一种筛选小鼠精子发生相关互作蛋白的方法及应用
文献发布时间:2023-06-19 19:33:46
技术领域
本发明涉及生物技术领域,具体涉及一种筛选小鼠精子发生相关互作蛋白的方法、一种小鼠单倍体生精细胞酵母双杂交cDNA文库的构建及应用。
背景技术
不孕不育症是一种多因素、复杂的病理生理过程,研究表明异常精液参数是近50%非自愿无子女的不育男性中主要的相关因素,主要表现为精子发生障碍及形成异常,其中精子发生障碍约占男性不育因素的85%。精子发生是雄性生殖系统中高度特异、严格管控的多阶段持续分化的生物学进程,精子发生异常将导致少弱畸精子症,形成雄性不育表型。精子发生涉及有丝分裂、减数分裂、遗传重组、染色质重塑、精子变形等复杂的生物学变化,这一高度有序的进程受到众多基因及其编码蛋白的严格调控,基因表达紊乱将导致精子发生障碍产生不育表型。精子发生过程包含多种睾丸特异性基因在不同时期的特异性表达,其产物蛋白的表达多与精子细胞RNA的表达丰度相应,超过80%的蛋白质是与其他蛋白质相互作用形成稳定或瞬时复合物结构而发挥作用。在精子发生中,各阶段特异性基因及其蛋白质产物的正确表达是精子形成的重要保障。
酵母双杂交(Yeast Two-hybrid System)是利用遗传学方法在酵母真核细胞体内研究蛋白质之间相互作用的高通量生物学技术,应用于蛋白与宿主细胞的互作机制、蛋白质组学、细胞信号转导和功能基因组学等领域。
目前,小鼠、大鼠、兔、羊和人类睾丸组织cDNA文库已成功构建,由于睾丸包括生精细胞、间质细胞及支持细胞等多种成分,因此利用睾丸cDNA文库筛选与精子发生相关蛋白其特异性及阳性率均不理想,同时,以往研究蛋白质互作的生化技术,如偶联、免疫共沉淀、以及串联亲和层析等体外实验易受环境、实验条件等诸多因素影响的问题。因此迫切需要进一步构建单倍体生精细胞cDNA文库进行精子发生的相关研究。目前小鼠单倍体生精细胞酵母双杂交cDNA文库构建未见商品化销售及文献报道,因而该文库的成功构建,将成为探究小鼠精子发生相关机制的基本工具。
发明内容
本发明针对上述现有技术存在的问题,提供一种筛选小鼠精子发生相关互作蛋白的方法,一种小鼠单倍体生精细胞酵母双杂交cDNA文库的构建及应用。
本发明的目的是通过以下技术方案来实现的:
本发明目的之一在于,提供一种小鼠单倍体生精细胞酵母双杂交cDNA文库的构建方法。本方法适用于构建某种单细胞酵母双杂交cDNA文库,可以显著提升对标研究相关领域互作蛋白筛选的阳性率及灵敏度,具有操作简便、成本低廉、适用范围广等特点。
一种小鼠单倍体生精细胞酵母双杂交cDNA文库的构建方法,所述构建方法如下:
步骤S1:小鼠单倍体生精细胞的分选纯度与鉴定;
步骤S2:提取步骤S1中筛选出的小鼠单倍体生精细胞RNA,并反转录成单链cDNA,将单链cDNA通过PCR扩增形成双链cDNA并纯化;
步骤S3:应用醋酸锂法将双链cDNA与线性载体pGADT7-Rec共转化至感受态酵母细胞Y187中构建酵母双杂交cDNA文库;
步骤S4:酵母双杂交cDNA文库的鉴定。
所述步骤S1具体如下:取成年野生型小鼠睾丸经酶消化后获取睾丸单细胞悬液,使用活细胞DNA荧光染料Hochest33342处理细胞,于流式细胞仪分选小鼠单倍体生精细胞;Hochest33342+PNA双荧光鉴定小鼠单倍体生精细胞分选纯度。
优选的,所述步骤S2具体如下:采用酵母双杂交cDNA文库构建试剂盒的方法,以1μg小鼠单倍体生精细胞总RNA为模板反转录成单链cDNA,再通过PCR扩增单链cDNA形成双链cDNA并对双链cDNA进行纯化,使用1.2%的琼脂糖对每个样品的PCR产物进行分析。
优选的,所述反转录过程中应用的引物为SMARIII及CDSIII,SMARIII如SEQ IDNO:1所示,CDSIII如SEQ ID NO:2所示;所述通过PCR扩增单链cDNA形成双链cDNA中应用的引物为5’PCR Primer及3’PCR Primer,5’PCR Primer如SEQ ID NO:3所示,3’PCR Primer如SEQ ID NO:4所示。
优选的,所述步骤S3具体如下:将双链cDNA、pGADT7-Rec、变性鲑鱼精DNA转化至感受态酵母细胞Y187后涂于SD/-Leu平板上,30℃培养3-5天直至克隆出现;洗脱下所有克隆,收集所有的液体,调整细胞密度约为2×10
本发明的目的之二在于,提供一种筛选小鼠精子发生相关互作蛋白的实验方法,为深入研究精子发生相关蛋白的功能及分子机制提供可能。
本发明构建的酵母双杂交cDNA文库在筛选小鼠精子发生相关互作蛋白中的应用。
本发明目的之三在于,提供一种小鼠单倍体生精细胞酵母双杂交cDNA文库的应用实例。
应用的方法,所述方法如下:
步骤(1):目的蛋白编码基因酵母双杂交诱饵质粒pGBKT7-X的构建及鉴定(以X替代基因名称,以下相同);
根据NCBI数据库小鼠X基因序列设计特异性PCR引物并扩增X的开放阅读框序列;1%琼脂糖凝胶电泳鉴定并割胶回收PCR产物,采用pGEMT-easy试剂盒将PCR产物插入pGEM-T-easy载体内,经序列测定确认正确无误后,使用特定的限制性内切酶双酶切该质粒及pGBKT7载体,转化至大肠杆菌,经双酶切鉴定筛选阳性克隆。
步骤(2):采用醋酸锂法分别将诱饵质粒pGBKT7-X及空载质粒pGBKT7转化至感受态酵母细胞AH109及Y187中;
各挑取一个新鲜的AH109及Yl87单克隆菌落至两管10mL YPDA液体培养基中30℃培养过夜。次日取过夜酵母培养液加入100mL-200mL新鲜YPDA培养液中调0D600至0.3左右,30℃培养2h-3h左右,测0D600约0.6,收集酵母培养液室温700g离心5min,弃上清。用超纯水洗涤沉淀后离心弃上清,用1mL新鲜配制的1×TE/LiAc重悬细胞,即为酵母感受态细胞。
取pGBKT7-X及pGBKT7、pGBKT7-p53(阳性对照质粒)各1μg,分别与变性鲑鱼精DNA20μL、PEG/LiAc 500μL,50μL AH109及Y187酵母感受态细胞共转化,30℃孵育30min;加入70μL DMSO,42℃孵育15min后离心细胞,弃去上清液,加入1mL YPD Plus Medium重悬沉淀,于30℃摇床孵育90min,离心细胞弃去上清,1mL生理盐水重悬沉淀,每200μL涂布于营养缺失培养基SD/-Trp上,30℃培养3-5天直至克隆出现。
步骤(3):诱饵质粒pGBKT7-X对
分别挑取转化了诱饵质粒pGBKT7-X及空载质粒pGBKT7的AH109及Y187单克隆,加入5mL SD/-Trp液体培养基中30℃培养16h;次日取过夜酵母培养液检测OD600值,如果0D600大于0.8,说明诱饵质粒对酵母细胞无毒性作用,若OD600远小于0.8,说明诱饵质粒对酵母细胞有毒性作用。
步骤(4):诱饵质粒pGBKT7-X自激活活性分析;
将转化了pGBKT7-X、pGBKT7-p53及pGBKT7的酵母细胞AH109及Yl87分别铺板于营养缺失培养基SD/-Trp/X-a-gal、SD/-His/-Trp/X-a-gal、SD/-Ade/-Trp/X-a-gal三种培养基上,30℃培养3-5d,pGBKT7-X仅在SD/-Trp培养基上生长,其它两种营养缺陷培养基中未见任何克隆,说明诱饵质粒pGBKT7-X没有自激活活性,可以用于进一步筛选。
步骤(5):应用酵母交配法进行cDNA文库的筛选;
将转化有诱饵质粒pGBKT7-X的酵母细胞AH109过夜培养细胞与小鼠单倍体生精细胞cDNA文库加入培养液中,30℃低速震荡培养20-24小时。将交配后的酵母细胞离心后,铺板于营养缺失型的培养基SD/-Leu/-Trp/-His/-Ade,30℃培养5-8天,直至克隆出现。
步骤(6):阳性克隆的表型验证;
挑取酵母交配法筛选出的阳性克隆,采用划线法将其划至营养缺失型培养基SD/-Leu/-Trp/-His/-Ade/X-α-gal上,30℃培养3-5天,将实验重复3次;对经过验证的阳性克隆进行PCR扩增、测序及生物信息学分析,观察插入片段是否含有正确的开放阅读框架,经过对比分析其在NCBI中是否能找到其同源序列。
步骤(7):直接酵母双杂交对阳性克隆进行酵母中相互作用进行验证;
采用醋酸锂转化法(同步骤(2)),将阳性克隆的质粒分别与诱饵质粒pGBKT7-X共同转化入酵母细胞AH109中,将转化后的酵母细胞铺到营养缺失的培养基SD/-Leu/-Trp上,30℃培养3-5天,挑取5-10个克隆混合后,划线到营养缺失型培养基SD/-Leu/-Trp/-His/-Ade/X-α-gal上,30℃培养3-5d,观察培养的克隆(含Y蛋白)如果变蓝且没有自激活活性,则二者在酵母中能够直接相互作用。
步骤(8):应用免疫共沉淀技术及双重免疫荧光染色技术对X与Y是否相互作用进行验证;
在哺乳动物细胞内,应用CO-IP技术及免疫荧光共定位验证X编码蛋白与Y蛋白之间是否能够相互作用。具体步骤为:分别构建pCMV-Myc-X与pCMV-HA-Y重组质粒,将两种重组质共转染至HEK293T细胞中,48h后收取细胞总蛋白,分别加入Myc与HA抗体进行免疫共沉淀分析,最后对沉淀物分别用HA及Myc抗体进行Western blot检测,如果结果显示沉淀中含目的蛋白则表明Y能与X编码蛋白相互作用;将重组质粒pCMV-Myc-X与pCMV-HA-Y共同转染入Hela细胞中,48小时后,以Myc与HA为一抗进行双重免疫荧光染色分析,通过荧光显微镜观察X与Y蛋白在Hela细胞中是否共定位。
通过流式分选高纯度小鼠单倍体生精细胞,提取细胞总RNA并反转录合成单链cDNA,通过PCR扩增将单链cDNA合成双链cDNA并纯化,将纯化的双链cDNA产物与pGADT7载体,感受态酵母细胞Y187共转化构建酵母双杂交cDNA文库,通过检测文库滴度,重组效率,插入片段大小等鉴定cDNA文库是否构建成功。同时扩增目的基因Ccdc189并构建诱饵质粒PGBKT7-Ccdc189,确定诱饵质粒无毒性及自激活活性。将诱饵质粒pGBKT7-Ccdc189与cDNA文库共培养后接种于营养缺失型培养基SD/-Leu/-Trp/-His/-Ade及SD/-Leu/-Trp/-His/-Ade/X-α-gal上,排除假阳性后挑取蓝色单克隆菌落进行PCR鉴定及测序,确认相互作用蛋白。
本发明具有以下有益效果:
(1)本发明提供了一种利用酵母双杂交技术筛选小鼠精子发生相关互作蛋白的实验方法,同等条件下,这种方法优于其他体外实验筛选精子发生相关蛋白的传统技术,可以同时大量获取目的蛋白的相互作用蛋白及其编码基因序列,所筛选的互作蛋白与精子发生密切相关,为深入研究精子发生相关蛋白的功能及分子机制提供可能。
(2)同时本方法的成功应用,为单细胞类型及特定生物进程互作蛋白的筛选提供可能。本方法可推广至其他细胞类型、组织器官以及不同物种类型酵母双杂交cDNA文库的构建及应用,可以极大优化蛋白质互相作用的筛选,为不同领域蛋白功能的科学研究提供理论基础及技术支持;可以利用相应的抗体以及可与相应诱饵蛋白结合的配体筛选文库,从中筛选出与已知蛋白互作的蛋白克隆,并获取该蛋白基因表达cDNA序列。该方法的成功应用将为小鼠精子发生蛋白相互作用提供重要的研究手段,可以从分子水平揭示精子发生相关蛋白的表达、调控及相互作用关系,为精子发生及不孕不育相关基因的作用机制研究提供新的研究思路及理论支持,也为男性不育症的诊断及治疗提供理论基础及可能的靶标。
(3)本发明提供了一种小鼠单倍体生精细胞酵母双杂交cDNA文库,可应用于不同目的蛋白大批量、多次筛选小鼠精子发生相关蛋白,具有实用性强、成本低廉、适用范围广等优势。相较于其他传统的蛋白质互相作用筛选方法如串联亲和层析、免疫共沉淀等,酵母双杂交体内筛选系统避免了外界环境、实验条件等诸多因素影响,在核酸水平上就可以筛选出蛋白之间微弱的相互作用,不仅灵敏度高,特异性强,可以批量筛选互作蛋白,一个酵母双杂交cDNA文库的构建可以满足上百次蛋白筛选实验,具有极低的成本及较大的经济效益;这种利用酵母体内同源重组构建cDNA文库的方法,克服了传统的cDNA文库构建过程中的缺陷,如cDNA合成不佳、连接效率较低及在限制性内切酶使用过程中造成的克隆缺口或者断裂等问题,因而保证了cDNA文库中含有高质量的完整的全长基因。
附图说明
图1:野生型雄性小鼠单倍体生精细胞高纯度流式分选(图1A:野生型雄性小鼠睾丸单细胞悬液Hochest33342染色结果;图1B:野生型雄性小鼠单倍体精子细胞流式分选纯度为99.3%);
图2:PNA-Hochest33342双荧光鉴定流式分选小鼠单倍体生精细胞,阳性率为99.8%(图2A:分选小鼠单倍体生精细胞Hochest33342荧光染色结果;图2B:分选小鼠单倍体生精细胞PNA荧光染色结果;图2C:分选小鼠单倍体生精细胞PNA-Hochest33342荧光染色Merge结果;Bar=10μm);
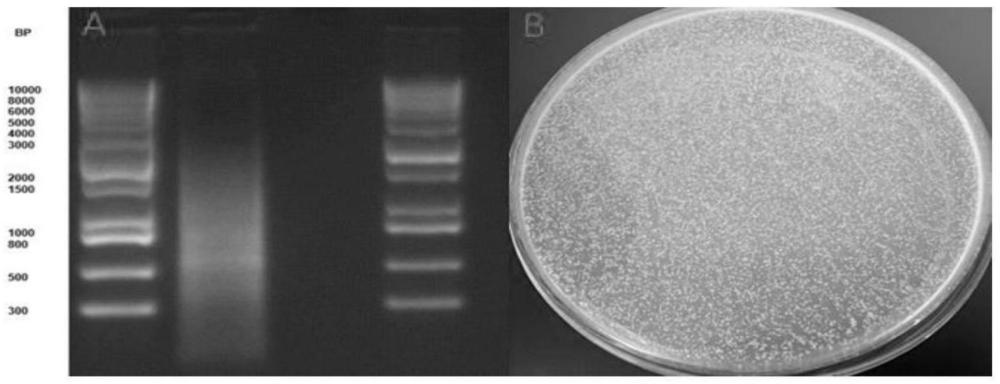
图3:成功纯化小鼠单倍体生精细胞cDNA,成功构建小鼠单倍体生精细胞cDNA文库克隆质量为4.26μg,符合建库标准(2-5μg)(图3A:小鼠单倍体生精细胞cDNA纯化结果;图3B:小鼠单倍体生精细胞cDNA文库克隆);
图4:小鼠单倍体生精细胞cDNA文库鉴定,文库滴度为7.86*10
图5:PCR鉴定cDNA文库重组效率为92%,插入片段大小集中于1000bp;
图6:pGBKT7-Ccdc189诱饵质粒的构建及鉴定;
图7:pGBKT7-Ccdc189诱饵质粒毒性分析;
图8:pGBKT7-Ccdc189诱饵质粒自激活活性分析;
图9:Ccdc189相互作用蛋白同源序列;
图10:细胞免疫荧光与Co-IP验证Ccdc189蛋白与筛选互作蛋白Pabpc2共定位并且相互作用(图10A:HA-Pabpc2与c-Myc-Ccdc189在Hela细胞中共定位;图10B:Ccdc189蛋白与Pabpc2蛋白Co-IP互作验证)。
具体实施方式
下面结合附图以及附图说明书对本发明作进一步说明。
第一部分:小鼠单倍体生精细胞酵母双杂交cDNA文库的构建与鉴定
1.小鼠单倍体生精细胞的分离与鉴定
(1)取成年野生型小鼠睾丸经酶消化后获取睾丸单细胞悬液,使用活细胞DNA荧光染料Hochest33342处理细胞,于FACSAria SORP流式细胞仪分选小鼠单倍体生精细胞,并鉴定分选细胞纯度为99.3%(图1)。
(2)Hochest33342+PNA双荧光鉴定小鼠单倍体生精细胞分选纯度为99.8%(图2)。
2.小鼠单倍体生精细胞cDNA的合成
据Takara公司Make Your Own“Mate&Plate”Library System酵母双杂交cDNA文库构建试剂盒的方法,以小鼠单倍体生精细胞总RNA为模板,SMARIII及CDSIII为引物,将RNA合成单链cDNA。引物序列为SMARIII(SEQ ID NO:1)5’-AAGCAGTGGTATCAACGCAGAGTGGCCATTATGGCCGGG-3’;CDS III(SEQ ID NO:2)5’-ATTCTAGAGGCCGAGGCGGCCGACATG-3’。PCR反应体系为:小鼠单倍体生精细胞总RNA1μg,CDSⅢ引物1μL,去离子水补足4μL,72℃孵育2min,冰上孵育2min;瞬时高速离心后与以下组分混合:5×First-Strand Buffer 2μL,DTT(100mM)1μL,dNTP Mix(10mM)1μL,SMART MMLV Reverse Transcriptase 1μL,42℃孵育10min;加入1μLSMARIII混匀,42℃孵育1小时后于75℃孵育10min终止反应。冷却至室温后加入1μLRNase H,37℃温育20min,进一步进行双链cDNA合成。
以单链cDNA为模板,5’PCR Primer(SEQ ID NO:3)及3’PCR Primer(SEQ ID NO:4)为引物合成双链cDNA并纯化。引物序列为:5’PCR Primer:5’-TTCCACCCAAGCAGTGGTATCAACGCAGAGTGG-3’,3’PCR Primer:5’-GTATCGATGCCCACCCTCTAGAGGCCGAGGCGGCCGACA-3’。反应体系为:单链cDNA2μL,去离子水70μL,10×Advantage 2PCR Buffer 10μL,50×dNTP Mix 2μL,5’PCR Primer 2μL,3’PCR Primer 2μL,10×Melting Solution 10μL,50×Advantage2Polymerase Mix 2μL,体系为100μL。反应条件为:95℃30s,95℃10s,68℃6min(每个循环延伸时间增加5s),22个循环。使用试剂盒CHROMA SPIN+TE-400Columns将合成的双链cDNA进行纯化,随后进行琼脂糖凝胶电泳检测(图3A),使用1.2%的琼脂糖对每个样品的PCR产物进行分析。
3.应用醋酸锂法将双链cDNA与线性载体pGADT7-Rec共转化至感受态酵母细胞Y187
取纯化后的双链cDNA 20μL、pGADT7-Rec载体6μL、变性鲑鱼精DNA 20μL、PEG/LiAc2.5mL共转化至600μL感受态酵母细胞Y187中,30℃孵育45min;加入160μL DMSO,42℃孵育20min后离心细胞,弃去上清液,加入3mL YPD Plus Medium重悬沉淀,于30℃摇床孵育90min,离心细胞弃去上清,15mL生理盐水重悬沉淀,每200μL涂布于SD/-Leu营养缺失型培养基上,30℃培养3-5天直至克隆出现(图3B)。
4.收集转化子克隆构建cDNA文库
将SD/-Leu营养缺失型培养基于4℃放置3h,每个培养基使用5ml冻存液洗脱下所有克隆,收集所有洗脱克隆,调整酵母细胞密度约为2×10
5.小鼠单倍体生精细胞酵母双杂交cDNA文库的鉴定
(1)文库滴度鉴定:
将10μL cDNA文库加入1mL YPDA培养基中得到1:10稀释液,取10μL 1:10稀释液加入1mL YPDA培养基中得到1:100稀释液,取10μL 1:100稀释液加入1mL YPDA培养基中得到1:1000稀释液,将三种稀释液分别涂布于营养缺陷型培养基SD/-Leu上,30℃温箱培养3-5天统计克隆数,计算文库滴度,计算公式为Cfu=(colonies on plate*dilution facter)/plating volume(mL)。小鼠单倍体生精细胞酵母双杂交cDNA文库滴度为7.86*10
(2)文库重组效率及插入片段大小鉴定:
从SD/-Leu营养缺陷型培养基中随机挑取100个单克隆酵母细胞,应用PlatiumTaq酶,进行酵母细胞PCR,以5’PCR Primer及3’PCR Primer为引物(序列同前),反应条件为:95℃20min;95℃30s,60℃1min,72℃90s,35个循环;72℃5min。PCR产物用1.5%琼脂糖凝胶电泳分析,判断重组效率(重组效率%=含插入片段酵母细胞数量/总酵母细胞数量)及插入片段的大小(图5)。
第二部分:Ccdc189酵母双杂交诱饵载体的构建及自激活活性分析
1.Ccdc189酵母双杂交诱饵载体的构建及鉴定
根据小鼠Ccdc189基因序列设计上、下游引物并进行PCR扩增,引物序列为:mCCDC189PGBKT7F(SEQ ID NO:5):
5’-GGCATATGATGATTACCCCCAGTTCCAGC-3’、mCCDC189PGBKT7R(SEQ ID NO:6):5’-GGGAATTCCTTGGTCTTGGTCTTGCCTTTG-3’,PCR反应体系为:Ccdc189 cDNA1μL,去离子水34μL,10×LAPCR Buffer 5μL,dNTP Mix 8μL,Primer mix 1μL(设计的上、下游引物的混合物),LATaq 1μL体系为50μL。反应条件为:95℃5min,95℃50s,60℃45s,72℃1min,35个循环;72℃延10min,使用1%琼脂糖疑胶电泳鉴定并切胶回PCR产物,采用pGEMT-easy试剂盒将PCR产物插入pGEMT-easy载体内,经序列测定确认正确无误后,以EcoR I和Nde I双酶切后,插入到同样双酶切的pGBKT7载体上,转化大肠杆菌DH5a利用酶切方法筛选阳性克隆(图6)。
2.采用醋酸锂法制备感受态酵母细胞AH109,将诱饵质粒pGBKT7-Ccdc189及空载质粒pGBKT7转化至感受态酵母细胞AH109及将诱饵质粒pGBKT7-Ccdc189及空载质粒pGBKT7转化至感受态酵母细胞Y187。
各挑取一个新鲜的AH109及Yl87单克隆菌落至两管10mL YPDA液体培养基中30℃培养过夜。次日取过夜酵母培养液加入100mL-200mL新鲜YPDA培养液中调0D600至0.3左右,30℃培养2h-3h左右,测0D600约0.6,收集酵母培养液室温700g离心5min,弃上清。用超纯水洗涤沉淀后离心弃上清,用1mL新鲜配制的1×TE/LiAc重悬细胞,即为酵母感受态细胞。
取pGBKT7-X及pGBKT7、pGBKT7-p53(阳性对照质粒)各1μg,分别与变性鲑鱼精DNA20μL、PEG/LiAc 500μL,50μL AH109及Y187酵母感受态细胞共转化,30℃孵育30min;加入70μL DMSO,42℃孵育15min后离心细胞,弃去上清液,加入1mL YPD Plus Medium重悬沉淀,于30℃摇床孵育90min,离心细胞弃去上清,1mL生理盐水重悬沉淀,每200μL涂布于营养缺失培养基SD/-Trp上,30℃培养3-5天直至克隆出现。
3.诱饵质粒pGBKT7-Ccdc189对酵母细胞AH109及Y187的毒性分析
分别挑取转化了诱饵质粒pGBKT7-Ccdc189和转化了空载质粒pGBKT7的酵母细胞AH109及Y187单克隆,放入5mL SD/-Trp液体培养基中30℃震摇培养16h。次日取过夜菌检测OD600的值,如果OD600大于0.8,则说明诱饵质粒对酵母细胞无毒性作用,如果OD600远小于0.8,则说明诱饵质粒对酵母细胞有毒性作用(图7)。
4.诱饵质粒pGBKT7-Ccdc189自激活活性分析
分别将转化了诱饵质粒pGBKT7-Ccdc189和转化了空载质粒pGBKT7的酵母细胞AH109及Y187铺板于SD/-Trp/X-α-gal、SD/-His/-Trp/X-α-gal、SD/-Ade/-Trp/X-α-gal 3种培养板上,30℃培养3~5d,转化有pGBKT7-Ccdc189及空载pGBKT7的酵母细胞AH109及Y187在SD/-Trp/X-α-gal培养基上长出白色克隆,但在SD/-His/-Trp/X-α-gal、SD/-Ade/-Trp/X-α-gal培养基上没有长出任何克隆,说明诱饵蛋白Ccdc189与空载质粒pGBKT7一样没有自激活活性(图8),可以用于进一步筛选。
第三部分:采用酵母直接双杂交法,从小鼠单倍体生精细胞cDNA文库筛选与Ccdc189相互作用的蛋白
1.应用酵母交配法进行cDNA文库的筛选
将转化有诱饵质粒pGBKT7-Ccdc189的酵母细胞AH109过夜培养细胞与小鼠单倍体生精细胞cDNA文库加入培养液中,30℃震荡培养20-24小时。将交配后的酵母细胞离心后,铺板于营养缺失型的培养基SD/-Leu/-Trp/-His/-Ade,30℃培养5-8天,直至克隆出现。
2.阳性克隆的表型验证
挑取酵母交配法筛选出的阳性克隆,采用划线法将其划至营养缺失型培养基SD/-Leu/-Trp/-His/-Ade/X-α-gal上,30℃培养3-5天,将实验重复3次。对经过验证的阳性克隆进行PCR扩增、测序及生物信息学分析,观察插入片段是否含有正确的开放阅读框架,经过对比分析其在NCBI中是否能找到其同源序列。
3.直接酵母双杂交对阳性克隆进行酵母中相互作用进行验证
采用醋酸锂转化法,将阳性克隆的质粒分别与诱饵质粒pGBKT7-Ccdc189共同转化入酵母细胞AH109中,将转化后的酵母细胞铺到营养缺失的培养基SD/-Leu/-Trp上,30℃培养3-5天,挑取5-10个克隆混合后,划线到营养缺失型培养基SD/-Leu/-Trp/-His/-Ade/X-α-gal上,30℃培养3-5d,观察两者在酵母中是否能相互作用,如果变蓝且没有自激活性,则二者在酵母中能够直接相互作用。在哺乳动物细胞内,应用CO-IP技术及免疫荧光共定位验证Ccdc189与相互作用蛋白之间是否能够相互作用(图9,图10)。
图9:上面方框为筛选出的Ccdc189的相互作用蛋白Pabpc2开放阅读框蛋白全长示意图,最下方方框为酵母双杂交筛选出的编码氨基酸序列片段,由图中可以看出,筛选出的相互作用蛋白片段为Pabpc2全长编码蛋白羧基端部分编码氨基酸。
图10A所示:pCMV-HA-Pabpc2与pCMV--Myc-Ccdc189质粒共同转染Hela细胞并涂片,通过荧光拍照分析,Pabpc2与Ccdc189在Hela细胞中共定位;图10B所示:将质粒pCMV-Myc-Ccdc189及pCMV-HA-Pabpc2共转染至HEK293T细胞48h后收取细胞蛋白,分别加入Myc与HA抗体进行IP,并用HA及Myc抗体进行Western blot检测,结果显示Ccdc189与Pabpc2可以相互作用。
4.应用免疫共沉淀技术及双重免疫荧光染色技术对Ccdc189与Pabpc2是否相互作用进行验证
在哺乳动物细胞内,应用CO-IP技术及免疫荧光共定位验证Ccdc189编码蛋白与Pabpc2蛋白之间是否能够相互作用。具体步骤为:分别构建pCMV--Myc-Ccdc189与pCMV-HA-Pabpc2重组质粒,将两种重组质共转染至HEK293T细胞中,48h后收取细胞总蛋白,分别加入Myc与HA抗体进行免疫共沉淀分析,最后对沉淀物分别用HA及Myc抗体进行Western blot检测,如果结果显示沉淀中含目的蛋白则表明Pabpc2能与Ccdc189编码蛋白相互作用;将重组质粒pCMV--Myc-Ccdc189与pCMV-HA-Pabpc2共同转染入Hela细胞中,48小时后,以Myc与HA为一抗进行双重免疫荧光染色分析,通过荧光显微镜观察Ccdc189与Pabpc2蛋白在Hela细胞中是否共定位。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,依据本发明技术实质对以上实施例所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。