阿霉素和紫草素共递送纳米胶束及其制备方法与应用
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于生物医药技术领域,具体涉及一种可以同时递送阿霉素和紫草素的阿霉素和紫草素共递送纳米胶束及其制备方法与应用。
背景技术
目前,乳腺癌仍然是女性中发病率最高的肿瘤,其中,三阴性乳腺癌(TNBC)由于缺乏雌激素受体(ER)、孕激素受体(PR)和人表皮生长因子受体2(HER-2)等,使用经典药物(如阿霉素)治疗的疗效有限,预后较差;同时,TNBC的治疗还存在易发生耐药的问题。
阿霉素(Doxorubicin,DOX)作为抗乳腺癌的一线小分子药物,主要通过上调上皮间充质转化(Epithelial-Mesenchymal Transition,EMT)途径发挥治疗作用。临床研究表明,DOX具有剂量相关的心脏毒性,且其用于治疗乳腺癌时容易引起耐药。通过将DOX包封于脂质体内以脂质体纳米药物的形式用于给药,可以有效降低DOX的心脏毒性,但该脂质体用于治疗乳腺癌时的疗效,仅与游离阿霉素疗效相当,疗效仍有待提高。
紫草素(Shikonin,SKN)是一种存在于紫草科植物紫草(Lithosperraumerythrorhizon Sieb.et Zucc.)的根、新疆紫草[Arnebia euchroma(Royle)Johnst]等植物中的天然生物活性物质,临床上主要用于治疗急、慢性肝炎。中国专利CN113181117B公开了一种紫草素和蒽环类化疗药共载脂质体,其通过将紫草素和蒽环类药物米托蒽醌或阿霉素同时包封于脂质体内制得的共载药脂质体用于治疗黑色素瘤时具有增效减毒的效果。然而,由于缺乏药物的靶向性,其达到有效剂量所需的剂量仍然很高,且其用于治疗乳腺癌的效果未知。此外,该共载脂质体用于治疗黑色素瘤时促进肿瘤细胞摄取的效果不明显,其对于肿瘤的治疗效果仍有待进一步提高。
发明内容
本发明的第一个目的在于提供一种阿霉素和紫草素共递送纳米胶束,以解决上述技术问题中的至少一个。
本发明的第二个目的在于提供上述阿霉素和紫草素共递送纳米胶束的制备方法,以解决上述技术问题中的至少一个。
本发明的第三个目的在于提供上述阿霉素和紫草素共递送纳米胶束在制备治疗乳腺癌的药物中的应用,以解决上述技术问题中的至少一个。
根据本发明的第一个方面,提供了一种阿霉素和紫草素共递送纳米胶束,其制备方法包括如下步骤:
将阿霉素、N,N’-羰基二咪唑(CDI)溶解于DMSO中,在温度为35-45℃的条件下搅拌2-6h,得混合液I;
将FA-PEG-COOH溶解在含有4-二甲基吡啶(DMAP)和1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDCI)的DMSO中,活化20-45min,得混合液II;
将混合液I和混合液II混合,在温度为35-45℃的条件下反应20-30h,反应产物在去离子水中透析(3000Da)4-8h,冷冻干燥,得FA-PEG-DOX;
将FA-PEG-DOX、紫草素溶解于DMSO中,在去离子水中透析(7000Da)3-6h,即得阿霉素和紫草素共递送纳米胶束。
药物释放实验结果表明,本发明提供的阿霉素和紫草素共递送纳米胶束在pH=7.4的PBS中药物释放速率较低,但在酸性条件下药物释放显著增加,具有pH敏感性,可以实现药物的定位释放。体外抗肿瘤药效测试结果表明,本发明提供的阿霉素和紫草素共递送纳米胶束对肿瘤的治疗效果显著高于游离双药,能有效抑制TNBC的活力和转移。此外,细胞摄取实验结果表明,本发明提供的阿霉素和紫草素共递送纳米胶束能够可显著增加细胞对DOX的摄取,并能维持肿瘤细胞质中DOX的毒性浓度,实现了持久的细胞摄取。
在一些实施方式中,阿霉素和紫草素的质量比可以为1:3~1:1。
在一些实施方式中,阿霉素和紫草素的质量比为1:1。由此,药物的协同作用最为显著,抗肿瘤效果最佳。
在一些实施方式中,阿霉素和N,N’-羰基二咪唑的质量比可以为1:3~1:2。
在一些实施方式中,FA-PEG-COOH和1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐的质量比可以为2:1~5:1;4-二甲基吡啶和1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐的质量比可以为1:2~1:1。
在一些实施方式中,阿霉素和FA-PEG-COOH的质量比可以为1:10~1:4。通过调节DOX与FA-PEG-COOH的比例可以调控纳米胶束的粒径,制备合适粒径的纳米胶束。
在一些实施方式中,阿霉素和FA-PEG-COOH的质量比可以为1:10。由此,制得的纳米胶束的流体力学尺寸较小,不大于200nm,制得的纳米胶束具有更强的肿瘤靶向性。
在一些实施方式中,FA-PEG-COOH的分子量可以为2000~3000Da。优选地,为2000Da。
在一些实施方式中,FA-PEG-COOH的制备方法可以包括如下步骤:
将叶酸(FA)和1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDCI)加入DMSO中,在温度为35-45℃的条件下活化20-45min;然后加入NH
本发明提供的阿霉素和紫草素共递送纳米胶束在相同的药物浓度下比游离药物具有更高的抗TNBC活性,能显著增加三阴性乳腺癌细胞对DOX的摄取,维持肿瘤细胞质中DOX的毒性浓度以及抑制三阴性乳腺癌细胞的活性和转移,具有显著TNBC靶向治疗效果,能够应用于制备治疗乳腺癌,尤其是三阴性乳腺癌的药物。
附图说明
图1为FA、NH
图2-4依次为不同浓度的游离DOX、游离CNK、游离DOX+游离CNK双联药物对MDA-MB-231细胞的毒性,其中,
图5为使用SynergyFinder 3.0计算协同效应得分进行组合协同得分分析的结果;
图6为FPD NMs和SKN@FPD NMs的表征图,其中,图(A)和图(B)分别为通过DLS测定FPD NMs的粒径和zeta电位结果图;图(C)和图(D)分别为通过DLS测定SKN@FPD NMs的粒径和zeta电位结果图;图(E)和图(F)依次为FPD NMs和SKN@FPD NMs的TEM图;
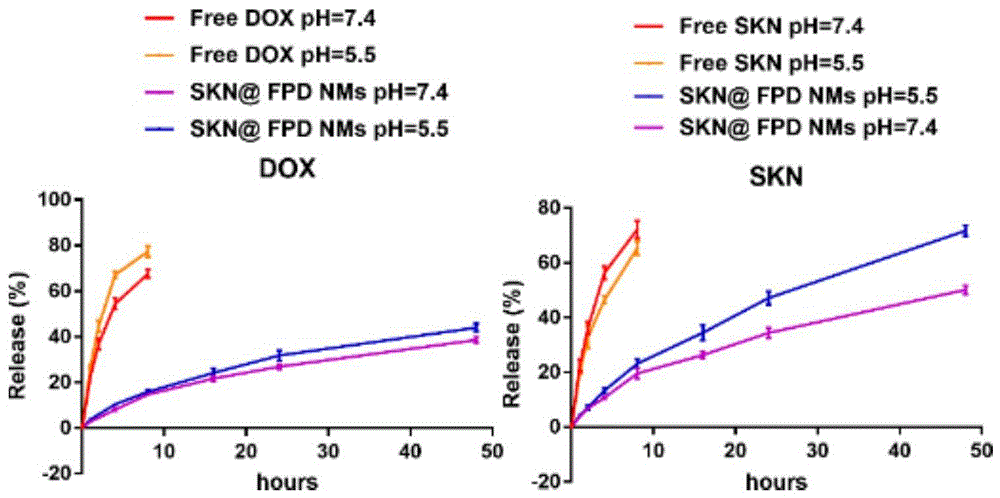
图7为体外生理条件(PBS,pH=7.4)和模拟肿瘤内体微环境(PBS,pH=5.5)的DOX和SKN的释放图;
图8为游离DOX、FA-PEG-DOX、游离DOX+游离SKN双联药物和SKN@FPD NMs对MDA-MB-231中的细胞毒性,其中,
图9-10为当DOX浓度为3μg/mL,DAPI浓度为1μg/mL时,细胞对游离DOX、游离DOX+游离SKN双联药物及SKN@FPD NMs的摄取效果图,其中,
图11-12为细胞迁移实验结果图,其中,
具体实施方式
下面结合具体实施例对本发明作进一步详细的说明,但本发明的实施方式不限于此。
下述实施例中涉及的材料,除特别注明的材料外,均可从商业渠道获得。对于未特别注明的工艺参数,可参照常规技术进行。
本发明中,阿霉素(DOX)、叶酸(FA)、二甲基亚砜(DMSO)、N,N’-羰基二咪唑(CDI)和4-二甲基吡啶(DMAP)购于阿拉丁(中国上海);NH
细胞培养:将三阴性乳腺癌细胞(MDA-MB-231)接种到25T培养瓶中,用含有10%胎牛血清和1%青霉素-链霉素的DMEM培养基培养。将培养瓶置于二氧化碳培养箱(37℃,5%CO
实施例1FA-PEG-COOH的制备
包括如下步骤:
将1g FA和1g EDCI加入到30mL的DMSO中,在40℃下活化30min。然后加入2g NH
实施例2FA-PEG-DOX纳米胶束的制备
包括如下步骤:
(1)将0.1g DOX和0.20g CDI溶解在10mL DMSO中,在40℃下搅拌4h,得混合液I;
(2)将1.0g实施例1制得的FA-PEG-COOH溶解在含有0.1g DMAP和0.2g EDCI的DMSO中,活化30分钟直到混合均匀,得混合液II;
(3)将混合液I和混合液II搅拌混合,在40℃下反应1天;最后,反应产物在去离子水中透析(3000Da)6小时,冷冻干燥,得FA-PEG-DOX;
(4)将10mg FA-PEG-DOX溶解于5mL DMSO中,在去离子水中透析(7000Da)4h,即得FA-PEG-DOX纳米胶束(FPD NMs)。
实施例3SKN@FA-PEG-DOX纳米胶束的制备
包括如下步骤:
(1)将0.1g DOX和0.20g CDI溶解在10mL DMSO中,在40℃下搅拌4h,得混合液I;
(2)将1.0g实施例1制得的FA-PEG-COOH溶解在含有0.1g DMAP和0.2g EDCI的DMSO中,活化30分钟直到混合均匀,得混合液II;
(3)将混合液I和混合液II搅拌混合,在40℃下反应1天;最后,反应产物在去离子水中透析(3000Da)6小时,冷冻干燥,得FA-PEG-DOX;
(4)将FA-PEG-DOX、0.1g SKN溶解于50mL DMSO中,在去离子水中透析(7000Da)4h,即得SKN@FA-PEG-DOX纳米胶束(SKN@FPD NMs)。
使用傅里叶变换红外光谱(FTIR)和核磁共振光谱(
对于FA-PEG-COOH,FITR光谱证明了其在1105cm
试验例1游离阿霉素(DOX)和紫草素(SKN)的细胞毒性和协同作用
将MDA-MB-231细胞接种到96孔板中(每孔5000个细胞),然后将DOX和SKN分散在完全培养基中,用等体积的含有不同浓度药物的完全培养基分别处理细胞2天。接着,用含10%CCK-8试剂的新鲜完全培养基替换上述培养基,处理细胞1h后检测490nm处的吸光度(OD)值。后续实验中,采用类似的方法来评价游离药物和纳米胶束的疗效。
细胞活性的计算方法如下:
其中,在无细胞的孔中加入含10%CCK-8试剂的培养基作为空白组,而在未经药物处理的细胞的孔中加入相同的含CCK-8试剂的培养基作为对照组。
此外,为探讨DOX和SKN在TNBC治疗中的协同作用疗效,通过两种药物不同浓度组合的处理,利用CCK-8试剂检测细胞活性来评价游离DOX和SKN的协同作用,并使用SynergyFinder 3.0计算协同效应得分。
结果如图2-5所示。
实验结果表明,随着药物浓度的增加,MBA-MD-231细胞的活性逐渐下降。通过计算可知,DOX的IC
将DOX和SKN共同处理MBA-MD-231细胞时,当DOX和SKN浓度分别为0.1μg/mL和0.1μg/mL时,细胞存活率降低到0.4328±0.068,而在0.2μg/mL DOX和0.3μg/mL SKN处理下,细胞存活率为0.2151±0.0267。如图5所示,DOX和SKN具有一定的协同效应,协同效应得分为3.75。当DOX和SKN的浓度比为1:1时,药物的协同作用更显著。
试验例2NMs的表征
使用动态光散射(DLS)测量实施例2和实施例3制得的纳米胶束的水动力尺寸和zeta电位,测量过程中的参数设置如下:
水动力尺寸:波长为658nm,温度为25±0.1℃,DLS角为90°;
zeta电位:1.4V/cm,13.0mA,25±0.1℃。
为了进行形态学测量,将NMs滴在铜网上,干燥后用透射电镜(TEM)扫描并拍照。
结果如图6所示。
FPD NMs的水动力尺寸为106.8±0.8nm,PDI为0.226±0.009;SKN@FPD NMs的水动力尺寸为121.8±1.1nm,PDI为0.216±0.020。与空白NMs相比,载药NMs的尺寸增大。
透射电镜图像显示两种NMs形状规则,大小均匀。同时,这两种NMs具有相似的zeta电位,其中,FPD NMs的zeta电位为-6.44±0.23mV,而SKN@FPD NMs的zeta电位为-6.33±0.16mV。
此外,FPD NMs和SKN@FPD NMs在低温下在3天内具有很高的稳定性。
试验例3NMs的载药量与药物释放的评价
为了检测载药量,首先检测了FA-PEG-DOX中DOX的取代度。制备浓度为2、4、6、8和10μg/mL的DOX盐酸溶液(pH=5.5),分别在紫外可见光吸收仪器上检测其在480nm处的吸光度,然后进行标准曲线绘制。检测20μg/mL的FA-PEG-DOX溶液(pH=5.5)在488nm处的吸光度,根据下述公式计算DOX的取代量:
采用双波长分光光度法测定了SKN@FPD NMs中的双药物浓度。通过检测双药在480nm和565nm的吸光度,绘制标准曲线,根据下述公式计算载药量:
根据上述公式,可以计算得到,实施例2制得的FA-PEG-DOX中DOX的含量为9.78±0.14%。同时,DOX和SKN在实施例3制得的SKN@FPD NMs中的载药量分别为8.86±0.21%和9.43±0.13%,SKN@FPD NMs的总载药量接近20%。
为了检测SKN@FPD NMs的药物释放情况,将5mL NMs放入透析袋(7000Da)中,在30mLPBS(pH=7.4或pH=5.5)中透析,在摇床上摇晃(37℃,75rpm)。在0、0.5、2、4、8、16、24和48h时用30mL新鲜PBS更换旧的PBS,调节旧的PBS的pH为5.5,然后在480nm和565nm处检测吸光度。双载药药物的释放量计算公式如下:
其中,Q
结果如图7所示,在前8h内,游离DOX和SKN在pH=7.4的PBS中快速释放,释放率分别为67.57±1.45%和72.12±2.61%;而游离DOX在pH=5.5的PBS中的释放率显著增加,为77.36±1.85%,游离SKN在pH=5.5的PBS中的释放率为65.32±2.08%。对于SKN@FPD NMs,在pH=7.4和pH=5.5的PBS中,DOX的释放率依次为38.56±1.05%、43.96±1.42%;SKN的释放率依次为50.11±1.33%、71.72±1.65%。结果表明,酸性环境会显著提高SKN的释放率,SKN@FPD NMs在生理条件下可以有效延缓药物的释放,而在肿瘤微环境或肿瘤内涵体中可以显著增加药物的释放。
试验例4NMs的体外细胞毒性
采用CCK-8试剂盒检测NMs和游离药物对TNBC的体外毒性。
结果如图8所示,在最大游离DOX浓度(0.094μg/mL)下,MBA-MD-231细胞存活率为0.5706±0.0283,而在最大FA-PEG-DOX浓度下,细胞存活率为0.6394±0.0202。PEG键似乎对DOX的毒性没有显著的影响。另外,MBA-MD-231细胞在最大浓度的双游离药物下的存活率为0.4252±0.0246;与之相比,细胞在对应药物浓度的SKN@FPD NMs下的存活率显著降低,为0.3730±0.0096。结果表明,本发明提供的的SKN@FPD NMs在相同的药物浓度下比游离药物具有更高的抗TNBC活性。
试验例5体外细胞摄取评估
将MDA-MB-231细胞接种到24孔板中(每孔2×10
结果如图9-10所示。游离DOX组细胞在4小时内持续摄取DOX,游离双药物组与游离DOX组有相似的DOX细胞摄取效果,游离DOX主要进入细胞核。与游离药物相比,SKN@FPD NMs在4小时内有更高的细胞连续摄取效果。此外,SKN@FPD NMs中的DOX部分分布在细胞核附近的细胞质中,在细胞核周围形成微弱的红光。以上结果表明,SKN@FPD NMs可显著增加细胞对DOX的摄取,并能维持肿瘤细胞质中DOX的毒性浓度。
试验例6细胞迁移试验
将MDA-MB-231细胞接种到24孔板中(每孔5×10
结果如图11-12所示。对照组细胞在6小时和12小时有明显的转移。而经游离DOX处理的细胞在12小时内无明显转移。同时,双游离药物组和NMs组在12h内与DOX组或对照组相比,几乎无转移。此外,在协同药物和SKN@FPD NMs处理6h或12h后,MDA-MB-231的细胞形态发生了明显的变化。
以上所述的仅是本发明的一些实施方式。对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。