一种基于双链环化的MGI测序平台测序文库的构建方法
文献发布时间:2023-06-19 09:35:27
技术领域
本发明涉及高通量测序技术领域,具体涉及一种基于双链环化的MGI测序平台测序文库的构建方法。
背景技术
自20世纪解析出DNA分子双螺旋结构之后,生物学的发展进入了分子生物学的时代。为了更深入地探索生命的奥秘,对其复杂的密码解读便是第一步。随着人类基因组计划(HGP)的开展,开始尝试对物种进行大规模的生命密码的解读,在此过程中也催生了一些测序方法的发展,实现了对人类和一些重要的物种在基因组水平上遗传密码的测序,对生命密码的解读进入了基因组学的发展阶段。
在基因组学研究中,测序技术起着不可代替的作用。对于不同的测序方法中根据其发展过程将其大致分为三代。第一代DNA测序技术主要包括Maxam-Gilbert化学降解法和Sanger双脱氧终止法。第一代测序技术对人类基因组计划以及初期基因组测序起到了巨大的作用,但存在成本高,通量低,速度慢等不足。进入21世纪后,以Roche公司的454测序技术,Illumina公司的Solexa测序技术和ABI公司的SOLiD测序技术为代表的第二代测序技术发展起来,又被称为高通量测序技术,在保持了高准确度的同时,较大程度上降低了测序成本,提高了测序速度和通量,为基因组学的发展起到了巨大的推动作用。近年来,对于单分子测序技术的发展形成了第三代测序技术,主要代表有Pacific Biosciences公司的SMRT技术和Oxford Nanopore Technologies公司的纳米孔单分子测序技术,相对于前两代技术,第三代测序技术最大特点是可以实现单分子测序,另外还具有样品无需扩增,无需荧光标记,读长更长,后期数据处理更加方便等特点。
目前在进行基因组学方面的研究时主要应用的测序技术还是第二代高通量测序技术,相对一代和三代具有通量高,速度快,成本低等优势,非常适合大规模的基因组学水平的研究,短时间内不易被淘汰。而目前主流的商业平台大部分被illumina公司的各种型号的测序仪占据着,虽然测序的价格降了很多,但测大规模的样本时以及加上试剂盒的费用对大部分实验室开展相关研究时来说仍然是一个很大的限制。另外,测序仪是测序产业的上游平台,也是基因检测、编辑以及合成产业中的决定性设备,但长久以来测序核心技术大部分被国外企业把持,并有严格的专利进行保护,对国内研究开展和产业发展形成了巨大的障碍。
而国内华大集团旗下华大智造目前已成功开发了基于DNBseqTM测序技术的测序平台,并推出了BGISEQ-500等一系列测序仪,拥有完全的自主知识产权,相对于illumina测序平台,有着通量更大,成本更低等优势,在一定程度上打破了国外企业在国内测序产业垄断的限制,为国内研究和产业的发展打开了新的局面。
华大智造开发的MGISEQ系列测序仪主要拥有以下核心技术:1)DNA纳米球(DNB)技术,通过滚环扩增的方式实现对DNA片段的扩增,线性扩增方式能够确保每条扩增链始终扩增原始模板,其扩增错误不会像桥式PCR那样累积成指数型放大,最大程度还原了原始序列信息。2)规则阵列芯片(Patterned array),通过在硅片表面形成结合位点阵列和对准标记,实现DNB的规则排列,信号均匀,密度高,且彼此互不干扰,保证了测序准确度,而且提高了测序芯片的利用效率。3)优化的联合探针锚定聚合技术(cPAS),将DNA分子锚和荧光探针聚合在DNB上,利用连接测序(SBL)的方式读取序列信息,相对于合成测序(SBS)的方式碱基读取准确度更高。
MGI测序平台相对于illumina平台具备一定的优势,其文库构建与测序流程如图1所示。由于其建库与测序的一些固有特性,目前在文库构建过程中仍存在进一步改进和优化的空间。
(1)由于利用滚环的方式进行目的片段的扩增,所以需要以单链环状DNA为上机扩增起始样品文库,文库构建相对于常规流程多了产生环状单链的环化过程,使得整个文库构建过程变得繁琐和耗时;
(2)单链环化方式在效率上只能使一小部分PCR产物发生成环反应产生有效数据,对文库均一化和信息原始性造成了一定的影响。
有鉴于此,需要对现有的MGI测序平台文库的构建方法进行改进,以使建库整个过程更简单、快捷;另外杜绝由于扩增产生的信息改变。
发明内容
本发明所要解决的技术问题是现有的MGI测序平台文库的构建方法,繁琐、耗时,并且对文库均一化和信息原始性有一定影响的问题。
为了解决上述技术问题,本发明所采用的技术方案是提供一种基于双链环化的MGI测序平台测序文库的构建方法,包括以下步骤:
构建双链结构的接头骨架,所述双链结构的接头骨架具有MGI测序平台要求的接头序列,且可供TA连接,在中间部分保留有一段单链结构;
对样本DNA进行片段化并进行末端修复加A;
以构建好的所述双链结构的接头骨架和末端修复加A的样本DNA片段为底物,利用TA连接成环,生成双链连接产物;
使用外切酶对所述双链连接产物进行消化,只保留成环的那条单链环状结构产物,然后对消化产物进行纯化,从而获得兼容MGI测序平台的最终文库。
在上述方法中,所述双链结构的接头骨架的3端均有T碱基突出,用于和加A之后的样本DNA片段进行互补配对。
在上述方法中,所述双链结构的接头骨架的序列为:
在上述方法中,通过超声或者酶切打断的方式将所述样本DNA片段化,片段化后的样本DNA分布在200bp-700bp范围内。
在上述方法中,文库构建全过程包括样本DNA打断、DNA片段筛选、修复加A、双链环化、单链化以及文库纯化6个反应阶段,总耗时3-4小时。
在上述方法中,样本DNA片段化的时间为20min、DNA片段筛选的时间为30min、修复加A的时间为50min、双链环化的时间为35min、单链化的时间为35min、文库纯化的时间为35min。
在上述方法中,利用磁珠进行样本片段DNA的筛选和纯化。
与现有技术相比,本发明在环化过程中利用TA连接的原理在双链基础上进行环化,然后获得可以进行直接上机的单链环状DNA文库;整个建库过程变得更简单、快捷,而且提高了一定的环化效率;另外在建库中间过程由于未经过片段PCR扩增而保留了样本DNA序列的原始信息,杜绝了由于扩增产生的信息改变。
附图说明
图1为MGI测序平台常规的文库构建与测序流程示意图;
图2为本发明方法的流程示意图;
图3为本发明中构建的连接接头骨架产物电泳图;
图4为本发明实施案例中的文库产物RCA产物电泳图。
具体实施方式
本发明提供了一种基于双链环化的MGI测序平台测序文库的构建方法,测序文库的构建过程简单、快捷,并且杜绝了由于扩增产生的样本DNA信息改变。下面结合说明书附图和具体实施方式对本发明做出详细说明。
为了对本发明的技术方案和实现方式做出更清楚地解释和说明,以下介绍实现本发明技术方案的几个优选的具体实施例。显然,以下所描述的具体实施例仅为本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
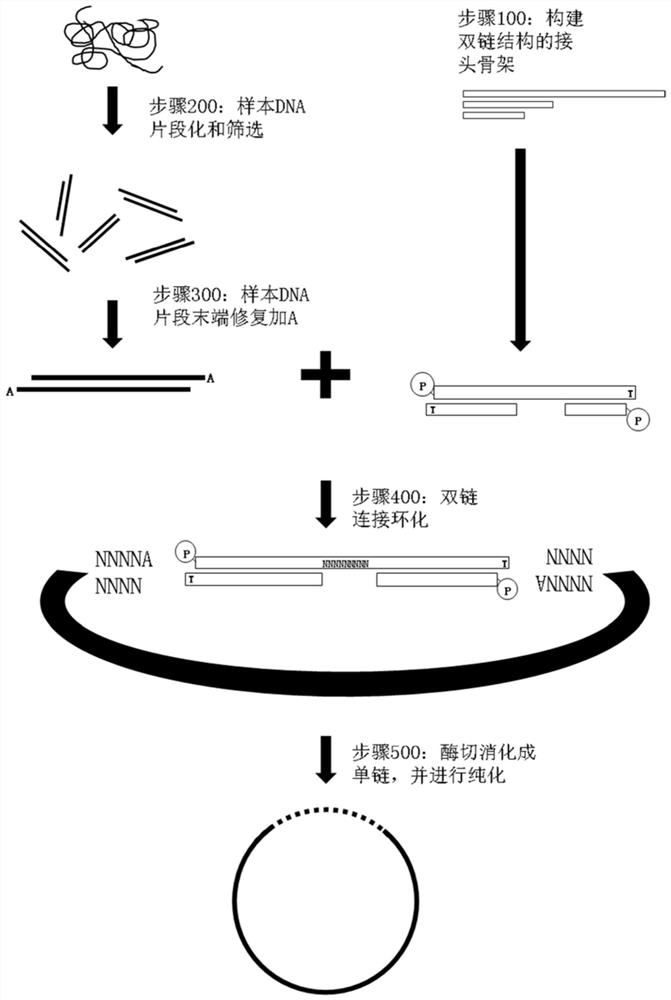
图2为本发明提供的基于双链环化的MGI测序平台测序文库的构建方法流程图,在环化过程中利用TA连接原理在双链基础上进行环化,然后获得可以直接上机的单链环状测序文库。如图2所示,本发明方法包括以下步骤:
步骤100:构建双链结构的接头骨架,该接头骨架可供TA连接且兼容下游文库生成。
由于是在双链的基础上利用TA连接的原理进行环化,所以需要提前构建上述可供TA连接,并且兼容下游文库生成的双链结构的接头骨架,此双链结构的接头骨架满足以下要求:
(1)连接部分的DNA片段为双链结构,以使连接酶进行高效识别和连接;
(2)3端均有T碱基突出,以能和加A之后的样本DNA片段进行互补配对;
(3)双链结构的接头骨架序列为MGI测序平台所用的接头序列,以保证最终文库与传统文库序列结构完全一致;
(4)双链结构的接头骨架的中间部分片段为单链结构DNA形式,以实现环化后形成单链环状结构。
一种优选的双链结构的接头骨架的序列为:
步骤200、对样本DNA打断,进行片段化,并筛选。
步骤300、筛选出的样本DNA片段末端修复加A。
步骤400、以构建好的双链结构的接头骨架和末端修复加A的样本DNA片段为底物,利用TA连接成环,生成双链连接产物。
该步骤利用TA连接的原理进行连接成环反应,反应过程和常规连接反应一致,但反应体积需要增大,反应Buffer需要调整,以减少接头骨架与样本DNA片段多重连接的线性结构产物,提高环状产物的产生和积累。
步骤500、使用外切酶对所述双链连接产物进行消化,只保留成环的那条单链环状结构产物,然后对消化产物进行纯化,从而获得兼容MGI测序平台的最终文库。
在以上方法中,步骤100的详细步骤如下:
步骤110、将构建双链连接的三段骨架单链DNA片段用水稀释至100μmol;
其中,DNA片段1序列为:
AGTCGGAGGCCAAGCGGTCTTAGGAAGACAANNNNNNNNNNCAACTCCTTGGCTCACAGAACGACATGGCTACGATCCGACTT;
DNA片段2序列为:
TTGTCTTCCTAAGACCGCTTGGCCTCCGACTT;
DNA片段3序列为:
AGTCGGATCGTAGCCATGTCGTTCTGTGAGCC。
步骤120、在PCR管中配置如下反应体系:
步骤130、涡旋混匀,瞬时离心;
步骤140、把PCR管放置于PCR仪上,反应程序如下:
其中,从95℃降温至25℃,采用0.1℃/s缓慢降温的方式,这样可以使反应的效果最好。
步骤150、反应产物用水稀释至50ul,置于4℃待用,或者置于-20℃储存。
在以上方法的步骤200中,将样本DNA打断进行片段化(约20min)的详细步骤如下:
通过超声或者酶切打断的方法将样本DNA片段化,片段化后DNA分布在200bp-700bp范围内效果较好;
将DNA片段化产物补水至总体积50μL,全部转移到新的1.5mL离心管中,进行磁珠片段筛选。
在以上方法的步骤200中,DNA片段筛选与纯化(约30min)的详细步骤如下:
取出磁珠置于室温平衡,充分涡旋混匀;
吸取30μL磁珠至上述DNA片段化产物中,吸打至完全混匀;
室温孵育5min,将离心管置于磁力架上;
静置2-5min至液体澄清,用移液器小心吸取上清至新的1.5mL离心管中;
用移液器吸取10μL纯化磁珠至上述80μL上清中,吸打至完全混匀;
室温孵育5min,将离心管置于磁力架上;
静置2-5min至液体澄清,用移液器小心吸取并丢弃上清;
保持离心管置于磁力架上,加入500μL新鲜配制的80%乙醇,静置30s后吸取并丢弃上清;
重复上一步,用移液器将管内残留液体吸干净;
保持离心管置于磁力架上,打开离心管管盖,室温干燥~5min,避免过度干燥;
将离心管从磁力架上取下,加入40μL洗脱液,用移液器轻轻吸打至完全混匀;
室温孵育5min,瞬时离心,将离心管置于磁力架上;
静置2-5min至液体澄清,将40μL上清液转移到新的0.2mL PCR管中,得到筛选与纯化后的样本DNA片段。
在以上方法的步骤300中,样本DNA片段末端修复与加A(约50min)的详细步骤如下:
在冰上配置反应液如下:
吹打混匀,瞬时离心;
将PCR管置于PCR仪上,按照如下程序进行反应:
取出稍微离心,放在冰上,以进行下一步的双链环化操作。
在上述方法的步骤400中,双链环化(约35min)的详细步骤如下:在冰上配置连接反应液如下:
涡旋震荡混匀,瞬时离心;
将PCR管置于PCR仪上,按照如下程序进行反应;
取出稍微离心,放在冰上,以进行下一步单链化操作。
在上述方法中,步骤500中的单链化(约35min)的详细步骤如下:在冰上配置酶切消化反应液如下:
涡旋震荡混匀,瞬时离心;
将PCR管置于PCR仪上,按照如下程序进行反应;
取出瞬时离心;
向上述反应产物中加入10ul酶切反应终止液,涡旋混匀,瞬时离心,然后将全部反应液转移至新的1.5mL离心管中。
在上述方法中,文库纯化(35min)的详细步骤如下:
提前取出纯化磁珠置于室温平衡,并充分震荡混匀;
吸取200μL纯化磁珠至上述酶切消化产物中,用移液器轻轻吸打至完全混匀;
室温孵育10min;
将离心管置于磁力架,静置2-5min至液体澄清,吸取并丢弃上清;
保持离心管置于磁力架上,加入500μL新鲜配制的80%乙醇,静置30s后小心吸取并丢弃上清;
重复上一步,吸干管内残留液体;
保持离心管固定于磁力架上,打开离心管管盖,室温干燥2~5min,避免过度干燥;
将离心管从磁力架上取下,加入20μL洗脱液,用移液器轻轻吸打至完全混匀;
室温下孵育5min;
将离心管置于磁力架上,静置2-5min至液体澄清,将20μL上清液转移到新的1.5mL离心管中;
上述产物即为最终文库,可进行下一步质检和上机测序,也可放置于-20℃冰箱储存。
整个建库过程,准备时间与反应时间加起来总耗时约3-4个小时。
对本发明方法中的几个关键步骤进行了可行性验证,以证明本技术方案可以实现上述目的,主要验证结果如下:
(1)对双链结构的接头骨架构建成双链的验证。
双链结构的接头骨架主要通过合成的单链DNA片段,经退火复性以及磷酸化形成,一个长单链和与其互补的两条短单链在合适的浓度下通过PCR仪缓慢降温,从而复性成一个长的双链结构,即为双链结构的接头骨架。以没经过复性的单链片段为对照,经凝胶电泳分析后,构建的双链结构的接头骨架形成了预期的长度,电泳胶图如图3所示,泳道1为双链结构的接头骨架反应产物,泳道2为未经过退火反应的片段对照,根据电泳所示大小对比证明了双链接头构建成功。
(2)以双链形式进行成环反应的验证。
双链成环主要以前期构建成功的接头骨架以及片段化后修复加A的样本DNA片段为反应物,以TA连接的方式将一个接头骨架的两端与一个样本DNA片段的两个分别连接形成双链环状结构。然后使用外切酶对连接产物中未成环的接头骨架、样本DNA片段以及双链环状结构中含有缺口的那条链进行酶切消化,以最终获得单链环状文库。为了验证双链连接的成功以及环状结构的形成,以酶切消化纯化后的产物为模板,进行滚环扩增反应,同时以使用华大建库试剂盒所形成的单链环状产物作为对照。对扩增产物进行凝胶电泳,结果如图4所示,泳道2样品为根据MGI建库方法所得文库,泳道1样品为根据本发明方法接头所得文库,两组文库样品经过RCA反应都产生了滚环扩增产物,从而验证了在双链形式上进行成环反应的可行性。
(3)对上述产生的环状产物进行了纯化和浓度测定,然后在华大测序平台进行上机测序,最终在测序数据中产生了目的基因组的有效数据,即证明了本发明可用于DNA文库的构建和测序。
本发明方法,与MGI提供的常规方法在操作时间和环化效率方面对比如下,以验证本发明的优越性和价值性。
在操作时间上,从基因组片段化为反应起始过程,得到最终环状纯化的文库为反应结束过程,本发明所设计的方法主要经过6个反应阶段(样本DNA打断约20min、DNA片段筛选约30min、修复加A约50min、双链环化约35min、单链化约35min、文库纯化约35min),总耗时约3-4个小时,半天即可完成整个文库的构建;而MGI目前官方提供的方法需主要经过11个反应阶段(样本DNA打断约20min、DNA片段筛选30min、修复加A约50min、接头连接约35min、产物纯化约30min、PCR约35min、产物纯化约30min、双链变性约10min、单链环化约35min、产物消化约35min、文库纯化约35min),总耗时约6-7个小时,在时间消耗上,本发明方法将近缩短了一半。
在环化效率上,以同一份样品DNA为起始材料,在环化反应过程中投入等物质的量的片段化的双链DNA,然后分别进行本发明所用的双链环化以及MGI目前提供的单链环化,对环化产物进行酶切消化和磁珠纯化以去除未环化的片段和接头,接着使用Qubit荧光定量试剂和仪器对最终产物进行浓度测序,以最终环化产物的质量与起始投入DNA片段的质量的比值作为环化效率的评估。使用本发明方法得到的比值可达20%~30%,使用MGI目前提供的单链环化方法的得到在比值在10%左右。
综上,本发明主要针对目前MGI测序平台高通量测序文库构建的方法进行新的设计和优化,提供了一个新的文库构建流程,与目前MGI提供的全基因组常规文库的构建方法相比主要有以下优势:
(1)操作过程更简单,整个过程所需时间更短;
(2)所用酶试剂种类更少,建库成本更低;
(3)建库过程中片段未经过PCR扩增,杜绝了由于扩增而产生的序列改变;
(4)双链环化效率更高,使更多的DNA片段形成最终有效测序文库。
以上描述的本发明方法,以样本DNA为起始样品类型构建MGI测序平台全基因组测序文库,但本发明方法同样适用于甲基化文库和以RNA为样品起始类型的转录组等相关兼容文库的构建。
本发明并不局限于上述最佳实施方式,任何人应该得知在本发明的启示下做出的结构变化,凡是与本发明具有相同或相近的技术方案,均落入本发明的保护范围之内。