一种Flavagliness天然产物不对称多样性导向合成方法
文献发布时间:2023-06-19 12:27:31
技术领域
本发明属于化学制备技术领域,涉及一种天然化合物的方法,具体涉及一种Flavagliness天然产物不对称多样性导向合成方法。
背景技术
Flavagliness是从树兰植物中分离得到的、具有特殊结构骨架和重要生理活性的一类天然产物。主要的生理活性包括:抗癌、消炎、神经和心脏保护作用等。其中,Flavagliness显著的抗癌活性引起了广大科学家的研究兴趣。在结构上,Flavagliness大都具有苯并呋喃并环戊烷的多环核心骨架。其中,环戊烷具有五个连续手性中心,给这类天然产物的不对称合成带来一定挑战。
1990年,Trost课题组报道了rocaglamide的第一例不对称全合成,通过底物诱导的、钯催化[3+2]环加成构建Flavagliness手性环戊烷骨架。2006年,Porco课题组结合手性辅剂与光催化作用,实现此类天然产物的高效仿生合成。2015年,Tius课题组利用钯与配体催化的Nazarov环化反应,完成Flavagliness家族天然产物的第一例催化不对称合成。尽管具有潜在的抗癌细胞活性和已报道的多例合成策略,Flavagliness类天然产物在其药物开发方面却一直未达到临床试验要求。同时,目前的工作和已知合成方法,都局限于芳环上取代基的改变,未能涉及烷基取代的Flavagliness的合成(R
因此,我们开展了Flavagliness类天然产物不对称多样性导向合成,通过合成多个类似天然产物建立化合物库,并进行深入的药物活性测试,以期筛选出最具癌细胞生长抑制性和选择毒性的新型抗癌药物。
发明内容
发明目的:本发明目的在于针对当下Flavagliness类天然产物临床试验的困境,提供一种多样性导向的不对称合成方法,本发明另一目的在于提供该方法制备的Flavagliness天然产物。
技术方案:本发明所述一种Flavagliness天然产物不对称多样性导向合成方法,由芳香性呋喃酮制备的烯丙基化前体发生钯催化的不对称脱羧烯丙基化得到第一中间体,再由第一中间体经过端位选择性Wacker氧化制得第二中间体,由第二中间体发生分子内苯偶姻缩合即得到第三中间体,第三中间体经脱氢、迈克尔加成构建三个连续的手性中心,得到第五中间体,最后由第五中间体合成Flavaglines天然产物,所述Flavaglines 天然产物如式(I)所示;
其中:
R
R
R
R
R
R
R
进一步地,作为较优实施方案,R
R
进一步地,作为较优实施方案,R
R
进一步地,作为较优实施方案,具体包括如下步骤:
(1)向反应瓶中加入芳香性呋喃酮衍生烯丙基碳酸酯,并加入溶剂、催化剂与配体,在-20~-15℃下反应40~50h,反应结束后除去溶剂,以正己烷/乙酸乙酯进行柱层析分离,得到第一中间体,如式(IV);
(2)向第一中间体中加入溶剂,并加入金属催化剂,在氧气氛围下室温反应20~30h,反应完成后加入淬灭剂,经萃取、干燥后,除去溶剂后经以正己烷/乙酸乙酯进行柱层析分离,得到第二中间体,如式(V);
(3)向第二中间体中加入溶剂,并加入卡宾前体、碱,室温反应4~5h。反应结束后除去溶剂,以正己烷/甲基叔丁基醚进行柱层析分离,得到第三中间体,如式(VI);
(4)向第三中间体中加入溶剂,并加入催化剂,在氧气氛围下70~90℃反应20~30h,反应完成后加入淬灭剂,经萃取、干燥后,除去溶剂后经以正己烷/甲基叔丁基醚进行柱层析分离,得到第四中间体,如式(VII);
(5)向第四中间体中加入溶剂,并加入催化剂、格式试剂,在-80~-75℃反应2~3h后升温至0℃继续反应1h。反应完成后加入淬灭剂,经萃取、干燥后,除去溶剂后经以正己烷/乙酸乙酯进行柱层析分离,得到第五中间体,如式(VIII);
(6)向第五中间体中加入溶剂,并加入酯化试剂,反应完成后加入淬灭剂。经萃取、干燥、除去溶剂后,向所得中间产物加入溶剂,并加入缩合剂、胺,在100℃反应3~5 h,反应完成后直接除溶剂。向所得中间产物酰胺继续加入溶剂,并加入还原剂,在 20~30℃反应3h,反应完成后加入淬灭剂,经萃取、干燥后,除去溶剂后经以正己烷/ 乙酸乙酯进行柱层析分离,得到Flavaglines天然产物,式(I)所示;
具体反应过程如下方程式:
进一步地,作为较优实施方案,步骤(1)中所述溶剂为四氢呋喃,催化剂为Pd
其中:Ar
所述环庚烷Trost配体的制备方法具体为:由环烯基甲酸甲酯与二芳基膦氧化合物发生加成反应得到第一中间体,再由第一中间体经一锅还原、硼化制得第二中间体,由第二中间体水解即得到第三中间体,最后由第三中间体与环庚烷手性二胺发生缩合反应得到新型Trost配体L1。
进一步地,作为较优实施方案,步骤(2)中所述溶剂为叔丁醇、硝基甲烷;催化剂为PdCl
进一步地,作为较优实施方案,步骤(3)中所述溶剂为四氢呋喃;缩合剂为氮杂卡宾试剂,所述氮杂卡宾试剂结构如式L2所示:
进一步地,作为较优实施方案,步骤(4)中所述溶剂为二甲基亚砜;脱氢试剂为醋酸钯;Lewis酸催化剂为三氟乙酸;淬灭剂为冰水混合液。
进一步地,作为较优实施方案,步骤(5)中所述溶剂为四氢呋喃;Lewis酸催化剂为CuBr·Me
步骤(6)中所述溶剂为N,N-二甲基甲酰胺,甲醇、甲苯、乙腈、乙酸;酯化试剂为Stile’s reagent、三甲基硅重氮甲烷,酰胺化试剂为4-二甲氨基吡啶、二甲基胺,还原剂为Me
本发明还提供了上述合成方法制备的Flavaglines天然产物,如式(I)所示;
其中:
R
R
R
R
R
R
R
有益效果:本发明开创性的研究了Flavaglines天然产物的合成方法,为这类天然产物的多样性合成提供了不对称合成策略,有效促进了Flavaglines类天然产物在药物活性测试方面的进一步发展;本发明中,通过发明人对原料特性的研究,针对性的设计新颖的配体,选择卡宾催化剂、氧化/还原剂、缩合剂等,配合相应的溶剂,不仅保证反应过程的顺利进行,还有助于提高产物的立体选择性和产率。
附图说明
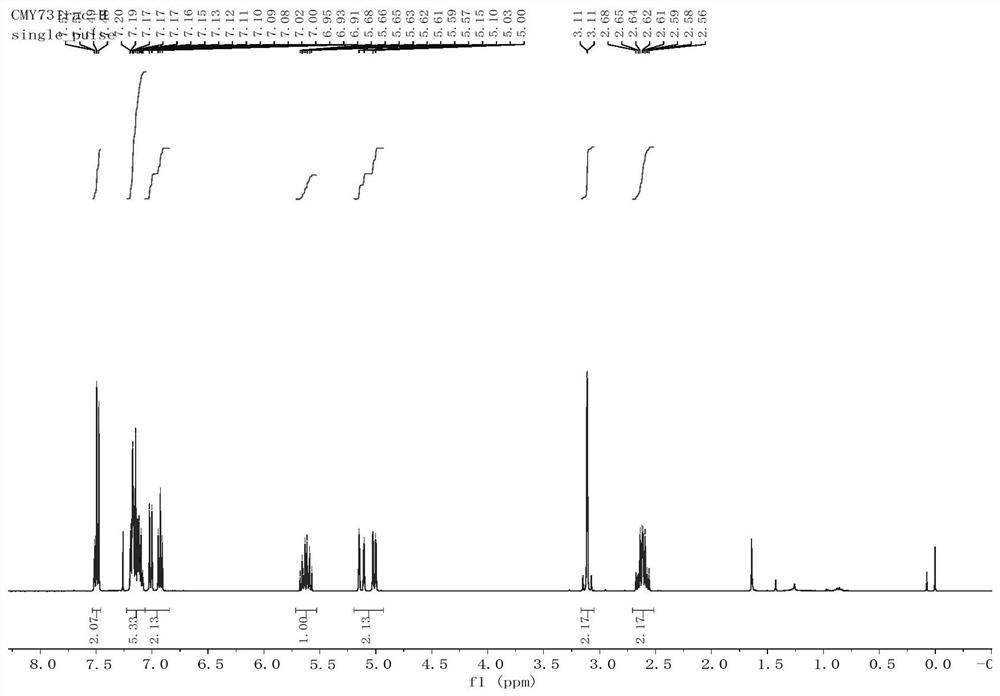
图1为实施例1中产物的
图2为实施例1中产物的
图3为实施例2中产物的
图4为实施例2中产物的
图5为实施例3中产物的
图6为实施例3中产物的
图7为实施例4中产物的
图8为实施例4中产物的
图9为实施例5中产物的
图10为实施例5中产物的
图11为实施例6中产物的
图12为实施例6中产物的
具体实施方式
下面通过附图对本发明技术方案进行详细说明,但是本发明的保护范围不局限于所述实施例。
实施例1:第一中间体的的制备
在氩气氛围保护下,将36.6mg Pd
实施例1所得产物的结构表征数据如下所示:
通过产物的核磁共振氢谱、碳谱,证明成功合成第一中间体。
式(IVa)化合物的比旋光度:[α]
HRMS:calculated for C
本实施例中环庚烷Trost配体具体结构为:
其制备方法为:(1)在室温及N
(2)在室温及N
在-10℃及N
在0℃及N
实施例2:第二中间体的制备
在室温及O
实施例2所得产物的结构表征数据如下所示:
通过产物的核磁共振氢谱、碳谱,证明成功合成第二中间体。
式(Va)化合物的比旋光度:[α]
HRMS:calculated for C
实施例3:第三中间体的制备
在N
实施例3所得产物的结构表征数据如下所示:
通过产物的核磁共振氢谱、碳谱,证明成功合成第三中间体。
式(VIa)化合物的比旋光度:[α]
HRMS:calculated for C
实施例4:第四中间体的制备
在氧气保护下,将136.9mg醋酸钯、45uL三氟乙酸、1.7g,6.1mmol第三中间体(VIa)加入到6.1mL二甲基亚砜中,并用氧气球鼓气10min。然后将反应升温至80℃反应 24h,反应结束后,将体系缓慢倒入30mL冰水混合物中,并用甲基叔丁基醚萃取三次。萃取得到的有机相用无水硫酸钠干燥,减压脱去溶剂得到粗产品,并进行柱层析,洗脱液:正己烷:乙酸乙酯=20:1~10:1,纯化粗产物,得到1.5g化合物第四中间体,产率 92%。
实施例4所得产物的结构表征数据如下所示:
通过产物的核磁共振氢谱、碳谱,证明成功合成第四中间体。
式(VIIa)化合物的比旋光度:[α]
HRMS:calculated for C
实施例5:第五中间体的制备
在氮气保护下,将4.1g溴化亚铜二甲硫醚络合物,2.78g,10mmol第四中间体(VIIa) 加入到100mL无水四氢呋喃中,然后将反应降温至-78℃。在此温度下搅拌5min,将3.5mL异丙基溴化镁(3mol/L in THF)逐滴加入反应体系中,滴加完毕在-78℃反应2 h后,升温至0℃继续反应1h。反应结束后,用饱和氯化铵溶液淬灭反应,水相用乙酸乙酯萃取三次。萃取得到的有机相用无水硫酸钠干燥,减压脱去溶剂得到粗产品,并进行柱层析,洗脱液:正己烷:乙酸乙酯=15:1~10:1,纯化粗产物,得到2.9g化合物第四中间体,产率90%。
实施例5所得产物的结构表征数据如下所示:
通过产物的核磁共振氢谱、碳谱,证明成功合成第五中间体。
式(VIIIa)化合物的比旋光度:[α]
HRMS:calculated for C
实施例6:Flavagliness天然产物(Ia)的制备
在氮气保护下,将322mg,1.0mmol第五中间体(VIIIa)加入到1mL N,N-二甲基甲酰胺中,逐滴加入1.5mL甲氧基镁碳酸甲酯(Stile’s reagent,2.0mol/L in DMF)。滴加完毕升温至100℃反应12h。反应结束后移至冰水浴中降温,加入5mL乙醚稀释后用冷的6mol/L盐酸溶液淬灭,并用乙醚萃取三次。萃取得到的有机相用无水硫酸钠干燥,低温条件下(0~5℃)减压除溶剂至3.0mL,得到粗产品的甲醇溶液。将0.5mL(三甲基硅烷基)重氮甲烷(2.0mol/L in hexane)加入到上述所得粗产品的甲醇溶液中。滴加完毕在0℃反应1h。反应结束后,直接减压脱去溶剂,得到酯化粗产品。
将上述所得粗产品、36.6mg 4-二甲氨基吡啶、5mL二甲基胺(2mol/L in THF)加入到12.55mL无水甲苯中,升温至100℃反应3h。反应结束后,减压除去溶剂,得到粗产品。
将上述所得粗产品、1.3g Me
实施例6所得产物的结构表征数据如下所示:
通过产物的核磁共振氢谱、碳谱,证明成功合成Flavagliness类天然产物(Ia)。
式(Ia)化合物的比旋光度:[α]
HRMS:calculated for C
本发明开创性的完成了多样性导向的Flavagliness类天然产物不对称合成研究,基于Pd催化不对称脱羧烯丙基化的关键策略,成功构建了烷基取代基的手性环戊烷,拓展了此类天然产物的药物活性测试范围,为Flavagliness类天然产物的合成研究及药物研发具有指导意义。
如上所述,尽管参照特定的优选实施例已经表示和表述了本发明,但其不得解释为对本发明自身的限制。在不脱离所附权利要求定义的本发明的精神和范围前提下,可对其在形式上和细节上作出各种变化。
- 一种Flavagliness天然产物不对称多样性导向合成方法
- 一种天然产物补骨脂酚及其对映体的不对称合成方法