突变酶
文献发布时间:2023-06-19 12:21:13
本申请是申请日为2008年10月8日、申请号为201510867081.X、发明名称为“突变酶”的分案申请的再次分案申请,其中,申请号为201510867081.X的分案申请是申请日为2008年10月8日的题为“突变酶”的中国专利申请No.200880119582.8的分案申请。
技术领域
本发明涉及具有增强性能的突变酶。
背景技术
生物酶催化剂,如P450
巨大芽孢杆菌(Bacillus megaterium)(1)的P450
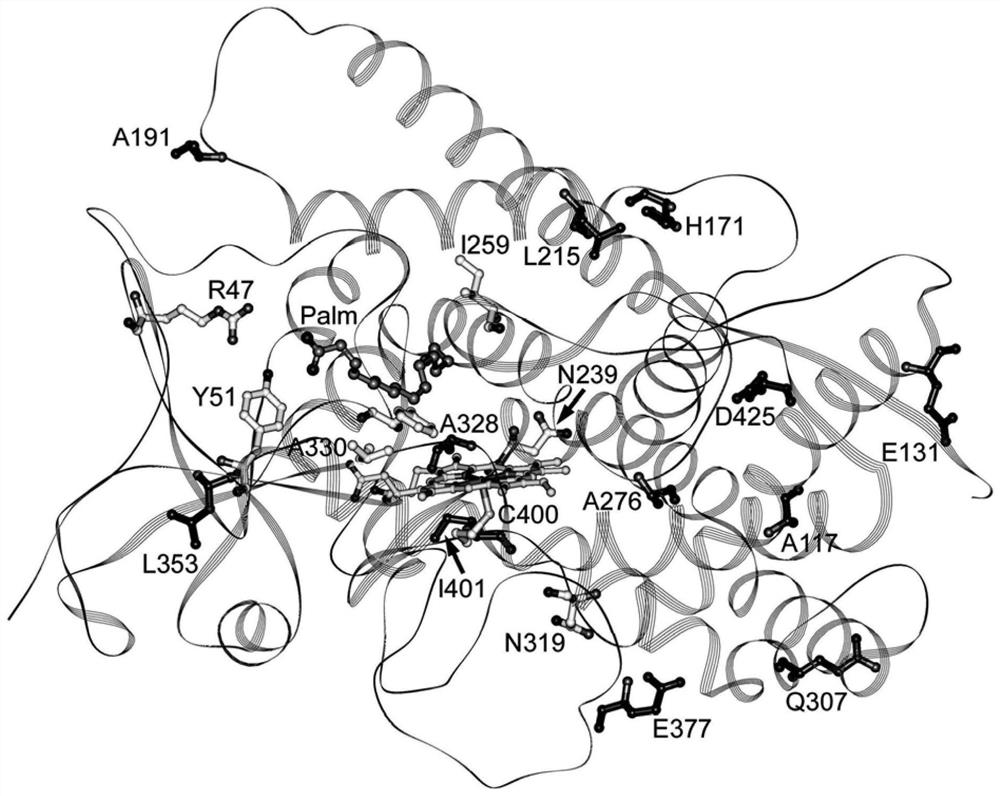
CYP102A1(1)是用于生物转化应用的受关注的酶,因为其是催化上自给自足的。与其他P450酶不一样,其中P450单加氧酶和电子转移辅助因子蛋白是独立的部分(entities),CYP102A1具有融合至二弗拉芬(diflavin)电子转移还原酶域的血红素单加氧酶域,其在单个多肽中同时含有FAD和FMN辅基。CYP102A1的天然底物被认为是直链或支链的中链脂肪酸(1,2)。CYP102A1血红素域的晶体结构在1993年(3)就可获得,其中揭示了活性位点结构并且存在底物进入通道(或底物接近通道,substrate access channel)。四年之后公布的具有结合底物的晶体结构,指出对于F87一旦底物结合后侧链构象的变化(4)。
对CYP102A1的蛋白质工程已经进行了综述(5~7)。早期的研究集中在活性位点残基F87,其中F87V、F87A、F87Y和F87G突变已经显示出对脂肪酸氧化的活性和选择性具有不同的影响(8~11)。F87上的突变已经发现有利于各种底物的氧化(7)。在底物进入通道入口处的残基如F42、R47和Y51也被标靶。虽然F42A突变降低了酶活性(10),但是中和或逆转在47位置处的电荷改变了底物的特异性(8,12),正如疏水性取代Y51A一样。WO0031273公开了突变的R47L/Y51F偶联体(或结合体,couplet)用来促进疏水性有机分子如聚芳香烃和类萜烃的进入、结合和氧化。该偶联体也与F87A、I263A、A264G和M354A突变组合以提供增强的底物氧化的活性和/或产物选择性(13,14)。R47L/Y51F组合,以及在其自身上的R47L和Y51F突变,现在通常用于CYP102A1工程(15~19)。
除了突变位点的合理选择之外,筛选技术已经用于识别对活性和选择性具有期望影响的其他突变和突变位点。随机或位点饱和诱变早在1997年就应用于CYP102A1(20)。NO20020380公开了使用经由吲哚氧化生成靛蓝作为发现具有新的活性的CYP102A1突变的筛选方法。饱和诱变应用于许多可能影响底物结合的残基,而突变体A74G/F87V/L188Q据报道相比于野生型能以增强的活性和改变的选择性氧化很宽范围的有机分子(21~25)。AT342351T公开了经由ω-对硝基苯氧基-羧酸氧化而采用光谱检测到对硝基苯酚的生成,在一组随机诱变实验中作为筛选程序。突变V26T、R47F、S72G、A74G、F87A&V、L188A,G,N,K,Q,R,S&W、M354T都已公开(26,27)。
对硝基苯酚筛选方法通过采用对硝基苯氧基辛烷作为替代底物(surrogatesubstrate)而得以扩展。WO2002083868、EP1470219和US2005202419(随后在WO2005017116、EP1660646和US2005037411中校正)公开了突变L52I、I58V、F87A、H100R、S106R、F107L、A135S、M145A&V、A184V、N239H、S274T、L324I、V340M、I366V、K434E、E442K、V446I。
WO2003008563和US2003100744公开了采用相同方法的更多轮随机诱变、基因重组(gene shuffling)和筛选的结果,并报道了突变M30I、E64A、V78A、F87A,D,G,H,I,K,N,R,V&W、H138Y、F162S、H171Q、T175I、V178I、A184V、N186D、D217V、I220T、K224I、S226I、D232G、T235A、H236Q、E252G、R255S、I258T、I259V、T268Q、A290V、A295T、L353V,D370Q,E380G、G396M、T411A、M416L。
WO2005017105、US2005059128和EP1639091公开了相同方法的使用并报道了突变R47C、L75I&W、V78A,F&T、A82L,F,G,I,S&T、F87I,L&V、T88C、K94I、P142S、T175I、A184V、F205C、S226R、H236Q、E252G、R255S、T260,L,N&S、A290V、A328V&M、L353V。
随后,WO2006105082公开了突变R47C、V78F、A82S、K94I、P141S、T175I、A184V、F205C、S226R、H236Q、E252G、R255S、A290V、A291V、A328F、L353V。
这些通过随机诱变产生的系列突变体显示出对于从乙烷到中链烷烃的烷烃氧化的增强的活性(28~30)。也存在选择性变化,尤其是在定向进化变异体与通过定点诱变引入到活性位点的突变组合时,例如,在辛烷氧化中,其中突变将氧化位点向末端碳转移(31),在末端烯烃的选择性环氧化中(32),以及对映选择性在环戊烷羧酸衍生物的氧化中(33)。值得注意的是,通过使定向进化与合理的重新设计进行组合经常可以获得更好的结果。
CYP102A3是与CYP102A1属于相同子族(sub-family)的P450酶。CYP102A3的随机诱变,接着烷烃氧化并在对末端醇特异性的醇脱氢酶存在的情况下监控NADH的形成,产生了由辛烷氧化生成50%的1-辛醇的突变。这是迄今为止通过设计的CYP102族P450酶所观察到的直链烷烃的末端C-H键氧化的最高比例(34)。
仍继续需要分离工业上有用的酶如CYP102A1酶的另外的突变,以进一步理解结构变化对它们的催化机理的影响,改进它们的催化转化(catalytic turnover),并扩大其底物和/或产物的范围。通常,进行P450酶如CYP102A1的设计以增强酶活性,其中控制产物选择性和底物特异性是第二重要的目标。可将选择性控制结合至增强的单加氧酶活性的突变和突变位点是显著缺乏的,以致化合物的酶转化可以是较快的但是没有足够的选择性,或虽有一定的选择性但反应很慢,或期望的产物并未形成。对于可以提供具有增强的活性和/或期望的选择性的突变体的筛选方法也存在需要。
发明内容
现在已经发现,根据本发明,CYP102A1特异性位置的取代突变在增强单加氧酶活性方面具有期望的作用,并且也提供了改变的选择性。这些突变位点通过使用提供增强的活性和增强/改变的选择性选择的新型筛选方法来识别。
本发明提供了一种突变CYP102A1酶,其具有增强的单加氧酶活性和/或改变的选择性,并含有在CYP102A1的位置117、131、191、215、276、307、330、377、401、403、425中的一个或多个处的取代。另外还提供了一种用于氧化作为有机化合物的底物的方法,该方法包括用本发明的突变CYP102A1酶来氧化所述有机化合物。
所要求的取代(替代,substitution)构成相同的本发明构思的一部分,因为它们共同具备增强单加氧酶活性的效应和/或改变CYP102A1的选择性。在位置330、401和403的取代也经由如下所概述的共同结构和功能机制发挥它们的作用。
SEQ ID NO:1是CYP102A1的序列。
附图说明
图1:相关残基在CYP102A1(P450
图2:在野生型CYP102A1和A330P突变体的底物进入通道中的关键残基和活性位点的比较,突出表明了由A330P突变产生的P329和A328处的结构摄动(structuralperturbations)。
具体实施方式
本发明可以适用于CYP102A1的天然和人工同源物(或同系物,homologues),例如,其包括与CYP102A1具有至少40%的氨基酸序列同一性的序列。这样的同源物典型地包括对应于(即与之具有同源性或相同于)CYP102A1的血红素单加氧酶域的氨基酸序列(由氨基酸位置1~480表示)。
本发明的酶包括与SEQ ID NO:1(CYP102A1的序列)具有至少40%同一性的序列(或由其构成)。在优选的实施方式中,该序列在至少20,优选至少30,例如至少40、60、100、200、300、400或更多个邻近氨基酸内,或甚至在同源物的整个序列内与其可能具有至少55%、65%、80%或90%,并且更优选至少95%、97%或99%的同源性。在一个实施方式中,本发明的酶在与CYP102A1的氨基酸残基1~480相比时具有任何指定的百分比同源性。邻近氨基酸可以包括活性位点。该同源性可以可替换地并不在邻近氨基酸内而是仅仅在活性位点的氨基酸内进行测定。因此,同源性基于氨基酸的同一性典型地至少40%同源于CYP102A1。本发明的酶可以与CYP102A1序列具有一定百分比同一性,这与以上提及的任何序列长度内任何具体百分比同源性值(即,其可以具有至少40%、55%、80%或90%以及更优选至少95%、97%或99%的同一性)相同。
同源序列可以表示CYP102A1序列的突变部分和/或可以以本发明酶的全长融合多肽的形式存在。
本文中所提及的任何同源蛋白(即,描述为与另一蛋白同源)典型地至少40%同源于相关的蛋白。同源性可以采用已知的方法进行测定。例如,UWGCG软件包提供了可以用于计算同源性的BESTFIT程序(例如,以其缺省设置进行使用)(Devereux et al(1984)Nucleic Acids Research 12,387-395)。
PILEUP和BLAST算法可以用于计算同源性或比对序列(典型地按照其缺省设置),例如在Altschul S.F.(1993)J MoI Evol36:290-300;Altschul,S,F et al(1990)J MoIBiol 215:403-10中所描述的。
用于进行BLAST分析的软件可以通过国家生物技术信息中心(National Centerfor Biotechnology Information)(http://www.ncbi.nhn.nih.gov/)公开获得。该算法首先涉及通过在与数据库序列中相同长度的字比对时匹配或满足一些正值阈值评分T的查询序列中识别长度W的短字来识别高评分序列对(HSP)。T称为邻近字评分阈值(Altschul etal,supra)。这些初始的邻近字命中(word hits)作为用于初始搜索以找到含有它们的HSP的种子(seeds)。字命中在沿着直至能够增加累积比对评分的每一序列的两个方向上延伸。当累积比对评分从其最大达到值下降量X;由于一个或多个负评分残基比对的累积使累积评分变成零或更低;或任一序列的末端到达时,对在每一方向上字命中的延伸就停止。BLAST算法参数W、T和X决定了比对的灵敏度和速度。BLAST程序使用11的字长(W),50的BLOSUM62评分矩阵(参见Henikoff and Henikoff(1992)Proc.Natl.Acad.Sci.USA 89:10915-10919)比对(B),10的期望值(E),M=5,N=4和双链对照作为缺省值。
BLAST算法进行两序列之间类似性的统计分析;参见例如,Karlin and A1tschul(1993)Proc.Natl.Acad.Sci.USA 90:5873-5787。通过BLAST算法提供的一种类似性度量是最小和概率(P(N)),其提供了一个概率指示,通过该概率指示两个核苷酸或氨基酸序列之间的匹配就会偶然出现。例如,如果在第一序列与第二序列的比较中最小和概率低于约1,优选低于约0.1,更优选低于约0.01,并且最优选低于约0.001,则该序列被认为类似于另一序列。
典型地,当比较所有蛋白或任何以上提及的邻近氨基酸(contiguous aminoacids)的长度时,同源蛋白不同于相关蛋白至少、或少于2、5、10、20、40、50或60个突变(其每一个都可以是取代、插入或缺失)。
本发明的CYP102A1酶的酶活性典型地在体外采用本文中所提及的任何底物或条件进行测定,并作为NADPH氧化速率,产物生成速率和结合效率(coupling efficiency)给出。速率是转化频率并以(nmol NADPH)(nmol CYPl 02A1)
本发明的突变CYP102A1酶显示出相对于相应的野生型CYP102A1酶的增强的单加氧酶活性和/或改变的选择性。增强的单加氧酶活性可以根据增加的结合效率或增加的产物生成速率采用一种或多种用于氧化的底物进行表征。增加的结合效率或增加的产物生成速率可以在或可以不在被突变CYP102A1酶利用的所有底物中共享。突变CYP102A1酶典型地表现出比该野生型酶的结合效率大至少10%、20%、50%、100%、500%、1000%或1500%的结合效率。突变CYP102A1酶也可以具有比该野生型酶的产物生成速率大至少50%、100%、150%、500%、1000%、2000%、5000%、10000%的产物生成速率。
应该理解到,本发明的突变CYP102A1酶也可以显示出相对于相应的野生型酶和文献中所公开的突变体的其他改变的特性,使得这些效应可以包括但也不限于增强的单加氧酶活性。例如,突变酶可以显示出改变的底物特异性,允许优先利用特异性底物,或可以显示出单加氧酶活性,其中野生型酶或已知的突变体并不能氧化底物有机化合物。
本发明的突变酶也可以显示出改变的产物选择性,其中由野生型以较小比例形成的产物成为突变体的优势产物,或以较小比例或根本不由野生型形成的新产物成为大多数或优势产物。下面描述突变酶和通过该突变酶实施的氧化过程的另外的改变的特性。
突变CYP102A1酶在CYPl02A1的位置117、131、191、215、276、307、330、377、401、403和425中的一个或多个处包括取代。典型地,它们可以在2个或更多个、3个或更多个、4个或更多个、5个或更多个、6个或更多个以上定义的位置处包括取代。在一个优选的实施方式中,其中在位置330处具有取代,存在小于5个其他取代,如小于3,或在一个实施方式中,其他位置没有被取代。
在描述CYP102A1的特异性突变的情况下,在CYP102A1的天然形式中存在的氨基酸残基的字母在位置之前,位置之后是突变体中的氨基酸。这些位置可以关联于SEQ ID NO:1中所示的编号。为了指示出相同蛋白中的多个突变,每一突变通过斜线分开而列出。而且,尤其优选的突变体可以采用以下概述的内部命名法进行描述。
虽然参照CYP102A1中的位置定义了突变,但是本发明也涵盖享有与SEQ ID NO:1至少40%氨基酸同一性的CYP102A1同源物的多肽链中同源的或相应位置的等价取代突变。等价位置(equinalent position)通过参照SEQ ID NO:1的氨基酸序列进行确定。基于序列之间的同源性,同源或相应位置可以通过对比CYP102A1(SEQ ID NO:1)的序列和同源物(或同系物,homologue)序列来容易地推导。PILEUP和BLAST算法可以用于比对这些序列。在所指的同源的或相应的氨基酸是活性位点残基的情况下,一般将处于如同本文中讨论的任何特定氨基酸的同源物活性位点中的类似位置。
对于本领域技术人员来说众所周知的是,P450酶超家族尽管具有高度保守的三级结构,但在蛋白和酶中具有低同源性的一级结构却是不常见的(35~37)。在基因组数据库中现在存在>6500CYP基因,而在蛋白数据库(Protein Data Bank)中存在>150的结构。迄今确定的所有P450结构表现出结合针对主β链域包装的富螺旋域的特性形态学,如Poulos及其合作者首次报道的P450酶(CYP101A1)的晶体结构中所描述的(38)。螺旋命名为A-L,而β链βl-β5,其中整个形态现在称为“P450折叠”(38-42)。在二级结构元件中,血红素远侧上的B和B'螺旋、BC环、F和G螺旋和FG环形成底物结合袋(substrate binding pocket)。序列比对很容易识别这些螺旋和环中的残基,但是根据氨基酸序列和结构排列,在这些一般框架内存在高度可变性,而且这也产生了P450催化的多种特异性、活性和选择性模式。
在不同家族中的P450酶具有低至20%的同源性(氨基酸同一性)(35~37)。在CYP102A1和结构表征的P450酶之间比对的样例示于表A中。直至近来,连续序列分析已经建议,在P450酶或域中典型的400~460中少至3个残基是绝对保守的:血红素铁的邻近半胱氨酸配体,和在血红素连接和结合中可能起作用的K螺旋中的EXXR基序(43)。然而,有关2006年公开的CYP157家族的结果表明,甚至EXXR基序也不是保守的,剩下邻近半胱氨酸作为整个P450超家族中仅有的保守残基。在P450超家族的系统分类中(44),刚好具有40%氨基酸同一性的酶因此而位于相同的家族中,而紧密相关的家族成员(>55%同一性)归成子族(参见,例如,表B)。
实际上详细的分子结构,底物特异性和产物选择性在家族内是保守的,而序列同一性却经常较低。最显著的实例是在生命王国中发现的固醇14α-去甲基酶的CYP51家族。这些在角鲨烯氧化物环化之后形成的中间体的C14甲基的氧化性去甲基化作用中起到关键作用。序列比对表明,整个生命王国的已知CYP51族基因之间的同源性平均为30%,在紧密相关的物种如哺乳动物中升高至95%,而在低级生物之间下降至23%(45)。越来越认识到,用于将酶分配给同一家族的40%临界值在一些情况下可能过高,而经常对于活性位点残基观察到的酶活性和更高的同源性可能需要在未来被更多地考虑。
因此,典型地基于“P450折叠”也可以容易地识别基于氨基酸同一性与CYP102A1至少40%同源的同源物,而在相应或同源位置引入等价突变的同源物序列的比对可以通过包含在整个酶家族中共享的P450折叠的α螺旋和β链排列的保守性质的知识进行辅助。
应该理解到,CYP102A1是电子转移还原酶域和血红素单加氧酶域的融合体。这些域可以通过蛋白酶解或通过全长基因的截断而切下。活性位点(底物结合袋)位于血红素域中。CYP102家族的一些成员并不是融合蛋白,但是与CYP102A1血红素域的序列同源性为40%。因此,序列同源性可以单一地在这些环境下在整个血红素域范围内进行测定。在这些酶中等价于本发明中所公开的CYP102A1的等价残基(equivalent residues)可以通过本领域技术人员已知的序列同源性和结构分析进行识别。
在活性位点中的氨基酸是排列或限定在催化期间结合有底物位点的那些氨基酸或排列或限定底物在到达催化位点之前必须经过的位点的那些氨基酸。因此,这样的氨基酸典型地在进入催化位点期间或催化期间与底物发生相互作用。这样的相互作用典型地通过静电相互作用(在带电的或极性基团之间)、疏水性相互作用、氢键或范德华力而发生。活性位点氨基酸可以通过序列比对并参照野生型CYP102A1的血红素域的已知晶体结构、或同源物的晶体结构进行识别。
在突变残基并不是活性位点残基的情况下,进行同源物和CYP102Al序列的计算机化或人工比对以推断出同源或相应的位置,这可以通过对CYP102A1中侧接突变位置的残基的了解进行辅助。因此,例如,在CYP102A1中将残基侧接至以下位置的10个N-端和C-端侧接残基是:
FSQQAMKGYH(A117)MMVDIAVQLV;
DIAVQLVQKW(E131)RLNADEHIEV;
LDEAMNKLQR(A191)NPDDPAYDEN;
FQEDIKVMND(L215)VDKIIADRKA;
HETTSGLLSF(A276)LYFLVKNPHV;
VLVDPAPSYK(Q307)VKQLKTVGMV;
EALRLWPTAP(A330)FSLYAKEDTV;
GDDVEEFRP(E377)RFENPSAIPQ;
KPFGNGQRAC(I401)GQQFALHEAT;
FGNGQRACIG(Q403)QFALHEATLV;
GMMLKHFDFE(D425)HTNYELDIKE。
2,3或多个N-和/或C-端侧接残基的保守可以允许推断被引入突变的同源或相应位置。
类似的分析可以对于在本描述中所指的CYP102A1中任何其他位置进行实施以识别在天然存在的CYP102A1同源物中的同源或相应的位点。
CYP102Al酶的功能性片段也涵盖在本发明内。这些片段因此可以仅仅包含对于氧化活性所需的那些氨基酸。因此,参照CYP102A1的多肽序列,还原酶域和/或单加氧酶域在N-端或C-端部分多至20个残基能够被缺失而不会显著影响活性位点的折叠或单加氧酶域的固有底物氧化能力。在CYP102A1的同源物中,类似的截断是可能的,而可能截断的程度可以通过本文中描述的用于监控氧化活性的方法进行测定。酶的截断形式在稳定性、表达水平和蛋白活性方面可能具有有利的性质。
在本文中描述的CYP102A1的位置(或如以上定义的等价位置)取代的氨基酸的性质主要通过表现出增强的单加氧酶活性的突变条件进行测定。因此,引入的氨基酸将典型地增强单加氧酶活性。在任何提及CYP102A1中的具体取代突变的情况下,应该理解,根据本发明涵盖相同位置处的另一氨基酸残基的任何取代,其所具有的影响比具体取代突变对CYP102A1酶的氧化活性的影响更多或与之类似。类似地,在具体取代突变也对CYP102A1酶的另一参数,如底物特异性、或在给定的底物氧化中获得的氧化产物的范围或比率具有影响的情况下,应该理解到,那些也引起更多或类似影响的其他氨基酸残基的取代也期待用于根据本发明的用途。
在一些实施方式中,取代引入保守变化,这是用类似化学结构、类似化学性质或类似侧链体积的另一氨基酸取代该氨基酸。引入的氨基酸可以具有其取代的氨基酸类似的极性、亲水性或疏水性。保守氨基酸变化在本领域内是众所周知的,并且可以根据表C中定义的变化进行选择。在氨基酸具有类似极性的情况下,这也可以参照氨基酸侧链的亲水尺度(hydropathy scale)(表D)进行确定。
保守氨基酸变化也可以参照对于氨基酸序列保守性的评分矩阵Point AcceptedMutation(PAM)或BLOcks Substitution Matrix(BLOSUM)家族进行确定。因此,保守氨基酸变化可以是等价组的成员,其是在选择用于参照和突变多肽链的比对中的评分矩阵的相似性描述中具有突变正分的一组氨基酸。
应该理解,在表C中提供的物理特性的定义并不被认为对本发明的限制,并且非极性氨基酸包括具有脂族侧链的氨基酸和具有芳族侧链的氨基酸。氨基酸脯氨酸归为非极性氨基酸,但是其也具有固定的性质并且可以在二级结构中发生变化。例如,脯氨酸经常在螺旋末端找到。而且,取决于给定氨基酸残基侧链的具体情况,例如,氨基酸酪氨酸,一般由于其芳香环而分类成非极性的,可以具有极性氨基酸残基(如经由其羟基的苏氨酸)的类似功能效应。因此,对于本发明的目的来说,酪氨酸可以被认为是非极性和极性氨基酸。而且,可以描述成极性或亲水性的氨基酸可以是不带电或带电的,并且也可以是碱性的或酸性的。氨基酸组氨酸众所周知具有接近7的pKa值,使得取决于蛋白环境在中性pH下,其可以在或可以不在其侧链上被质子化,并且因此可以携带或可以不携带电荷。因此,对于本发明的目的来说,组氨酸可以被认为是极性带电的或极性不带电的氨基酸残基。
对于CYP102A1中的位置117、131、215、307、330、401和403处(或其等价位置)的保守氨基酸变化的具体实例包括但不限于:
A117V,A117I,A117L,A117P,A117M,A117F,A117W,A117Y;
E131D;
L215I,L215V,L215P,L215F,L215W,L215Y;
Q307H,Q307N,Q307S,Q307T,Q307Y;
A330P,A330I,A330L,A330M,A330V,A330F,A330W,A330Y;
I401P,I401I,I401L,I401M,I401V,I401F,I401W,I401Y;
Q403N,Q403H,Q403A,Q403T,Q403Y。
在其他优选的实施方式中,氨基酸取代在野生型酶的给定位置引入极性氨基酸,典型地在现存残基是非极性残基的情况下,因此改变了极性。对于在CYP102A1中的位置191和276(或其等价位置)处的极性氨基酸取代的具体实例包括但不限于:
A191T,A191S,A191C,A191Y,A191H,A191K;A191R,A191N,A191Q;
A276T,A276S,A276C,A276Y,A276H,A276K,A276R,A276N,A276Q。
比较而言,在其他优选的实施方式中,氨基酸取代在野生型酶的给定位置引入了非极性氨基酸,典型地在现存残基是极性残基的情况下。例如,非极性氨基酸可以在位置377或403或其等价位置引入。具体实例包括但不限于:
E377A,E377V,E377L,E377I,E377P,E377F,E377Y,E377W;
Q403P,Q403W,Q403F,Q403Y;
在另外的实施方式中,氨基酸取代在野生型酶的给定位置导致失去带电的侧链基团。因此,这种取代在相关位置引入了不带电的氨基酸。这可以在或可以不在该位置导致极性损失,使得引入极性不带电的或非极性的(芳族或脂族)残基。例如,非极性或极性不带电残基可以在位置425或其等价位置引入。具体实例包括但不限于:
D425N,D425Q,D425H,D425S,D425T,D425A,D425L,D425V,D425I,D425P,D425W,D425Y,D425F。
在又一实施方式中,增加的侧链体积的氨基酸在本发明的位置处引入。在优选的实施方式中,增加的侧链体积的氨基酸,典型地是大体积非极性氨基酸在位置330、401、403或其等价位置处引入。对于位置330尤其优选的取代是A330P、A330V、A330L、A330I、A330W、A330F、A330Y。对于位置403尤其优选的取代是Q403P、Q403W、Q403F。在其他实施方式中,例如在位置377处,可能优选的是,待引入的氨基酸具有降低的侧链体积,如E377A或E377G。
本文中所讨论的突变一般通过采用本领域中的已知方法,如酶的定点诱变法、PCR和基因重组(gene shuffling)法或通过使用在定点突变法的循环中多个诱变寡核苷酸而引入到酶中。因此,突变可以以定向或随机方式引入。诱变方法因此产生编码一个或多个不同突变体的一个或多个多核苷酸。典型地,生成的突变基因库可以用于产生突变酶库。
除了以上指定的一个或多个突变,如取代、插入或缺失之外,该酶还可以具有1、2、3、4、5~10、10~20、20~40或更多个其他突变。这些附加的突变可以或不可以增强突变CYP102A1酶的单加氧酶活性。其他突变可以处于活性位点或在活性位点之外。例如,突变可以在第二球体之中,即,影响或接触活性位点中的一个或多个氨基酸的位置或取向的残基。插入将典型地在N和/或C端。因此,该酶可以含有高达20个氨基酸的短肽或融合至一端或两端的全长蛋白,例如,用于辅助亲合色谱或在固态基质上固定的蛋白纯化。缺失典型地包括并未参与催化的氨基酸的缺失,如在活性位点之外的那些氨基酸(因此,该酶是天然存在的酶的突变片段)。
在活性位点的其他突变典型地当其在活性位点中结合时改变底物的位置和/或构象。突变可以使该位点在待氧化的底物上更易于接近血红素基团。因此,突变可以是具有更小或更大、或者更多或更少极性的侧链的氨基酸的取代。
附加突变可以包括那些可以提高酶的稳定性的氨基酸残基变化。这些突变典型地防止蛋白的低聚作用,例如P450
优选地,附加突变选自一个或多个在CYP102Al中的以下突变:R47L、Y51F、A74G、A264G、N239H、I259V、L3531、F87A、F87L、F87G、H171L、L188Q、N319Y、I263A、A328P,或选自其等价位置处的相同突变。也应该理解,有关选择对于具体所列的那些具有更多或类似效应的其他氨基酸变化,以上参照列出的相同的考虑因素适用于位置117、131、191、215、276、307、330、377、401、403和425。因此,例如,取决于以上所列的具体附加突变是否是保守变化、是否改变极性或引入不带电氨基酸,类似范围的氨基酸将适合在每一附加位置处引入。
在特别优选的实施方式中,本发明的突变CYP102Al酶包含选自CYP102A1以下突变组中的一个或多个突变组:
i)A330P
ii)A191T/N239H/I259V/A276T/L353I;
iii)F87A/H171L/Q307H/N319Y;
iv)F87A/A330P/E377A/D425N;
v)F87A/A117V/E131D/L215I;
vi)I401P;
vii)R47L/Y51F/I401P;
viii)F87A/I401P;
ix)R47L/Y51F/F87A/I401P;
x)R47L/Y51F/A330P/I401P;
xi)Q403P;
xii)R47L/Y51F/Q403P;
xiii)R47L/Y51F/F87A/Q403P。
或包括其等价突变组。
如在i)中定义的A330P突变是在几个方面中的异常突变,由于不同于其他定向进化变异体(其影响可取决于共同作用的改变残基混合),其活性衍生自单点突变。在CYP102Al中,A330靠近位置329处的天然存在的脯氨酸残基,因此,A330P在β-折叠(β-sheet)内的主要底物接触点处并列了两个脯氨酸(3)。突变体的晶体结构(图2)表明,这压缩了进入通道并且使活性位点更不易接近,从而导致更封闭的活性位点并改变了底物的结合构象。如将在以下所看到的,这对单加氧酶活性和产物选择性具有特性效应。
突变组ii)和iii)增强CYP102A1的活性,同时广泛地反映由野生型酶表现出的特异性特征。除了在组iii)至v)中的残基87之外,在任何突变组中突变的位置无一是活性位点残基。
例如,在组ii)中,位置353靠近底物进入通道残基354,同时残基191、239、259、276和353都位于蛋白表面。根据晶体结构(4)A191在棕榈酸酯结合上被显著取代,并且处于进入通道的外缘(或外唇,outer lip)上。可以推断,突变该残基可以对底物诱引和/或捕获具有影响。
在组iii)中,位置171位于蛋白表面之上或与之靠近,同时残基307和319靠近于被认为是用于电子转移还原酶域的停泊位点的区域,并且因此可以潜在地通过对电子转移动力学的影响来调节其对单加氧酶活性的增强的作用。
突变组iv)包括在底物接触点处的A330P突变,和在外围位于酶结构中靠近蛋白表面的位置377和425处的突变,如在突变组v)中的位置117、131和215。
在另外的优选实施方式中,以上定义类型的本发明的突变CYP102A1酶为:
i)另外包括在CYP102A1中的一个或多个以下突变:R47L、Y51F、A74G、A264G或其等价突变(equivalent mutations);
ii)另外包括在CYP102A1中的一个或多个以下突变:R47L、Y51F、F87A、F87L;以及
iii)另外包括在CYP102A1中的一个或多个以下突变:F87A、F87G、I259V、I263A。
应该理解,除了特定突变或以上指定的特定附加突变之外,在本发明的突变CYP102Al酶的这些优选实施方式中也可以包括高达1、2、3、4、5至10、10至20或更多个其他突变。
用于氧化方法的底物是任意有机化合物,更典型地是任何能够通过单加氧酶氧化的有机化合物。通过单加氧酶氧化的任何有机化合物的合适性可以通过本文中描述的方法来常规地进行确定。
氧化方法导致在化合物中形成C-O键,一般是由碳氢键的氧化而形成为醇,但是由C=C键的氧化可以形成环氧化物。因此氧化可以引入醇、醛、酮或环氧基团。可替换地,氧化可以导致含氧基团的进一步氧化,如将醇基团转化成醛或酮。1、2或更多个碳原子可以在相同的底物分子中受到进攻。氧化也可以导致底物分子的N-和O-去烷基化。
氧化典型地产生1、2或更多种氧化产物。这些不同的产物可以源自受到进攻的不同碳原子和/或在给定的碳原子处发生的不同氧化程度。
氧化可以在环碳原子处或取代碳原子处发生或在两者处同时发生。至少初始的氧化将会涉及到可以是活化的或未活化的C-H键的进攻或在碳碳双键上的进攻(典型地给出环氧化物)。通常,活化的C-H键是其中碳原子处于苄基或烯丙基位置的情况。芳香环和烯烃双键通过稳定化自由基中间体或在反应路径期间产生的电荷的任何累积而活化C-H键进攻。C-H键中的碳可以是伯、仲或叔碳。氧化可以发生以导致脱氢,导致C=C双键形成而不是插入氧原子。这在烷基取代被支化或脱氢产生结合至芳香体系的C=C键或脱氢导致形成芳香体系时最可能发生。
底物可以是野生型CYP102Al酶的天然底物或并不是野生型酶通常底物但是能够在这种突变酶中利用的底物。CYP102A1酶的天然底物的实例是支链和直链脂肪酸,其通过野生型CYP102A1在次末端(sub-terminal)位置(ω-1~ω-3)羟基化。优选的实例是月桂酸、十一酸、癸酸、壬酸和辛酸。
在优选的实施方式中,底物是短链烷烃或中链烷烃或烷基苯。术语烷烃是指具有通式C
短链烷烃典型地具有1至约9个碳原子,更优选1~8,1~6或1~4个碳原子。C
烷基苯具有一个或多个在苄基芳环上的位置取代的烷基基团或部分。在烷基基团或部分中的碳原子数典型地可以为1~约8碳原子,更优选1~8、1~6或1~4个碳原子。
在一些实施方式中,在短链或中链烷烃或直接在苄基环上取代的骨架上,或在烷基苯的烷基取代上存在1,2,3或更多个取代。以下取代基的任何组合都可以存在。取代基典型地是卤原子或烷基或烯基基团,其一般具有1~6个碳原子,取代基可选地被一个或多个卤素取代。取代基也可以包括1、2或更多个氧原子、卤素或氮原子,而且例如可以是醇、醛、酮、醚、胺或环氧基团。
优选的短链烷烃底物的实例包括但不限于戊烷、3-甲基戊烷,2-甲基丁烷、丁烷、丙烷、乙烷和甲烷、辛烷和壬烷。优选的烷基苯底物的实例包括但不限于丙苯、乙苯、丁苯、枯烯、叔丁苯、邻二甲苯、间二甲苯、对甲基异丙基苯和乙基茴香醚。其他优选的芳香化合物是萘和芴。
应该注意到,有机化合物如丁烷、萘,并且尤其是丙烷,叔丁苯和邻二甲苯,广义上被归为野生型CYP102A1酶的“非天然”底物,但是能够通过本发明的突变CYP102A1酶氧化。非天然底物可以被定义为当用野生型CYP102A1酶培养时没有可检测的结合速率和/或产物生成的分子。非天然底物也可以包括这样的分子,其氧化速率<野生型CYP102Al酶天然底物速率的10%,使得它们不能被视为真正意义上的底物。
在本发明的其他实施方式中,底物是萜烯,例如单萜、或倍半萜。底物也可以是环烯烃。尽管在本发明中所用的萜烯一般具有式(C
单萜(其中n为2)将一般具有10个碳原子,典型地具有1~3个双键,尤其是1或2个环双键,并且典型地具有0~2个环。对于环中的一个环可以形成为含有典型地0或1个碳原子的桥。换句话说,其可以通过在现有环的2个碳原子之间直接连接或用中间亚甲基基团连接而形成。如果该萜是无环的,则将一般含有至少2个双键,并且一般包含3个双键。
倍半萜将一般含有14或15个碳原子,典型地具有0~2个双键并且一般具有1~3个环,并可能具有稠合环和/或桥接环。
在这种萜中可以存在的环将典型地具有3~9个碳原子,更特别的是5或6个碳原子。因此,特别地,该萜将会包含环己烷、或环己二烯环。
该萜一般含有总共3或4个环外甲基或亚甲基,例如,对于单萜,包含2个甲基和1个亚甲基或3个甲基,而对于倍半萜,包含3个甲基和1个亚甲基或4个甲基。
该单萜典型地是1,8-萜二烯如R-1,8-萜二烯、蒎烯如(+)-α-蒎烯、萜品烯、桧萜、崖柏烯、月桂烯、罗勒烯、橙花醇或香叶醇。
倍半萜一般通过三个异戊二烯单元的头至尾排列而形成。倍半萜一般是香木兰烯、石竹烯、长叶烯、朱栾倍半萜、异鞭苔烯(isobazzanene)、松香草烯(silphinene)、苡四环烷(ishwarane)、异广藿香-3-烯(isopatchchoul-3-ene)或二丙基甲戊烯基双环庚烯(isosesquicarene)。尤其优选的是倍半萜底物是朱栾倍半萜。
环烯烃一般含有高达9个环成员,例如其可以是5、6、7、8、9或更多元环。环烯烃典型地是环己烯。
也可以使用以上提及的任何萜或环烯烃的取代衍生物。典型地,存在1、2、3或更多个取代基。以下取代基的任何组合都可以存在。取代基典型地是卤原子或含氧或含氮基团或烷基或烯基基团,其一般具有1~6个碳,取代基可选地用一个或多个卤素取代。
取代基典型地具有式C
在本发明的另外的实施方式中,底物是卤代芳香化合物。该卤代芳香化合物典型地是苯或联苯化合物。苯环可选地被稠合并可以被取代。卤素典型地是氯。在许多情况下,分子中存在多于一个的卤原子,典型地有2~5个或6个,例如,3个卤原子。一般而言,卤原子中的2个将互为邻位或对位。化合物可以含有或可以不含有氧原子如羟基基团、芳氧基基团或羧基基团。化合物可以是或可以不是氯代苯酚或氯代苯氧乙酸化合物。
这种可以通过本发明的方法氧化的具体化合物包括1,2-;1,3-和1,4-二氯苯,1,2,4-;1,2,3-和1,3,5-三氯苯,1,2,4,5-和1,2,3,5-四氯苯,五氯苯,六氯苯,和3,3'-二氯联苯。
可以通过本发明的方法氧化的其他化合物包括难分解(damascones)的卤代芳香化合物,尤其是二氧芑(dioxins)和卤化二苯并呋喃类,以及其中一个或两个氧原子被硫取代的相应化合物,尤其是具有至少一个卤素取代基的二氧芑的化合物,如二氧芑本身,2,3,7,8-四氯二苯并二氧芑。
卤代芳香化合物的氧化典型地产生1、2或更多个氧化产物。被氧化的原子可以是环碳。这些氧化产物将一般含有1个或多个羟基基团。因此,一般而言,氧化产物是酚,其可以通过各种假单胞菌(Pseudomonads)和其他细菌降解,而未氧化的卤代芳香化合物是难以氧化的。如以下描述的,这使得本发明的酶适合于对卤代芳香化合物污染的位点去污。
设想用于本发明方法的另外的底物包括但不限于氯唑沙宗、苯胺、对硝基苯酚、硝苯地平、崖柏酮非对映体、烯烃(包括丙烯、1-己烯和苯乙烯)、吲哚、多环芳烃、心得安(propanolol)、烷氧基试卤灵(包括7-乙氧基试卤灵、7-甲氧基试卤灵、7-戊氧基试卤灵、7-苄氧基试卤灵)、丁螺环酮、睾酮、阿莫地喹、右美沙芬、醋胺酚、3,4-亚甲二氧基甲基安非他明(MDMA)。
采用本发明的突变CYP102A1酶实施的氧化方法,根据如上所定义的改进的结合速率或产物生成速率,可以区别于通过另一野生型或突变CYP102A1酶实施的氧化方法。本发明方法的特征还在于,由氧化的底物形成具体产物,典型地是通过野生型CYP102A1酶或另一突变CYP102A1酶并不能形成的产物、或以可以忽略的量形成,即低于产物总量的10%、8%、5%、2%、1%或更低的产物。例如,丙苯的氧化可以以高选择性来生成2-丙基苯酚、或1-苯基-2-丙醇,或者乙苯的氧化可以生成2-苯基乙醇和苯乙烯。
相比于通过野生型CYP102A1酶或其他突变CYP102A1酶实施的氧化方法,采用本发明的突变CYP102A1酶实施的方法也可以表现出氧化产物的改变的比率或数目。在存在产物的改变比率的情况下,对于具体氧化产物的产物生成速率,相比于通过野生型CYP102A1酶或其他突变CYP102A1酶实施的相应方法,典型地被提高。具体氧化产物的优势提高可以超过通过野生型CYP102A1酶或其他突变CYP102A1酶形成的产品混合物中所述氧化产物量的至少10%、20%、50%,更优选100%、200%、300%、500%或更高。
在本发明的方法中可以表现出优势提高的氧化产物的具体实例包括:i)1-苯基-2-丙醇,其中氧化的底物是丙苯;ii)2-乙基苯酚,其中氧化的底物是乙苯;iii)1-苯基-2-丁醇或4-苯基-2-丁醇,其中氧化的底物是丁苯;iv)苄醇,其中氧化的底物是甲苯;v)2-甲基-2-苯基丙-1-醇,其中氧化的底物是叔丁苯;vi)2-甲基苄醇,其中氧化的底物是邻二甲苯;vii)香芹酚或麝香草酚或4-异丙基苄醇,其中氧化的底物是对甲基异丙基苯;viii)香柏酮,其中氧化的底物是朱栾倍半萜;ix)2-壬醇,其中氧化的底物是壬烷;x)2-丁酮或2-丁醇,其中氧化的底物是丁烷;xi)3-甲基-3-戊醇,其中氧化的底物是3-甲基戊烷;xii)2-丙醇,其中氧化的底物是丙烷;xiii)反式-异胡椒醇(反式-异薄荷烯醇,trans-isopiperitenol),其中氧化的底物是R-1,8-萜二烯;xiv)2,3-蒎烯环氧化物、顺式-马鞭草烯醇、反式-马鞭草烯醇,其中氧化的底物是α-蒎烯;xv)9-芴醇,其中氧化的底物是芴;xvi)8-羟基十二酸和7-羟基十二酸,其中氧化的底物是十二酸(月桂酸)。
本方法典型地在CYP102A1酶、底物和该酶的天然辅助因子(它们是NADPH和分子氧)存在的情况下实施。在一个实施方式中,该方法采用酶如脱氢酶及其辅底物(共底物、或协同底物)来实施,以在脱氢酶的辅底物的伴随氧化下由NADP
应该理解到,增加的活性和改变的选择性源于在CYP102A1酶的血红素域中的取代。因此,本发明也提供了系统,在这类系统中底物氧化方法在该酶的血红素域(a)、底物、电子转移还原酶(b)、电子转移还原酶(redoxin)(c)、该酶的辅助因子和氧给体的存在下实施。在该系统中,电子流一般为:辅助因子→(b)→(c)→(a)。
(b)一般是能够介导电子从辅助因子至(c)转移的电子转移还原酶,如天然存在的还原酶或与天然存在的还原酶同源、典型地具有至少70%同源性的蛋白;或还原酶或同源物的片段。(b)典型地是在天然存在的P450酶系统中发现的任何电子转移链的还原酶,并且典型地是核黄素依赖性还原酶,如假单孢氧还蛋白还原酶。
(c)一般是电子转移还原酶,其能够介导电子从辅助因子经由(b)至(a)转移,(c)典型地是天然存在的电子转移还原酶或与天然存在的电子转移还原酶同源、典型地具有至少70%同源性的蛋白;或还原酶或同源物的片段。(c)典型地是在天然存在的P450酶系统中发现的任何电子转移链的还原酶,(c)典型地是2Fe-2S还原酶,如假单孢氧还蛋白,或黄素氧还蛋白。
典型地(a)、(b)和(c)作为独立的蛋白存在;然而,它们可以在相同的融合蛋白中存在。典型地,仅仅其中的两种,优选(b)和(c),存在于融合蛋白中。典型地,这些组分在融合蛋白中是相邻的而不存在连接肽(连接子肽,linker peptide)。
可替换地,连接子可以存在于组分之间。连接子一般包括,不具有庞大侧链的氨基酸,并且因此不会阻碍蛋白亚单元的折叠。优选地,连接子中的氨基酸是不带电的。优选的连接子中的氨基酸是甘氨酸、丝氨酸、丙氨酸或苏氨酸。在一个实施方式中,连接子含有序列N-Thr-Asp-Gly-Gly-Ser-Ser-Ser-C。连接子典型地具有至少5个氨基酸的长度,如至少10、30或50或更多个氨基酸的长度。
在该方法中,酶,(b)或(c)的浓度典型地为10
在该方法中,可以存在本发明的多于一种的不同突变CYP102A1酶。典型地,每一突变体能够氧化不同底物或可以比另一种酶能够更好地氧化给定的底物,因此采用突变CYP102A1酶的混合物使得能够氧化更宽范围的底物。该方法也可以包括野生型CYP102A1酶、其他P450酶或它们的同源物、任何单加氧酶、以及适用于所需合成或氧化反应中的任何其他酶。
在一个实施方式中,该方法在能够除去过氧化氢副产物的物质(例如,过氧化氢酶)的存在下进行。在另一实施方式中,该方法在全长酶或该酶的血红素域、底物和氧原子给体如过氧化氢或叔丁基过氧化氢的存在下,例如采用过氧化物分流(peroxide shunt)来实施。
在另外的实施方式中,该方法在电化电池(electrochemical cell)中存在全长酶或仅该酶的血红素域、底物和氧的情况下实施,使得通过电极提供氧活化和活性中间体生成所需的两个电子,其要么通过从电极进行直接电子转移,要么经由小分子调节剂间接实现电子转移。
该方法可以在细胞内或细胞外进行。细胞典型地在培养基中(in culture),在某个位点(at a locus),体内或原位(这些方面将在以下进行讨论)。该方法典型地例如在陆地(例如在土壤中)或在水中(例如淡水或海水)在某个位点(或地点,locus)进行。当该方法在培养基中实施时,培养基典型地含有本发明不同类型的细胞,例如表达本发明的不同突变CYP102A1酶的细胞。一般而言,这样的细胞在可吸收碳源和氮源的存在下进行培养。
典型地,在其中实施该方法的细胞是其中本发明突变CYP102A1或野生型CYP102A1并非天然产生的细胞。在另一实施方式中,突变CYP102A1酶在并非天然产生野生型CYP102A1的细胞中表达,但是其水平比天然存在的水平更高。该细胞可以产生1、2、3、4或更多个本发明的不同突变CYP102A1酶。这些突变CYP102A1酶能够氧化不同的有机化合物底物,不同的短链烷烃或不同的烷基苯。
这种细胞可以是原核或真核细胞,并且通常是本文提及的任何细胞或任何生物。优选的细胞是大肠杆菌(Escherichia coli)、假单胞菌属(Pseudomonas sp.)、粘金黄杆菌(flavobacteria)或真菌细胞(例如,曲霉(Aspergillus)和酵母,尤其是毕赤酵母属(Pichia sp.))。根据本发明的用途,也可以设想的是红球菌属(Rhodococcus sp.)和芽孢杆菌属(Bacillus sp.)。这种细胞可以是或可以不是能够氧化任何底物或产生任何本文提及的氧化产物的以其天然存在形式的细胞。典型地,这种细胞是以基本分离的形式和/或基本纯化的形式,在这种情况下,其一般含有至少90%,例如至少95%、98%或99%的细胞或制备物的干质量。
细胞典型地通过向细胞(即用其转化该细胞)中引入包含编码本发明的突变CYP102A1酶的多核苷酸的载体来产生。应该理解到,由于核苷酸密码的兼并,多于一种的多核苷酸可以编码本发明的每一种突变CYP102A1酶。也应该理解到,核苷酸序列可以被设计成展示出适合特定细胞或生物的密码子偏性(codon bias)。载体可以整合到细胞的基因组中或保持为染色体外(extra-chromosomal)。细胞可以发育成以下讨论的动物或植物。典型地,多核苷酸的编码序列可操作地连接至操控序列,其能够提供宿主细胞对编码序列的表达。操控序列一般是启动子,典型地是其中表达单加氧酶的细胞的启动子。
术语“可操作地连接”是指一种邻接(juxtaposition),其中描述的元件所处的关系容许它们以其所预想的方式发挥功能。“可操作地连接”至编码序列的操控序列以这种方式结合,使得编码序列的表达在与操控序列相容的条件下实现。
载体典型地是转座子、质粒、病毒或噬菌体载体。其典型地包含复制起点。其典型地包含一个或多个选择性的标记基因,例如,在细菌质粒情况下的耐受氨苄青霉素的基因。载体典型地采用传统的技术(包括磷酸钙沉淀、DEAE-葡聚糖转染或电穿孔)引入到宿主细胞中。
本发明进一步提供转基因动物或植物,其细胞是本发明的任何细胞。这种动物或植物对于一种或多种突变CYP102A1基因来说是转基因的。它们可以是这类基因的纯合子或杂合子,其典型地瞬时引入到细胞中、或被稳定整合(例如,在基因组中)。动物典型地是蠕虫(例如,蚯蚓)或线虫类。通过转化合适的细胞(例如,胚胎干细胞、愈伤组织或生殖细胞),如果需要培育该细胞,使该细胞发育成动物或植物,并且如果需要培育该动物或植物,从而可以获得这种植物或动物。动物或植物可以通过本发明的动物或植物或Fl生物(或从F1去除的任何后代,或从转化的细胞发育成的嵌合体)的有性或无性繁殖(例如,克隆)、增殖而获得。
如以上所讨论的,该方法可以在某个位点上实施。因此,本发明也提供了一种处理被本发明的底物,例如短链烷烃、烷基苯或卤代芳香化合物污染的位点的方法。该方法包括用本发明的突变CYP102A1酶、细胞、动物或植物接触该位点。这些有机体然后典型地容许氧化卤代芳香化合物。在一个实施方式中,这些用于处理该位点的有机体对该位点是固有的。因此它们可以从该位点(例如,在污染之后)获得,转化/转染(如以上讨论的)以表达突变CYP102A1(和可选地适当的电子转移还原酶(reductase)和/或还原酶(redoxin))。
在一个实施方式中,该位点用多于一种类型的本发明的有机体,例如用2、3、4或更多种表达不同单加氧酶(其氧化不同有机化合物底物,例如不同短链烷烃、烷基苯或卤代芳香化合物)的类型进行处理。在一个实施方式中,它们之间的这种有机体聚集能够氧化存在于污染区域中的特定组的所有底物,即,短链烷烃。
有机体(例如,以聚集(collection)的形式)可以在生物反应器中(例如其中它们以固定形式存在)实施本发明的方法。因此,待处理的水或土壤可以经过这样的生物反应器。土壤可以用补充有表面活性剂或乙醇的水冲洗,随后引入到生物反应器中。
迄今公开用于筛选通过随机诱变和基因重组产生的CYP102A1突变体文库的方法倾向于使用替代底物(surrogate substrates)如吲哚(靛蓝形成)和对硝基苯酚衍生物(释放的对硝基苯酚的检测)。对于替代底物氧化活性增强的一些所选突变体,已经发现对于不同结构的化合物具有增强的活性,但是产物选择性变化较少见。
关于对产物选择性的筛选,WO2006105082公开了一种将产物醇结合至可以进行光谱检测的化合物的方法。类似地,在通过CYP102A3的辛烷氧化中对1-辛醇的选择性提高是通过在脱氢酶筛选方法中靶向该产物而获得的(34)。这些方法偏向于对选择性的搜索,其中在活性方面提高较小。而且,仅仅发现这些促进特异性靶化合物形成的突变,同时在以不同和潜在期望的方式向更宽范围的化合物影响产物选择性的位点处的突变还没有揭示。
相反,用于分离根据本发明的突变体的筛选方法利用了经由吲哚氧化针对靛蓝形成筛选突变的随机文库然后搜索化学上感兴趣的产物的增强的活性和选择性这一组合。因此,来自吲哚氧化筛选的活性最强的突变进一步经由萘、丙苯和辛烷的体内氧化进行筛选。产物通过气-液相色谱(参见实施例部分)进行分析。萘比吲哚更具疏水性而更类似于P450催化中常常靶向的烃底物。丙苯比萘更小但是因为芳香环氧化、苄基氧化之间的竞争和在两个未活化脂族碳中心处的进攻而提出产物选择性变化的测试。辛烷表现出不同的挑战,其柔软且较不紧凑,并且具有可用于氧化的4组不同的C-H键,使得选择性偏向于端部和内部位置的突变。产物生成速率提高和/或产物分布改变的变异体被选择用于体外活性研究。
在这种多步骤程序中,约1500个菌落在初始靛蓝形成(活性)步骤中进行筛选,其数目在11个第一代突变体的基因重组之后降低至约800个菌落。从800个这些菌落之中选出130个菌落进行体内底物氧化筛选步骤。130个这些菌落之中,5个变异体选择用于进一步体外研究,其全部显示出对宽范围的有机化合物,从萘至戊烷的增加的活性和/或改变的产物选择性。因此,用于分离根据本发明的突变体的筛选方法容许活性和/或产物选择性增加的突变体的有效、严格的选择。在突变发现中筛选的初始文库的较小数量也表明,本发明人用于发现CYP102A1的变异体的方法尚未尽其潜力。
与先前公开的定向进化技术的另一不同之处在于采用靛蓝形成对CYP102Al的筛选涉及在CYP102A1血红素域中的特异性位点的诱变(例如位点饱和),而本筛选涉及全长血红素域基因的随机诱变。因此,这种筛选具有分离出增加数量的变异体的潜力。而且,很显然,位点饱和诱变可应用于根据本发明分离的特异性突变体,从而分离出另外有用的变异体。
分析级或更高质量的一般试剂和化学底物来自Alfa-Aesar,Fisher Scientific和Sigma-A1drich或其子公司。HPLC质量的溶剂来自Rathburn Chemicals(UK)以及Sigma-Aldrich和Merck的子公司。缓冲剂组分来自Anachem,UK。NADPH(四钠盐)来自ApolloScientific和Melford Laboratories。异丙基-β-D-硫代半乳糖吡喃糖苷(IPTG)来自Melford Laboratories。限制酶,T4 DNA连接酶和相关缓冲剂来自New England Biolabs。Taq和KOD聚合酶来自Merck Biosciences。感受态和超感受态的大肠杆菌菌株来自Stratagene。定点诱变采用在Stratagene Quik-Change诱变试剂盒中描述的PCR方法来实施。侧接诱变寡核苷酸中改变的密码子的寡核苷酸的合适长度按照生产商的说明书进行设计。寡核苷酸来自MWG Biotech。一般性分子生物学操作根据文献方法(Sambrook,J.,Fritsch,E.F.,and Maniatis,T.(1989)Molecular Cloning:A Laboratory Manual,2
SpeI限制位点采用寡核苷酸:5′
易错PCR在这个位点和血红素域-编码区域上游的EcoRI位点之间采用正向和反向引物:5′
在根据所采用的Stratagene GeneMorph方案设计成引入1~3个突变/1000bp的条件下,由野生型CYP102A1(WT)和突变体F87A模板来构建文库。基因扩增30个循环,每个循环为94℃下60s链分离、45℃下90s退火以及68℃下110s+2s延伸。在采用EcoRI和SpeI消化后,短片段采用T4 DNA连接酶重新引入pGLWl1(SpeI WT变异体),转化到大肠杆菌DH5α感受态细胞中并在Luria-Bertani(LB)琼脂板上培养36h。
筛选约1500个菌落。那些显示靛蓝形成(Gillam,Ε.M.J.et al(1999)Biochem.Biophys.Res.Commun.265,469-472;Li,Q.S.,et al(2000)Chem.Eur.J.6,1531-1536)的菌落被分离出来,转移到新板上并再培养36h以最小化测序前的假阳性。表现16个具有潜在关注性的新突变的11个变异体,随后通过随机引导重组(random-primingrecombination)而进行重组(Shao,Z.,et al(1998)Nucleic Acids Res.26,681-683)。
Volkov和Arnold(Volkov,A.A.,and Arnold,F.H.(2000)Methods Enzymol 328,447-456)所给出的方案在阶段9进行改良,其中采用Taq和KOD聚合酶和2μL MgSO
在约800个菌落中,约130个呈现出靛蓝形成。这些在5~10mL规模上生长,并对于萘和丙苯氧化活性采用气相色谱进行体内筛选。对相对于WT显示出最大产物峰值面积增加或产物分布改变的12个变异体进行测序,以较大规模生长并相对于相同的两个底物进行筛选。其中,5个选择用于体外研究。
我们也制备了各种单位点突变体及其与来自随机诱变筛选程序的5个突变体的组合,并采用气相色谱对靛蓝形成和对于萘、丙苯和辛烷氧化活性进行了体内检测。这些单位点突变是R47L、Y51F、E267V、I263A、A74G、L188Q、M177V/K、A399P、I401P、G402P、Q403P、V302I、A264G、A99T、S270I、R179H和F87L/A/G。这些大多数都是先前已知的突变(在R47、Y51、I263、A74、L188、M177、A264、F87处)或属于我们在随机诱变/筛选过程(在E267、V302、A99、S270、R179处)的早期轮次中发现的那些,我们已经基于其在结构中的位置而选择用于另外的轮次,其对于底物结合可能具有一些影响。在A399、I401、G402和Q403处的脯氨酸突变是基于在A330P突变体(图2)的晶体结构中所观察的结构变化进行选择的,其为5个选自筛选程序的突变体之一。这些突变体随后也通过体内筛选方法对于增加活性的证据而进行筛选。
R47L和Y51F突变,要么在其自身要么在与其他突变组合时,与R47L/Y51F偶联体(couplet)并不一样有效。其他单位点突变在靛蓝形成筛选中显示不同的有效性,而同时I401P和Q403P单位点突变相对于野生型在体内氧化筛选中显示出增加的产物生成,并且制备这些突变体并在体外对它们的活性进行研究。变异体R47L/Y51F(RLYF)和F87A如所描述的进行制备(13)。RLYF偶联体以标准克隆程序采用NcoI和AflII限制位点引入到变异体KT2和A330P(Sambrook,J.,Fritsch,E.F.,and Maniatis,T.(1989)Molecular Cloning:ALaboratory Manual,2
感兴趣的变异体采用NcoI和BamHI限制位点转移到pET28载体中,使得与tac启动子相比采用T7启动子在pGLW11载体中表达水平能够更紧密地得到控制。30ml.L
NADPH转化(turnover)(除了丁烷和丙烷)在于30℃下充氧并含有0.1或0.25μM酶、125μg牛肝过氧化氢酶和以DMSO中100mM原液加入的1mM底物的1250μL 50mM Tris(pH 7.4)中进行。蛋白浓度如所描述(13)或经由CO差光谱(Omura,T and Sato,R(1964)J.Biol.Chem.239,2379-85)进行检测。分析在NADPH以20mg·ml
在所有的转化中,监控在340nm处的吸收衰减(absorbance decay)并采用ε
对于除月桂酸之外的底物,在萃取进入400μL乙酸乙酯或氯仿中之前,将3μL的内标(在DMSO中的100mM)加入到1000μL的每种完全转化(completed turnover)之中。以21000g在1500-μL微型离心管中实施离心3
在GC产物分析中的校准(galibrants)、内标和炉温
在通过随机诱变产生的变异体中,其都对活性增强和产物选择性改变进行了体内筛选,选择5个特异性变异体用于体外研究。这些是:
(i)A330P
(ii)A191T/N239H/I259V/A276T/L353I(突变体KT2)
(iii)F87A/H171L/Q307H/N319Y(突变体KSK19)
(iv)F87A/A330P/E377A/D425N(突变体KT5)
(v)F87A/A117V/E131D/L215I(突变体LO25)
所有5个突变体易于表达并通过标准程序(13)进行纯化。在-20℃下于50%v/v甘油中储存至少15个月后,没有任何一个显示出生成惰性“P420”形式的证据。突变体LO25比其他4个变异体研究更少,但是在这些5个初始变突体(initial mutants)中其对于突厥酮(damascone)/紫罗酮类分子具有最高的活性和选择性,例如生成86%的羟基突厥酮产物。A330P、KT2、KSK19和KT5的活性采用宽范围的底物进行分析。对于这些中的某些的数据在表1~20中给出,其中NADPH氧化和产物生成速率(PFR)以单位nmol(nmol P450)
本发明的四种变异体(A330P、KT2、KT5、KSK19)增强了采用萘的WT的PFR(3.1min
WT以606min
R47L/Y51F(RLYF)偶联体,其已经证实提高CYP102A1对于几种底物的活性(13,14,17),被引入到变异体A330P和KT2中,其目的是进一步提高产物的生成速率。两种高度活性的第二代变异体得到,RLYF/KT2和RLYF/A330P。单位点突变也围绕血红素表面以及活性位点在各个残基处引入,而这些也与5个在第一轮中识别的突变进行组合。这些第二代突变体的活性通过靛蓝形成和体内氧化筛选程序进行筛选。I401P和Q403P突变体作为来自体内氧化程序的有前景的新变异体而被识别并制备它们。也制备了组合突变体R47L/Y51F/I401P、F87A/I401P和R47L/Y51F/A330P/I401P。
RLYF/KT2相似于GVQ变异体在萘转化中的PFR 496min
其他烷基苯也作为WT CYP102A1和新变异体的底物进行检测。显著的活性增强利用甲苯(表3),尤其是利用RLYF/KT2,RLYF/A330P和I401P进行观察。Q403P突变体的影响再次类似于变异体KT2的影响。较大的选择性变化也是显著的,虽然变异体相比于最快的突变体具有降低的产物生成速率。WT CYP102A1将甲苯主要氧化成邻甲酚(98%)。该环氧化是出乎意料的,因为苄基C-H键是高度活化的。例如,来自恶臭假单胞菌(Pseudomonas putida)的WT CYP101A1进攻苄基位置以生成>95%的苄醇。变异体RLYF/A330P将PFR提高了60~189min
NADPH速率和结合一般对于丁苯比对于丙苯低,对于WT和最快变异体,RLYF/KT2记录的PFR为229min
利用叔丁基苯,NADPH转化速率与丁苯对于大多数变异体是一致的,但是结合水平除了在F87A变异体的情况下都大大降低(表5)。GVQ变异体,F87A/KT2和KSK19相对于WT都提高PFR两个数量级,最高速率由新的变异体F87A7KT2以234min
对于乙苯(表6),本发明的变异体一般显示出比WT(60min
在紧接活化苄基碳的甲基处的优先进攻是很难实现的,因为如果这两种类型的键都从相同的距离等同地可接近于P450铁中间体(ferryl intermediate),那么更加活化的C-H键将会受到更迅速的进攻。因此,KT5可以在甲基C-H键比苄基C-H键更接近铁(ferryl)的取向上结合乙苯。在脱氢反应中,铁中间体夺取氢原子以形成Fe
天然存在的烃,对甲基异丙基苯(4-异丙基甲苯),是一种感兴趣的底物,因为其是四种香料化合物(flavouring compound)的前体。WT给出82%p-α,α-三甲基苄醇,其源自异丙基侧链的次甲基C-H键的氧化。也形成了少量的两种可能的芳香羟基化产物,麝香草酚(3%)和香芹酚(7%),以及仅仅2%的4-异丙基苄醇。相反,变异体A330P给出19%的麝香草酚、17%的香芹酚、22%的4-异丙基苄醇、和仅仅37%的p-α,α-三甲基苄醇(表7)。F87L/KT2给出74%的4-异丙基苄醇和21%的香芹酚,对于通常的大多数产物,p-α,α-三甲基苄醇仅仅占产物混合物的5%。观察到活性增强,其中变异体KSK19给出1442min
本发明的变异体显示出增强的枯烯氧化活性。单位点突变体A330P和Q403P,以及KSK19变异体相对于野生型表现出相似的活性增强,主要是由于提高的NADPH转化速率,其也高于KT5变异体。WT主要生成苄基氧化产物,而KT5由脱氢给出27%的1-甲基苯乙烯,并且也由进一步氧化给出1%的苯乙烯氧化物产物(表8a)。这些结果表明,在烷基取代基的2-位具有伯C-H键的芳香化合物可以发生脱氢。该反应路径可以构成用于合成作为聚合物前体的取代苯乙烯的方法(例如由乙基茴香醚制备乙烯基茴香醚)的基础。
KT2和A330P显示出相对于野生型对短链烷烃的活性大大增强,尤其是组合RLYF(表9-13)时更是如此。I401P突变体也证明是高度活性的。RLYF/KT2和RLYF/A330P显示出对于戊烷的类似产物生成速率(1206min
这些变异体的高活性在3-甲基戊烷(所有都>1000min
长链烷烃对于大多数突变体都是较差的底物,RLYF/KT2和RLYF/A330P给出对于辛烷的PFR分别为246min
本发明的变异体也显示出对于氧化氯代芳香化合物的活性提高,如通过1,4-二氯苯(1,4-DCB)所举例说明的。这种提高主要是由于更高的NADPH转化活性,而同时结合作用并不比WT高很多。最高的结合作用是A330P变异体的15%(表14)。通过气相色谱仅仅检测到一种产物,但是在所用条件下不可能确定其是否为2,4-或2,5-二氯苯酚。这些结果证实了本发明的变异体对于氯化苯和芳香氧化的效能。
我们先前已经报道了通过WT CYP102A1和突变体如F87A的倍半萜烯朱栾倍半萜氧化(14)。这种酶生成了各种产物,包括香柏醇(努特卡醇,nootkaytol)、香柏酮和环氧化产物。F87A突变已经显示出,产物选择性稍微转向香柏酮(约20%)。相比而言,变异体KSK19相对于WT提高了朱栾倍半萜氧化速率30倍(表15)。更重要的是,其相对于突变体F87A,葡萄柚香味的香柏酮生产速率加倍。变异体F87A/KT2和KT5将香柏酮的比例提高至约30%,同时其自身也比F87A提高了PFR。
WO0031273公开了通过CYP102A1的单萜氧化。新的变异体表现出比WT和先前报道的突变体更高的活性。新变异体的催化性能与WT对R-和S-1,8-萜二烯氧化的催化性能相比较(表16;16a)。尤其值得注意的是,两个对映体采用两种酶生成了不同的产物,但是转化活性和结合密切相似,这说明CYP102A1底物袋(substrate pocket)的不对称性质。1,8-萜二烯氧化活性在所有变异体中都提高,其中Q403P、I401P和R47L/Y51F/I401P三重突变显示出的活性相当于或高于野生型CYP102A1对脂肪酸天然底物(月桂酸)的氧化活性(1439min
芴是一种比萘更具立体严格性的底物,其用于体内筛选程序中。R47L/Y51F组合稍微提高了活性,并且加入A330P突变将NADPH转化速率提高至510min
月桂酸(十二酸)是CYP 102A1的已认知的天然底物,其中NADPH转化速率为2777min
这些发现表明,尽管现有技术的显著体现,但是CYP102A1体系仍维持定向进化和定点诱变技术应用的肥沃之地,而具有改进的活性和产物选择性的变异体可以被表征。本发明的所有变异体包含对于本发明人的认知而言先前并未公开的许多变异体。
A330P突变具有向较宽范围的化合物提高活性的广受欢迎的作用,同时当以其自身作用时以及与另一选择性改变突变(F87A)组合时也改变了产物分布。另一方面,I401P起到作为一般性速率加速剂的作用,其中对选择性的影响很小。I401在β-突出部(β-bulge)上靠近血红素并可以影响电子转移。A330P是不太可能的定向进化产物,对于其效能取决于衍生自单位点突变而不是常见的改变的残基的整体部分(concert party)的单独取代。脯氨酸直接引入在位置329处的现存脯氨酸旁边,底物在β-链的端部接触残基。所产生的骨架灵活性的损失是可预测的,并且由晶体结构(图2)看起来限制活性部位袋(active sitepocket),容许更紧密的底物结合并增强活性。这个解释与异常的和潜在有用的所观察的选择性效应是一致的,其典型地是由F87A产生的那些的逆转,其中活性部位袋比WT更敞开(open)。令人感兴趣的是,看到并列(juxtaposing)脯氨酸残基的策略是否能够在CYPl02A1中别的地方或实际上在其他酶体系的重新设计中有用地展开。
获得了A330P突变体的晶体结构,并且示于图2中。该突变体的Calpha位置对野生型的那些而言很大程度上是可叠加的,但是在位置328、329和330处还存在显著的重排。如图2中所示,引入的A330P突变诱导了Pro329的环显著向底物结合袋迁移,减小了活性位点体积并在关键区域压缩了底物进入通道。这些结构变化最可能导致利用A330P突变观察到的异常效应,如,非天然底物的增强的结合和改变的产物选择性,其由于Pro329环突出进入底物袋。然而,它们并未导致二级结构元件如螺旋的陡然终止。
基于这些不预期的发现,对于通过引入大体积残基如脯氨酸来重构环区域的潜势进行了进一步研究。具体而言,脯氨酸取代在A1a399-Gln403环中实施,这提供了最接近的血红素配体,Cys400。Arg398涉及到静电和氢键相互作用,并且可能在稳定化蛋白折叠中是很重要的,同时超过Gln403的残基可能从血红素中移除太远而不能具有任何效应。探究的突变因此有A399P、I401P、G402P和Q403P。如在实施例部分中所示出的,I401P和Q403P两种都对酶活性产生显著的增强作用。
对脯氨酸的突变残基401和403可以在最邻近的环中诱导构象变化,使得例如,通过改变Fe-S距离来改变Fe-S键相互作用的强度。这可以使得血红素铁更容易还原,电子转移的重组能垒(reorganisation energy barrier)将会降低,以此将便于血红素铁在催化循环中移动进入卟啉环的平面内。
环区域在CYP102A1中经由大体积残基如脯氨酸的引入的重组是本发明的A330P、I401P和Q403P取代突变异体共同的独特且特异性的结构机理。
变异体KT5,其中A330P和F87A一起出现,促进F87A而不是A330P的选择性变化特性。然而,所涉及的转化能够比通过其他含F87A的变异体产生的更为显著(例如,由甲苯产生95%的苄醇vs.F87A/KT2的48%、F87A的23%和WT的2%),并且经常以提高的产物生成速率发生,产生一定范围的互补(complementary)可能性。将令人感兴趣的是看到,A330P如何结合其他已知的选择性定向突变,如A264G、A82L和A328V。
变异体KT2(A191T/N239H/I259V/A276T/L353I)和F87A/KT2给出的产物分布分别密切类似于WT和变异体F87A。类似地,I401P加速与野生型具有相同产物生成的底物范围的速率。因此,KT2和I401P起到速率加速剂的作用。在KT2中的组分突变N239H和1259V先前已经报道(55)。然而,变异体KSK19,其包含初始的F87A,具有与F87A/KT2相同的选择性模式,并且尽管仅仅含有三种其他突变,但是在整个底物范围内活性稍微更高,这表明,当制备其时,可证实KSK19的无F87A衍生物比KT2作为速率加速剂更具有效能。
在KT2和KSKl9(H171L、A191T、N239H、I259V、A276T、Q307H、N319Y和L353I)看起来起到速率加速剂作用的这些突变中,Gln307和Asn319(图1)接近于被认为是还原酶域停泊位点的区域,自此血红素域接收电子(56),并且能够影响电子转移动力学。Leu353靠近底物进入通道残基,Met354,而A1a191(位于进入通道的外缘上)当棕榈酸酯结合时显著地位移(57),并能够在底物诱引和/或捕获中起到作用。其余四个突变(Hisl71、Asn239、Ile259、A1a276),都是在蛋白表面之上或与之靠近,并且其作用的详细机制仍是不得而知。可以推测,至少一些突变的作用可以根据其在CYP102A1结构中的背景方面而进行合理化。P450酶的转化活性是通过引发催化循环的第一电子转移步骤的速率来进行速率限制的。电子转移反应的速率通常根据马库斯理论(Marcus theory)进行讨论,这种理论陈述了电子转移的活化能取决于热力学驱动力(反应的自由能变化)和重组能(需要用于变形反应物状态以与产物状态一致的能量输入)。底物结合通过改变血红素的电子性能(大多数情况下是血红素铁的第六配体被取代),由此使反应热力学更为有利(更高驱动力)并降低电子转移的重组能垒(需要反应物状态的更小变形)来起主要作用(58)。如果活性位点结构被改变,例如通过活性位点取代,非天然底物的结合可以被增强,并观察到更快的底物氧化。
用于改变底物结合的另一机制(因此是血红素的电子性质)可以是在围绕底物袋(pocket)的二级结构元件中的变化的诱导。在P450酶中,底物袋通常通过B和B'螺旋、BC环、F/G环、G螺旋和I螺旋的残基限定。在远离底物袋的残基处的氨基酸取代可以通过在这些二级结构元件的位置诱导变化来改变底物结合。His171处于F螺旋的起始位置并在L215处接触G螺旋。N239处于H螺旋中,并且这个螺旋接触I螺旋的N-端末端。在H171和N239处的取代将会分别影响G和I螺旋的定位,并且可以改变底物结合。Ile259和A1a276都在I螺旋中。尽管这些残基都不会接触底物,但是氨基酸取代可能例如通过诱导定位于活性位点中的干扰残基,1263中的结构变化来影响活性位点结构,并在本发明人的早期工作中已经证明改变CYP102A1的活性(13)。
特别令人感兴趣的是注意到,变异体LO25包含L215I突变,其可以在H171/L215邻近通路(close approach)处影响F和G螺旋之间的接触。整体上而言,当活性位点没有突变时,它们中的所有都处于在二级结构元件之间的包装/相互作用中起到一定作用的残基处。通过A330P引入的串联式脯氨酸排列是独特的,并显示出非常出乎意料,但是高度有益的效应。
在本发明中公开的突变可以引入到现有的变异体(例如,那些包括L188Q、R47L、Y51F突变的变异体)中,用于方法开发,而且还作为用于进一步进化的起点。
1.Miura,Y.,and Fulco,A.J.(1975)Biochim.Biophys.Acta 388,305-317.
2.Cryle,M.J.,Espinoza,R.D.,Smith,S.J.,Matovic,N.J.,and De Voss,J.J.(2006)Chem Commun,2353-2355.
3.Ravichandran,K.G.,Boddupalli,S.S.,Hasemann,C.A.,Peterson,J.A.,andDeisenhofer,J.(1993)Science 261,731-736.
4.Li,H.,and Poulos,T.L.(1997)Nature Struct.Biol.4,140-146.
5.Munro,A.W.,Leys,D.G.,McLean,K.J.,Marshall,K.R.,Ost,T.W.,Daff,S.,Miles,C.S.,Chapman,S.K.,Lysek,D.A.,Moser,C.C.,Page,C.C.,andDutton,P.L.(2002)Trends Biochem.Sci.27,250-257.
6.Urlacher,V.B.,Lutz-Wahl,S.,and Schmid,R.D.(2004)Appl.Microbiol.Biotechnol.64,317-325.
7.Bell,S.G.,Hoskins,N.,Whitehouse,C.J.C.,and Wong,L.-L.(2007)MetalIons Life Sci.3,437-476.
8.Graham-Lorence,S.,Truan,G.,Peterson,J.A.,Falck,J.R.,Wei,S.,Helvig,C.,and Capdevila,J.H.(1997)J.Biol.Chem.272,1127-1135.
9.Oliver,C.F.,Modi,S.,Sutcliffe,M.J.,Primrose,W.U.,Lian,L.Y.,andRoberts,G.C.K.(1997)Biochemistry 36,1567-1572.
10.Noble,M.A.,Miles,C.S.,Chapman,S.K.,Lysek,D.A.,Mackay,A.C.,Reid,G.A.,Hanzlik,R.P.,and Munro,A.W.(1999)Biochem.J.339,371-379.
11.Cowart,L.A.,Falck,J.R.,and Capdevila,J.H.(2001)Arch.Biochem.Biophys.387,117-124.
12.Oliver,C.F.,Modi,S.,Primrose,W.U.,Lian,L.Y.,and Roberts,G.C.K.(1997)Biochem.J.327,537-544.
13.Carmichael,A.B.,and Wong,L.L.(2001)Eur.J.Biochem.268,3117-3125.
14.Sowden,R.J.,Yasmin,S.,Rees,N.H.,Bell,S.G.,and Wong,L.L.(2005)Org.Biomol.Chem.3,57-64.
15.Lussenburg,B.M.,Babel,L.C.,Vermeulen,N.P.,and Commandeur,J.N.(2005)Anal Biochem 341,148-155.
16.Wong,T.S.,Wu,N.,Roccatano,D.,Zacharias,M.,and Schwaneberg,U.(2005)J Biomol Screen 10,246-252.
17.Urlacher,V.B.,Makhsumkhanov,A.,and Schmid,R.D.(2006)Appl.Microbiol.Biotechnol.70,53-59.
18.van Vugt-Lussenburg,B.M.,Damsten,M.C.,Maasdijk,D.M.,Vermeulen,N.P.,and Commandeur,J.N.(2006)Biochem.Biophys.Res.Commun.346,810-818.
19.van Vugt-Lussenburg,B.M.,Stjernschantz,E.,Lastdrager,J.,Oostenbrink,C.,Vermeulen,N.P.,and Commandeur,J.N.(2007)J.Med.Chem.50,455-461.
20.Maves,S.A.,Yeom,H.,McLean,M.A.,and Sligar,S.G.(1997)FEBS Lett.414,213-218.
21.Li,Q.S.,Schwaneberg,U.,Fischer,P.,and Schmid,R.D.(2000)Chem.Eur.J.6,1531-1536.
22.Appel,D.,Lutz-Wahl,S.,Fischer,P.,Schwaneberg,U.,and Schmid,R.D.(2001)J.Biotechnol.88,167-171.
23.Li,Q.S.,Ogawa,J.,Schmid,R.D.,and Shimizu,S.(2001)Appl.Environ.Microbiol.67,5735-5739.
24.Schulze,H.,Schmid,R.D.,and Bachmann,T.T.(2004)Anal.Chem.76,1720-1725.
25.Sulistyaningdyah,W.T.,Ogawa,J.,Li,Q.S.,Maeda,C.,Yano,Y.,Schmid,R.D.,and Shimizu,S.(2005)Appl.Microbiol.Biotechnol.67,556-562.
26.Schwaneberg,U.,Schmidt-Dannert,C.,Schmitt,J.,and Schmid,R.D.(1999)Anal.Biochem.269,359-366.
27.Li,Q.S.,Schwaneberg,U.,Fischer,M.,Schmitt,J.,Pleiss,J.,Lutz-Wahl,S.,and Schmid,R.D.(2001)Biochim.Biophys.Acta 1545,114-121.
28.Farinas,E.T.,Bulter,T.,and Arnold,F.H.(2001)Curr.Opin.Biotechnol.12,545-551.
29.Glieder,A.,Farinas,E.T.,and Arnold,F.H.(2002)Nat.Biotechnol.20,1135-1139.
30.Meinhold,P.,Peters,M.W.,Chen,M.M.,Takahashi,K.,and Arnold,F.H.(2005)ChemBioChem 6,1-4.
31.Peters,M.W.,Meinhold,P.,Glieder,A.,and Arnold,F.H.(2003)J.Am.Chem.Soc.125,13442-13450.
32.Kubo,T.,Peters,M.W.,Meinhold,P.,and Arnold,F.H.(2006)Chemistry 12,1216-1220.
33.Munzer,D.F.,Meinhold,P.,Peters,M.W.,Feichtenhofer,S.,Griengl,H.,Arnold,F.H.,Glieder,A.,and de Raadt,A.(2005)Chem.Commun.,2597-2599.
34.Lentz,O.,Feenstra,A.,Habicher,T.,Hauer,B.,Schmid,R.D.,and B.,U.V.(2005)ChemBioChem 7,345-350.
35.Ortiz de Montellano,P.R.(1986)Cytochrome P450:Structure,Mechanism,and Biochemistry,Plenum Press,New York.
36.Ortiz de Montellano,P.R.(ed)(1995)Cytochrome P450:Structure,Mechanism,and Biochemistry,2
37.Ortiz de Montellano,P.R.(ed)(2005)Cytochrome P450:Structure,Mechanism,and Biochemistry 3rd Ed.,Kluwer Academic/Plenum Press,NewYork.
38.Poulos,T.L.,Finzel,B.C.,and Howard,A.J.(1987)J.Mol.Biol.195,687-700.
39.Poulos,T.L.(2005)Biochem.Biophys.Res.Commun.338,337-345.
40.Poulos,T.L.(2005)Drug Metab Dispos 33,10-18.
41.Poulos,T.L.(2003)Proc.Natl.Acad.Sci.USA 100,13121-13122.
42.Poulos,T.L.(2003)Biochem.Biophys.Res.Commun.312,35-39.
43.Hasemann,C.A.,Kurumbail,R.G.,Boddupalli,S.S.,Peterson,J.A.,andDeisenhofer,J.(1995)Structure 2,41-62.
44.Rupasinghe,S.,Schuler,M.A.,Kagawa,N.,Yuan,H.,Lei,L.,Zhao,B.,Kelly,S.L.,Waterman,M.R.,and Lamb,D.C.(2006)FEBS Lett.580,6338-6342.
45.Lepesheva,G.I.,and Waterman,M.R.(2007)Biochim.Biophys.Acta 1770,467-747.
46.Li,Q.S.,Ogawa,J.,Schmid,R.D.,and Shimizu,S.(2001)FEBS Lett.508,249-252.
47.Capdevila,J.H.,Wei,S.,Helvig,C.,Falck,J.R.,Belosludtsev,Y.,Truan,G.,Graham-Lorence,S.E.,and Peterson,J.A.(1996)J.Biol.Chem.271,22663-22671.
48.Skiles,G.L.,and Yost,G.S.(1996)Chem.Res.Toxicol.9,291-297.
49.Lanza,D.L.,and Yost,G.S.(2001)Drug Metab Dispos 29,950-953.
50.Reilly,C.A.,Ehlhardt,W.J.,Jackson,D.A.,Kulanthaivel,P.,Mutlib,A.E.,Espina,R.J.,Moody,D.E.,Crouch,D.J.,and Yost,G.S.(2003)Chem.Res.Toxicol.16,336-349.
51.Kassahun,K.,Skordos,K.,McIntosh,I.,Slaughter,D.,Doss,G.A.,Baillie,T.A.,and Yost,G.S.(2005)Chem.Res.Toxicol.18,1427-1437.
52.Sun,H.,Ehlhardt,W.J.,Kulanthaivel,P.,Lanza,D.L.,Reilly,C.A.,andYost,G.S.(2007)J Pharmacol Exp Ther 322,843-851.
53.Di Nardo,G.,Fantuzzi,A.,Sideri,A.,Panicco,P.,Sassone,C.,Giunta,C.,andGilardi,G.(2007)J Biol Inorg Chem 12,313-323.
54.Lentz,O.,Feenstra,A.,Habicher,T.,Hauer,B.,Schmid,R.D.,andUrlacher,V.B.(2006)ChemBioChem 7,345-350.
55.Cirino,P.C.,and Arnold,F.H.(2003)Angew.Chem.Int.Ed.42,3299-3301.
56.Sevrioukova,I.F.,Hazzard,J.T.,Tollin,G.,and Poulos,T.L.(1999)J.Biol.Chem.274,36097-36106.
57.Paulsen,M.D.,and Ornstein,R.L.(1995)Proteins:Struct.,Funct.,Genet.21,237-243.
58.Honeychurch,M.J.,Hill,H.A.O.,and Wong,L.L.(1999)FEBS Lett.451,351-353.
表A.CYP102A1血红素域(氨基酸残基1-470)与各种结构表征的细胞色素P450酶之间的序列相似性
*CYP505(P450foxy)未进行结构表征并且比对仅使用了血红素域
表B.CYP102A1整个序列与各种细胞色素P450酶之间的序列相似性(相对于Swissprot蛋白数据库中的蛋白进行比对)。可以注意到,尽管属于相同的子族,CYP102A2和CYP102A3仅59%和58%同源于CYP102A1。
表C.氨基酸的物理特性
表D.亲水性尺度(Hydropathy scale)
表1:CYP102A1变异体对萘的体外氧化活性、结合效率和选择性。速率以nmol·min
表2:CYP102A1变异体对丙苯的体外氧化活性、选择性和自旋迁移(spin shift)。速率以nmol·min
表3:CYP102A1变异体对甲苯的体外氧化活性和选择性。速率以nmol·min
表4:CYP102A1变异体对丁苯的体外氧化活性和选择性。速率以nmol·min
表5a:CYP102A1变异体对叔丁苯的体外氧化活性、选择性和自旋迁移性。速率以nmol·min
表5b:CYP102A1变异体对乙苯的体外氧化活性/选择性和自旋迁移性。速率以nmol·min
在检测器响应重新校准后对于F87A、F87A/KT2、KSK19和GVQ也获得了NB/结果。NADPH转化是相同的。结合效率为23(F87A)、21(F87A/KT2)、25(KSK19)、26(GVQ)。产物生成速率为32(F87A)、120(F87A/KT2)、178(KSK19)、572(GVQ)。1-醇为99%(F87A)、96%(F87A/KT2)、95%(KSKl9)、99%(GVQ)。苯乙烯现在对于这些变异体在反应中检测到为2%(F87A)、4%(F87A/KT2)、5%(KSK19)、1%(GVQ)。
表6a:CYP102变异体对邻二甲苯(o-xylene)的体外氧化活性和选择性。速率以nmol·min
表6b:CYP102A1变异体对间二甲苯的体外氧化活性和选择性。速率以nmol·min
表7:CYP102A1变异体对对甲基异丙基苯的体外氧化活性/选择性和自旋迁移性。速率以nmol·min
表8:一些CYP102A1变异体对枯烯的体外氧化活性和选择性。速率以nmol·min
表8a:在进一步GC分析之后,一些CYP102A1变异体对枯烯的体外氧化活性和选择性。速率以nmol·min
表9:CYP102A1变异体对戊烷的体外氧化活性、选择性和自旋迁移性。速率以nmol·min
表10:CYP102A1变异体对3-甲基戊烷的体外氧化活性/选择性和自旋迁移性。速率以nmol·min
表11:CYP102A1变异体对2-甲基丁烷的体外氧化活性和选择性。速率以nmol·min
表12:一些CYP102A1变异体对丁烷和丙烷的体外氧化活性和选择性。速率以nmol·min
表13:CYP102A1变异体对辛烷的体外氧化活性和选择性。速率以nmol·min
表14:一些CYP102A1变异体对1,4-二氯苯的体外氧化活性和选择性。速率以nmol·min
表15:CYP102A1变异体对朱栾倍半萜的体外氧化活性和选择性。速率以nmol·min
表16:一些CYP102A1变异体对R-和S-1,8-萜二烯的体外氧化活性和选择性。速率以nmol·min
表16a:CYP102A1变异体对R-1,8-萜二烯的体外氧化活性和选择性。N=NADPH转化速率。C=偶合。PFR=产物生成速率。速率以nmol·min-1·(nmol P450)-1为单位。产物为顺-1,2-1,8-萜二烯环氧化物(顺-1,2)、反-1,2-1,8-萜二烯环氧化物(反-1,2)、未鉴定产物(U)、顺-异胡椒醇(顺-3-醇)、反-异胡椒醇(反-3-醇)、香芹醇(2异构体)(6-醇(A)和(B))、香芹酮(6-酮)和紫苏子醇(10-醇)。
表17:一些CYP102A1变异体对(+)-α-蒎烯的体外氧化活性和选择性。速率以nmol·min-1·(nmol P450)-1给出。产物为2,3-蒎烯环氧化物(环氧化物),顺式-和反式-马鞭草烯醇(马鞭草烯醇)、马鞭草烯酮和桃金娘烯醇。由于在底物中的杂质,数据夸大偶合。
表17a:一些CYP102A1变异体对(+)-α-蒎烯的体外氧化活性和选择性。速率以nmol·min-1·(nmol P450)-1给出。N=NADPH转化速率。C=偶合。PFR=产物生成速率。速率以nmol·min-1·(nmol P450)-1给出。产物为(-)-2,3-蒎烯环氧化物((-)-2,3)、(+)-2,3-蒎烯环氧化物((+)-2,3)、顺-马鞭草烯醇(顺-4-醇)、反-马鞭草烯醇(反-4-醇)、马鞭草烯酮(4-酮)和桃金娘烯醇(10-醇)。
表18:CYP102A1变异体对芴的体外氧化活性和选择性。N=NADPH转化速率。C=偶合。PFR=产物生成速率。所有速率以nmol·min-1·(nmol P450)-1给出。产物为9-芴醇(9-醇)和2-芴酮(9-酮)。
表19:CYP102A1变异体对β-紫罗酮的体外氧化活性和选择性。N=NADPH转化速率。C=偶合。PFR=产物生成速率。速率以nmol·min-1·(nmol P450)-1给出。产物为4-羟基-β-紫罗酮(4-醇)。
表20:CYP102A1变异体对月桂(十二)酸的体外氧化活性和选择性。N=NADPH转化速率。C=偶合。PFR=产物生成速率。速率以nmol·min-1·(nmol P450)-1给出。产物为11-羟基十二酸(ω-1)、10-羟基十二酸(ω-2)、9-羟基十二酸(ω-3)、8-羟基十二酸(ω-4)和7-羟基十二酸(ω-5)。
- 活性型突变酶的制造方法及活性型突变酶、以及可溶性化突变蛋白的制造方法
- 活性型突变酶的制造方法及新型活性型突变酶、以及可溶性化突变蛋白的制造方法