烷(5mL)溶解,在搅拌状态下加入TFA(0.7mL,9.3290mmol),反应在室温下搅拌过夜。反应结束后,先将反应液浓缩蒸干除去二氯甲烷。先将反应液转移到1L分液漏斗中,加入饱和氯化钠溶液(300mL)和乙酸乙酯(200mL),振摇,萃取。再向有机相中继续加入饱和氯化钠溶液(300mL),振摇,萃取。然后,向有机相中加入去离子水(300mL),振摇,萃取。最后,有机相浓缩蒸干。将其用20%甲醇/二氯甲烷(30mL)混合溶剂溶解,加入30mL硅胶粉,蒸干,干法上样,柱层析。用洗脱剂(1%-8%甲醇:99%-92%二氯甲烷)进行洗脱,收集液体浓缩蒸干。得到产品25-280:0.7586g,产率:51.43%。
MALDI-TOF MS:[M+H
25-283
将25-280(0.7497g,0.3161mmol),HBTU(0.2451g,0.6462mmol),HOBT(0.0873g,0.6462mmol)和4ARM-NH
44-10
将25-283(1.9752g,0.0407mmol)用THF(35mL)溶解;取四丁基氟化铵三水合物(TBAF·3H
实施例十三化合物42-7的合成
25-256
将28-65(1.8784g,1.3796mmol,按31-149方法合成)投入氢化反应釜中,加入100mg钯炭,再加入DMF(30mL)使之溶解,最后通入氢气,压力P=1.6MPa,在室温下搅拌反应过夜。反应结束后,将反应釜取出,将反应液通过硅藻土过滤,用DMF(20mL×3)清洗反应釜。得到产品25-256:1.3852g,产率:100%。
25-257
将25-256(1.3852g,1.3796mmol),HBTU(3.1390g,8.2776mmol),HOBT(1.1180g,8.2776mmol)和25-254(5.8g,7.0562mmol按30-33方法合成)加入到500mL圆底烧瓶中,用DMF(60mL)溶解后,在将反应置于-5℃条件下搅拌约30分钟,然后缓慢滴加DIEA(4.1mL,24.8328mmol),滴加完毕后,继续在-5℃搅拌反应3小时。反应结束后,向反应液中加入正己烷(150mL)和甲基叔丁基醚(30mL)沉降5次,倾倒上层清液。然后用二氯甲烷溶解下层油状物,加入甲基叔丁基醚(100mL)沉降,呈粉末状固体,过滤,用甲基叔丁基醚(20mL×3)洗涤滤饼,滤饼置于真空烘箱中干燥。得到产品25-257:5.8g,产率:100%。
25-267
将25-257(5.8176g,1.3796mmol)投入到500mL圆底烧瓶中,加入DMF(50mL)溶解,在搅拌状态下加入吗啉(1.8mL,20.6940mmol),置于室温下搅拌反应3小时。反应结束后,向反应液中加入正己烷(150mL)和甲基叔丁基醚(30mL)沉降5次,倾倒上层清液。然后用二氯甲烷溶解下层油状物,加入甲基叔丁基醚(100mL)沉降,呈粉末状固体,过滤,用甲基叔丁基醚(20mL×3)洗涤滤饼,滤饼置于真空烘箱中干燥。得到产品25-267:3.1g,产率:56.26%。
25-270
将25-267(3.1g,0.7761mmol)用二氯甲烷(50mL)溶解后,加入三乙胺(0.55mL,3.8800mmol),并将其置于-5℃条件下搅拌约20分钟,然后缓慢滴加氯甲酸苯酯(0.3mL,2.3280mmol),滴加完毕后,反应继续在-5℃搅拌反应3小时。反应结束后,先将蒸掉反应液中的大部分二氯甲烷,然后加入甲基叔丁基醚(100mL)沉降,成粉末状固体,过滤,并用甲基叔丁基醚(20mL)洗涤滤饼3次,滤饼置于真空烘箱中干燥。得到产品25-270:3.1934g,产率:100%。
25-277
将25-270(3.1g,0.7761mmol)和25-132(0.6919g,0.7761mmol,按3-16方法合成)投入到250mL圆底烧瓶中,加入DMF(30mL)溶解,将反应置于80℃油浴锅中机械搅拌反应过夜。反应结束后,向反应液中加入正己烷(150mL)和甲基叔丁基醚(30mL)沉降5次,倾倒上层清液。然后用二氯甲烷溶解下层油状物,加入甲基叔丁基醚(100mL)沉降,呈粉末状固体,过滤,用甲基叔丁基醚(20mL×3)洗涤滤饼。滤饼用20%甲醇/二氯甲烷混合溶剂(60mL)溶解,加入40mL硅胶粉,蒸干,干法上样,柱层析。用洗脱剂(1%氨水:1%-8%甲醇:98%-91%二氯甲烷)进行洗脱,收集液体浓缩蒸干。得到产品25-277:2.0245g,产率:80.76%。
42-5
将25-277(2.0245g,0.4121mmol)投入到100mL圆底烧瓶中,加入二氯甲烷(10mL)溶解,在搅拌状态下加入TFA(0.6mL,6.1822mmol),反应在室温下搅拌过夜。反应结束后,先将反应液浓缩蒸干除去二氯甲烷。然后加入甲基叔丁基醚(100mL)沉降,呈粉末状固体,过滤,用甲基叔丁基醚(20mL×3)洗涤滤饼。滤饼用20%甲醇/二氯甲烷混合溶剂(60mL)溶解,加入40mL硅胶粉,蒸干,干法上样,柱层析。用洗脱剂(1%氨水:1%-8%甲醇:98%-91%二氯甲烷)进行洗脱,收集液体浓缩蒸干。得到产品42-5:1.3568g,产率:68.41%。
42-7
将42-5(0.9995g,0.2077mmol)和M-SCM-20K(2.0g,0.1888mmol)加入到500mL圆底烧瓶中,用DMF(20mL)溶解后,置于室温下避光反应一周。反应结束后,向反应液中加入正己烷(100mL)和甲基叔丁基醚(10mL)沉降3次,倾倒上层清液。然后加入二氯甲烷(5.0mL),再加入甲基叔丁基醚(60mL)沉降,呈粉末状固体,过滤。用甲基叔丁基醚(20mL)洗涤滤饼3次。滤饼用20%甲醇/二氯甲烷混合溶剂(50mL)溶解,加入40mL硅胶粉,蒸干,干法上样,柱层析。用洗脱剂(1%氨水:4%-10%甲醇:95%-89%二氯甲烷)进行洗脱,收集液体浓缩蒸干。得到产品42-7:1.3454g,产率:46.66%。
实施例十四化合物24-266的合成:
33-124
将19-107(1g,0.8mmol),30-33(0.7g,0.88mmol),HBTU(0.45g,1.2mmol)和HOBT(0.16g,1.2mmol)置于250mL圆底烧瓶中然后加入DMF(20mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。再缓慢滴加DIEA(0.59mL,3.6mmol)继续反应,反应2h后提出在室温下搅拌。反应结束后,取出反应液加入去离子水(200mL),用乙酸乙酯萃取三次(100mL×3),合并有机相,有机相加入饱和氯化钠溶液(200mL)清洗两次,浓缩蒸干.干法上样,柱层析,洗脱剂用1%氨水:3%甲醇/二氯甲烷-1%氨水:5%甲醇/二氯甲烷梯度洗脱收集产品,浓缩,蒸干得到产物。实际产量0.8g,产率50%。
33-127
将33-124(0.8g,0.39mmol)置于250mL的圆底烧瓶中,用DMF(20mL)溶解,然后加入吗啉(1.01mL,11.7mmol)室温下搅拌反应1小时。反应结束后,在反应液中加入甲基叔丁基醚(100mL),加入正己烷(200mL),沉降得粉末。实际产量0.5g,产率71.4%。
33-130
将33-127(0.5g,0.27mmol),Boc-Gly-OH(0.049g,0.28mmol),HBTU(0.15g,0.4mmol)和HOBT(0.054g,0.4mmol)置于250mL圆底烧瓶中然后加入DMF(20mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。再缓慢滴加DIEA(0.19mL,1.21mmol)继续反应,反应2h后提出在室温下搅拌。反应结束后,在反应液中加入甲基叔丁基醚(100mL),加入正己烷(200mL),沉降得粉末。实际产量0.6g,产率100%。
33-131
将33-130(0.5g,0.27mmol)于250mL的圆底烧瓶中,用二氯甲烷(20mL)溶解,然后加入TFA(0.6mL,8.1mmol)室温下搅拌反应过夜,反应停止,将反应液减压浓缩蒸干,加入甲基叔丁基醚(100mL),加入正己烷(200mL),沉降得粉末。实际产量0.5g,产率100%。
24-262
将33-131(0.5g,0.2657mmol)置于250mL的圆底烧瓶中,用二氯甲烷(15mL)超声溶解,加入TEA(0.1494mL,1.0628mmol),0℃条件下搅拌反应30分钟。再缓慢滴加氯甲酸苯酯(0.0668mL,0.5314mmol)低温反应结束。2小时后反应结束,蒸干反应液,除去二氯甲烷,加甲基叔丁基醚(20mL),超声2分钟,加正己烷(100mL),抽滤得产品0.5g,产率100%。
24-205
称取反应物15-91(31-149路线合成,0.33g,0.1503mmol)投入微型反应釜中,加入10%Pd/C(30mg),加DMF(30mL)溶解,通入H
41-7
将41-6(0.84g,0.44mmol),24-205(0.091mmol),HBTU(0.07g,0.546mmol)和HOBT(0.2g,0.546mmol)置于250mL圆底烧瓶中然后加入DMF(20mL),使其溶解,将混合液置于-5℃条件下搅拌30分钟。再缓慢滴加DIEA(0.27mL,1.638mmol)继续反应,反应2h后在室温下搅拌过夜。
41-9
反应物41-7(0.7g,0.082mmol),用DMF(30mL)溶解,加入吗啉(0.214mL,2.46mmol),搅拌反应,反应结束,加入甲基叔丁基醚(100mL),加入正己烷(200mL)沉降得粉末。柱层析,干法上样,用1%氨水:5%甲醇/二氯甲烷--1%氨水:12%甲醇/二氯甲烷梯度洗脱。产品重0.5克。
24-261
称取41-9(0.5g,0.0602mmo)、24-262(0.1446g,0.0722mmol)投入250mL反应瓶中,加DMF(20mL)溶液,加DIEA(0.1mL),在100℃条件下搅拌反应至结束。反应结束,在反应液中加甲基叔丁基醚(150mL),超声5分钟,抽滤烘干得产品0.5g,产率100%。
24-264
取24-261(0.5g,0.0489mmol),加入二氯甲烷(10mL),再加入TFA(0.1mL,1.4654mmol),超声使完全溶解,用磨口玻璃塞,常温搅拌反应。反应结束,在反应液中直接加甲基叔丁基醚(150mL)与正己烷(100mL),抽滤,固体加20%甲醇/二氯甲烷(50mL),超声溶解,加硅胶粉(3g),用旋转蒸发仪蒸干,柱层析。用1%氨水+7%-10%甲醇/二氯甲烷梯度洗脱得产品0.2g,产率46%。
24-266
称取24-264(0.2g,0.0198mmol)加入DMF(20mL)溶解,再加M-SCM-40K(0.799g,0.0189mmol),超声溶解。低速避光反应。反应结束,在反应液中加甲基叔丁基醚(150mL),正己烷(70mL)析出固体,抽滤,加20%甲醇/二氯甲烷溶解,加硅胶粉(3g),蒸干,柱层析。用1%氨水+7%甲醇/二氯甲烷洗脱,收集产品蒸干,加无水乙醇(3mL),超声均匀,加甲基叔丁基醚(150mL),加正己烷(50mL),抽滤,继续加溶解无水乙醇(3mL),甲基叔丁基醚与正己烷沉降,反复溶解沉降三次,抽滤,烘干,得产品0.54g,产率56%。
实验例一本发明化合物对人结肠癌COLO-205细胞BALB/c裸鼠皮下移植瘤模型的体内抗肿瘤药效试验
一、实验目的
建立人结肠癌COLO-205细胞的BALB/c裸鼠皮下移植瘤模型,考察供试品对皮下移植瘤的抗肿瘤作用。
二、供试品和对照品信息
1、供试品
化合物22-278
2、对照品:
成分1:SB7 HCL(南京药石科技股份有限公司)
成分2:帕布昔利布(PCB,纯度:99.5%,天津法莫西医药科技有限公司)
3、溶媒/阴性对照品
氯化钠注射液(山东齐都药业有限公司,批号:4B19030803,浓度:0.9%,规格:100mL:0.9g)
三、供试品/对照品配制
1、供试品:
称取适量体积的22-278,加入适量的生理盐水,配制成浓度为9.47mg/mL的溶液。
3、小分子对照品:
称取适量的SB7(折算系数93.4%),加入一定体积的乙醇(5%,V/V)溶解,待完全溶解后,再加入适量的氯化钠注射液,配制成浓度为0.4mg/mL的溶液。
称取适量的PCB,加入一定体积的0.5%的CMC-Na溶液,在磁力搅拌器上搅拌均匀,配制成浓度为0.35mg/mL的溶液。
4、阴性对照:直接使用生理盐水。
四、实验系统
1、肿瘤细胞株信息
人结肠癌细胞COLO-205:来源于中国医学科学院基础所细胞资源中心,培养条件为RPMI1640+10%FBS,37℃、5%CO
2、实验动物
SPF级雄性BALB/c裸鼠,4周龄,购入动物体重:11.0~14.9g
实验动物来源:北京维通达生物技术有限公司,动物生产许可证号:SCXK(京)2016-0011,订购动物合格证编号:No.1100111911049640。
3、动物饲养和管理
饲养环境:SPF级环境动物房
动物使用许可证号:SYXK(京)2016-0029
饲料:动物给予合格的鼠料(提供单位:北京科澳协力饲料有限公司,批号:19063113、19063123、19073113,生产许可证号:SCXK(京)2014-0010),动物自由摄食。
饮水:检疫期间和试验过程中,动物给予纯化水(二级反渗透RO膜过滤水),用饮水瓶供应,自由摄水。
饲养条件:动物饲养于独立送风系统(IVC)中,每笼饲养3~5只,环境条件控制在20℃~26℃,相对湿度40%~70%,实际温度为22℃~26℃,实际相对湿度为42%~59%,光照约12小时明暗交替。
五、实验设计
复苏COLO-205细胞,进行细胞传代扩增,待扩增至足量细胞数时,收集处于对数生长期的细胞,进行细胞接种。调整细胞浓度在5×10
依据瘤体积大小随机分为5组,每组6只。各组给药剂量和动物号如表1所示。阴性对照、SB7、各供试品静脉注射,PCB灌胃给药。给药频次为1次/周,连续2周。
表1动物分组和给药剂量
六、检测指标
1、给药期间每天2次一般临床观察,每周进行2次体重和瘤径测量,安乐死后剥取肿瘤,称量肿瘤重量。动物安乐死后,进行大体解剖,观察主要脏器变化。
2、瘤径的测量
所有动物分组(即首次给药当天为D1)、首次药后每周2次、安乐死前一天,游标卡尺测量并记录肿瘤长、短径,计算肿瘤体积,并根据肿瘤体积绘制肿瘤生长曲线。按照以下公式计算肿瘤体积:V=1/2×长径×短径
3、根据肿瘤体积进行疗效评价
按照以下公式计算相对肿瘤体积(RTV)和相对肿瘤增殖率T/C%:
RTV=V
V
V
T/C%=给药组的RTV平均值/对照组的RTV平均值×100%
如果T/C%≤40%,实验组RTV与模型组RTV经统计学处理P≤0.05,为有肿瘤增长抑制作用;反之,如果T/C%>40%,则为对肿瘤增长无抑制作用。
4、根据肿瘤重量进行疗效评价
D16实验结束后,剥离肿瘤结节并进行称重,比较各组间肿瘤重量的差异以进一步计算肿瘤抑制率IR
IR
七、统计分析
使用统计学软件SPSS13.0对数据进行处理,计量资料以平均值±标准误来表示。具体分析过程如下:用单因素方差分析(ANOVA)进行统计分析,如果ANOVA有统计学意义(P≤0.05)且方差齐性,用Tukey test进行组间比较分析,若方差不齐性,则用Dunnett’sT3test进行组间比较分析。
八、结果
1、体重和临床观察:
试验中的动物未见明显异常。整个实验过程中,阴性对照组体重较分组前明显降低,其他组动物体重较分组前略有升高。各组动物体重见表2。
表2体重统计数据表
注:a表示与1组比较P≤0.05
2、肿瘤体积
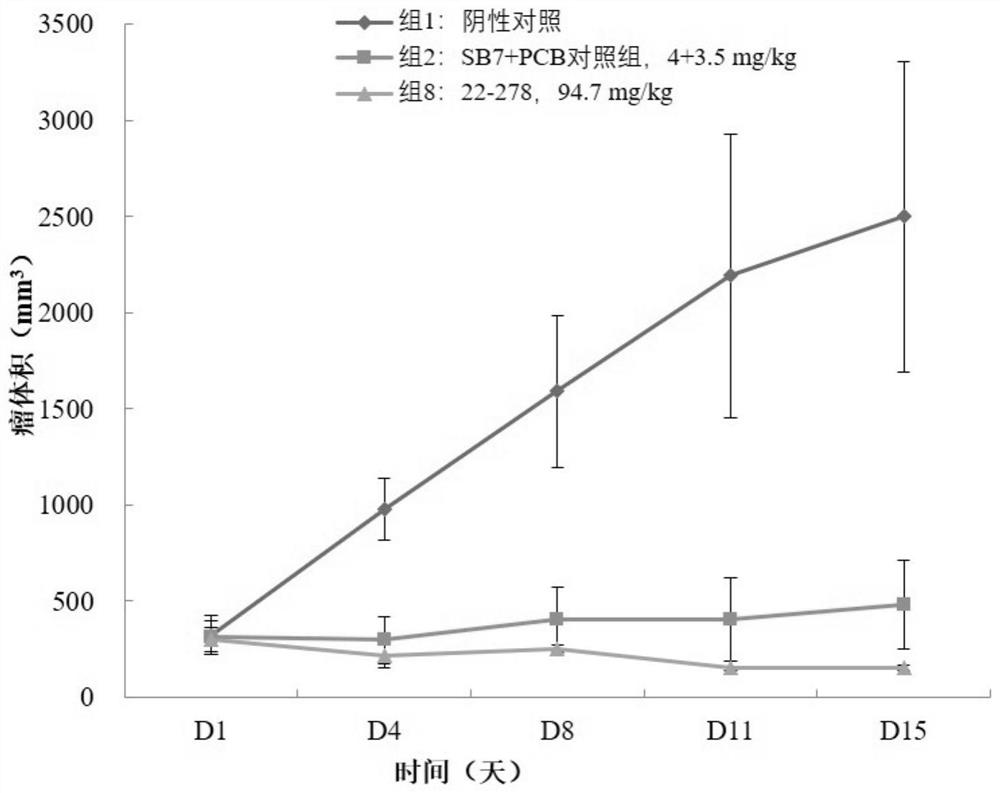
D1分组时各组间瘤体积未见明显差异(P>0.05),2-10组瘤体积均值、RTV均值在给药后各时间点均低于阴性对照组。阴性对照组肿瘤结节保持稳定增长,各组瘤体积及RTV随时间变化及各组间统计差异见表3。各组平均肿瘤体积增长趋势图见图1。
表3肿瘤体积和相对增殖体积(RTV)统计数据表
注:a表示与1组比较P≤0.05
从表3和图1可以看出,SB7+PCB、22-278组均有明显的肿瘤生长抑制作用。22-278抗肿瘤作用优于SB7+PCB联合组。
3、相对肿瘤增殖率T/C%
肿瘤T/C%、IR
表4肿瘤相对增殖率(T/C%)统计数据表
注:“*”表示T/C%≤40%,且RTV与1组比较P≤0.05。
表5瘤体积抑制率(IR
注:“*”表示T/C%≤40%,且RTV与1组比较P≤0.05。
从表4和表5可以看出,至安乐死前一天D15,37-26、28-126、28-206组T/C%值分别为17.22%、2.87%、6.40%、9.58%,其IR
4、瘤重
D16动物安乐死称量瘤重,瘤重及抑制率统计数据见表6,肿瘤重量抑制率示意图见图2。
表6瘤重
注:1.IR
2.“▲”表示IR
3.a表示与1组比较P≤0.05
从表6和图2可以看出,SB7+PCB、22-278组的瘤重显著低于阴性对照组且IR
结论:在本实验条件下,化合物22-278在94.7mg/kg剂量下尾静脉注射给药对人结肠癌细胞COLO-205皮下移植瘤模型有明显的肿瘤生长抑制作用,PCB在3.5mg/kg剂量下灌胃与SB7在4.0mg/kg剂量下尾静脉注射联合给药对肿瘤模型也有明显的生长抑制作用。化合物22-278抗肿瘤作用优于SB7+PCB联合组。
尽管本发明的具体实施方式已经得到详细的描述,但本领域技术人员将理解:根据已经公开的所有教导,可以对细节进行各种修改和变动,并且这些改变均在本发明的保护范围之内。本发明的全部范围由所附权利要求及其任何等同物给出。
- 具有协同抗癌活性的中间体药物和聚乙二醇偶联协同抗癌药物、及其制备方法和应用
- 具有协同抗癌活性的中间体药物和聚乙二醇偶联协同抗癌药物、及其制备方法和应用