FUT3基因作为靶点在提高猪对大肠杆菌抗性中的应用
文献发布时间:2023-06-19 11:50:46
技术领域
本发明属于基因工程技术领域,具体涉及FUT3基因作为靶点在提高猪对大肠杆菌抗性中的应用。
背景技术
CRISPR/Cas9技术是来源于细菌和古生菌中的一种由sgRNA(small guide RNA)介导的适应性免疫系统。其中sgRNA用于识别、结合靶点DNA,当Cas9蛋白活性被激活后可结合区域下游的DNA并对其进行切割,生成双链DNA断裂,诱发由非同源末端连接修复机制造成的移码突变,从而实现对靶基因的编辑。CRISPR/Cas9技术因其简便和高效的特性,近几年来作为真核基因进行定点编辑的重要遗传手段在人类及动植物基因功能研究领域得到广泛应用。猪小肠上皮细胞(IPEC-J2)分离自新生仔猪空肠中段的柱状上皮细胞,具有研究猪病原体感染的物种特异性,是研究猪多种疾病研究的理想体外模型,尤其在产肠毒素大肠杆菌和病毒性腹泻抗性基因功能鉴定研究中广泛应用。目前尚未有猪FUT3基因的敲除载体及FUT3基因敲除的小肠上皮细胞模型。
FUT3是岩藻糖转移酶基因家族中的一员,能够编码α-(1,3/4)岩藻糖转移酶,在组织血型抗原(ABO抗原和Lewis抗原)形成中发挥重要的调控作用。研究发现,FUT3基因与人类病毒感染以及弓形虫感染等多种疾病密切相关,并且FUT3基因还参与机体的免疫应答及炎症反应。迄今为止,大多数研究集中在探讨基因的遗传多态性和表达与人类疾病的关系,关于猪FUT3基因的研究较少,尤其关于猪FUT3表达与大肠杆菌感染的关系研究还鲜有报道,因此在仔猪组织和细胞水平上开展猪FUT3对大肠杆菌抗性的调控机制和功能验证工作,对于深入研究FUT3基因具有重要的突破意义。
发明内容
本发明的目的在于提供FUT3基因作为靶点在制备用于提高猪对大肠杆菌或细菌性腹泻抗性的试剂中的应用。
本发明的另一目的在于FUT3基因在作为提高猪细菌性腹泻抗性功能基因中的应用。
本发明的另一目的在于FUT3基因在仔猪抗大肠杆菌腹泻病的分子选育中的应用。
本发明的另一目的在于干扰、抑制、沉默或敲除FUT3基因的物质或者以FUT3基因为靶点降低FUT3基因表达的化学药物在制备用于提高猪对大肠杆菌或细菌性腹泻抗性的试剂中的应用。
本发明的又一目的在于提供一组基于CRISPR/Cas9技术靶向敲除猪FUT3基因的sgRNA向导序列、敲除载体和细胞系及其应用。
本发明的目的可以通过以下技术方案实现:
FUT3基因作为靶点在制备用于提高猪对大肠杆菌或细菌性腹泻抗性的试剂中的应用。
FUT3基因在作为提高猪细菌性腹泻抗性功能基因中的应用。
FUT3基因在仔猪抗大肠杆菌腹泻病的分子选育中的应用。
干扰、抑制、沉默或敲除FUT3基因的物质或者以FUT3基因为靶点降低FUT3基因表达的化学药物在制备用于提高猪对大肠杆菌或细菌性腹泻抗性的试剂中的应用。
上述的应用中以FUT3基因作为靶点采用基因干扰、抑制、沉默或敲除技术或者采用化学药物降低猪FUT3基因的表达,提高猪对大肠杆菌或细菌性腹泻的抗性,或者培育抗大肠杆菌腹泻病的仔猪品种。
上述的用于提高猪对大肠杆菌抗性的试剂中包含能够干扰、抑制、沉默或敲除FUT3基因的物质。
上述的干扰、抑制、沉默或敲除FUT3基因的物质,它包含以下(1)~(7)中的至少一种:
(1)针对FUT3基因的干扰序列;
(2)用于干扰的FUT3基因的干扰载体;
(3)包含有FUT3基因干扰载体的转基因细胞系;
(4)基于CRISPR/Cas9技术靶向敲除猪FUT3基因的sgRNA向导序列;
(5)基于CRISPR/Cas9技术靶向敲除猪FUT3基因的敲除载体;
(6)基于CRISPR/Cas9技术特异性靶向敲除猪FUT3基因的细胞系。
(7)以FUT3基因为靶点降低FUT3基因表达的化学药物。
所述的针对FUT3基因的干扰序列为RNAi-1~4中的任意一组:
所述的基于CRISPR/Cas9技术靶向敲除猪FUT3基因的sgRNA向导序列为sgRNA1、sgRNA2、sgRNA3和sgRNA4中的至少一种:
sgRNA1:caccGGCGCCGCTGTGGTCTGGCAG
sgRNA2:caccGGCAGCAGAAGCAGGGGTGGG
sgRNA3:caccGGGTGGCTGTGTCTCGCTGCT
sgRNA4:caccGCCAGCTGACTGACAACCGCG。
上述的基于CRISPR/Cas9技术靶向敲除猪FUT3基因的sgRNA向导序列与各自的反向互补序列退火形成多组双链DNA,各组所述的sgRNA向导序列与其各自的反向互补序列的核苷酸序列如下所示:
所述的用于干扰的FUT3基因的干扰载体的制备方法为:将所设计的针对FUT3基因的干扰序列的正义链和反义链退火形成双链DNA,将双链DNA产物和线性化的shRNA表达载体进行连接得到干扰载体;
所述的基于CRISPR/Cas9技术靶向敲除猪FUT3基因的敲除载体的制备方法为:将所设计的sgRNA向导序列与其各自的反向互补序列退火形成双链DNA,将双链DNA与线性化载体进行T4 DNA Ligase连接反应得到敲除载体。
所述的包含有FUT3基因干扰载体的转基因细胞系为将上述的干扰载体转染靶细胞IPEC-J2得到;
所述的基于CRISPR/Cas9技术特异性靶向敲除猪FUT3基因的细胞系为将上述的敲除载体转染靶细胞IPEC-J2得到。
本发明通过qPCR技术检测FUT3基因在仔猪上的组织表达谱以及大肠杆菌抗性与敏感群体之间的表达差异,并通过免疫组织化学技术(Immunohistochemistry,IHC)检测FUT3在抗性与敏感群体肠道组织的定位以及表达情况;同时利用F18大肠杆菌刺激和脂多糖诱导处理猪肠上皮细胞系(IPEC-J2)检测处理前后FUT3基因的表达水平和蛋白水平,剖析F18大肠杆菌侵染和LPS诱导条件下FUT3基因的表达规律。同时设计并构建靶向猪FUT3基因的慢病毒过表达载体、慢病毒干扰载体以及敲除载体,转染猪小肠上皮细胞系后利用qPCR和Western blot方法检测干扰效率和过表达效率,同时通过PCR技术和Western blot技术检测敲除效率。选择效率最高的转染细胞进行下一步功能验证,通过细菌计数、菌毛蛋白基因定量、间接免疫荧光以及革兰氏染色法综合分析FUT3基因过表达、沉默及敲除对小肠上皮细胞黏附F18大肠杆菌能力的影响。本研究从mRNA、蛋白质以及细胞水平上系统探讨了FUT3基因在仔猪抗F18大肠杆菌感染过程中发挥的重要作用,以期确定其可以作为细菌性腹泻重要的抗性候选功能基因,同时也将为之后在生猪生产中开展仔猪抗大肠杆菌腹泻病的分子选育提供科学根据。
本发明的有益效果:
猪腹泻病原体—大肠杆菌F18具有小肠亲嗜性,建立FUT3基因敲除的小肠上皮细胞系,并系统验证其与大肠杆菌F18抗性密切相关,有望确定断奶仔猪大肠杆菌F18病抗性的候选功能基因,以期解决中国地方猪种大肠杆菌病抗性育种的关键科学问题,为今后制订抗仔猪细菌性腹泻病分子选育策略奠定基础和提供依据。
附图说明
图1为FUT3基因在苏太猪11个检测组织中的表达情况。
图2为FUT3基因在敏感型和抗性型断奶仔猪肠道组织中的表达情况。
图3为FUT3基因在敏感型和抗性型肠道组织中免疫组织IHC定位分析;
注:S表示敏感型仔猪十二指肠组织(n=3),R表示抗性型仔猪十二指肠组织(n=3)。
图4为FUT3基因在LPS诱导IPEC-J2细胞后的mRNA表达水平。
图5为FUT3基因在LPS诱导IPEC-J2细胞后的蛋白表达水平。
图6为FUT3基因在大肠杆菌侵染细胞后的mRNA表达水平。
图7为FUT3基因在大肠杆菌侵染细胞后的蛋白表达水平。
图8为FUT3基因慢病毒重组干扰载体测序结果;
注:A、B、C、D依次为4个干扰载体的测序结果
图9为FUT3基因敲除载体构建验证结果;
注:A为敲除载体的PCR验证结果,B为测序结果;其中KO-1、KO-2、KO-3、KO-4依次为4个敲除载体的结果。
图10为慢病毒滴度检测(24h,40×);
注:1~4:病毒液10倍梯度稀释处理,分别加入浓缩病毒液为1×10
图11为病毒液侵染和敲除载体转染IPEC-J2细胞的荧光表达(40×);
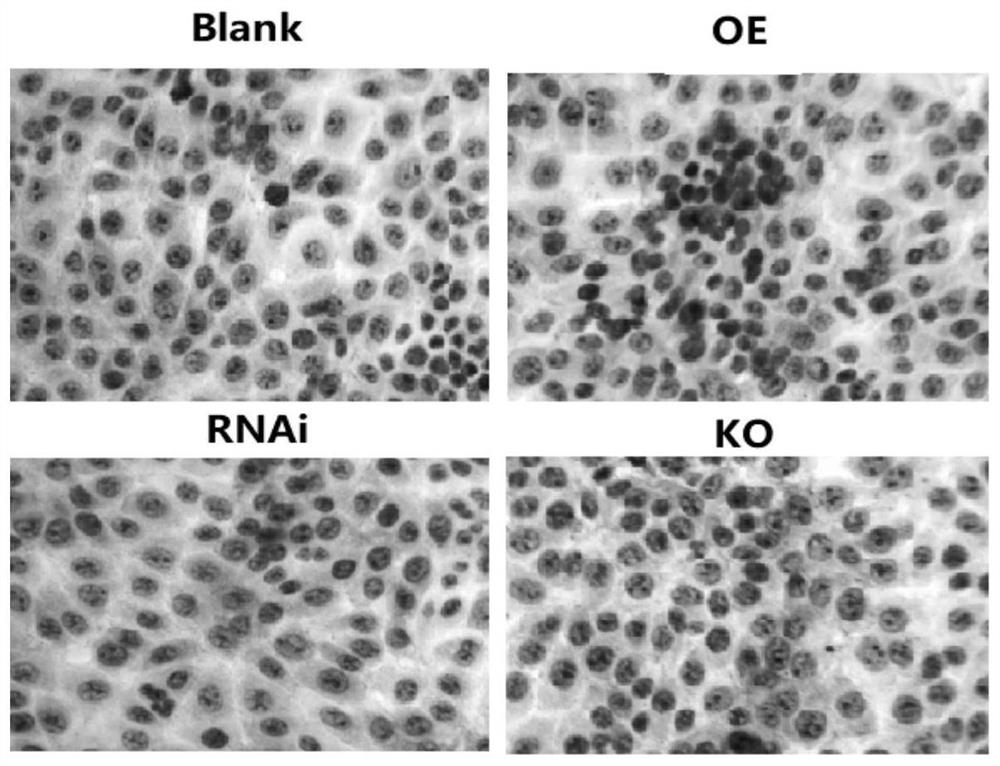
注:RNAi表示FUT3基因干扰,OE表示FUT3基因过表达,KO表示FUT3敲除细胞。左列是荧光显微镜下观察到的细胞,右列是普通光镜下同一视野的对照。
图12为干扰组和过表达组细胞中FUT3基因mRNA表达水平;
注:A为干扰组细胞中FUT3基因的mRNA表达水平,其中RNAi-1~4和Control依次为4个干扰质粒载体与阴性对照载体转染IPEC-J2细胞处理;B为过表达组细胞中FUT3基因mRNA表达水平,其中FUT3-OE、Control分别为过表达载体和阴性对照载体转染IPEC-J2细胞处理。
图13为FUT3基因敲除效果分析;
注:A为敲除载体转染细胞PCR结果,其中Blank为空白细胞,KO-1~4为不同敲除载体转染细胞;B为敲除载体转染细胞PCR产物酶切结果;C为敲除载体转染细胞PCR测序结果;D为敲除单细胞PCR结果,其中1为空白细胞,2~6为敲除单细胞;E为敲除载体转染细胞PCR测序结果,其中最后一行为空白未处理细胞。
图14为不同处理组FUT3蛋白表达水平;
注:A为不同处理组FUT3的Western Blot结果,其中Blank、OE、RNAi和KO分别代表空白细胞、FUT3过表达细胞、FUT3干扰细胞和敲除细胞;B为FUT3不同处理组的灰度值半定量结果。
图15为细菌黏附IPEC-J2细胞的检测结果;
注:A为细菌计数的统计结果,单位为10
图16为间接免疫荧检测F18大肠杆菌对IPEC-J2细胞的黏附水平(100×)。
图17为革兰氏染色法检测F18大肠杆菌对IPEC-J2细胞黏附能力的影响(100×);
注:紫色部分为细胞核,红色部分为细菌。
具体实施方式
以下结合具体实施例对本发明做出详细的描述。根据以下的描述和这些实施例,本领域技术人员可以确定本发明的基本特征,并且在不偏离本发明精神和范围的情况下,可以对本发明做出各种改变和修改,以使其适用各种用途和条件。
一、靶基因FUT3表达水平与F18大肠杆菌抗性的关系研究
1.材料与方法
1.1试验材料
试验猪来自于前期建立的苏太猪F18大肠杆菌敏感型和抗性型资源群体(吴圣龙,原志伟,鞠慧萍,等.苏太仔猪FUT1基因M307位点多态性与F18大肠杆菌抗病相关性的体外鉴定.中国预防兽医学报,2007,29(10):783-787.),通过受体结合试验和V型分泌系统展呈功能性黏附素试验(葛兆宏,陆广富,朱军,等.F18大肠杆菌黏附素受体结合域鉴定新方法[J].扬州大学学报(农业与生命科学版),2010,31(02):1-5.),从中严格选取苏太抗性型以及敏感型断奶仔猪各4头,屠宰后收集仔猪心脏、肝脏、脾脏、肺脏、肾脏、胃、肌肉、胸腺、淋巴结、十二指肠和空肠组织样,现场液氮冷冻保存,然后移至实验室-70℃冰箱备用。猪小肠上皮细胞系IPEC-J2由本实验室冻存和培养;人胚肾脏上皮细胞293T购自中国科学院细胞库;大肠杆菌F18ab、F18ac菌株由扬州大学兽医学院朱国强教授馈赠。
1.2试验试剂
DMEM/F12(1:1)培养基、DMEM培养基、Opti-MEM培养基、胎牛血清、Trypsin-EDTASolution均购自Gibco BRL(美国);青链霉素混合液(100×)、DAPI溶液购自Solarbio公司(中国北京);LPS购自Sigma(美国);Lipofectamine
1.3引物设计与合成
根据GenBank数据库提供的猪FUT3基因序列(AF130972.1),利用Invitrogen RNAiDesigner软件设计并合成4个针对FUT3基因的短发夹RNA(short hairpin RNA,shRNA)干扰序列(RNAi-1~4)和1条阴性对照序列(Negative control,NC);同时利用CRISPR Design软件在FUT3基因第一外显子上设计4个gRNA序列(KO-1~4),位置尽量在基因CDS的前1/3,并在靶点上下游设计一对敲除验证引物,用于后续鉴定敲除阳性克隆,引物序列为F:CAGGACTCGTGAAGATTGACCAT,R:TCAGGTTGAAGTACCCGTCCA,扩增产物长度581bp。以ACTB基因(内参基因)和FUT3基因为扩增模板设计实时荧光定量PCR引物,荧光定量引物片段长度均在100~200bp左右且都跨外显子设计,以避免扩增时基因组DNA污染;同时以大肠杆菌菌毛蛋白基因PILIN为模板设计检测细菌黏附的荧光定量引物,以β-actin为内参。所有引物均由上海生工生物工程有限公司合成。引物的详细信息如表1和表2中所示。
表1寡聚单链DNA序列
注:shRNA干扰序列的斜体部分为引入的酶切位点,小写字母部分为干扰序列及其互补序列,下划线部分为loop序列;敲除oligo序列的大写字母部分为其靶点序列。
表2 Real-time PCR引物信息
1.4 FUT3的免疫组化(IHC)分析
1.4.1制备肠道组织石蜡切片
(1)取材与固定:取2~3cm的十二指肠组织,冲洗干净肠道内壁,立即放入40mL的4%多聚甲醛中室温固定24h。
(2)脱水:脱水前用流水进行冲洗,并切取合适大小的组织;然后从低浓度乙醇溶液到高浓度乙醇溶液依次进行脱水,用30%、50%、70%乙醇溶液依次处理1h,85%、95%、100%、100%乙醇溶液依次处理30min。
(3)透明:将组织块分别经过50%二甲苯的乙醇溶液、二甲苯Ⅰ和二甲苯Ⅱ,每次浸泡时间分别为15min、30min和30min。
(4)浸蜡:将组织块放入二甲苯和石蜡(熔点为52~56℃)等量的混合溶液中浸泡15min,再分别经过两次石蜡溶液浸渍,每次30min。浸蜡步骤均在55℃恒温箱中进行。
(5)包埋:先在包埋框中倒入熔化的石蜡,再用加热后的镊子夹取组织块放入石蜡中,待石蜡稍凝后可将其移入冰箱中加快凝固。
(6)切片:修整蜡块便于切片,再利用切片机对石蜡块进行切片,切下合适的蜡带在45℃温水中进行展片,用毛笔捞出放置在载玻片上;将玻片放置于预热(37℃)的烤片仪上干燥或者自然干燥。
1.4.2免疫组织化学分析
采用三步法进行免疫组化分析,其主要步骤如下:
(1)组织固定:将石蜡切片于60℃恒温箱中放置1h。
(2)脱蜡:将切片分别经过两次二甲苯浸泡,每次10min。
(3)水化:将切片分别经过100%、95%、85%、70%的乙醇溶液浸泡5min,最后用双蒸水冲洗2次(每次5min)。
(4)抗原修复:将玻片放入3%H
(5)血清封闭:擦干玻片上的液体,加入血清封闭液(稀释10倍),37℃孵育30min,弃去液体。
(6)添加一抗:吸干玻片上的液体,加入100μL的一抗稀释液(1:100),37℃环境下于湿盒中孵育2h或4℃过夜;用PBS缓冲液清洗切片3次(每次5min)
(7)添加增强剂:添加50μL的增强剂,于湿盒中室温孵育20min;用PBS缓冲液清洗切片3次(每次5min)。
(8)添加酶标二抗:在切片组织上滴加50μL的辣根过氧化物酶标记(HRP标记)的二抗,37℃环境下于湿盒中孵育30min;用PBS清洗切片3次(每次5min)。
(9)DAB显色:取出玻片,擦干上面的液体,在每个切片组织上滴加3滴新配制好的DAB溶液进行显色。染色完成后,将切片用双蒸水温和冲洗15min。
(10)复染:将玻片置于苏木精染液中进行染色,时间为3min左右,之后使用双蒸水对切片进行冲洗。将切片放入1%盐酸的乙醇溶液中数秒,快速用双蒸水洗涤干净。
(11)脱水和透明:将切片分别在70%、85%、95%、100%和100%无水乙醇中依次浸泡5min,再将切片用二甲苯Ⅰ和二甲苯Ⅱ溶液分别浸泡10min。
(12)封片和观察:玻片晾干后,添加中性树胶进行封片,之后使用光学显微镜进行观察。
1.5大肠杆菌F18ab、F18ac菌体和LPS刺激IPEC-J2细胞
当IPEC-J2细胞长至90%时,将其消化铺板至6孔细胞培养板,当覆盖率达到80%时进行细菌侵染试验。将已经培养好的大肠杆菌F18ab和F18ac菌株分别接种于LB液体培养液中(1:1000),220rpm培养12h;4000rpm离心5min,收集细菌菌体沉淀,用PBS缓冲液重悬沉淀混匀并离心,重复洗涤3次;最后用DMEM/F12培养液将细菌菌体沉淀稀释成1.0×10
1.6靶基因FUT3表达检测
1.6.1反转录和荧光定量检测:
以细胞RNA作为模板,严格按照
1.6.2 Western blot蛋白表达检测:
1.6.2.1细胞总蛋白的提取及浓度测定
(1)用细胞刮刮下细胞,将细胞及培养液移至离心管中,1000rpm离心10min,弃上清。加入5mL的PBS洗涤2次,每次摇晃几次以尽量去除残留培养液。
(2)每1mL冷的Lysis Buffer中加入5μL 100mM PMSF、10μL磷酸酶抑制剂和1μL蛋白酶抑制剂,混匀,置于冰上数分钟待用。
(3)细胞清洗后,转至新的预冷离心管中,加入1mL配好的冷Lysis Buffer。
(4)置于4℃剧烈振荡30s,冰上放置4min,重复5次。
(5)4℃条件下离心(12000rpm)5min收集上清,上清即为提取的全蛋白,-70℃保存备用。
(6)BCA蛋白浓度测定试剂盒对所提取的全蛋白进行检测,使蛋白水平标准化。
1.6.1.2 Western blot技术检测蛋白水平
(1)十二烷基硫酸钠-聚丙烯酰胺凝胶(sodium dodecyl sulfate-polyacrylamide gel,SDS-PAG)的制备:按照说明书分别配置好5%的浓缩胶和10%的分离胶。
(2)电泳检测:将各组蛋白样品通过细胞裂解缓冲液稀释到一样的浓度水平,吸取相等体积的蛋白溶液(约70μg)与蛋白上样缓冲液混合后加入到离心管中,95℃孵育5min后迅速放置在冰上进行冷却,再于凝胶中上样。电泳条件:浓缩胶80V跑胶20min;分离胶100V跑胶90min。
(3)湿电转膜:取出凝胶放置在事先提前配制好的电转移缓冲液中室温平衡15min,另外将聚偏二氟乙烯(polyvinylidene-fluoride,PVDF)膜和滤纸分别放入到去离子水和电转移缓冲液中,液体要保持浸没状态;将底部电极(阳极)水平放置,上面分别放滤纸、PVDF膜、蛋白凝胶以及滤纸,挤压排出各层气泡后放置上方电极(阴极)于上层滤纸上,以200mA恒流进行电转移1h。
(4)封闭:将转移后的PVDF膜浸入含5%脱脂奶粉的封闭液进行封闭处理1h(室温),倒掉封闭液。
(5)抗体与靶蛋白特异性结合:在盒中加入合适体积的封闭液和一抗Histone H4(1:500)、H4K16ac mAb(1:400)、H4K12ac mAb(1:400)、H4K8ac mAb(1:400)、H4K5ac mAb(1:400)、FUT3(1:600)以及内参一抗β-actin(1:4000)稀释液,4℃摇床振荡孵育过夜后,用PBST缓冲液将PVDF膜漂洗4次(每次5min)。将PVDF膜和辣根过氧化酶(HRP)标记的二抗(1:5000)室温条件下振荡孵育1-2h,最后用PBST缓冲液洗涤4次(每次5min)。
(6)显影和成像:按每cm
1.7 FUT3基因慢病毒干扰载体、过表达载体以及敲除载体构建
1.7.1 FUT3基因慢病毒干扰载体构建
1.7.1.1 FUT3基因干扰载体构建
(1)Oligo退火:将表1中的5对shRNA寡聚单链DNA用ddH
(2)连接:稀释双链DNA至10nmol·L
(3)转化:将DH5α高效感受态细胞从-80℃冰箱取出,置于冰上融化;取上述连接产物5μL加入到50μL感受态细胞中,轻弹混匀置于冰上冰浴30min,42℃热激90s,最后冰上静置3min;转化产物加入450μL不含氨苄的LB液体培养基中37℃振荡培养1h后,直接涂布于LB固体培养基(含Amp 100ng·mL
(4)阳性单克隆筛选与验证:每个干扰载体各挑取多个圆润透明的单菌落将其接种至1mL含氨苄抗性的LB液体培养基,37℃摇床振荡培养12h,将菌液送到公司进行测序,验证重组载体单克隆中的插入序列是否与目的Oligo序列碱基数目以及位置完全一致。若验证正确后继续菌液培养,用于提取无内毒素的重组质粒。
1.7.1.2重组质粒抽提
(1)柱平衡:向吸附柱CP4中加入500μL的平衡液BL,12000rpm室温离心1min,弃去收集管中的液体,将吸附柱重新放回收集管中。
(2)取10mLLB液体培养基中过夜培养的重组载体菌液于离心管中,12000rpm室温离心1min,弃尽上清。
(3)向离心管中加500μL溶液P1(确认溶液P1中已加入RNase A),使用涡旋振荡器彻底悬浮细菌沉淀。
(4)加入500μL溶液P2,轻轻地上下颠倒混合8次使菌体能够充分裂解,此时溶液会变得清亮粘稠。混匀时间不应该超过5min,以免让质粒受到损坏。
(5)加500μL溶液P4,立即轻轻地上下颠倒混合8次,室温放置10min,12000rpm室温离心10min。
(6)将步骤5中的上清液转移至过滤柱CS(置于收集管中),12000rpm室温离心1min,收集滤液。
(7)加入0.3倍体积的异丙醇于滤液中,上下颠倒混匀转至吸附柱CP4管。12000rpm室温离心1min后,弃去收集管中废液。
(8)向吸附柱CP4中加入500μL的去蛋白液PD,12000rpm室温离心1min后,弃去收集管中废液。
(9)向管中加600μL漂洗液PW,室温静置数分钟,12000rpm室温离心1min,弃滤液。
(11)将吸附管放置回收集管中,加600μL漂洗液PW,12000rpm室温离心1min,弃滤液。
(12)将吸附管放置回收集管中,12000rpm室温离心2min,去除残余液。
(13)将吸附管转移至新的1.5mL离心管中,室温放置数分钟自然风干,在吸附管的膜中央加入90μL去离子水,室温放置2~3min。12000rpm室温离心1min,收集质粒溶液。
1.7.1.3慢病毒载体包装、病毒液及滴度测定
293T细胞在10cm培养皿中培养至汇合度达到60%,按照RNAi-Mate转染步骤将构建成功的重组慢病毒载体与包装质粒(pRev、pGag/Pol、pVSV-G,上海吉玛制药技术有限公司)共转染293T细胞。72h后收集培养上清液,4℃4000rpm离心4min,离心收集上清,弃去细胞以及碎片,并用0.45μm的滤器过滤。将滤液在20000rpm下超速离心2h收集浓缩病毒液。
293T细胞按照3×10
1.7.2 FUT3基因慢病毒过表达载体构建
(1)FUT3基因CDS片段的合成
由金开瑞公司合成猪源FUT3基因完整的CDS序列(如序列1所示),并在C端添加3×Flag标签和酶切位点。
atggatccccttgactcggccaagataaaatgttcctggcgtcactatctcccttggctgtttttgcagctgcttttggctctgtgctttttctcctacctgcatgtgtcccaggacgcgcccacatgggcccctgaagcaaaggccccctgccagaccacagcggcgccctccagccgcccacccctgcttctgctgctgtggacgtggcccttccatgtccgggtggctgtgtctcgctgctcggagctgcggcccggcacggccgactgccagctgactgacaaccgcggggagtaccctcgggcagatgcagtcctcgtgcatcaccgggaggtcagctcccacccccagaggcagctcccgccttcccctcggccccccggccagcgctgggtctggttcagcatggaatcgcccagccactgcccgcagctcaaggccctggacgggtacttcaacctgaccatgtcctaccgcagggactctgatatcttcatgccatacgggtggctcgagccatggcccggccagcctgtggacgtgcagctcaacatctcggccaagaacaagctggtggcctgggtggtgtccaactggagggaggactcggtcagggtgcgctactaccggcagctgcaggcccacctccaagtggacgtgtatgggcgctcccacaagccactgccctcgtcggccatgaccacgcagctgtcccagtacaagttttacctggcttttgagaattccttgcatactgactacatcaccgagaagctatggagaaacgccctgctggcctgggccgtgcccgtggtgctgggccccagccggaggaactacgaacagttcctgccccccgaagccttcatccacatcaacgacttccagagccccaaggaactggcccagtatctgctggcactggacaaggacgacgcccgctatctgggctacttccgctggcgggagaccctgcggcctcaattcttcagctgggccttgatgttctgtaaggcctgctggcggctgcagcaggagcccagctaccagacagtgcccagcatcgcgtcttggttcacataa
(2)PCDH-CMV-MCS-EF1-GFP+Puro慢病毒过表达载体和目的片段双酶切
利用限制性内切酶XbaI和NotI对慢病毒过表达载体(上海吉玛制药技术有限公司)和目的片段进行双酶切处理,双酶切体系(50μL)如下:载体和基因片段各2μg,10×buffer5μL,XbaI和NotI酶各1μL,加水补足至50μL;酶切程序:37℃孵育3h。最后酶切产物通过DNA纯化试剂盒进行纯化回收。
(3)连接和转化
取酶切后的基因片段与载体进行T4 DNA Ligase连接反应,20μL反应体系如下:载体50ng,目的产物150ng,T4 DNA Ligase 1μL,10×T4 DNA Ligase Buffer 2μL,ddH
(4)筛选阳性克隆及鉴定
培养12-14h后,从中挑取多个圆润的透明单个菌落摇菌;将菌液送到生物公司进行测序,验证插入序列是否与目的序列一致。若验证正确后继续菌液培养,提取制备纯化的重组质粒DNA。
(5)慢病毒载体包装、病毒液及滴度测定
293T细胞在10cm培养皿中培养至覆盖率达到60%,按照RNAi-Mate转染步骤将构建成功的慢病毒重组载体与包装辅助质粒(pMDLg-pRRE、PMD2.G、pRSV-Rev,上海吉玛制药技术有限公司)共转染293T细胞。72h后收集培养上清液,4℃4000rpm离心4min,去除细胞和碎片,将上清液并用0.45μm的滤器过滤。将滤液在20000rpm下超速离心2h收集浓缩病毒液。将不同梯度慢病毒稀释液加入96孔板的细胞中于培养箱中培养,根据荧光细胞的数目结合稀释梯度来计算病毒液的病毒滴度。
1.7.3 FUT3基因敲除载体构建
(1)oligo退火
用ddH
(2)pGK1.2(EGFP+Puror)vector线性化处理
利用限制性内切酶BbsI对pGK1.2(EGFP+Puror)vector(上海吉玛制药技术有限公司)进行酶切,50μL酶切体系如下:2μg质粒,5μL 10×buffer,2μLBbsI酶,加水补足至50μL;酶切程序:37℃孵育3h。最后酶切产物通过DNA纯化试剂盒进行纯化回收。
(3)连接和转化
取1μL的杂交后的dsDNA与线性化载体进行T4 DNA Ligase连接反应,10μL反应体系如下:pGK1.2 linear vector 50ng,Hybridized dsDNA 1μL,T4 DNA Ligase 0.5μL,10×T4DNA Ligase Buffer 1μL,ddH
(4)筛选阳性克隆
培养12~14h后,从中挑取5个单个的、圆润的透明菌落摇菌;按照无内毒素质粒中量小提试剂盒步骤提取质粒DNA进行PCR或测序验证。测序引物为VSP primer:5’-CATATGCTTACCGTAACTTGAAAG-3’,PCR验证时通过上游VSP primer(Tm=56.7℃)与下游负链Oligo引物进行扩增,阳性克隆PCR产物正确长度应为100bp,再对筛选到的阳性单克隆进行测序验证。
1.8 FUT3基因慢病毒干扰、过表达以及敲除细胞系的建立
1.8.1 FUT3基因干扰载体干扰效率验证
将IPEC-J2细胞铺板至12孔板,待细胞生长至指数生长期时进行慢病毒侵染;取慢病毒滴度为1×10
1.8.2 FUT3基因过表达载体过表达效率验证
将IPEC-J2细胞铺板至12孔板培养,待细胞生长至指数生长期时进行慢病毒侵染;取慢病毒滴度为1×10
1.8.3 FUT3基因敲除载体敲除效率验证
1.8.3.1细胞转染及初步效率验证
按照lipofectamine
(1)DNA提取
严格按照血液/细胞/组织基因组DNA提取试剂盒步骤提取敲除细胞的DNA。
1)将贴壁细胞消化处理为细胞悬液,之后12000rpm离心1min,弃去上清;往管中加入200μL的GA缓冲液,振荡混匀细胞沉淀。
2)往管中加入20μL的Proteinase K溶液,晃动混匀;在样品管中加入200μL的GB缓冲液,充分颠倒混匀,70℃处理10min,快速离心去除管壁液体。
3)然后往管中加入200μL的无水乙醇,涡旋振荡15s;用移液枪将上述混合液(包含部分沉淀)均加入CB3吸附柱中,吸附柱置于2mL的收集管中,12000rpm的转速离心30s,弃去废液。
4)向CB3吸附柱中加入500μL的GD缓冲液,12000rpm离心30s后,弃去收集管中废液。
5)向CB3吸附柱中加600μL漂洗液PW,室温静置数分钟,12000rpm离心30s,弃滤液。
6)将CB3吸附柱放置回收集管中,加600μL漂洗液PW,12000rpm离心30s,弃滤液;再次重复此步骤。
7)将吸附管放置回收集管中,12000rpm离心2min,除去残余液。
8)将吸附管移入新的1.5mL离心管中,室温静置数分钟,在吸附管膜中央加50μL去离子水,室温静置2~3min。12000rpm离心1min,收集质粒溶液。
(2)普通PCR扩增
PCR扩增总体系为25μL:2×Taq Master Mix(Dye Plus)12.5μL,上下游引物各1.25μL,细胞DNA溶液1.25μL,ddH
(3)T7E1 Enzyme酶切
PCR产物退火:反应体系(10.5μL)为PCR产物5μL,T7E1 Reaction buffer 1.1μL,ddH
1.8.3.2敲除细胞阳性单克隆挑选与测序验证
将上述测序验证成功的敲除细胞继续培养,将敲除细胞在96孔板中经过有限梯度稀释后进行培养(Corning公司的Single Cell Cloning by Serial Dilution)。2~3天后观察贴壁细胞把单克隆细胞孔做上标记,待96孔板长至1/4面积时,可转移至48孔板扩大培养,待48孔板长满后,可取48孔板一半的细胞提基因组DNA,另一半继续培养。然后通过PCR和测序筛选阳性克隆。
当FUT3基因过表达、沉默及敲除载体转染细胞均验证有效率后,仔细按照说明书提取这3组和空白无处理组细胞的总蛋白,检测FUT3基因过表达、沉默及敲除对FUT3蛋白翻译水平的影响。
1.9 FUT3基因过表达、沉默及敲除对F18大肠杆菌黏附能力的影响
建立FUT3基因过表达、沉默及敲除的IPEC-J2细胞系,F18ab和F18ac大肠杆菌菌株分别接种至LB培养液,通过细菌计数法、菌毛蛋白基因定量法、间接免疫荧光法、革兰氏镜检法以及扫描电镜法进行检测和观察大肠杆菌与IPEC-J2细胞黏附能力,具体方法参考公开文献(Dai et al,Regulation and molecular mechanism of TLR5 on resistance toEscherichia coli F18 in weaned piglets[J].Animals(Basel),2019,9(10):735.)。
1.10数据处理与分析
采用2
2.结果与分析
2.1猪FUT3基因mRNA组织表达谱分析
通过qPCR技术检测FUT3基因在35日龄苏太断奶仔猪中11个不同组织(心、肝、脾、肺、肾、胃、肌肉、胸腺、淋巴结、十二指肠和空肠)中的表达水平。结果显示,FUT3基因在所检测的所有组织中均有表达,在心、肝、脾、肾、肌肉和淋巴结中表达水平都较低,其次在肺、肾、胃、胸腺中表达水平较高,但十二指肠和空肠中表达水平最高,其表达极显著高于其他组织(P<0.01,图1)。
2.2 FUT3基因在F18敏感型和抗性型肠道组织间的表达差异分析
检测FUT3基因在敏感型和抗性型个体肠道组织(十二指肠、空肠)中的差异表达水平。结果显示,FUT3基因在敏感组十二指肠、空肠组织中的表达均极显著高于抗性组(P<0.01,图2)。
2.3 FUT3在F18敏感型和抗性型肠道组织中免疫组化IHC分析
为了进一步探究FUT3在肠道组织的定位表达情况,本研究在大肠杆菌敏感型和抗性型个体十二指肠组织进行免疫组化IHC分析。结果显示(图3),FUT3主要分布于小肠组织上皮黏膜表面,并且敏感型个体明显高于抗性型。
2.4 LPS诱导IPEC-J2细胞后的FUT3表达水平变化
同时以诱导0h时FUT3基因的表达量为1,对FUT3基因的相对定量结果进行统计,在1μg·mL
2.5大肠杆菌菌体侵染IPEC-J2细胞后的FUT3表达水平变化
本试验通过大肠杆菌菌体侵染IPEC-J2细胞系,以空白对照组细胞FUT3基因的表达量为1,检测发现F18ab和F18ac大肠杆菌菌体分别侵染IPEC-J2细胞后,细胞FUT3基因的表达量均呈现极显著上升状态(P<0.001)(图6)。同时利用Western blot检测大肠杆菌刺激后FUT3的表达水平,结果显示,相对于空白细胞,F18ab和F18ac大肠杆菌菌体侵染肠上皮细胞后其蛋白水平也显著上升(图7),这与相对定量的结果基本一致。
2.6 FUT3基因干扰、过表达及敲除载体构建
通过对所挑取的重组质粒克隆进行测序验证,发现序列大小、碱基顺序完全一致,最终获得连接正确的干扰和过表达慢病毒重组质粒载体以及敲除载体。构建成功的干扰载体分别命名为FUT3-RNAi-1、FUT3-RNAi-2、FUT3-RNAi-3、FUT3-RNAi-4载体,序列结果如图8;过表达载体命名为FUT3-OE载体,经测序验证载体插入序列与FUT3基因CDS区完全一致;构建成功的敲除载体分别为FUT3-KO-1、FUT3-KO-2、FUT3-KO-3、FUT3-KO-4,序列如图9。
2.7干扰和过表达载体慢病毒液滴度测定
慢病毒重组载体和包装载体共转染293细胞24h后,视野下观察到大量绿色荧光,说明慢病毒重组载体病毒包装成功,可用于后续的病毒液收集及浓缩处理。
慢病毒液梯度稀释后侵染96孔细胞用于病毒液的滴度检测,如图10可见,观察到每孔均有带绿色荧光的细胞,说明所包装的慢病毒颗粒能够有效地侵染进细胞。并按照最小梯度处理孔中观察到的少量表达绿色荧光的细胞进行病毒滴度(TU·mL
2.8干扰、过表达和敲除效率验证
由图11可知,慢病毒液侵染IPEC-J2细胞和敲除载体转染细胞后,细胞均能表达绿色荧光蛋白,这表明载体已成功转染细胞并稳定表达。对于干扰和过表达组处理细胞提取总RNA进行实时荧光定量检测FUT3基因转录水平表达情况,由图12A可知,4号干扰载体处理的IPEC-J2细胞中猪FUT3基因相对表达水平为0.131,干扰效率为86.9%,即RNAi-FUT3-4干扰效率最高,用于后续试验;由图12B可知,FUT3过表达慢病毒液侵染细胞后,相对于阴性对照组,FUT3基因表达极显著上升(P<0.01),约为140倍,表明此过表达载体效果显著,可用于继续试验。对于敲除细胞提取DNA进行PCR、T7E1酶切和测序进行分析(如图13),敲除载体转染进细胞并药筛完成后提取DNA进行RCR扩增,发现4组敲除载体转染组细胞的PCR产物片段大小没有明显变化,但经过T7E1酶酶切都有3条带,说明靶点位置发生了突变;同时测序结果显示4组敲除载体转染组细胞产物在靶点附近开始均有很明显的杂峰,再次说明有的细胞发生了敲除;选取KO-1敲除载体转染组细胞进行单细胞克隆,将初步得到的单细胞克隆提DNA后PCR验证发现有的细胞其扩增产物与目的片段稍微有点偏差,进行测序分析发现,这些细胞均发生了不同程度的缺失和突变。
2.9 FUT3基因过表达、沉默及敲除对FUT3蛋白翻译水平的影响
对FUT3基因过表达、沉默及敲除细胞提取总蛋白进行Western blot检测(如图14),结果显示,相比于空白细胞,过表达细胞FUT3蛋白条带明显且阳性体积大,而干扰和敲除细胞FUT3蛋白条带则稍微较弱;灰度值半定量分析结果显示,过表达细胞FUT3蛋白水平极显著上升(P<0.01),而干扰和敲除细胞FUT3蛋白水平极显著下调(P<0.01)。
2.10 FUT3基因过表达、沉默及敲除对大肠杆菌黏附能力的影响
细菌黏附IPEC-J2细胞后进行菌毛蛋白基因荧光定量和细菌计数检测,结果显示(图15),以空白未处理组为对照,FUT3基因过表达后IPEC-J2细胞对F18大肠杆菌的黏附能力显著上升(P<0.01),FUT3基因沉默或敲除后IPEC-J2细胞对F18大肠杆菌的黏附能力均显著下降(P<0.01)。同时利用间接免疫荧光和革兰氏镜检方法检测大肠杆菌的黏附状态,间接免疫荧光结果表明(图16),FUT3过表达组的细菌表达量比对照组多,而FUT3干扰和敲除组的细菌表达明显比对照组少;革兰氏镜检结果表明(图17),FUT3过表达组的黏附细菌量比对照组多,并且有些细胞出现了明显的皱缩和脱落,而FUT3干扰和敲除组的黏附细菌量明显比对照组少。
3.讨论
本研究检测了FUT3基因在35日龄苏太仔猪中的组织表达谱,FUT3基因在所检测的所有仔猪的11个组织中均有表达,表明其在动物机体中发挥着广泛的作用。但在呼吸、消化道系统组织尤其是肠道的表达水平最高,在其他组织中表达水平较低。在研究FUT3基因组织表达谱的基础上,针对FUT3高表达的肠道,本试验进一步检测了FUT3基因在35日龄苏太仔猪敏感组与抗性组肠道组织的表达,发现敏感组FUT3的表达均显著或极显著高于抗性组。同时大肠杆菌侵染和LPS诱导试验结果显示F18ab和F18ac大肠杆菌菌体分别侵染IPEC-J2细胞后,细胞FUT3基因的表达量均呈现极显著上升状态;同时LPS诱导4-8h后FUT3基因呈阶梯状增长;且LPS诱导8h后FUT3基因的表达量极显著地高于空白对照和4h。LPS是大肠杆菌黏附肠上皮细胞所释放的内毒素,在介导炎症中起着至关重要的作用。其诱导细胞后FUT3基因的显著上调与大肠杆菌感染所引起的基因变化基本一致,综合说明FUT3基因的表达与机体抵抗大肠杆菌的感染有关,可能参与F18的抗性调控。本研究的结果表明了FUT3基因及其所参与产生的HBGA在大肠杆菌入侵时发挥的重要作用。当大肠杆菌入侵肠道时,HBGA识别并介导病原菌的入侵,会使肠道菌群结构变化发生紊乱,破坏肠道黏膜屏障,刺激肠上皮相关基因和FUT3的改变,最终引起肠道炎症反应。本研究初步推断FUT3基因的过表达与大肠杆菌侵染有着重要的联系,但是其作用机制尚不明确。
此外,为了对FUT3基因与大肠杆菌抗性的关系进行更深入的功能验证,本研究构建了FUT3基因过表达、沉默及敲除稳定细胞系,并通过细菌计数、菌毛蛋白基因荧光定量、细胞间接免疫荧光以及革兰氏染色法等4种方法综合分析FUT3基因过表达、沉默及敲除对小肠上皮细胞黏附E.coli F18能力的影响。黏附实验发现,FUT3基因过表达后IPEC-J2细胞对F18大肠杆菌的黏附能力均显著上升,反之则黏附水平下降。
4.本发明小结
(1)靶基因FUT3主要在肠道组织(十二指肠和空肠)黏膜表面中高度表达,并且F18大肠杆菌敏感型个体显著高于抗性型。
(2)LPS诱导和F18大肠杆菌菌体侵染猪小肠上皮细胞后,靶基因FUT3 mRNA和蛋白质表达水平均显著上调。
(3)建立了靶基因FUT3干扰、过表达以及敲除的猪小肠上皮细胞系(IPEC-J2),系统验证了FUT3基因表达水平与大肠杆菌抗性密切相关。
序列表
<110> 扬州大学
<120> FUT3基因作为靶点在提高猪对大肠杆菌抗性中的应用
<160> 1
<170> SIPOSequenceListing 1.0
<210> 1
<211> 1086
<212> DNA
<213> 人工序列(Artificial Sequence)
<400> 1
atggatcccc ttgactcggc caagataaaa tgttcctggc gtcactatct cccttggctg 60
tttttgcagc tgcttttggc tctgtgcttt ttctcctacc tgcatgtgtc ccaggacgcg 120
cccacatggg cccctgaagc aaaggccccc tgccagacca cagcggcgcc ctccagccgc 180
ccacccctgc ttctgctgct gtggacgtgg cccttccatg tccgggtggc tgtgtctcgc 240
tgctcggagc tgcggcccgg cacggccgac tgccagctga ctgacaaccg cggggagtac 300
cctcgggcag atgcagtcct cgtgcatcac cgggaggtca gctcccaccc ccagaggcag 360
ctcccgcctt cccctcggcc ccccggccag cgctgggtct ggttcagcat ggaatcgccc 420
agccactgcc cgcagctcaa ggccctggac gggtacttca acctgaccat gtcctaccgc 480
agggactctg atatcttcat gccatacggg tggctcgagc catggcccgg ccagcctgtg 540
gacgtgcagc tcaacatctc ggccaagaac aagctggtgg cctgggtggt gtccaactgg 600
agggaggact cggtcagggt gcgctactac cggcagctgc aggcccacct ccaagtggac 660
gtgtatgggc gctcccacaa gccactgccc tcgtcggcca tgaccacgca gctgtcccag 720
tacaagtttt acctggcttt tgagaattcc ttgcatactg actacatcac cgagaagcta 780
tggagaaacg ccctgctggc ctgggccgtg cccgtggtgc tgggccccag ccggaggaac 840
tacgaacagt tcctgccccc cgaagccttc atccacatca acgacttcca gagccccaag 900
gaactggccc agtatctgct ggcactggac aaggacgacg cccgctatct gggctacttc 960
cgctggcggg agaccctgcg gcctcaattc ttcagctggg ccttgatgtt ctgtaaggcc 1020
tgctggcggc tgcagcagga gcccagctac cagacagtgc ccagcatcgc gtcttggttc 1080
acataa 1086
- FUT3基因作为靶点在提高猪对大肠杆菌抗性中的应用
- CDPK18L基因作为负调控因子在提高番茄细菌性叶斑病抗性和高温抗性中的应用