一种基底上原位生长表面等离激元金属催化剂的方法及应用
文献发布时间:2024-04-18 20:00:50
技术领域
本发明属于先进材料技术,具体涉及一种基底上原位生长表面等离激元金属催化剂的方法及应用。
背景技术
目前针对电催化剂的制备方法主要有液相合成法、水热法、气相沉积法等,但往往这些方法存在制备周期长、涉及到高温高压环境,同时制备的催化剂还需要进一步转移到电极上、操作繁琐等问题。比如CN111293323B公开了一种多孔铂钯纳米立方材料及其制备方法及其在催化甲醇氧化中的应用,将制备得到的多孔铂钯纳米立方材料加入到水、炭黑的N,N-二甲基甲酰胺溶液和Nafion溶液,制成墨水,超声30分钟,将4微升墨水滴在玻碳电极上,自然晾干。现有技术利用磁控溅射技术或原子层沉积技术以预设质量比将过渡金属的单质或化合物沉积到碳粉表面;在保护气氛下,将表面沉积有过渡金属的单质或化合物的碳粉在预设温度下进行热处理;将热处理后的碳粉在纯水中分散,并加入铂的可溶性盐前驱体以及预设金属的可溶性盐前驱体,形成混合溶液;将混合溶液升温至反应温度并恒温搅拌,调节溶液pH值以生成碱性沉淀物溶液,过滤、烘干,得到碱性沉淀物前体;将碱性沉淀物前体在惰性气氛或还原性气氛下进行热处理,获得铂与预设金属构成的铂基合金催化剂。因此,开发一种简便快捷的催化剂制备方法仍然是一项的重要的挑战。
发明内容
本发明旨在开发一种简便快捷的表面等离激元电催化剂制备方法,可设计出具有不同元素比例、元素组成以及粒子尺寸的表面等离激元催化剂,制备方法简便,能够有效的缩短制备周期,避免高温高压等危险的实验环境,同时制备的电催化剂能够直接负载在碳纸基底上,可以直接作为工作电极应用于光辅助电催化反应中。通过本发明制备的表面等离激元电催化剂应用于光辅助电催化氧化反应和光辅助电催化还原反应,实现光辅助下性能的显著提升。
本发明采用如下技术方案。
一种基底上原位生长表面等离激元金属催化剂的方法,包括以下步骤:
(1)将带有金属前驱体的基底还原处理,得到复合基底;
(2)将复合基底、表面活性剂溶液、金属前驱体溶液混合,再加入还原剂,然后进行生长反应,实现基底上原位生长表面等离激元金属催化剂,得到表面等离激元金属催化剂及表面生长表面等离激元金属催化剂的基底。
本发明中,步骤(1)中,金属前驱体为一种金属前驱体,或者多种金属前驱体的组合,金属为等离激元金属中的一种或几种,比如金、银、铜中的一种或多种,或者金属为等离激元金属与其他金属的组合,比如金、银、铜的一种或多种与铂、钯、钌、钴、铑的一种或多种的组合。优选的,金属为金与另一金属,进一步优选的,金、另一金属的摩尔比为1:1~1:8,优选的摩尔比为1:1~1:5。优选的,每个金属对应一种金属前驱体。
本发明中,步骤(1)中,基底为柔性基底或者刚性基底,比如碳纸、碳布、硅片、石英片、玻璃、导电玻璃,比如氧化铟锡(ITO)导电玻璃、氟掺杂氧化铟锡(FTO)导电玻璃等,也可以为镍电极、气溶胶或者石墨烯电极等三维结构基底。
本发明中,步骤(1)中,还原处理利用还原剂进行,还原剂包括无机还原剂或者有机还原剂,比如硼氢化钠、抗坏血酸等。优选的,还原剂以浓度为0.01~1 M优选为0.05~0.5 M的还原剂溶液形式存在。还原处理的时间为5~60 s,优选10~30 s。
本发明中,步骤(1)中,将金属前驱体溶液滴在基底上,干燥后,得到带有金属前驱体的基底;优选的,金属前驱体溶液的浓度为1~30 mM,优选5~20 mM,进一步优选5~15mM;当金属前驱体为多种金属前驱体时,各金属前驱体的浓度独立存在。
优选的,步骤(1)中,金属前驱体溶液、基底的表面积的比例为(5~50 μL)∶1 cm
本发明中,步骤(2)中,金属的种类与步骤(1)一致。金属前驱体溶液的浓度为1~30 mM,优选5~20 mM,进一步优选5~15 mM;当金属前驱体为多种金属前驱体时,各金属前驱体的浓度独立存在。金属前驱体溶液的加入量、基底的表面积的比例为(0.1~2 mL)∶1cm
本发明中,步骤(2)中,还原剂包括无机还原剂或者有机还原剂,比如硼氢化钠、抗坏血酸等。优选的,还原剂以浓度为0.01~1 M优选为0.05~0.5 M的还原剂溶液形式存在。
本发明中,步骤(2)中,表面活性剂包括有机表面活性剂,比如十六烷基二甲基溴化铵;优选的,表面活性剂溶液的浓度为0.01~1 M,优选为0.05~0.5 M。
本发明中,步骤(2)中,生长反应的时间为20 s~20 min,优选30 s~10 min。
本发明公开了上述表面等离激元金属催化剂或者表面生长等离激元金属催化剂的基底作为催化剂的应用,比如在电催化醇类氧化反应中的应用或在电催化氮还原反应中的应用等。优选的,在光辅助电催化醇类氧化反应中的应用,在光辅助电催化氮还原反应(NRR)中的应用。
一种电催化醇类氧化的方法,以上述表面等离激元金属催化剂或者表面生长等离激元金属催化剂的基底作为电催化剂,优选为光辅助电催化醇类氧化。
一种电催化氮还原的方法,以上述表面等离激元金属催化剂或者表面生长等离激元金属催化剂的基底作为电催化剂,优选为光辅助电催化氮还原。
本发明旨在开发一种简便快捷的表面等离激元电催化剂制备方法,可设计出具有不同元素比例、元素组成以及粒子尺寸的表面等离激元催化剂;制备方法简便,能够有效的缩短制备周期,避免高温高压等危险的制备环境;同时制备的电催化剂负载在碳纸等基底上,可以直接作为工作电极应用于多种光/电催化反应中。通过本发明制备的表面等离激元电催化剂应用于光辅助电催化氧化反应和光辅助电催化还原反应,实现光辅助下电催化性能的显著提升。在7200 s后,AuPt/CP(原位生长在碳纸基底上的AuPt双金属催化剂,简称“AuPt/CP”)在光辅助MOR和EOR中仍能保持较高的电流密度,其活性仍显著高于商用Pt/C催化剂,体现了本发明合成的AuPt双金属催化剂在MOR和EOR中具有良好的稳定性。在连续测试5次后,AuCu/CP催化剂在光辅助下的电催化氮转氨反应性能均显著优于无光条件,尤其是仍能保持较好的稳定性,这进一步证明本发明表面等离激元电催化剂在光辅助氮转氨反应中具有较好的性能。
附图说明
图1为在碳纸基底上不同生长时间下的AuPt纳米粒子的SEM图和对应时间下的粒径分布图,生长30 s~10 min。
图2为AuPt/CP催化剂的形貌示意图,其中,(a)为低倍SEM图;(b)为AuPt/CP的TEM图,(α)、(β)为对应的高分辨TEM图;(c)为能量色散X射线元素分析图。
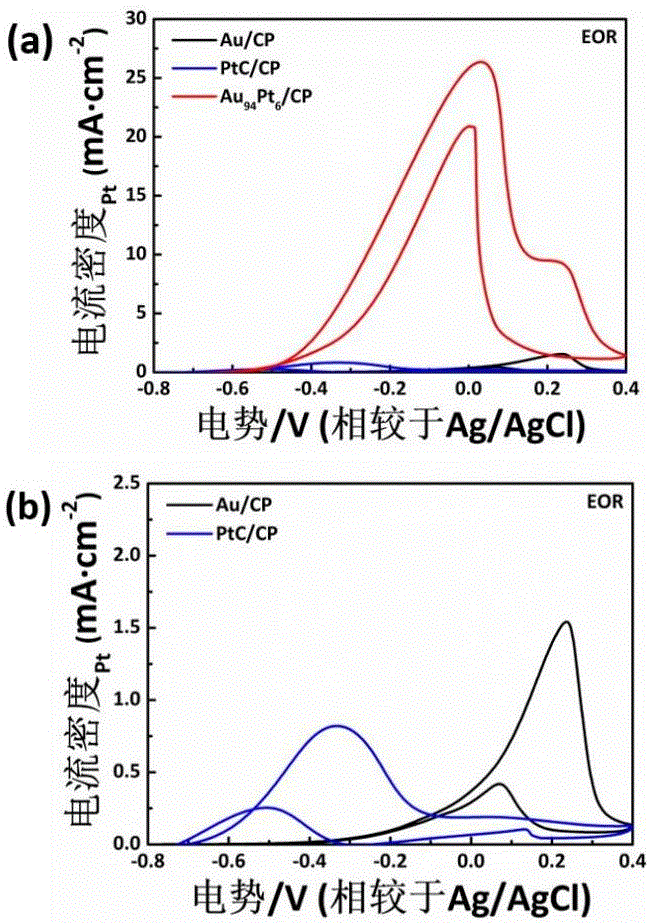
图3为不同Au、Pt元素比例的AuPt/CP在电催化氧化反应性能,其中,(a)为MOR图;(b)为EOR图。
图4为Au
图5为AuPt/CP在1 W·cm
图6为AuPt/CP和PtC/CP在不同电催化氧化反应中的恒电位时间-电流(I-t)曲线,其中,(a)为MOR图;(b)为EOR图。
图7为在碳纸基底上不同生长时间下的AuCu纳米粒子的SEM图和对应时间下的粒径分布图,生长3~10 min。
图8为AuCu/CP催化剂的形貌示意图,其中,(a)为Au
图9为在-0.3 V电压下AuCu/CP的光辅助电催化氮还原反应性能,其中(a)为在不同光功率密度下的氨产率和法拉第效率;(b)为氨产率与635 nm激光功率密度的关系。
图10为AuCu/CP催化剂在无光/光照条件下的电催化氮还原反应性能,其中,(a)为黑暗和AM 1.5G光辅助电催化循环测试5次的氨产率和法拉第效率;(b)为光辅助条件下,连续5次电催化测试的时间-电流曲线。
图11为对比样品纯Au/CP连续测试5次的光辅助电催化氮还原反应性能。
图12为AuPt/石英片催化剂的形貌示意图,其中,(a)为低倍SEM图;(b)为高倍SEM图。
具体实施方式
本发明将带有金属前驱体的基底还原处理,得到复合基底;将复合基底、表面活性剂溶液、金属前驱体溶液混合,再加入还原剂,进行二次生长反应,实现在基底上原位生长表面等离激元金属催化剂,得到表面生长表面等离激元金属催化剂的基底。
上述技术方案中,将金属前驱体溶液滴在基底上,干燥后,得到带有金属前驱体的基底;金属前驱体溶液的用量为(5~50)μL·cm
作为示例,以金与另一金属组成双金属,催化剂的制备方法如下:
(1)复合基底(AuX-CP)的制备:将两种金属前驱体盐溶液等体积混合;随后取混合前驱体盐溶液滴在碳纸上,室温下干燥;待溶液完全干燥后,将含有前驱体盐的碳纸浸入硼氢化钠溶液中,还原后用去离子水冲洗并用氮气枪吹干,得到AuX-CP复合基底;两种金属前驱体盐溶液浓度相同,为5~20 mM;
(2)AuX/CP的制备
取去离子水和十六烷基二甲基溴化铵(CTAB)于玻璃瓶中搅拌均匀,放入制备的AuX-CP并继续搅拌1 min,随后依次加入等量的氯金酸溶液和X前驱体溶液,待搅拌均匀后,加入抗坏血酸溶液,待溶液被还原为无色后开始计时,待反应结束后取出碳纸,并用去离子水清洗以除去多余的CTAB,用氮气枪吹干,得到AuX/CP。
根据上述方法,在碳纸上得到AuX电催化剂,优选的,X=Cu、Pt。
本发明采用的原料为市售产品,具体制备操作以及性能测试为常规方法;除有特殊说明,本发明的方法在室温、常压下进行。还原剂溶液的体积没有特别限定,可以没入基底即可。
实施例一
以金与铂组成双金属,催化剂的制备方法如下:
(1)复合基底(AuPt-CP)的制备:将浓度均为10 mM的氯金酸溶液、氯铂酸溶液等体积混合;随后取20 µL混合前驱体盐溶液滴在1 cm×1 cm的碳纸基底上,室温下干燥;待溶液完全干燥后,将含有前驱体盐的碳纸浸入0.1 M的硼氢化钠溶液中,还原20 s,后用去离子水冲洗并用氮气枪吹干;
(2)AuPt/CP的制备
取3 ml去离子水和6 mL 0.1 M十六烷基二甲基溴化铵(CTAB)溶液于玻璃瓶中搅拌均匀,转速设置为400 rpm;放入制备的AuPt-CP并继续搅拌1 min,随后依次加入等量(0.5 ml)的10 mM氯金酸溶液和10 mM氯铂酸溶液,待搅拌均匀后,加入1 mL 0.1 M的抗坏血酸溶液,待溶液被还原为无色后开始计时,生长时间为10 min,待反应结束后取出碳纸,用去离子水清洗以除去多余的CTAB,用氮气枪吹干。
在步骤(2)可以通过调控金前驱体盐、铂前驱体盐的比例以及生长时间获得不同元素比例和粒子尺寸的AuPt电催化剂AuPt/CP。本发明在碳纸电极基底上设计并制备了具有尺寸均匀、高催化活性、高密度的AuPt合金纳米粒子,不同生长时间的形貌与粒径分布如图1所示。通过透射电子显微镜进一步表征了AuPt纳米粒子在生长10 min时的形貌和晶格间距(即:Au
通过调整反应过程中前驱体氯金酸溶液和氯铂酸溶液的比例,制备了不同元素比例的AuPt/CP催化剂。具体地,通过电感耦合等离子体发射光谱仪(ICP-OES)对反应之后碳纸电极上的Au、Pt的元素含量进行定量分析,得到Au、Pt的元素比例,如表1所示,Au和Pt下标数据为两者摩尔比。
表1 不同比例的AuPt/CP中的Au和Pt的ICP-OES结果
实施例二
将催化剂成功应用到电催化甲醇氧化反应(MOR)和电催化乙醇氧化反应(EOR)中,并成功实现了光辅助电催化醇类氧化反应性能的显著提升。
本实验是在上海辰华的CHI 660 E电化学工作站上完成的,整个实验过程均在常温常压下进行。采用三电极体系:工作电极采用AuPt/CP,对电极采用铂丝,参比电极采用含有饱和氯化钾溶液的银/氯化银电极(Ag/AgCl)。电解液为1 M KOH和1 M CH
在进行测试前,将N
采用计时安培法分别在有/无光时测试催化剂在电催化甲醇氧化(或电催化乙醇氧化)性能的稳定性,时间设置为7200 s。
对比样品:纯Au/CP的制备方法遵循上述在碳纸上原位还原双金属纳米粒子的方法,区别在于省略氯铂酸。PtC/CP为市售的商用PtC充分研磨后分散在水溶液中并滴涂在1cm×1 cm的碳纸基底上,随后覆以萘酚溶液固定,以此制备而成,Pt含量为10微克。电化学测试方法与上述完全一致。
分别测试不同元素比例催化剂在MOR和EOR反应中的性能,其结果如图3所示,其中,(a)为MOR图;(b)为EOR图,不同Au、Pt比例的Au/CP催化剂在电催化醇类氧化反应中均表现出优异的催化性能;且Au
在635 nm的单波长激光照射的光辅助MOR测试中,Au
上述图中,EOR反应更复杂,产物更多;不同电位下的氧化峰代表EOR在不同反应路径生成不同产物的过程。
催化剂的稳定性是衡量催化剂性能的一个重要指标。将Au
以上实验可以说明,本发明采用Pt含量很低的催化,取得的催化性能显著高于现有Pt含量高的催化剂,出乎人们意料。
进一步的,本发明与现有电催化甲醇氧化反应(MOR)和电催化乙醇氧化反应(EOR)催化剂的性能比较见表2以及表3,可以确定,本发明制备的催化剂较现有技术公开了的同类产品具有显著的技术进步。
表2 本发明合成的催化剂与目前已报道的催化剂的MOR性能对比
表3 本发明合成的催化剂与目前已报道的催化剂的EOR性能对比
针对电催化醇类氧化反应,本发明制备的催化剂在电催化醇类氧化反应中具有高催化活性且具有等离激元性质,同时采用碳纸作为基底,能够保留催化剂的催化优势,同时可以借助等离激元性质实现光辅助下电催化性能的显著提升。
实施例三
以金与铜组成双金属,催化剂的制备方法如下:
(1)复合基底(AuCu-CP)的制备:将浓度均为10 mM的氯金酸溶液、硫酸铜溶液等体积混合;随后取20 µL混合前驱体盐溶液滴在1 cm×1 cm的碳纸基底上,室温下干燥;待溶液完全干燥后,将含有前驱体盐的碳纸浸入0.1 M的硼氢化钠溶液中,还原20 s,后用去离子水冲洗并用氮气枪吹干;
(2)AuCu/CP的制备
取3 ml去离子水和6 mL 0.1 M十六烷基二甲基溴化铵(CTAB)水溶液于玻璃瓶中搅拌均匀,转速设置为400 rpm;然后放入制备的AuCu-CP并继续搅拌1 min,随后依次加入等量(0.5 mL)的10 mM氯金酸溶液和10 mM硫酸铜溶液,待搅拌均匀后,加入1 mL 0.1 M的抗坏血酸溶液,待溶液被还原为无色后开始计时,反应时间为10min,待反应结束后取出碳纸,并用去离子水清洗以除去多余的CTAB,用氮气枪吹干,得到负载金与铜的碳纸,同样地,通过ICP-OES定量分析金、铜的含量,最终确定在此条件下Au:Cu的摩尔比为3∶1,即Au
在步骤(2)可以通过调控金前驱体盐、铜前驱体盐的比例以及生长时间获得不同元素比例和粒子尺寸的AuCu/CP电催化剂;具有较宽光谱响应、高密度、尺寸均匀的Au
实施例四
将催化剂应用到电催化氮还原反应中,成功实现光辅助氮还原反应性能的显著提升。
本实验的电化学测试也是在CHI 660 E电化学工作站进行的。电解液采用1M KOH溶液每次测试前需要提前通入N
图9为在-0.3 V电压下Au
作为对比,本实验制备了Au/CP。将0.1 mL Nafion D-521分散液加入到2 mL浓缩后的金纳米棒溶液中,并滴涂在1 cm×1 cm的碳纸基底上晾干使用,电化学测试方法与上述一致,其连续测试5次的实验结果如图11所示。AuCu/CP的法拉第效率(5.33%)显著优于纯Au/CP(金含量为0.2mg)的法拉第效率(3.98%),表明AuCu/CP具有更好的光辅助电催化氮还原性能,进一步证实通过本方法合成的双金属催化剂具有好的电催化性质。
上述循环测试为了获取氨的产率和法拉第效率,相当于是五次独立实验,为了不影响每次数据真实性,需要测试后更换新的电解液、通气30min除干扰、获取LSV曲线、测试I-t曲线。连续测试证明本催化剂的稳定性,在每次7200 s的I-t测试结束后不用更换电解液,直接进行下一个I-t测试。
进一步的,本发明与现有催化氮还原反应(NRR)催化剂的性能比较见表4,可以确定,本发明制备的催化剂较现有技术公开了的同类产品具有显著的技术进步。
表4 本发明合成的催化剂与目前已报道的催化剂的NRR性能对比
实施例五
通过改变纳米粒子生长的基底,证实本发明方法合成双金属纳米粒子的可行性。以AuPt纳米粒子为例,参照实施例一,使用石英片基底(替换碳纸),同样成功在石英基底上合成了尺寸均匀、高密度的AuPt合金纳米,具体步骤同上述一致,具体形貌如图12所示,生长时间为5 min,其中,(a)为低倍SEM图;(b)为高倍SEM图。
目前所报道的所有方法中,大多采用溶液法直接合成或通过高温高压条件合成,其合成步骤繁琐,且在溶液中合成的催化剂往往需要提纯,炭黑负载并进一步转移到催化电极表面(如玻碳电极、碳布、碳纸、镍网等)参与电催化反应,过程复杂且由于催化剂分散性差等因素,很难实现对催化剂的高效利用和优异的催化活性。本发明在常温常压下即可制备,通过调整前驱体的元素浓度和比例即可制备不同元素组成和元素比例的催化剂,操作简单,避免了催化剂提纯、二次转移、增加负载造成的催化剂损失的问题,也有效避免了传统催化剂在电极表面堆积导致的利用率低、活性差等问题,并增大了进一步商业应用的可行性。通过本方法制备的催化剂电催化性能显著优于目前通过其他方法报道的催化剂,如表2~表4所示,表现出了本发明非常显著的制备优势、应用优势与性能优势。
综上所示,本发明在碳纸基底上成功设计并合成了两种均具有表面等离激元性质的AuX合金纳米粒子催化剂,并分别应用到了电催化醇类氧化反应和电催化氮还原反应中,具有非常好的催化性能。