具有网格尺寸可逆调节的聚合物网络及其制备方法
文献发布时间:2024-04-18 19:58:53
技术领域
本发明属于高分子材料领域,具体涉及一种网格尺寸可调节的聚合物网络以及该聚合物网络的合成方法。
背景技术
作为聚合物网络的重要组成部分,链之间的交联点在确定材料特性(如弹性、网络动力学和缺陷容忍度)方面发挥着至关重要的作用。随着先进合成方法的出现,聚合物网络中新结构组成和化学成分已被报道,例如纳米粒子网络水凝胶、金属有机笼作为连接点的聚合物网络中,以及超分子聚合物网络;其中,使用纳米粒子作为构建网络的关键构件的聚合物纳米粒子网络(PNN)成为过去十年中最有前景的聚合物网络。越来越多的证据表明,当通过以共价交联或物理相互作用而结合在纳米粒子(NP)上的聚合物链进行桥接时,能够给材料带来各方面性能的改进,例如提升其模量、刚度、电导率或PNN的光学性能。
最近,大量的NP(Fe
另外,本领域人员均知晓,聚合物网络的形成过程中的微观结构变化将直接影响其物理化学性质和应用。已有研究通过机械性能实验或使用X射线/中子散射或显微镜(电子显微镜、偏振光学显微镜)等技术深入探索PNN结构和性能,但由于缺乏能够在合适的长度和时间尺度上提供直接动态信息的实验方法,现有技术中对网络形成过程中的动力学和微观结构知之甚少。然而,动力学和微观结构信息,尤其是粘弹性行为,对调控聚合物网络性能具有很好的反馈价值。因此,拓扑可转换网络的微观结构、动力学和性能增强之间的综合相关性仍然是一项艰巨的挑战。
发明内容
与现有技术中的NP不同,单链纳米粒子(SCNP)具有独特的交联纳米域,其能够表现出与其它聚合物纳米粒子(例如大环、树枝状聚合物、支化聚合物和无机纳米粒子)所不同的性能。进一步,基于可控聚合技术与合成策略的改进,SCNP(5-20nm)的结构和尺寸都可以通过调整线形前体中的反应性侧基的类型和数量来有效地控制;此外,可逆的单链折叠可以通过动态共价/非共价化学来实现,例如Diels-Alder反应和动态超分子相互作用。
由此,本发明的发明人设想,通过外部刺激驱动实现SCNP和线形前体(LP)的可逆拓扑可转换性是能够实现的,而将其应用于聚合物网络的制备则可以使得聚合物网络的性能也能够实现外部刺激调节。基于此,本发明提出了一种具有网格尺寸可逆调节的聚合物网络的制备方法,其通过拓扑结构可逆的单链内交联纳米粒子构建了网格尺寸可调节的聚合物网络,从而实现网络的多重性能,如网络的弹性性能及拉伸强度等的可逆转换。
由于本发明利用拓扑结构可逆转换的SCNPs来驱动聚合物网络的形成,而SCNPs表面有许多活性基团,当与双官能的线形前驱体反应时,其可作为交联点存在网络中,这种网络制备的方法不同于传统网络制备过程,所得到的聚合物网络也不同于传统的聚合物网络。
此外,本发明利用微流变技术确定了网络形成的动力学、微观结构(即网格尺寸(ξ)、终端弛豫时间(T
具体地,本发明提供了一种具有网格尺寸可逆调节的聚合物网络的制备方法,该方法首先合成一种单分散四嵌段共聚物(线形共聚物,即SCNP的前体),其包含甲基丙烯酸甲酯(MMA)、2-(乙酰乙酰氧基)甲基丙烯酸乙酯(AEMA)、甲基丙烯酸糠酯(FMA)和马来酰亚胺官能化甲基丙烯酸酯单体(FMIMA)重复单元;通过可逆加成-断裂链转移(RAFT)聚合,可以合成得到具有β-酮酯,呋喃和马来酰亚胺反应官能团的聚合物;该聚合物的合成反应路线如图11所示。
随后,在稀溶液条件下,通过呋喃和马来酰亚胺之间的DA反应,即分子内Dies-Alder反应制备SCNP,反应路线如图12所示。
然后,通过SCNP中所含的β-酮酯与聚(乙二醇)二丙烯酸酯在碱性条件下发生分子间迈克尔加成反应,构建得到聚合物网络(PSNN),其反应路线如图13所示。该PSNN的构建过程中,采用扩散波光谱(DWS)测量对网络形成的动力学过程及网络的微观结构进行分析。
更具体地,本发明的网格尺寸可逆调节的聚合物网络的制备方法包括如下步骤:步骤1,采用多种单体通过RAFT反应合成线形聚合物;步骤2,将步骤1得到的线形聚合物进行分子内Dies-Alder反应,得到单链内交联聚合物纳米粒子;步骤3,采用步骤2得到的单链内交联聚合物纳米粒子进行分子间迈克尔加成反应,得到聚合物纳米粒子网络。
进一步,上述具有网格尺寸可逆调节的聚合物网络的制备方法还可以具有这样的技术特征,其中,步骤1中所采用的多种单体包括甲基丙烯酸甲酯、2-(乙酰乙酰氧基)甲基丙烯酸乙酯、甲基丙烯酸糠酯和马来酰亚胺官能化甲基丙烯酸酯。
另外,上述具有网格尺寸可逆调节的聚合物网络的制备方法中,步骤1的具体操作为:先在聚合反应瓶中加入多种单体以及RAFT试剂,抽真空除氧,维持预定时间后加入偶氮二异丁腈,继续除氧后置于油浴锅中反应,然后用乙酸乙酯溶解,将溶解所得的溶液滴入到乙醇中沉降,得到沉降后的聚合物,置于常温真空干燥箱干燥至恒重,得到线形聚合物。其中,RAFT试剂可以是2-氰基-2-丙基苯并二硫。
上述具有网格尺寸可逆调节的聚合物网络的制备方法中,步骤2的具体操作为:将步骤1所得到的线形聚合物溶于过量的N,N-二甲基甲酰胺中配置成极稀溶液,将该极稀溶液缓慢升温除去保护的呋喃环,最后缓慢降低至室温反应,反应结束后除去溶剂,将得到的溶液滴入到甲醇,得到单链内交联聚合物纳米粒子。
上述具有网格尺寸可逆调节的聚合物网络的制备方法中,步骤3的操作为:将步骤2所制备的单链内交联聚合物纳米粒子溶于四氢呋喃中,加入催化剂,搅拌均匀后加入聚(乙二醇)二丙烯酸酯溶液,室温条件下搅拌,得到聚合物纳米粒子网络。
进一步,上述催化剂可以是KOH。
本发明还提供了一种具有网格尺寸可逆调节的聚合物网络,其特征在于,采用如上任一项的制备方法制备得到。
发明的作用与效果
根据本发明提供的网格尺寸可逆调节的聚合物网络及其制备方法,由于单链内交联聚合物纳米粒子是通过线形聚合物进行分子内Dies-Alder反应而得到的,因此,其制备得到聚合物纳米粒子网络后,能够通过不同温度下的处理来使其中的DA键被破坏或者重新形成,从而让网格尺寸变大或缩小,进而使聚合物网络的弹性模量、拉伸强度等发生改变,能够表现出不同的性能,且这种改变以及不同性能表现是可逆的,所以,该聚合物网络具有良好的应用潜力。
附图说明
图1是本发明实施例1的线形聚合物和单链内交联聚合物纳米粒子的核磁图。
图2是本发明实例1的交联尺寸5nm的网络尺寸中单链内交联聚合物纳米粒子的粒径分布图。
图3是本发明实施例1的线形聚合物和单链内交联聚合物纳米粒子的SEC流出曲线图。
图4是本发明实施例1的线形聚合物和单链内交联聚合物纳米粒子的粒径分布图。
图5是本发明实施例1的单链内交联聚合物纳米粒子形成凝胶状态的实物照片。
图6是本发明实施例1的聚合物纳米粒子网络形成过程中的自相关函数曲线图。
图7是本发明实施例1的聚合物纳米粒子网络形成前后弹性模量随时间变化曲线图。
图8是本发明实施例1的PSNN网络尺寸可逆调节原理示意图。
图9是本发明实施例1的PSNN网络尺寸变化前后弹性模量的变化图。
图10是本发明实施例1的PSNN网络尺寸变化前后拉伸强度的变化图。
图11是本发明的线形共聚物的合成反应路线图。
图12是本发明的分子内Dies-Alder反应的反应路线图。
图13是本发明的聚合物网络构建反应路线图。
具体实施方式
以下结合实施例说明本发明的具体实施方式。以下各实施例中所使用的聚(乙二醇)二丙烯酸酯(Mw~10000)、甲基丙烯酸甲酯(AR,≥99%)、甲基丙烯酸糠酯(AR,≥99%)、2-(乙酰乙酰氧基)甲基丙烯酸乙酯(AR,≥99%)、无水乙醇(AR,≥99%)、N,N-二甲基甲酰胺(DMF AR,≥99%)、四氢呋喃均购买于阿拉丁试剂有限公司;未提及来源的试剂和材料的来源均为常规市售,未提及的操作均采用本领域的常规操作。
<实施例1>
本实施例提供了一种交联点尺寸大小为5nm且网格尺寸能够可逆地改变聚合物网络,其制备方法包括如下步骤:
步骤1,采用四种单体,即甲基丙烯酸甲酯、2-(乙酰乙酰氧基)甲基丙烯酸乙酯、甲基丙烯酸糠酯和马来酰亚胺官能化甲基丙烯酸酯单体,通过RAFT反应合成单分散四嵌段共聚物(即线形聚合物),具体操作为:
按照MMA:AEMA:FMA:FMIMA:AIBN:ECPDB=40:20:20:20:1:1的比例(物质的量比例),先在放有磁力搅拌子的100mL的聚合反应瓶中加入四种单体(四种单体总量为5g)及RAFT试剂2-氰基-2-丙基苯并二硫(0.107g),抽真空除氧,维持40min后加入偶氮二异丁腈(AIBN)(0.082g),继续除氧15min后置于油浴锅中60℃条件下反应12h,得到聚合物。反应结束后,用乙酸乙酯溶解聚合物,将溶解所得的溶液滴入到乙醇中沉降,得到沉降后的聚合物,置于常温真空干燥箱干燥24h至恒重,得到线形聚合物(LP),即聚合物P1。
步骤2,将步骤1得到的单分散四嵌段共聚物进行分子内Dies-Alder反应,得到单链内交联聚合物纳米粒子,具体操作为:
将步骤1所得到的聚合物P1溶于过量的N,N-二甲基甲酰胺中(具体比例为每1g聚合物P1溶于1000mL以上的N,N-二甲基甲酰胺),配置成极稀溶液;将该极稀溶液缓慢升温至120℃,除去保护的呋喃环,最后缓慢降低至室温,反应24h。反应结束后,通过旋蒸仪除去溶剂,将得到的溶液滴入到甲醇,得到单链内交联聚合物纳米粒子(SCNP)。
步骤3,采用步骤2得到的单链内交联聚合物纳米粒子进行分子间的迈克尔加成反应,制备得到聚合物纳米粒子网络,具体操作为:
将步骤2所制备的SCNP(0.3g)溶于1mL四氢呋喃中,并加入10mg KOH作为催化剂。搅拌均匀后,加入0.05g聚(乙二醇)二丙烯酸酯溶液,室温条件下搅拌1h,最终得到聚合物纳米粒子网络(PSNN)。
图1是本发明实施例1的线形聚合物和单链内交联聚合物纳米粒子的核磁图。
如图1所示,相较于LP,SCNP的核磁图谱中,马来酰亚胺官能化甲基丙烯酸甲酯单体(FMIMA)上保护基的m和n峰消失,呋喃环的g和h质子峰消失,并在SCNP谱图中新出现了质子峰h和g,表明形成了DA加合物。
图2是本发明实施例1的交联尺寸的粒径分布图
如图2所示,通过DLS可以测量得出,本实施例的单链内交联聚合物纳米粒子的尺寸为5nm,也就是说,单链内交联聚合物纳米粒子作为交联点形成聚合物网络时,其交联点的尺寸就是5nm。
图3是本发明实施例1的线形聚合物和单链内交联聚合物纳米粒子的SEC流出曲线图。
如图3所示,LP经过分子内交联形成SCNP后,其流出体积减少,在凝胶色谱柱中有着较长的流出时间,说明其表观分子量有所降低。
图4是本发明实施例1的线形聚合物和单链内交联聚合物纳米粒子的粒径分布图。
如图4所示,LP经过分子内交联形成SCNP后,其在溶液中表现为流体力学半径减少,由15.4nm降低为12.2nm。
图5是本发明实施例1的单链内交联聚合物纳米粒子形成凝胶状态的实物照片,图中,左侧为单链内交联聚合物纳米粒子溶液,右侧为加入交联剂(聚(乙二醇)二丙烯酸酯)及催化剂(KOH)、室温搅拌1h所形成的凝胶状态。
如图5所示,在加入交联剂几分钟后,SCNP溶液由原先流体状态变成凝胶状态。
本实施例还采用扩散波谱(DWS)对步骤3中的聚合物纳米粒子网络的形成过程进行了动力学检测,具体操作为:
将步骤3的反应体系置于DWS测试皿中,同时加入示踪粒子TiO
其中,DWS具有高达10
根据布朗粒子的速度自相关函数(下式1),可以数值化计算粒子的均方位移<Δr
图6是本发明实施例1的聚合物纳米粒子网络形成过程中的自相关函数曲线图。
如图6所示,在PSNN形成之前,SCNP溶液表现为牛顿流体行为,自相关函数曲线显示出从1到0的快速衰减,这与粘性液体状态下样品结构能够快速运动一致;随着反应时间的增加,自相关函数曲线在很长一段时间内从基线开始变得不再降低,这意味着网络正在形成,反应体系的粘度显著增加。
图7是本发明实施例1的聚合物纳米粒子网络形成前后弹性模量随时间变化曲线图。图中,G’表示弹性模量,G”表示损耗模量。
如图7所示,对于粘性的SCNP溶液,其在溶液中的流变行为表现为损耗模量大于弹性模量;当反应体系形成弹性的PSNN后,弹性模量就大于损耗模量。
图8是本发明实施例1的PSNN网络尺寸可逆调节原理示意图。
如图8所示,PSNN(阶段1)可以通过温度激发其在刚性状态与柔性状态的可逆切换。对于合成得到的PSNN,通过加热即可破坏PSNN所含有的SCNP中的DA键,使PSNN的网格尺寸变大,转变成普通的聚合物网络,即、形成了PSNN的软弹性体(PSNN-s,阶段2);对于PSNN-s,可以通过快速冷却至室温后在较低温度下维持一段时间,使DA键重新形成,交联点恢复到SCNP结构,网格尺寸再次缩小,从而恢复到刚性状态(即PSNN-r,阶段3)。
本实施例中,通过120℃加热12h的处理方式让PSNN转变为PSNN-s,通过45℃维持24h的方式让PSNN-s转变为PSNN-r。
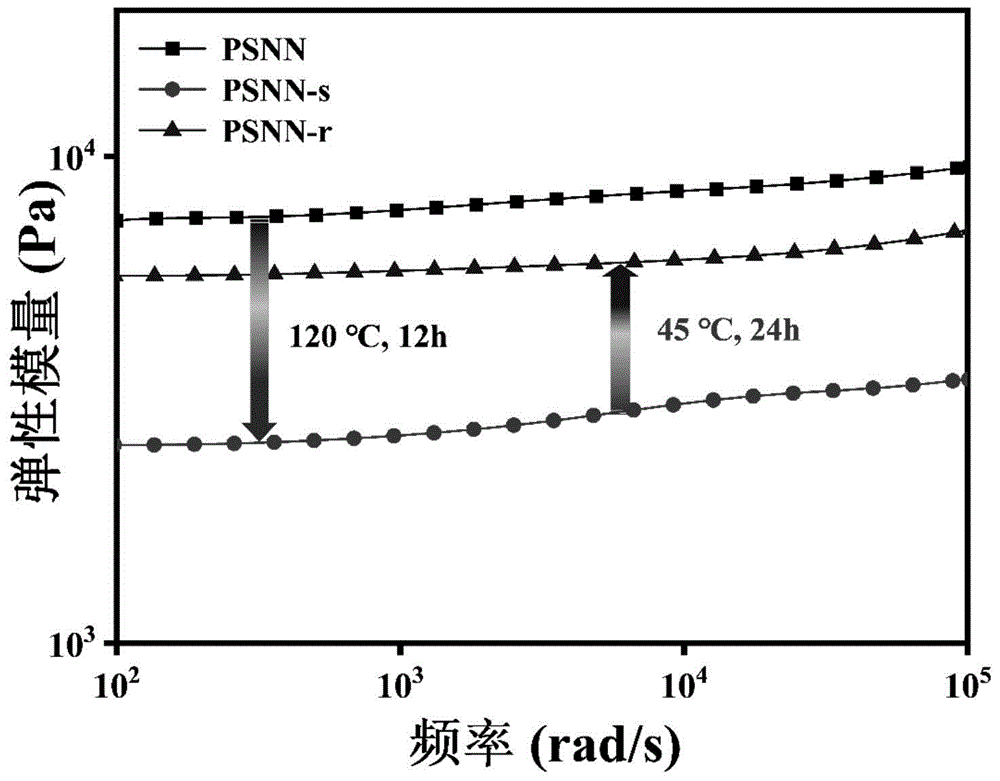
图9是本发明实施例1的PSNN网络尺寸变化前后弹性模量的变化图。
如图9所示,合成的PSNN的弹性模量较高,说明其中网格尺寸较小;在经过120℃加热12h形成PSNN-s后,弹性模量明显降低,说明其网格尺寸变大,转变为柔性状态;迅速冷却至室温并在45℃下维持24h后,其弹性模量增加,说明其中的网格恢复至较小的尺寸,也就是回复到刚性状态。
图10是本发明实施例1的PSNN网络尺寸变化前后拉伸强度的变化图。
如图10所示,合成的PSNN的拉伸强度较大,说明其中网格尺寸较小;在经过120℃加热12h形成PSNN-s后,弹性模量明显降低,说明其网格尺寸变大,转变为柔性状态;迅速冷却至室温并在45℃下维持24h后,其弹性模量增加,说明其中的网格恢复至较小的尺寸,也就是回复到刚性状态。
<实施例2>
本实施例提供了一种交联点尺寸大小为10nm且网格尺寸能够可逆地改变聚合物网络及其制备方法。
与实施例1相比,本实施例的步骤2、步骤3操作完全相同,但步骤1的具体操作有所不同,如下:
步骤1中,按照MMA:AEMA:FMA:FMIMA:AIBN:ECPDB=40:20:20:20:0.5:0.5的比例(物质的量比例),先在放有磁力搅拌子的100mL的聚合反应瓶中,加入单体(总量5g)及RAFT试剂2-氰基-2-丙基苯并二硫(0.0535g),抽真空除氧,40min后加入AIBN(0.041g),继续除氧15min后置于油浴锅中60℃条件下反应12h。反应结束后,用乙酸乙酯溶解聚合物,将溶解所得的溶液滴入到乙醇中沉降得到聚合物,置于常温真空干燥箱干燥24h至恒重,得到聚合物P1 3.9g。
通过与实施例1相同的步骤2、3,本实施例制备得到了PSNN。经温度激发测试,该PSNN也表现出了与实施例1相同的尺寸可逆调节的性能。
<实施例3>
本实施例提供了一种交联点尺寸大小为20nm且网格尺寸能够可逆地改变聚合物网络及其制备方法。
与实施例1相比,本实施例的步骤2、步骤3操作完全相同,但步骤1的具体操作有所不同,如下:
步骤1中,按照MMA:AEMA:FMA:FMIMA:AIBN:ECPDB=40:20:20:20:0.3:0.3的比例(物质的量比例),先在放有磁力搅拌子的100mL的聚合反应瓶中,加入单体(总量5g)及RAFT试剂2-氰基-2-丙基苯并二硫(0.032g),抽真空除氧,40min后加入AIBN(0.025g),继续除氧15min后置于油浴锅中60℃条件下反应12h。反应结束后,用乙酸乙酯溶解聚合物,将溶解所得的溶液滴入到乙醇中沉降得到聚合物,置于常温真空干燥箱干燥24h至恒重,得到聚合物P1 3.9g。
通过与实施例1相同的步骤2、3,本实施例制备得到了PSNN。经温度激发测试,该PSNN也表现出了与实施例1相同的尺寸可逆调节的性能。
实施例的作用与效果
根据上述实施例可以看出,通过将单体聚合形成单分散四嵌段共聚物,再通过DA反应制备得出SCNP、通过分子间迈克尔加成反应将SCNP构建为PSNN,所得到的PSNN具有能够通过温度激发改变网格尺寸大小的特性,基于此,该PSNN可以在不同温度处理后表现出不同的性能,即、具有传统聚合物网络所不具备的拓扑结构可逆性改变的性能,极具应用潜力。
- 一种具有可逆共价结合功能的分子印迹聚合物及其制备方法和应用
- 一种基于可逆动态相互作用的超拉伸聚合物网络及其制备方法
- 一种低温热可逆型热塑性聚氨酯聚合物的制备方法及聚合物
- 一种具有阻燃性能的线性热可逆聚氨酯聚合物的制备方法及聚合物
- 光响应聚合物及其制备方法、表面粗糙度的可逆调节方法