一种靶标基因富集建库方法
文献发布时间:2023-06-19 10:41:48
技术领域
本发明涉及基因测序技术领域,具体涉及一种靶标基因富集建库方法。
背景技术
高通量测序靶标基因测序文库的制备流程一般分为两套流程,主流的捕获建库方法需要经历文库构建(包括新型建库方法单链建库)、捕获前扩增、杂交捕获、捕获后扩增四个必需步骤,全流程一般长达2到3天。另一种常见方法称为扩增子建库,一般先做多重PCR,后对PCR产物建库,有的商业化试剂盒会在做多重PCR时,在引物的5′端外侧加上对应NGS平台的接头序列,以将上述两步整合为一步(即一步法多重PCR试剂盒)。
第一种主流技术路线必须将文库构建和杂交捕获严格分开,步骤繁多周期长,各模块之间衔接有一定技术难度,且杂交捕获所需试剂和捕获专用探针合成严重依赖进口。第二种技术路线虽然流程较前者更简洁,但因其基于多重PCR,有如下诸多问题:1、因其基于PCR扩增,使文库中存在大量扩增所得同样的拷贝分子,导致测序所得结果的冗余率高,浪费测序数据量,且使有效数据率降低,导致超低频的基因突变难以检出或无法准确检出;2、由于PCR引物之间互相干扰,同一反应体系里PCR重数(即多少个单一PCR混合在多重PCR反应)无法过多(一般不超过1000重),导致较大panel的基因检测很难通过单管反应完成,只能分成多个单管反应,然后合并产物来实现,大大增加了成本,延长了操作时间,不利于推广;3、PCR需要两端引物配对,导致其无法检测未知的融合基因(novel fusion)和病毒插入位点等基因组结构性变异;4、PCR的指数性扩增使不同基因间的拷贝数没有得到等比例的线性放大,导致其无法检测基因拷贝数变异;5、多重PCR不可避免的扩增偏好性导致均一度低,导致panel中部分区域不能很好覆盖(这些区域的突变无法准确检出),而部分区域过多覆盖(造成局部测序数据量浪费)。
发明内容
本发明提供一种靶向富集建库方法
根据第一方面,一种实施例中提供一种靶标基因富集建库方法,包括:
磷酸化步骤,包括在模板分子的5’端修饰磷酸基团;
延伸步骤,包括加入含与靶标基因互补配对序列的第一引物,所述第一引物的5’端修饰有标记分子,在模板分子的靶标区域退火并延伸,获得双链靶核苷酸分子,所述双链靶核苷酸含有由5’端修饰有标记分子的第一引物延伸得到的延伸链;
第一测序接头连接步骤,包括将第一测序接头连接至前一步骤延伸处理后的双链靶核苷酸分子;
解链步骤,包括将前一步骤所得产物解链处理,去除5’端修饰有标记分子的第一引物延伸链,得到连接有第一测序接头的原始模板分子,即为单链靶核苷酸分子;
第二测序接头连接步骤,包括加入发夹式第二测序接头,使单链靶核苷酸分子连接所述发夹式第二测序接头;
扩增步骤,包括加入含与第一测序接头反向链互补配对序列的第二引物、含与第二测序接头反向链互补配对序列的第三引物,PCR反应,得到完整文库。
依据上述实施例的靶标基因富集及建库方法,将建库和靶标基因富集两步整合为一个流程,显著缩短实验所需时间。
附图说明
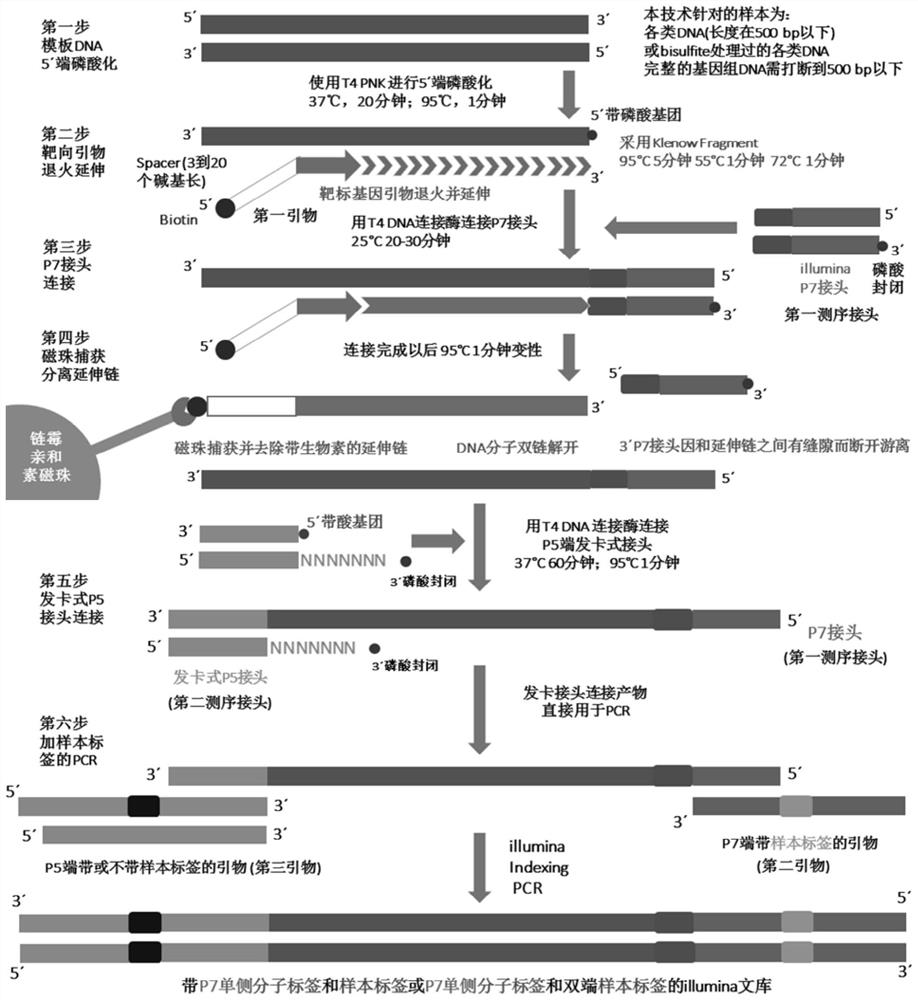
图1显示为一种实施例的流程图。
具体实施方式
下面通过具体实施方式结合附图对本发明作进一步详细说明。其中不同实施方式中类似元件采用了相关联的类似的元件标号。在以下的实施方式中,很多细节描述是为了使得本申请能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他元件、材料、方法所替代。在某些情况下,本申请相关的一些操作并没有在说明书中显示或者描述,这是为了避免本申请的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,他们根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
另外,说明书中所描述的特点、操作或者特征可以以任意适当的方式结合形成各种实施方式。同时,方法描述中的各步骤或者动作也可以按照本领域技术人员所能显而易见的方式进行顺序调换或调整。因此,说明书和附图中的各种顺序只是为了清楚描述某一个实施例,并不意味着是必须的顺序,除非另有说明其中某个顺序是必须遵循的。
本文中为部件所编序号本身,例如“第一”、“第二”等,仅用于区分所描述的对象,不具有任何顺序或技术含义。而本申请所说“连接”、“联接”,如无特别说明,均包括直接和间接连接(联接)。
现有技术中,NGS靶向文库的制备流程(包括新型建库方法单链建库)一般分为两套流程,主流的捕获建库方法需要经历文库构建、捕获前扩增、杂交捕获、捕获后扩增四个必需步骤,全流程一般耗时长达2到3天。另一种常见方法称为扩增子建库,一般先做多重PCR,后对PCR产物建库,有的商业化试剂盒会在做多重PCR时在引物的5’端外侧加上对应NGS平台的接头序列,以将上述两步整合为一步。
第一种主流技术路线必须将文库构建和杂交捕获严格分开,步骤繁多,周期长。第二种技术路线虽然流程较前者更简洁,但因其基于多重PCR,有如下诸多问题:1、建库起始投入量需要较高;2、同一反应体系里靶标位点数(plex数)无法过多,导致较大探针库(panel)的基因检测很难通过单管反应完成,只能分成多个单管反应,然后合并产物来实现,大大升高了成本和操作时间,限制了单管反应检测通量,不利于推广;3.PCR需要两端引物配对,导致其无法检测未知基因融合(novel fusion)和病毒插入位点等结构性变异;4.PCR的指数性扩增导致基因拷贝数变异无法检测;5、多重PCR不可避免地扩增偏好性导致均一度低,导致panel的靶标区域中部分区域不能很好覆盖,而部分区域过多覆盖。
相比而言,在一些实施例中,本发明将建库和靶标基因富集这两个步骤整合为一个流程,这一革命性的创新不仅克服了主流的建库加杂交捕获流程的步骤繁多成本高昂的缺点,同时也通过线性扩增规避了扩增子建库所固有的单管反应检测通量小、均一性差、无法有效检测基因组结构性变异和基因拷贝数变异等缺陷。
根据第一方面,在一实施例中,提供一种靶标基因富集建库方法,包括:
磷酸化步骤,包括在模板分子的5’端修饰磷酸基团;
延伸步骤,包括加入含与靶标基因互补配对序列的第一引物,所述第一引物的5’端修饰有标记分子,在模板分子的靶标区域退火并延伸,获得双链靶核苷酸分子,所述双链靶核苷酸含有由5’端修饰有标记分子的第一引物延伸得到的延伸链;
第一测序接头连接步骤,包括将第一测序接头连接至前一步骤延伸处理后的双链靶核苷酸分子;
解链步骤,包括将前一步骤所得产物解链处理,去除5’端修饰有标记分子的第一引物延伸链,得到连接有第一测序接头的原始模板分子,即为单链靶核苷酸分子;
第二测序接头连接步骤,包括加入发夹式第二测序接头,使单链靶核苷酸分子连接所述发夹式第二测序接头;
扩增步骤,包括加入含与第一测序接头反向链互补配对序列的第二引物、含与第二测序接头反向链互补配对序列的第三引物,PCR反应,得到完整文库。
在一实施例中,磷酸化步骤中,所述模板分子为单链核苷酸分子和/或由双链核苷酸分子解链处理得到的单链核苷酸分子。解链方法可以是本领域常用的热变性等方法,通常是将目标分子加热到至少80℃保持至少1min。
在一实施例中,所述模板分子为亚硫酸氢盐转化处理后的DNA分子。
在一实施例中,所述模板分子为单链DNA分子或由RNA转录得到的单链DNA分子。
在一实施例中,磷酸化步骤中,使用的酶包括但不限于T4多聚核苷酸激酶(T4polynucleotide kinase,又称T4磷酸激酶)。
在一实施例中,磷酸化步骤的反应条件依次如下:55-65℃,1分钟,37℃,20分钟;95℃,1分钟,然后对产物进行冰浴处理。
在一实施例中,冰浴处理时间为2-60min。
在一实施例中,延伸步骤中,所述第一引物的5’端修饰的标记分子包括但不限于生物素。
在一实施例中,延伸步骤中,在DNA聚合酶作用下,第一引物在模板分子的靶标区域退火并延伸。
在一实施例中,第一测序接头连接步骤中,所述第一测序接头选自于Illumina测序平台的P7端测序接头、MGI测序平台的P1端测序接头、或其他高通量测序平台右侧测序接头中的任意一种。
在一实施例中,所述第一测序接头为双链测序接头,所述第一测序接头串联有可串联至模板分子的分子标签。
在一实施例中,所述分子标签为随机核苷酸序列。
在一实施例中,所述分子标签的长度为4-19bp。
在一实施例中,所述第一测序接头包括互补配对连接的正向链、反向链,所述正向链的3’端修饰有封闭基团。
在一实施例中,所述封闭基团选自磷酸基团、氨基C7、双脱氧胞嘧啶中的至少一种。
在一实施例中,解链步骤中,解链后,用链霉亲和素包被的磁珠结合第一引物单链延伸产物5’端的标记分子,并用磁力架将其收集于容器内壁,将反应体系的上清液转移至另一容器中,即为单链靶核苷酸分子,然后加入发夹式第二测序接头进行连接反应。
在一实施例中,第二测序接头连接步骤中,所述发夹式第二测序接头包括互补配对连接的正向链、反向链,所述正向链的3’端串联有随机核苷酸序列。
在一实施例中,所述发夹式第二测序接头正向链的3’端串联的随机核苷酸序列长度为4-19nt。
在一实施例中,所述第二测序接头包括但不限于Illumina测序平台的P5端测序接头、MGI测序平台的P2端测序接头或其他高通量测序平台左侧测序接头中的任意一种。
在一实施例中,发夹式第二测序接头连接步骤中,所采用的酶为DNA连接酶。
在一实施例中,所述DNA连接酶为T4 DNA连接酶。
在一实施例中,连接发夹式第二测序接头时,25-37℃下孵育30-120分钟。
在一实施例中,解链步骤步骤中,解链方法为热变性。
在一实施例中,所述热变性处理具体是将目标分子加热到至少80℃保持至少1min。
在一实施例中,所述第二引物含有内接头序列、外接头序列,所述内接头序列的5’端串联连接至所述外接头序列的3’端,所述内接头序列可与所述第一测序接头反向互补配对连接,所述内接头序列、外接头序列之间串联有或未串联有第一样本标签。
在一实施例中,所述第三引物含有内接头序列、外接头序列,所述内接头序列的5’端串联连接至所述外接头序列的3’端,所述内接头序列可与所述第二测序接头反向互补配对连接,所述内接头序列、外接头序列之间串联有第二样本标签。
在一实施例中,所述第一样本标签、第二样本标签的长度独立地为4-19nt。
在一实施例中,扩增结束后,进行纯化处理,得到文库。
在一实施例中,所述纯化处理方法为磁珠法。
本发明针对的样本为各类DNA(长度在500bp以下)或bisulfite处理过的各类DNA,完整的基因组DNA需打断到500bp以下。通常情况下,若为片段完整的基因组DNA则需要打断为200至500bp之间,并纯化。
在一实施例中,本发明可用于基因组特定区域的靶标测序,还可用于全外显子测序。
在一实施例中,本发明的另一大优点是将起始样本中原始模板链(图1中第一步的深色模板链)构建入测序文库,入库的核苷酸链不是第一引物的延伸产物,也即是说,现有方法入库的是与原始模板互补配对的扩增产物,而非原始模板链本身。由于原始模板链构建入库后,文库的PCR扩增可以使用高保真酶进行,而第一引物的延伸所用的酶的保真性要低很多,所以现有技术中引物延伸过程有可能会产生错误,而本发明的保真性更高。本发明的高保真性可以更好地反应样本的真实面貌,在数据分析鉴定突变时降低背景噪音,从而实现超低频突变的准确检测。从本发明的两个具体实施例,分别实现了万分之三和万分之五的超低频突变检出,这对于临床检测这类准确度要求很高的应用至关重要,尤其对于肿瘤的早筛、早诊和微小病灶残留监测等方面的应用(在这些临床场景下,基因突变的频率都很低,对检测技术的超低频突变检测性能要求很高),本发明具有显著优势。
以下实施例以国际常用的Illumina测序平台的文库为例,本发明也可兼容其他NGS平台,只需更换对应测序平台的接头即可。
在一实施例中,本方法针对的样本为:各类DNA(长度在500bp以下)或bisulfite处理过的各类DNA,完整的基因组DNA需打断到500bp以下,如图1所示为主要流程图,靶向富集及建库方法如下:
1、使用T4磷酸激酶对DNA进行进行5’端磷酸化处理,在PCR仪中进行如下反应:55到65℃,1分钟,37℃,20分钟;95℃,1分钟,并立即对产物进行冰浴处理,冰浴处理时间为2分钟至1小时。
2、5’端带生物素且3’端带靶标基因序列的引物在靶标基因分子上引物退火并延伸,此步反应采用Klenow Fragment,在PCR仪中进行,反应条件为:37℃,15分钟;75℃,10分钟。
3、在PCR仪中采用T4 DNA连接酶连接P7接头,反应条件为:20至25℃,15至60分钟。P7接头连接,此P7接头靠近模板分子的一端带有6至12bp的分子标签,且外侧的3’端带有磷酸封闭基团。
4、连接完成以后将产物继续在PCR仪中,于95℃下变性1分钟,文库分子双链解开,然后立即用链霉亲和素磁珠抓取带生物素的引物延伸产物并去除,单链文库分子在上清中,将上清液转移到另一个新的PCR管内,向其中加入内侧5’端带磷酸基团的发夹式P5接头,用T4 DNA连接酶,配T4 DNA连接酶缓冲液和终浓度为20%(体积百分比)的PEG8000,连接P5端发卡接头,25-37℃下孵育30-120分钟,然后95℃下热变性1分钟,使得T4 DNA连接酶变性失活,以彻底终止接头连接反应。
5、直接在连接反应产物中加入DNA聚合酶及其配套缓冲液、P5端带或不带样本标签的引物、P7端带样本标签的引物、dNTP,配成100微升反应体系,做加样本标签的PCR,反应条件如下:95℃3分钟;6-30个循环,每个循环如下:95℃30秒,60℃10-30秒,72℃10-30秒;最后72℃,1-10分钟。
6、反应完成后,采用诺唯赞磁珠纯化反应,得到带P7单侧分子标签和样本标签的Illumina文库,或带P7单侧分子标签和双端样本标签的Illumina文库。
实施例1
本实施例制备突变频率为万分之三的突变游离核酸(cfDNA)标准品,取三等份30ng该cfDNA标准品,各用于三个独立的文库构建实验,分别采用本实施例的方法、现有的杂交捕获建库方法(对比例1)、现有的扩增子建库方法(对比例2)作为测序文库的文库制备,且三组实验所设计的靶标基因区域基本一致,然后在同样的高通两测序平台上上机测序,并测序相同数据量,最后采用同样的数据分析流程,检查同样的8个靶标基因位点的突变检测情况,以评估三种高通量测序靶标基因文库构建方法的性能差别。
本实施例以国际常用的Illumina测序平台的文库为例,其他高通量测序平台也适用于本发明,只是测序接头序列需做相应替换。
本实施例的标准品购自菁良基因科技(深圳)有限公司,具体为肺癌ctDNA标准品套装GW-OCTM009,其中含有野生型DNA标准品和突变频率为0.1%的ctDNA标准品,将两者按照7:3的质量比混合,得到突变频率为0.03%的稀释标准品。
靶标检测位点如表1所示。
表1
所需全部寡聚体(oligo)如下表2、表3所示(由南京金斯瑞生物科技有限公司合成、HPLC纯化)。
表2
表3
P7-index-1的结构中,下划直线标记的序列“TGATAG”为样本标签。
含有表3所示探针库的混合物,命名为睿法2基因panel。
对表2、表3中的符号说明如下:(1)“Biotin”代表生物素标记。
(2)“N”代表随机碱基。
(3)Biotin-GSP序列中,“X”代表与靶标基因区域互补配对的序列,长度为20个核苷酸,在靶标基因区域每10个核苷酸向前排布一个该种序列,即2×瓦片式覆盖。
(4)“Pho”代表磷酸基团。
(5)IS2和IS2-RC退火成双链即为第一测序接头,IS1-splint和IS1-RC退火成双链即为第二测序接头。
试剂及仪器说明如下:
1、对各类DNA模板做5’端磷酸化修饰所采用的T4 Polynucleotide Kinase(即T4多核苷酸激酶,10U/μL)购自英潍捷基(上海)贸易有限公司,货号EK0031。
2、第一引物延申反应所采用的DNA polymerase I Klenow Fragment(即DNA聚合酶I Klenow片段,5U/μL)购自南京诺唯赞生物科技股份有限公司,货号:N104-01。
3、各接头连接反应均采用T4 DNA Ligase(Rapid),即T4 DNA连接酶,购自南京诺唯赞生物科技股份有限公司,货号:N103-01。
4、文库扩增反应所采用的VAHTS HiFi Amplification Mix(货号:N616-01)购自南京诺唯赞生物科技股份有限公司。
5、PCR产物纯化磁珠VAHTS DNA Clean Beads(货号:N411-01)购自南京诺唯赞生物科技股份有限公司。
6、DNA样本磷酸化反应所需T4 Polynucleotide Kinase(10U/μL)购自英潍捷基(上海)贸易有限公司,货号:EK0031。
7、结合单链连接产物所用链霉亲和素磁珠为Dynabeads
8、各步实验所用超纯水均采用ULtraPure
9、仪器:ABI veriti96型PCR仪(英潍捷基(上海)贸易有限公司出品),恒温混匀仪(杭州佑宁仪器有限公司,货号HC-100),四维旋转混合仪(海门市其林贝尔仪器制造有限公司,BE-1100),磁力架(无锡百格生物科技有限公司,货号BMB16-1.5-2),Qubit
10、本实施例的TE缓冲液组成如下:10mmol/L Tris-HCl、1mmol/L EDTA,pH=8.0。
如图1所示,实验步骤如下:
1、取菁良基因-肺癌ctDNA标准品套装-GW-OCTM009,其中含有野生型DNA标准品和突变频率为0.1%的ctDNA标准品,按照野生型DNA标准品:突变频率为0.1%的ctDNA标准品=7:3的质量比混合,形成突变频率为0.03%的cfDNA样本60ng,95℃变性处理2分钟。
2、制备第一测序接头和第二测序接头
2.1在200μL PCR管中配置以下反应体系:
表4第一测序接头退火体系
表5第二测序接头退火体系
2.2退火反应条件:95℃,10秒;加入RAMP 4%,以0.1℃/s的速率降温至14℃。
2.3在上述反应产物体系(50μL)中加入50μL的TE缓冲液,所得的体系中第二测序接头的终浓度为100μM。制备好产物体系可存放在-20℃长期保存,或者4℃保存8小时。
3、在200微升PCR管里配置如下反应体系,用于将cfDNA分子5'端磷酸化:
表6
涡旋混匀并短暂离心,置于PCR仪中做如下反应:37℃,20分钟;95℃,1分钟。
4、第一引物的退火和延伸:
对每个单管反应,检测的靶标基因位点数可从1到1万个,每个位点对应一个带有特定靶标基因结合区的引物,因此对每个单管反应最多可混合1万个该类探针。本实施例的靶标基因检测位点数为7个,具体如表1所示,靶标这8个基因位点的第一引物如表3所示,每个位点2条引物,共16条。将这16条第一引物等摩尔数混合,得到本实施例所需的第一引物混合液,终浓度为200μM。
在200微升PCR管里配置如下反应体系:
表7
涡旋混匀并短暂离心,置于PCR仪中做如下反应:
多重靶标基因位点第一引物混合物在基因组的靶标区域退火和延伸,在PCR仪中进行,反应条件如下:
表8
5、在200微升PCR管里配置如下反应体系以连接第一测序接头:
表9
总反应体积为50μL,在PCR仪内,37℃下反应半小时做连接反应,然后95℃下反应1到10分钟,将连接产物变性成单链,然后将PCR管置于冰上。
6、加入10微升Dynabeads
7、在上一步的200微升PCR管里配置第二测序接头连接反应体系:
表10
总反应体积120微升,在PCR仪内37℃、1小时做链接反应,然后95℃下反应2分钟,将T4 DNA Ligase变性失活,再将PCR管置于冰上。
8、将第二测序接头连接反应产物等分成两份(每份各自做PCR和纯化,最后将纯化产物合并),每份40微升,加入含与第一测序接头反向链互补配对序列的第二引物、含与第二测序接头反向链互补配对序列的第三引物,PCR反应,得到完整文库(illumina indexingPCR)。
表11
按照表11配成100微升反应体系做PCR,反应条件如下:
表12
反应完成后,采用VAHTS DNA Clean Beads磁珠纯化产物,按该磁珠纯化PCR产物的标准操作进行,用22微升超纯水洗脱最终产物,并将两份洗脱产物合并,建成P7端带样本标签的Illumina靶标基因文库,即为可上机测序的文库。
对比例1
本对比例提供杂交捕获对照实验。
取菁良基因-肺癌ctDNA标准品套装-GW-OCTM009,其中含有野生型DNA标准品和突变频率为0.1%的ctDNA标准品,按照野生型DNA标准品:突变频率为0.1%的ctDNA标准品=7:3的质量比混合形成突变频率为0.03%的DNA样本,所得样本的总质量为60ng,采用可捕获NRAS、KRAS、PIK3CA、EGFR四个基因全部外显子区域的panel(即探针库)作为靶标基因组区域捕获探针(完全涵盖了实施例1中全部探针覆盖的基因组区域)。具体是按前述基因清单在南京金斯瑞生物科技股份有限公司下单合成杂交捕获探针(按照杂交捕获探针的通用设计理念设计并合成,覆盖下表所列基因的全部外显子区域,此为定制产品,无货号),采用购自南京金斯瑞生物科技有限公司的GenFisher建库和杂交捕获试剂盒(货号为NGS02409(24次反应)+NGS02406i(24次反应universal blocker)),按照标准操作流程进行文库构建,包括依次进行捕获前扩增、杂交捕获、捕获后扩增,获得可用于上机测序的文库。
对比例2
本对比例提供扩增子建库对照实验(基于多重PCR技术)。
取菁良基因-肺癌ctDNA标准品套装GW-OCTM009,其中含有野生型DNA标准品和突变频率为0.1%的ctDNA标准品,按照野生型DNA标准品:突变频率为0.1%的ctDNA标准品=7:3的质量比混合,形成突变频率为0.03%的DNA样本60ng,采用可捕获NRAS、KRAS、PIK3CA、EGFR四个基因全部外显子区域的panel(即探针库)作为靶标基因组区域捕获探针(完全涵盖了实施例1中全部探针覆盖的基因组区域)。具体是按前述基因清单在南京金斯瑞生物科技股份有限公司下单合成杂交捕获探针(按照多重PCR的通用设计理念设计并合成,覆盖下表所列基因的全部外显子区域,此为定制产品,无货号),采用南京金斯瑞生物科技有限公司所提供的扩增子建库试剂盒,按照标准操作流程进行扩增子建库,获得可用于上机测序的文库。
上机测序
取实施例1、对比例1、对比例2制得的文库产物,均用Qubit4.0测定浓度,并各取20ng,上机测序。测序仪器型号为Illumina HiSeq 4000,测序策略为PE150,每个样本数据量为1Gb。
测序数据质控和分析流程
原始数据处理采用fastp软件,基因组比对采用BWA软件(即Burrows-Wheeler-Alignment Tool,算法为BWA-MEM),参考基因组采用GRCh38(亦称hg38,为国际通用人类参考基因组序列),使用sambamba软件进行标记(markdup)。
分析结果如下:
实施例1的文库的测序结果为10个index拆分的读段数(reads数)的合集,具体见下表:
表13
由上表可见,各index间reads数分布偏好性低(各index所拆分的reads数相近),且无法列入index的reads数仅占总reads数的千分之一,说明本实施例1使用的P7端带样本标签的indexing扩增系统已经可以精准的对多个样本进行混合靶标基因建库和测序。
突变检测结果如下表:
表14
表14中,raw base是指原始数据量。
GC含量是指鸟嘌呤(Guanine)和胞嘧啶(Cytosine)所占的比率。
Q30代表正确率在99.9%的reads占总reads数的比例。
depth in target是指靶标位点的测序深度。
ref_reads表示人类参考基因组上对应的reads数。
alt reads表示突变(variant)的reads数。
MAF(Mutation Allele Frequency)为突变频率,具体为alt reads与ref_reads的比值。
由上表可见,实施例1构建的文库的测序数据质量相对于其他两项现有技术所构建文库的测序结果更高,具体地,Q30比例更高;且基于实施例1的文库所检测得到的靶标基因突变的频率更接近真实值,即MAF(Mutation Allele Frequency)更接近万分之三的预设值。因此,实施例1的文库构建方法在对人类等复杂基因组的特定靶标基因做测序检测时的性能更优,且耗时更短,对比例1的杂交捕获建库需72-80小时,对比例2的扩增子建库需24-32小时,实施例1的建库方法仅需11小时,且实施例1所需步骤少,所需各种试剂和耗材少,因此成本更低。综上,实施例1的建库方法在临床检测、医学研究和基因组科学研究中有更广阔的应用前景。
实施例2
本实施例将福尔马林固定和石蜡包埋(FFPE)组织标准品(购自菁良基因科技(深圳)有限公司,具体为肿瘤野生型FFPE标准品和肿瘤SNV 5%FFPE标准品)所提取DNA制备成突变频率为万分之五的肿瘤突变标准品,取三等份,各300ng,该DNA标准品各用于三个独立的文库构建实验,分别采用本实施例的方法、杂交捕获建库方法、扩增子建库方法作为测序文库的文库制备方法,且三组实验所设计的靶标基因区域基本一致,然后在同样的高通两测序平台上上机测序,并测序相同数据量,最后采用同样的数据分析流程,检查同样的7个靶标基因突变位点(这7各位点分布于4个基因的外显子区域,这4个基因分别为NRAS、KRAS、PIK3CA、EGFR,这也是被睿法四基因panel的检测内容完全涵盖)的检测情况,以评估三种高通量测序靶标基因文库构建技术的性能差别(本实施例、杂交捕获建库和扩增子建库)。
本实施例以国际常用的Illumina测序平台的文库为例,其他高通量测序平台也适用,只是测序接头序列需做相应替换。
DNA标准品为购自菁良基因科技(深圳)有限公司的肿瘤野生型FFPE标准品(突变频率为0,货号GW-OPSM005)和肿瘤SNV 5%FFPE标准品(货号GW-OPSM003)。
FFPE标准品的DNA提取采用广州美基生物科技有限公司磁珠法石蜡包埋组织DNA提取试剂盒(货号:D6323-02B)。
FFPE总DNA片段化(即将10kb以上的长片段总DNA打断成200-500bp长的短片段)采用美国KAPA Biosystem公司生产的KAPA Frag Kit for Enzymatic Fragmentation试剂盒(货号KK8600)做酶切打断。
靶标检测位点如下表所示。
表15
所需全部寡聚体(oligo)如下表16、表17所示(由南京金斯瑞生物科技有限公司合成、HPLC纯化)。
表16
表17带第一测序接头的靶标基因探针
对表16、17的说明如下:
(1)“Biotin”代表生物素标记
(2)IS2和IS2-RC退火成双链即为第一测序接头,IS1-splint和IS1-RC退火成双链即为第二测序接头。(3)N代表随机碱基。
(4)X代表和靶标基因区域互补配对的序列,20个核苷酸长,在靶标基因区域每10个核苷酸向前排布一个该种序列,即2x瓦片式覆盖
(5)Pho代表磷酸基团。
(6)带样本标签的P端7引物-标签1中,下划直线标记的序列为样本标签。
对各类DNA模板做5’端磷酸化修饰所采用的T4 Polynucleotide Kinase(10U/μL)购自英潍捷基(上海)贸易有限公司,货号EK0031。
第一引物延伸反应采用DNA polymerase I Klenow Fragment(5U/μL)(南京诺唯赞生物科技股份有限公司出品,货号:N104-01)。
各接头连接反应均采用T4 DNA Ligase(Rapid)(南京诺唯赞生物科技股份有限公司出品,货号:N103-01)。
文库扩增反应采用VAHTS HiFi Amplification Mix(南京诺唯赞生物科技股份有限公司出品,货号:N616-01)。
PCR产物纯化磁珠VAHTS DNA Clean Beads(货号:N411-01)(南京诺唯赞生物科技股份有限公司出品)。
对比例采用国际通用的甲基化建库试剂盒(for illumina),美国SwiftBiosciences公司出品,货号Catalog No30024。
结合第一引物延伸产物所用链霉亲和素磁珠为Dynabeads
仪器:ABI veriti96型PCR仪(英潍捷基(上海)贸易有限公司出品),恒温混匀仪(杭州佑宁,货号HC-100),四维旋转混合仪(海门市其林贝尔仪器制造有限公司,BE-1100),磁力架(无锡百格生物科技有限公司,货号BMB16-1.5-2),Qubit
实验步骤如下:
1、采用广州美基生物科技有限公司出品的磁珠法石蜡包埋组织DNA提取试剂盒(货号:D6323-02B),对购自菁良基因科技(深圳)有限公司出品肿瘤野生型FFPE标准品(突变频率为0,货号GW-OPSM005)和肿瘤SNV 5%FFPE标准品(货号GW-OPSM003)做总DNA提取,按照该试剂盒标准操作流程进行,最终按50微升体积洗脱获得DNA提取物。
2、用Qubit4.0测定浓度野生型和5%SNV的FFPE DNA浓度分别为15.54ng/μL和14.78ng/μL,总量分别为777ng和739ng,取297ng肿瘤野生型FFPE标准品DNA和3ng肿瘤SNV5%FFPE标准品DNA混合(即按质量比99比1混合),形成突变频率为0.05%的FFPE DNA样本300ng,涡旋充分混匀。
3、将上一步产物放入一个200微升PCR管中,采用美国KAPA Biosystem公司出品的KAPA Frag Kit for Enzymatic Fragmentation试剂盒做酶切打断。用0.8体积即44微升VAHTS DNA Clean Beads(货号:N411-01)按标准操作流程做纯化,最后用22.5微升纯水洗脱纯化产物。
4、制备第一测序接头和第二测序接头
4.1在200μL PCR管中配置以下反应体系:
表18第一测序接头退火体系
表19第二测序接头退火体系
4.2退火反应条件:95℃,10秒;加入RAMP 4%,以0.1℃/s的速率降温至14℃。
4.3在上述反应产物(50μL)中加入50μL的TE buffer,所得溶液中第二测序接头终浓度为100μM;制得的溶液可存放在-20℃长期保存,或者4℃保存8小时。
5、取上一步纯化产物(仍置于原200微升PCR管中)做磷酸化反应:在上一步反应产物的原200微升PCR管中配置如下反应体系:
表20
涡旋混匀并短暂离心,置于PCR仪中做如下反应:37℃,20分钟;95℃,1分钟。
6、第一引物的退火和延伸:
对每个单管反应,检测的靶标基因位点数可从1到1万个,每个位点对应一个带有特定靶标基因结合区的引物,因此对每个单管反应最多可混合1万个该类探针。本实施例的靶标基因检测位点数为7个,具体如表13所示,靶标这7个基因位点的第一引物如表17所示,每个位点2条引物,共14条。将这14条第一引物等摩尔数混合,得到本实施例所需的第一引物混合液,终浓度为200μM。
在200微升PCR管里配置如下反应体系:
表21
涡旋混匀并短暂离心,置于PCR仪中做如下反应:
多重靶标基因位点第一引物混合物在基因组的靶标区域退火和延伸,在PCR仪中进行,反应条件如下:
表22
7、在200微升PCR管里配置如下反应体系以连接第一测序接头:
表23
总反应体积80微升,在PCR仪内37℃、半小时做连接反应,然后95℃下反应1到10分钟,将连接产物变性成单链,并将PCR管置于冰上。
8、加入10微升Dynabeads
9、在上一步的200微升PCR管里配置第二测序接头连接反应体系:
表24
总反应体积100微升,在PCR仪内37℃、1小时做连接反应,然后95℃下反应2分钟,将T4 DNA Ligase变性失活,再将PCR管置于冰上。
10、将第二测序接头连接反应产物等分成两份(每份各自做PCR和纯化,最后将纯化产物合并),每份50微升(上一步产物经过95℃加热后实际体积已少于100微升),加入含与第二测序接头反向链互补配对序列的第二引物、含与第一测序接头反向链互补配对序列的第三引物,PCR反应,得到完整文库(Illumina indexing PCR):
表25
按照表25配成102微升反应体系做PCR,反应条件如下:
表26
反应完成后,采用VAHTS DNA Clean Beads磁珠纯化产物,按该磁珠纯化PCR产物的标准操作进行,用22微升超纯水洗脱最终产物,并将两份洗脱产物合并,建成P7端带样本标签的Illumina靶标基因文库,即为可上机测序的文库。
对比例3
本对比例提供杂交捕获对照实验。
取购自菁良基因科技(深圳)有限公司的肿瘤野生型FFPE标准品和肿瘤SNV 5%FFPE标准品,按照肿瘤野生型FFPE标准品:肿瘤SNV 5%FFPE标准品=99:1的质量比混合,得到突变频率为0.05%的DNA样本300ng,并按与实施例2相同的酶切方式做片段化和磁珠纯化后用于后续实验步骤。采用可捕获NRAS、KRAS、PIK3CA、EGFR四个基因全部外显子区域的panel(即探针库)作为靶标基因组区域捕获探针(完全涵盖了实施例2中全部探针覆盖的基因组区域)。具体是按前述基因清单在南京金斯瑞生物科技股份有限公司下单合成杂交捕获探针(按照杂交捕获探针的通用设计理念设计并合成,覆盖下表所列基因的全部外显子区域,此为定制产品,无货号),采用购自南京金斯瑞生物科技有限公司的GenFisher建库和杂交捕获试剂盒(货号为NGS02409(24次反应)+NGS02406i(24次反应universalblocker)),按照标准操作流程进行文库构建,包括依次进行捕获前扩增、杂交捕获、捕获后扩增,获得可用于上机测序的文库。
对比例4
本对比例提供扩增子建库对照实验(基于多重PCR技术)。
取购自菁良基因科技(深圳)有限公司的肿瘤野生型FFPE标准品和肿瘤SNV 5%FFPE标准品,按照肿瘤野生型FFPE标准品:肿瘤SNV 5%FFPE标准品=99:1的质量比混合,得到突变频率为0.05%的DNA样本300ng,并按与实施例2相同的酶切方式做片段化和磁珠纯化后用于后续实验步骤。采用可捕获NRAS、KRAS、PIK3CA、EGFR四个基因全部外显子区域的panel(即探针库)作为靶标基因组区域捕获探针(完全涵盖了实施例2中全部探针覆盖的基因组区域)。具体是按前述基因清单在南京金斯瑞生物科技股份有限公司下单合成杂交捕获探针(按照多重PCR的通用设计理念设计并合成,覆盖下表所列基因的全部外显子区域,此为定制产品,无货号),采用南京金斯瑞生物科技有限公司所提供的扩增子建库试剂盒,按照标准操作流程进行扩增子建库,获得可用于上机测序的文库。
上机测序
取实施例2、对比例3、对比例4制得的文库产物,均用Qubit4.0测定浓度,并各取20ng,上机测序。测序仪器型号为Illumina HiSeq 4000,测序策略为PE150,每个样本数据量为1Gb。
测序数据质控和分析流程
原始数据处理采用fastp软件,基因组比对采用BWA软件(即Burrows-Wheeler-Alignment Tool,算法为BWA-MEM),参考基因组采用GRCh38(亦称hg38,为国际通用人类参考基因组序列),使用sambamba软件进行标记(markdup)。
分析结果如下:
实施例2的文库所得测序结果为10个index拆分的读段数(reads数)的合集,具体见下表:
表27
由上表可见,各index间reads数分布偏好性低(各index所拆分的reads数相近),且无法列入index的reads数仅占总reads数的千分之一,说明本专利技术的P7端带样本标签的indexing扩增系统已经可以精准地对多个样本进行混合靶标基因建库和测序。
突变检测结果如下表:
表28
由上表可见,实施例2构建的文库的测序数据质量相对于其他两项现有技术测序结果更高,具体地,Q30比例更高,Q30代表正确率在99.9%的reads占总reads数的比例;且基于实施例2的文库所检测得到的靶标基因突变的频率更接近真实值,即MAF(MutationAllele Frequency)更接近万分之五的预设值。因此,实施例2的文库构建方法在对人类等复杂基因组的特定靶标基因做测序检测时的性能更优,且耗时更短,对比例3的杂交捕获建库需72-80小时,对比例4的扩增子建库需24-32小时,实施例2的建库方法仅需11小时,且实施例2所需步骤少,所需各种试剂和耗材少,因此成本更低。综上,实施例2的建库方法在临床检测、医学研究和基因组科学研究中有更广阔的应用前景。
一般的NGS的靶向测序样本制备需要经历建库、捕获前扩增、杂交捕获和捕获后扩增四大步骤,这几部分彼此串联缺一不可,整个流程加起来需要约2天时间,不仅费时费钱,而且四个步骤之间的衔接有一定的难度,在一实施例中,本发明可将这四个步骤整合为一体,流程短,操作简易,将常规的建库和捕获分开的步骤整合为一个流程。
在一实施例中,本发明使用自配试剂,可完全摆脱试剂进口。
在一实施例中,本发明的建库起始模板以单链形式,可适用于严重降解的样本和微量样本。
在一实施例中,基于单向引物延伸并直接抓取原始的DNA链,可实现基因组结构性变异检测(包括基因拷贝数变化,融合基因和病毒插入序列等)。
在一实施例中,RNA样本逆转录为cDNA后可自动兼容本发明,且无需二链合成,节省物料和时间,且避免常规的二链合成过程中随机引物带来的一系列错误和偏好性。
在一实施例中,本发明将建库和靶标基因富集两步整合为一个流程,这一革命性的创新不仅克服了现有的建库加杂交捕获流程的步骤繁多、成本高昂的缺点,同时也通过线性扩增规避了扩增子建库所固有的单管反应检测通量小,均一性差,无法有效检测基因组结构性变异和基因拷贝数变异,另外因为建成的文库的P5端带分子标签,可有效矫正PCR和测序错误从而实现超低频检测。
在一实施例中,本发明适用于一链合成的单链cDNA、亚硫酸氢盐处理后的DNA、cfDNA、打断后的基因组DNA等样品的建库。
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。
SEQUENCE LISTING
<110> 深圳市睿法生物科技有限公司
<120> 一种靶标基因富集建库方法
<130> 20I30443
<160> 31
<170> PatentIn version 3.3
<210> 1
<211> 34
<212> DNA
<213> 人工序列
<400> 1
gtgactggag ttcagacgtg tgctcttccg atct 34
<210> 2
<211> 34
<212> DNA
<213> 人工序列
<400> 2
agatcggaag agcacacgtc tgaactccag tcac 34
<210> 3
<211> 40
<212> DNA
<213> 人工序列
<220>
<221> misc_feature
<222> (34)..(40)
<223> n is a, c, g, or t
<400> 3
acactctttc cctacacgac gctcttccga tctnnnnnnn 40
<210> 4
<211> 33
<212> DNA
<213> 人工序列
<400> 4
agatcggaag agcgtcgtgt agggaaagag tgt 33
<210> 5
<211> 60
<212> DNA
<213> 人工序列
<400> 5
caagcagaag acggcatacg agattgatag gtgactggag ttcagacgtg tgctcttccg 60
<210> 6
<211> 60
<212> DNA
<213> 人工序列
<400> 6
caagcagaag acggcatacg agattatacg gtgactggag ttcagacgtg tgctcttccg 60
<210> 7
<211> 60
<212> DNA
<213> 人工序列
<400> 7
caagcagaag acggcatacg agatcgatca gtgactggag ttcagacgtg tgctcttccg 60
<210> 8
<211> 60
<212> DNA
<213> 人工序列
<400> 8
caagcagaag acggcatacg agatatacac gtgactggag ttcagacgtg tgctcttccg 60
<210> 9
<211> 60
<212> DNA
<213> 人工序列
<400> 9
caagcagaag acggcatacg agatatagcg gtgactggag ttcagacgtg tgctcttccg 60
<210> 10
<211> 60
<212> DNA
<213> 人工序列
<400> 10
caagcagaag acggcatacg agattgttca gtgactggag ttcagacgtg tgctcttccg 60
<210> 11
<211> 60
<212> DNA
<213> 人工序列
<400> 11
caagcagaag acggcatacg agatagatac gtgactggag ttcagacgtg tgctcttccg 60
<210> 12
<211> 60
<212> DNA
<213> 人工序列
<400> 12
caagcagaag acggcatacg agattagctg gtgactggag ttcagacgtg tgctcttccg 60
<210> 13
<211> 60
<212> DNA
<213> 人工序列
<400> 13
caagcagaag acggcatacg agatgtatgt gtgactggag ttcagacgtg tgctcttccg 60
<210> 14
<211> 60
<212> DNA
<213> 人工序列
<400> 14
caagcagaag acggcatacg agatggctca gtgactggag ttcagacgtg tgctcttccg 60
<210> 15
<211> 51
<212> DNA
<213> 人工序列
<400> 15
aatgatacgg cgaccaccga gatctacact ctttccctac acgacgctct t 51
<210> 16
<211> 32
<212> DNA
<213> 人工序列
<400> 16
caaggacatc cgtgatttgt agtggagaag ga 32
<210> 17
<211> 32
<212> DNA
<213> 人工序列
<400> 17
caaggacatc cgtggcctgg cttgcttacc tt 32
<210> 18
<211> 32
<212> DNA
<213> 人工序列
<400> 18
caaggacatc cggcatctgc ctcacctcca cc 32
<210> 19
<211> 32
<212> DNA
<213> 人工序列
<400> 19
caaggacatc cgtccaggag gcagccgaag gg 32
<210> 20
<211> 32
<212> DNA
<213> 人工序列
<400> 20
caaggacatc cgggaaactg aattcaaaaa ga 32
<210> 21
<211> 32
<212> DNA
<213> 人工序列
<400> 21
caaggacatc cggaccttac cttatacacc gt 32
<210> 22
<211> 32
<212> DNA
<213> 人工序列
<400> 22
caaggacatc cggaaataaa tacagatctg tt 32
<210> 23
<211> 32
<212> DNA
<213> 人工序列
<400> 23
caaggacatc cgaaaaggaa ttccataact tc 32
<210> 24
<211> 32
<212> DNA
<213> 人工序列
<400> 24
caaggacatc cggacgatac agctaattca ga 32
<210> 25
<211> 32
<212> DNA
<213> 人工序列
<400> 25
caaggacatc cgacaagttt atattcagtc at 32
<210> 26
<211> 32
<212> DNA
<213> 人工序列
<400> 26
caaggacatc cgtgagagac caatacatga gg 32
<210> 27
<211> 32
<212> DNA
<213> 人工序列
<400> 27
caaggacatc cgtatgtcca acaaacaggt tt 32
<210> 28
<211> 32
<212> DNA
<213> 人工序列
<400> 28
caaggacatc cgagaaggtg agaaagttaa aa 32
<210> 29
<211> 32
<212> DNA
<213> 人工序列
<400> 29
caaggacatc cgtcacatcg aggatttcct tg 32
<210> 30
<211> 32
<212> DNA
<213> 人工序列
<400> 30
caaggacatc cgccctccct ccaggaagcc ta 32
<210> 31
<211> 32
<212> DNA
<213> 人工序列
<400> 31
caaggacatc cgaggcagat gcccagcagg cg 32