混合样品中淋巴细胞丰度的确定
文献发布时间:2024-05-31 01:29:11
技术领域
公开内容涉及基于来源于包含多种细胞类型的混合样品的读取深度谱(readdepth profile,读取深度分布)来评估样品(例如肿瘤样品或血液样品)中淋巴细胞(例如T淋巴细胞或B淋巴细胞)的分数的方法,并且涉及用于实施这种方法的系统和相关产品。还描述了至少部分基于来自癌症患者的肿瘤样品的评估的肿瘤浸润性淋巴细胞分数为该患者提供预后的方法。
背景技术
淋巴细胞是人体系统的必不可少的部分,在人健康和疾病中发挥至关重要的作用。例如,在肿瘤的情况下,免疫系统被广泛认为是影响肿瘤发生和随后癌症演变的关键因素之一。T细胞尤其通过清除含有新抗原的癌细胞而在癌症免疫微环境中发挥重要作用,因此肿瘤样品内T细胞的数目是重要的临床因素。事实上,临床表现的肿瘤免疫微环境状态既可以预后又可以预测对免疫治疗的响应。近年来,免疫治疗,并且特别是检查点抑制剂(checkpoint inhibitor,CPI)已经成为许多癌症类型的革命性治疗。事实上,在抗CTL4或抗PDL1治疗后,黑素瘤和非小细胞肺癌(non-small cell lung cancer,NSCLC)在无病存活和总存活方面均表现出显著的改善(Robert et al.,2011;Schadendorf et al.,2015;Topalian et al.,2012)。然而,对免疫治疗的响应并不是普遍的,其中临床有益响应仅发生在一部分患者中(Goodman et al.,2017)。因此,确定哪些患者将受益于CPI是至关重要的。
新兴数据表明,对CPI的响应主要由两个特征决定;免疫刺激,例如可由下一代测序(next generation sequencing,NGS)预测的新抗原的存在,以及能够对刺激响应的T细胞的存在。浸润性T细胞的存在长期以来一直与癌症类型,例如卵巢癌(Zhang et al.,2003)和结直肠癌(Galon et al.,2006)的存活提高有关。最近的研究集中在新抗原的预测潜力上,其中肿瘤突变负荷(tumour mutational burden,TMB)提高成为对免疫治疗响应的最佳预测因素之一(Samstein et al.,2019)。由Litchfield等人(2020)的最近研究系统地调查了对CPI响应的不同潜在生物标志物在泛癌CPI治疗的数据组中表现的程度。鉴定的两个最具预测性的特征是克隆TMB,其反映了每个癌细胞中存在的突变和T细胞的RNA测序来源的转录组特征(例如CD8A RNA表达),表明存在肿瘤浸润性淋巴细胞(tumourinfiltrating lymphocyte,TIL)。
因此,评估TIL的存在和丰度二者在癌症治疗中是至关重要的。然而,目前还没有用于评估TIL的通用的最先进的方法。微阵列特征例如CIBERSORT(Newman et al.,2015)以及RNA-seq特征例如RNA-seq的CIBERSORTx(Newman et al.,2019)、ESTIMATE(Yoshiharaet al.,2013)和由Danaher等人汇编的那些,已经被用于量化肿瘤样品中TIL的转录组特征。或者,可以基于苏木精和伊红(H&E)组织病理学切片或适当的细胞特异性染色(最常见的是免疫组织化学)来确定免疫浸润的存在。如果需要更多的T细胞表型知识,可以在编码TCR的高度多样化的VDJ重组CDR3区域的那些序列的富集之后,进行T细胞受体(T cellreceptor,TCR)测序(Bolotin et al.,2012)。出于鉴定混合样品(例如肿瘤样品)中的免疫细胞的目的,所有这些方法都需要采集专用数据。最近,McGrath等人(2017年)提出了这样的方法:通过分离T细胞重排读取(跨越高变CDr3并包括V和J区段二者的读取),使用WES数据来表征样品中的TCR库。然而,这种方法需要极高的测序深度,不能处理非T细胞群体中非整倍体的存在(如例如在癌症的情况下是常见的),并且仅适合于表征TCR库。
因此,存在用于确定混合样品中淋巴细胞丰度的新方法以减轻现有方法的一个或更多个缺点的需求。
发明内容
广义地说,本发明人认识到,经常对肿瘤样品进行全外显子组测序(whole exomesequencing,WES)或甚至全基因组测序(whole genome sequencing,WGS)来计算肿瘤突变负荷并鉴定用于靶向治疗的可操作突变。同样地,这种类型的数据被越来越频繁地在其他疾病背景,例如新生儿护理下收集。然而,目前WES不能用于准确推断免疫浸润水平。因此,本发明人开发了使用在VDJ重组,例如如T细胞受体α(T cell receptor alpha,TCRA)基因的VDJ重组期间来源于T细胞受体切除环(T cell receptor excision circle,TREC)的信号,由WES/WGS或类似数据评估T细胞(或B细胞)分数的方法。该评分直接测量样品中T细胞(或B细胞,视情况而定)的比例。发明人进一步证明了,该评分与基于TRACERx100组群中RNA测序和组织病理学TIL评分的正交免疫浸润评分显著相关。使用接受免疫治疗的患者组群的荟萃分析,本发明人证实了该评分是对癌症免疫治疗响应的预测,提供了突变负荷之外的预测价值。发明人通过证明T细胞分数还可以由血液来源的种系样品计算,进一步示出了该方法的广泛适用性。因此,该方法提供了进入到WES(或WGS)肿瘤样品的免疫微环境中的窗口,这在正交数据不可用的情况下特别有用。
因此,在第一方面,本公开内容提供了用于确定包含来自多种细胞类型的基因组物质的混合样品中的淋巴细胞分数的方法,所述方法包括:获得样品的读取深度谱,其包括沿着目标的预定基因组区域的读取深度,其中目标的预定基因组区域包含经历VDJ重组的基因组基因座的至少一部分;通过参考来源于目标区域的子集的基线读取深度来归一化沿着目标的预定区域的读取深度来获得多个读取深度比(r
该方法可具有下列任选特征中的任意一个或更多个。
混合样品可以是细胞或组织样品(在这种情况下,样品可包含多种细胞类型,所述每种细胞类型包含各自的基因组物质),或者来源于其的核酸样品。
经历VDJ重组的基因组基因座可选自TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座。因此,淋巴细胞分数可以是T淋巴细胞分数,B淋巴细胞分数,αβT淋巴细胞分数,γδT淋巴细胞分数,包含TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座中任何基因区段的B细胞或T细胞的分数,或者包含表8中列出的任何基因区段或另外的物种中相应基因区段的B细胞或T细胞的分数(其中表8中列出的坐标仅是指示性的,并且特别是可以使用另一参考序列中的相应坐标)。经历VDJ重组的基因组基因座可选自TCRA、TCRB、TCRG或IGH基因座。目标的预定区域可包含来自TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座中的一个或更多个外显子。目标的预定区域可包含来自TCRA、TCRB、TCRG或IGH基因座中的一个或更多个外显子。目标的预定区域可包含来自TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座的多个外显子,其中多个外显子包含至少一个与各自基因座的V、D和C区段中的每一个对应的外显子。多个外显子可包含至少一个对应于各自基因座的V、D、J和C区段的每一个的外显子。多个外显子可包含与各自基因座的多个V区段对应的外显子。目标的预定区域可包含编码来自TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座的多个V、D、J和/或C区段的区域。目标的预定区域可包含位于来自TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座的V、D、J和/或C区段之间的一个或更多个区域。目标的预定区域可包含来自TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座的所有外显子。目标的预定区域可包含所有的TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座(即包含外显子和内含子二者)。
预定区域可包含来自TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座的外显子的子集。预定区域可包含TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座的子集。例如,预定区域可排除任何已被确定为与系统性偏差相关的外显子或区域,所述系统性偏差例如如由于用于收集读取数据的平台而产生的偏差、与外显子或区域中GC含量相关的偏差、或与外显子或区域中覆盖相关的偏差。例如,一些外显子捕获试剂盒已被证明与导致某些外显子中的覆盖偏离预期的偏差相关。类似地,例如由于测序或比对过程中的人为因素,某些区域可具有比全基因组测序中所预期的更低或更高的覆盖。这样的外显子或区域可以通过比较预期不经受相同系统性偏差(例如如使用不同平台(例如不同的捕获试剂盒)获得的)数据集之间的读取覆盖来鉴定。例如,可以针对第一组样品和第二组样品(其中预期两组样品不经受相同的系统性偏差)计算多个候选外显子或区域的平均logR比率,并且可以针对多个候选外显子或区域中的每一个比较每组样品的中位数,以鉴定两组样品之间显著不同的那些。例如,高于阈值(例如如0.5)的中位数logR之间的差异可用作排除可能经受偏差的外显子/区域的标准。具有比所预期的更低或更高覆盖的区域可以通过以下来鉴定:将模型拟合至多个样品的平均读取深度比数据,例如广义线性模型,并去除与不在拟合模型的预定距离内的概括值(例如平均值或中位数值)相关的预定大小(例如如100bp、200bp、500bp、1000bp)的任何区域。替代地或除此之外,具有比所预期的更低或更高覆盖的区域可以通过以下来鉴定:确定多个样品的淋巴细胞分数并进行全基因组相关性研究以鉴定与所确定的淋巴细胞分数相关的单核苷酸多态性,其中相关性的存在指示包含单核苷酸多态性的预定大小的区域中的覆盖偏差。因此,该方法可包括通过以下来鉴定全基因组测序中具有比所预期的更低或更高覆盖的区域:确定多个样品中的淋巴细胞分数,进行全基因组相关性研究以鉴定与所确定的淋巴细胞分数相关的单核苷酸多态性,选择包括单核苷酸多态性的样品,将模型拟合至所选样品的平均读取深度比数据,并去除与不在拟合模型的预定距离内的概括值(例如,平均值或中位数值)相关的预定大小(例如如100bp、200bp、500bp、1000bp)的任何区域。
可以处理读取深度或读取深度比,以校正与所使用的读取深度数据采集平台相关的偏差。例如,可以对读取深度/读取深度比进行GC校正。可以使用在每个位置处捕捉GC含量对读取深度的影响的线性模型来进行读取深度数据的GC校正,其中该模型的残差表示归一化的读取深度值。GC含量对读取深度的影响可由以下表示:反映外显子或区域水平处的GC含量的项(GC
概括读取深度值可以是概括对数读取深度比。该对数可以是底数为2的对数。确定作为概括读取深度比值(r
其中γ是常数。确定样品中的淋巴细胞分数(f)可包括如下确定f的值:
其中γ是常数。可以根据用于获得读取深度数据的分析平台来调整参数γ。例如,当使用Illumina Hiseq时,γ的值可以是=1(即该参数可以从所有方程式中去除)。不希望被理论所束缚,认为参数γ捕获了与理论预期值相比的log R谱的与平台相关的“压缩”(也称为“r”,在当前情况下,logR是目标区域相对于参考区域中读取深度的对数比)。例如,当比较预期具有相同拷贝数的区域时,r应该等于零。因此,通过与控制设置中的预期值进行比较,可以针对特定平台设置该参数。如本领域技术人员所理解的,如果ψ
可能通过VDJ重组被缺失的目标区域的子集可包含位于经历VDJ重组的基因组基因座的最后一个V区段的末端与经历VDJ重组的基因组基因座的J区段的起点之间的区域。可能通过VDJ重组被缺失的目标区域的子集可包含经历VDJ重组的基因组基因座的一个或更多个D区段(例如如,所有D区段)。可能通过VDJ重组被缺失的目标区域的子集优选不包含任何V或C区段。替代地或除此之外,可能通过VDJ重组被缺失的目标区域的子集优选不包含任何J区段。可能通过VDJ重组被缺失的目标区域的子集可包含以下、由以下组成、或包含在以下中:最后的V基因区段与第一区段之间的间隙,所述第一区段编码经历VDJ重组的基因组基因座的J区段,或者所述第一区段在TCRA的情况下编码TCRδ链的一部分;或者另一个目标的预定区域中的任何相应区段。例如,当观察TCRA基因座时,本发明人已发现将最大VDJ重组的位置限定为最后的TCRAV区段与TCRδ基因的第一区段之间的间隙是合适的,该间隙被四舍五入为大小为80000bp,并起始于第22800000位(通向区域chr14:22800000至22880000)。其他限定是可能的,并且该区域大小的微小变化预计不会对该方法的性能产生负面影响。最大VDJ重组的区域可以使用类似的方法针对其他VDJ基因座限定,即通过限定至少部分位于最后的V基因区段与第一J基因区段之间的区域的周围或内部的区域。使用表7中的基因组坐标、或其他物种或参考基因组的相应坐标,可以限定各个VDJ基因座的最大VDJ重组区域。可能通过VDJ重组被缺失的目标区域的子集可包含对应于经历VDJ重组的基因组基因座的任何V或J区段的区域。这可以在以下情况下使用:当淋巴细胞分数是包含TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座中的任何基因区段的B细胞或T细胞的分数时,或者是包含表8中列出的任何基因区段或另外的物种中相应基因区段的B细胞或T细胞的分数时。
该方法还可以包括将模型拟合至多个读取深度比,以及获得可能通过VDJ重组被缺失的目标区域的子集的概括读取深度比值(r
一般而言,概括值可以是多个值的中位数、平均值、修整平均值或任何其他已知的中心性统计度量。区域的概括读取深度比值可以是该区域内多个读取深度比值的中心性统计度量。在将模型(例如广义加性模型)拟合至读取深度比谱的实施方案中,可以获得区域的概括读取深度比值作为该区域内拟合模型的值的中心性统计度量。例如,可以使用整个区域的拟合模型的平均值。在将分段常数模型拟合至读取深度比谱的实施方案中,可以获得区域的概括读取深度比值作为对应于该区域的区段或对应于该区域的多个区段的拟合模型的值,或者该区域内的区段的模型的值。例如,使用如上所述约束的分段常数模型,可选择可能通过VDJ重组被缺失的目标区域的子集的概括读取深度比值作为模型的最小值(其也等于模型的相对于0的最大偏差)。
当淋巴细胞分数是包含TCRA、TCRB、TCRG、TCRD、IGH、IGL或IGK基因座中任何基因区段的B细胞或T细胞的分数,或者是包含表8中所列出的任何基因区段或另外的物种中相应基因区段的B细胞或T细胞的分数时,可能通过VDJ重组而被缺失的目标区域的子集可对应于所述基因区段。基因区段的概括读取深度比值可以是基因区段的概括读取深度比值与先前基因区段的概括读取深度比值之间的差值或差值的绝对值。概括读取深度比值可以是概括的对数读取深度比。概括的读取深度比可以从拟合至读取深度比的模型的值中获得。任何对数都可以是底数为2的对数。
该方法还可包括获得拟合至多个读取深度比的模型的可能性,其中低于预定阈值的可能性指示样品中的过度噪声。该方法还可包括获得多个候选模型以及与多个候选模型的拟合相关的一个或更多个统计度量(例如可能性和/或置信区间),并基于统计度量选择候选模型。例如,可以选择置信区间在预定范围内的可能性最高的模型。获得多个候选模型可包括获得预定数量的模型,例如如10、30、50或100个模型。
该方法还可以包括从目标区域的子集获得基线读取深度。基线读取深度可以是目标区域的所述对象的概括读取深度值。例如,基线读取深度可以是在目标区域的预定子集上的中位数读取深度。用于获得基线读取深度的目标区域的子集可以是不太可能通过VDJ重组被缺失的目标区域的子集。不太可能通过VDJ重组被缺失的目标区域的子集可包含经历VDJ重组的基因组基因座的前n个V区段。不太可能通过VDJ重组被缺失的目标区域的子集可包含经历VDJ重组的基因组基因座的C区段。优选地,用于获得基线读取深度的目标区域的子集不包含经历VDJ重组的基因组基因座的任何D或J区段。实际上,根据本文中所公开的方法,可以使用正在研究的VDJ基因座的起点、末端(而不是二者)或者起点和末端二者、或者可以假定具有不受VDJ重组影响的拷贝数值的其他邻近区域。可以使用表7中的基因组坐标、或其他物种或参考基因组的相应坐标,针对各个VDJ基因座限定用于获得基线读取深度的目标区域的子集。
不希望被理论所束缚,认为使用更长的归一化区域(例如,使用起始和终止区域二者,和/或增加起始区域的长度)有利地补偿了读取深度数据中典型的噪声。例如,起始区域可以延伸至包括许多不太可能经常丢失的V区段,或者如果该数据是可获得的(例如如当使用全基因组测序数据时),甚至包括基因外的区域。此外,认为使用基因起始处的信号是有益的,因为与VDJ重组相关的信号更靠近基因的末端而不是其起始处(由于J区段聚集在基因末端附近,而V区段更分散)。此外,也有可能使用研究中的VDJ基因座之外的区域,例如如在该基因座之前或之后。然而,增加距目标基因座的距离提高了所见信号将对应于癌细胞中不同拷贝数事件的可能性,并因此不再反映研究中的VDJ位点处的肿瘤拷贝数。可使用适当的训练数据来确定参数n,例如,当针对淋巴细胞分数的正交度量进行比较时,通过选择在训练数据中产生最准确的淋巴细胞分数评估的n的值。例如,可以基于转录组数据和/或组织病理学数据获得淋巴细胞分数的正交度量。参数n可以是8至16,10至14,例如如10、11或12。
获得读取深度谱可包括获得目标的预定区域中的多个原始读取深度值,并平滑读取深度值。平滑读取深度值可包括用预定宽度的窗口上的相应的滚动中位数来替换原始读取深度值。可以选择大约20b至大约200bp,或者大约50bp至大约150bp的窗口的预定宽度。例如,读取深度谱可包含通过计算沿着目标的预定区域的50bp窗口中的滚动中位数而获得的值。对于特定的数据集、数据类型或背景,通过将使用本方法在多种候选窗口大小下获得的淋巴细胞分数评估值与淋巴细胞分数的正交度量(例如Danaher评分)进行比较,可以凭经验确定合适的窗口大小。
混合样品可以是血液样品或肿瘤样品。该方法还可包括从对象中获得包含来自多种细胞类型的基因组物质的混合样品。该方法还可包括通过一个或更多个体外步骤从对象中获得包含来自多种细胞类型的基因组物质的混合样品的读取深度数据。一个或更多个体外步骤可包括核酸提取、文库制备、靶序列捕获、测序和/或与拷贝数阵列杂交。如技术人员所理解的,在通过例如测序获得了指示样品中核酸存在的数据之后,可以应用如本领域已知的或如本文中所述的另外步骤,例如如质量控制步骤、参考基因组比对步骤(也称为映射)、归一化步骤等。该方法还可包括对一种或更多种进一步混合的样品重复本方面的任何实施方案的方法,其中第一和进一步混合的样品已经从同一对象中获得,并且其中第一和进一步混合的样品是肿瘤样品。该方法还可包括基于第一和进一步混合的样品的淋巴细胞分数(f)获得一个或更多个概括淋巴细胞分数。可以获得概括淋巴细胞分数,即第一和进一步混合的样品中的平均分数、第一和进一步混合的样品中的最小分数、第一和进一步混合的样品中的最大分数、和/或第一和进一步混合的样品中的最小分数与第一和进一步混合的样品中的最大分数之间的倍数差。在可获得对象的多个混合样品的情况下,可使用一种或更多种概括的淋巴细胞分数来代替单一样品的单个淋巴细胞分数,以提供如下文将进一步所描述的预后。例如,多个样品中的最小分数、多个混合样品中的最小和最大分数、和/或第一和进一步混合的样品中的最小分数与第一和进一步混合的样品中的最大分数之间的倍数差可用于为对象提供预后。优选地,使用多个样品中的最小分数来提供预后。
该方法还可包括例如通过用户界面向用户提供所确定的淋巴细胞分数和/或从中推导出的任何值。从中推导出的值可包含诊断或预后指示。
如技术人员所理解的,本文所述操作的复杂性(至少由于通常由基因组DNA测序产生的数据量)使得它们超出了智力活动的范围。因此,除非上下文另有指示(例如,在描述样品制备或采集步骤的情况下),否则本文中描述的方法的所有步骤都是计算机实施的。
本公开内容的方法可用于多种临床情况,其中不同的诊断和/或预后可与来自对象的样品中不同水平的淋巴细胞相关。因此,本公开内容还涉及根据对象的淋巴细胞分数状态(例如肿瘤浸润淋巴细胞TIL状态)对对象群体进行分组的方法,该方法包括对来自该群体中每个对象的一个或更多个样品(例如肿瘤样品)实施本公开内容第一方面的方法。例如,根据对来自患者的一个或更多个肿瘤样品评估的淋巴细胞分数,可以将对象分为“免疫冷性”组和“免疫热性”组。这可以特别应用于个性化药物和/或临床试验的患者的鉴定。分类为免疫冷性的患者与分类为免疫热性的患者相比,预计对(例如使用检查点抑制剂的)免疫治疗的响应较低。因此,后者更可能受益于免疫治疗,而前者可更可能受益于替代治疗方案。替代地或除此之外,被分类的患者可比被分类为免疫热性的患者具有更差的生存预后。因此,本公开内容的方法还可用于对已被诊断患有癌症的对象在至少两个组之间进行分类,所述至少两个组具有不同的预后和/或对一种或更多种免疫治疗的预测敏感性。
因此,根据另一方面,本公开内容提供了为已被诊断为患有癌症的对象提供预后的方法,该方法包括使用第一方面的任何实施方案的方法确定来自对象的一个或更多个肿瘤样品中的淋巴细胞分数。还描述了监测已被诊断为患有癌症的对象的方法,该方法包括使用第一方面的任何实施方案的方法,确定在第一时间点获得的来自对象的一个或更多个肿瘤样品中的淋巴细胞分数,以及在另外的时间点获得的来自对象的一个或更多个肿瘤样品中的淋巴细胞分数。例如,第一时间点可以在治疗之前,并且另外的时间点可以在治疗之后。替代地,第一时间点和另外的时间点可以都在治疗之后。治疗可以采用任何抗癌治疗,包括免疫治疗。提供预后或监测对象的方法还可包括根据样品中的淋巴细胞分数(例如如T淋巴细胞分数)将样品分类为免疫热性或免疫冷性。例如,淋巴细胞分数高于阈值(例如,约0.1,即约10%)的样品可被分类为免疫热性,而淋巴细胞分数处于或低于阈值的样品可被分类为免疫冷性。该方法还可包括根据来自被分类为免疫冷性的对象的样品数量,将对象在良好预后组与不良预后组之间分类。例如,当分析来自不同肿瘤区域的样品时,被分类为免疫冷性的样品的数量处于或高于阈值(例如2)可将对象分类为不良预后组。与良好预后组相比,不良预后组可与降低的无复发存活和/或降低的总存活相关。淋巴细胞分数和/或免疫冷性样品数量的阈值可以通过以下来确定:使用适当的训练组群,例如通过评估多种阈值在与显著不同的预后相关的组之间对患者进行分类的能力(基于已知的响应状态)。还描述了确定已被诊断为患有癌症的对象是否可能对免疫治疗有响应的方法,该方法包括使用第一方面的任何实施方案的方法确定来自对象的一个或更多个肿瘤样品中的淋巴细胞分数。该方法还可包括对被诊断为可能对免疫治疗有响应的对象施用免疫治疗。该方法可包括推荐已被诊断为可能对免疫治疗有相应的对象用免疫治疗进行治疗。该方法可包括施用替代治疗(例如,常规化学治疗、放射治疗等)和/或推荐对象用替代治疗进行治疗,其中所述对象已被诊断为不太可能对免疫治疗有响应。
该方法还可包括将对象在可能对免疫治疗有响应的组与不太可能对免疫治疗有响应的组之间进行分类。事实上,本发明人已经证明,根据本公开内容确定的肿瘤以及血液样品中的T细胞分数可预测对免疫治疗的响应。例如,该方法可包括根据样品中的淋巴细胞分数(例如如T淋巴细胞分数)将样品分类为免疫热性或免疫冷性。例如,淋巴细胞分数高于阈值(例如,约0.1,即约10%)的样品可被分类为免疫热性,而淋巴细胞分数处于或低于阈值的样品可被分类为免疫冷性。如果来自对象的一个或更多个样品是免疫冷性的,则对象可被分类为不太可能对免疫治疗有响应的组。或者,如果来自对象的样品具有低于阈值的淋巴细胞分数,则对象可被分类为不太可能对免疫治疗有响应的组,否则可被分类为可能对免疫治疗有响应的组。
免疫治疗可以是调节免疫系统功能和/或利用现有免疫功能来治疗癌症的任何治疗。免疫治疗可选自T细胞转移治疗(肿瘤浸润淋巴细胞(或TIL)治疗和CART细胞治疗)、治疗性抗体、癌症治疗疫苗、检查点抑制剂治疗和免疫系统调节剂(例如干扰素和白介素)。该方法可包括基于来自对象的一个或更多个样品中的淋巴细胞分数和来自对象的评估肿瘤突变负荷二者,将对象在可能对免疫治疗(例如CPI治疗)有响应的组和不太可能对免疫治疗(例如CPI治疗)有响应的组之间进行分类。这样的分类可以使用多变量分类模型(例如,使用经过训练的广义线性模型的分类方法)来获得。分类模型的参数(例如阈值,多变量模型的参数)可以使用适当的训练组群,例如使用一组具有已知免疫治疗响应状态的对象来确定。
还描述了确定对象是否患有T细胞淋巴细胞减少症的方法,该方法包括使用第一方面的任何实施方案的方法确定来自对象的一个或更多个样品中的淋巴细胞分数。一个或更多个样品可以是血液样品。对象可以是新生儿对象。确定对象是否患有T细胞淋巴细胞减少症的方法可以在用于筛查严重联合免疫缺陷的方法的背景下进行。该方法还可包括治疗对象的淋巴细胞减少症,或推荐对象治疗淋巴细胞减少症。
还描述了诊断对象为患有与异常淋巴细胞计数相关的疾病、障碍或病症的方法,该方法包括使用第一方面的任何实施方案的方法确定来自对象的一个或更多个样品中的淋巴细胞分数。还描述了监测已被诊断为患有与异常淋巴细胞计数相关的疾病、障碍或病症的对象的方法,该方法包括使用第一方面的任何实施方案的方法确定来自对象的一个或更多个样品中的淋巴细胞分数。与异常淋巴细胞计数相关的疾病、障碍或病症可以是自身免疫性疾病、骨髓障碍或感染。
根据另一个方面,本公开内容提供了一种系统,其包括:
至少一个处理器;和至少一个包括指令的非暂时性计算机可读介质,所述指令在由至少一个处理器执行时使得至少一个处理器执行包括以下的操作:获得包含沿着目标的预定基因组区域的读取深度的样品的读取深度谱,其中目标的所述预定基因组区域包含经历VDJ重组的基因组基因座的至少一部分;通过参考来源于目标区域的子集的基线读取深度来归一化沿着目标的预定区域的读取深度,从而获得多个读取深度比(r
根据本方面的系统可以被配置为实施前述方面的任何实施方案的方法。具体地,所述至少一个非暂时性计算机可读介质可包含这样的指令:所述指令在由至少一个处理器执行时,使得至少一个处理器执行包括关于第一方面描述的任何操作的操作。该系统还可包括与处理器可操作连接的以下中的一者或更多者:用户界面,其中指令进一步使处理器向该用户界面提供至少淋巴细胞分数的评估值或从中推导出的值以输出给用户;一个或更多个序列读取深度数据采集装置(例如如测序装置);一个或更多个数据存储器,例如如序列读取深度数据存储器。
根据另一个方面,提供了非暂时性计算机可读介质或包含指令的介质,所述指令在由至少一个处理器执行时,使得至少一个处理器执行本文中所述的任何方面的任何实施方案的方法。
根据另一个方面,提供了包含代码的计算机程序,当在计算机上执行该代码时,该代码使得计算机执行本文中所述的任何方面的任何实施方案的方法。
本发明包括所描述的方面和优选特征的组合,除非其中这样的组合是明显不允许的或被指出要明确避免的。本发明的这些和另外的方面和实施方案在下文中并且参考所附实施例和附图更详细地描述。
附图说明
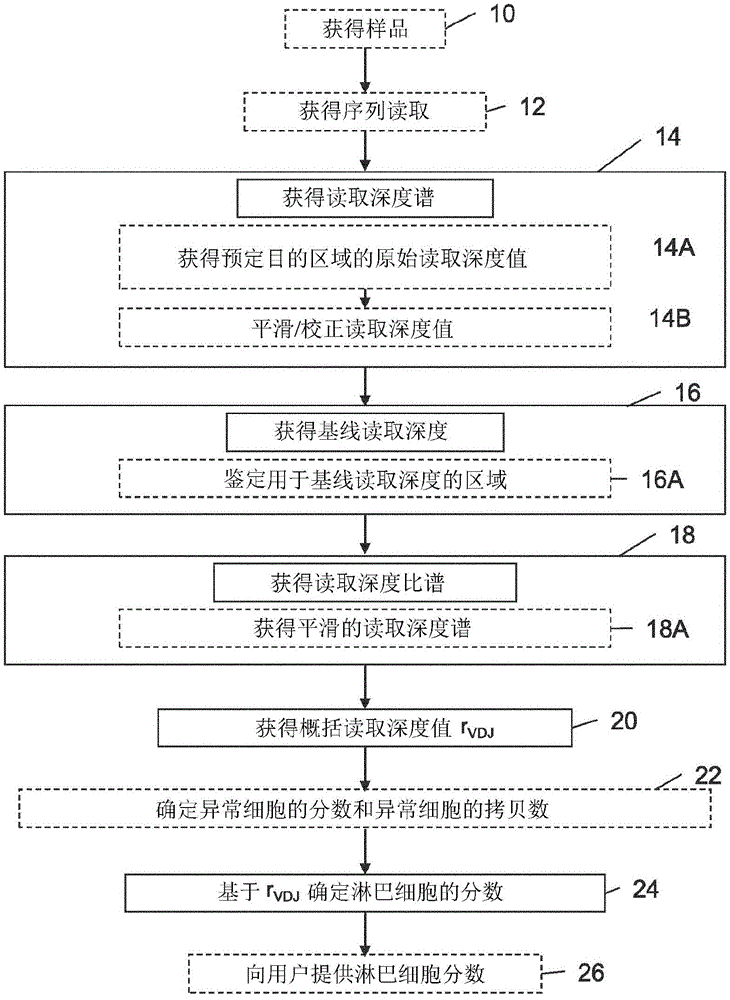
图1是示意性地示出了确定混合样品中的淋巴细胞分数的方法的流程图。
图2示出了用于确定混合样品中的淋巴细胞分数的系统的实施方案。
图3示意性地示出了使用VDJ重组信号确定本文中所述混合样品中的淋巴细胞分数背后的生物学概念。A.表明读取深度比信号如何用于检测标准肿瘤和匹配种系分析中的体细胞拷贝数改变增益或损失事件的示意图。在该分析中,细胞由三种不同的细胞类型组成:肿瘤细胞、T细胞和所有其他基质细胞。B.当聚焦于与VDJ重组和TREC的去除相关的TCRA基因时,该相同过程如何工作的示意图,左下图表明在TCRA基因内的TRACERx100数据集中检测到的断点数量相对于14q的周围区域增加,这表明TREC信号被捕获。
图4是示出了在两个TRACERx100区域中读取深度比的实例的图,表明与匹配的肿瘤相比血液中T细胞含量的水平提高(A)或与匹配的血液相比肿瘤中T细胞含量的水平提高(B)。VDV区段是指TCRα和TCRδ基因座二者中的可变区段。
图5示意性地示出了根据本公开内容的实施方案,如何可以将图3所示的信号转化成T细胞分数评估值。
图6示出了:A.一系列局部拷贝数和肿瘤纯度值的原初TCRA T细胞分数的理论值。点和线由实际的T细胞分数着色,其中水平直线表示点应该在哪里。B.TCRA基因的局部拷贝数在TRACERx100组群中的分布。C.肿瘤纯度在TRACERx100组群中的分布。D.TRACERx100组群中拷贝数的精确评分相对于初始评分,如可以看出的,在拷贝数为1的情况下,初始评分被高估,而在拷贝数高于2的其他情况下,初始评分被低估。
图7示出了使用细胞系数据验证本文中所述方法的结果。示出了由T细胞来源的细胞系或非T细胞来源的细胞系的WES计算的TCRA T细胞分数的饼图。
图8示出了使用模拟数据验证本文中所述的方法的结果。A.在背景TCRA拷贝数为2且T细胞分数值范围为0.01至0.99的模拟数据集中,计算的T细胞分数相对于实际的T细胞分数。B.来自由24% T细胞和75%肿瘤组成的样品的模拟对数读取深度比(TCRA拷贝数=1)。C.对于肿瘤拷贝数和纯度范围,计算的初始T细胞分数与实际分数之间的差异。D.对于一系列肿瘤拷贝数和纯度,TCRAT细胞分数与实际分数之间的差异。
图9示出了通过与TRACERx100数据中免疫浸润的正交度量进行比较来验证本文中所述方法的结果。A.对于CD8+、T细胞和总TIL的评估的T细胞分数与基于Danaher RNA的评分之间的关系。B.基于RNA-seq(Danaher、Davoli、EPIC、TIMER、CIBERSORT和xCell)或DNA(TIL ExTRECT和CDR3 VDJ评分)的CD8+T细胞含量的测量值和病理学TIL评分之间的相关性。C.评估的TCRA T细胞分数与来自Danaher、Davoli、EPIC、TIMER、CIBERSORT和xCell方法的不同免疫相关细胞类型的基于RNA的评分之间的相关性,评分通过根据由Spearman rho系数评估的TCRA T细胞分数的相关性强度来排序。D.基于iDNA方法的CDR3 VDJ读取评分与TCRA T细胞分数的相关性。
图10示出了通过与免疫浸润的正交度量进行比较来验证本文中所述方法的结果。由甲基化数据计算的TRACERx100数据中的TCGALUAD和LUSC样品的淋巴细胞分数相对于CIBERSORT计算的CD8+T细胞分数(B)或TCRAT细胞分数(A)。
图11示出了根据读取覆盖(A至B)或潜在的FFPE相关的改变(C)对评估的TCRAT细胞分数的影响来验证本文中所述方法的结果。A.将5个TRACERx100区域降采样至不同深度。B.将模拟数据降采样至不同的深度水平。C.CPI 1000+组群中膀胱肿瘤和黑素瘤肿瘤的FFPE和新鲜冷冻样品的TCRAT细胞分数(非GC校正的)值。D.将具有最高CDR3读取计数的5个TRACERx100区域降采样至不同深度,并得到CDR3读取计数。
图12示出了在TRACERx100组群中使用TCRAT细胞分数获得预后指标的结果。A.示出了高风险和低风险患者之间的生存率差异的Kaplan-Meier曲线,所述高风险和低风险患者如通过由TCRAT细胞分数评估推断的T细胞含量<10%的区域数所限定的。B至C.TRACERx100中LUAD和LUSC组织学亚型的Kaplan-Meier曲线。
图13示出了TCGA组群中TCRAT细胞分数的分析结果。示出了TCGALUAD和LUSC(A),单独LUAD(B)和单独LUSC(C)的总存活的Kaplan-Meier曲线,其中患者由在每组中的中位数TCRA T细胞分数的分类。
图14示出了TRACERx100组群中TCRAT细胞分数的分析结果。示出了TRACERx100中血液与肿瘤之间TCRAT细胞分数评估差异的箱线图。
图15示出了TRACERx100组群中TCRAT细胞分数的分析结果。按组织学划分的血液T细胞分数相对于肿瘤T细胞分数。来自同一患者的区域通过线连接,其中线的类型取决于当与TRACERx100中匹配的血液T细胞分数相比时,所有肿瘤区域是否具有较高(长虚线)、较低(实线)或不均匀(短虚线)的T细胞分数。
图16示出了TRACERx100组群中TCRAT细胞分数的分析结果。A.示出了TRACERx100组群中血液与肿瘤样品之间TCRA T细胞分数差异的箱线图。B.TRACERx100组群中血液TCRAT细胞分数的预测因子。C.由LUAD(顶部)和LUSC(底部)分隔的多区域TRACERx100组群除以肿瘤中存在的冷区域的数目(从左至右增加)的Kaplan Meier曲线。通过使用所有肿瘤区域的平均值(0.08095)作为阈值来限定热性区域和冷性区域。在每条Kaplan Meier曲线中,纳入的患者限于具有大于用于限定阈值的冷区域数的总区域数的患者。D.在整个TRACERx100族群中以及分别在LUAD和LUSC患者中,无病存活与不同的多区域测量值(与TCRA T细胞分数相关)相关的独立Cox回归模型的风险比。将相对区域免疫逃逸评分限定为最大区域TCRA评分除以最小区域(95% CI上限)TCRA评分。E.整个TRACERx100组群的Cox比例风险模型,其包括最小和最大区域TCRA T细胞分数、肿瘤分期、年龄和性别。F.由LUAD(顶部)和LUSC(底部)分隔的多区域TRACERx100组群除以肿瘤中存在的冷区域的数目(从左到右增加)的Kaplan Meier曲线。通过使用所有肿瘤区域的中位数(0.0736)作为阈值来限定热区域和冷区域。在每条Kaplan Meier曲线中,纳入的患者限于具有大于用于限定阈值的冷区域数的总区域数的患者。
图17示出了TCGA组群中TCRAT细胞分数的分析结果。示出了TCGA组群中LUAD(A)和LUSC(B)的血液与肿瘤样品的TCRA T细胞分数的差异的箱线图。示出了TCGA组群中血液(C)和肿瘤样品(D)中LUAD与LUSC的TCRA T细胞分数的差异的箱线图。E.TCGALUAD和LUSC组群中血液TCRAT细胞分数的预测因子。F.使用TCGALUAD组群的平均值(0.109)作为阈值将组群分为免疫热性组和免疫冷性组的TCGALUAD组群中总存活和无进展存活的Kaplan Meier曲线。G.TCGALUAD和LUSC组群的TCRAT细胞分数对总存活(overall survival,OS)和无进展存活(progression free survival,PFS)二者的独立Cox回归模型的风险比。H.基于0至0.16的不同阈值(以0.0025为步长)将肿瘤样品分为热性和冷性的TCGA的总存活和无进展存活的Kaplan Meier图的Log2(风险比)。I.使用TCGALUAD组群的平均值(0.109)作为区分热肿瘤和冷肿瘤的阈值,TCGALUSC和TCGA LUAD&LUSC族群的总存活和无进展存活的KaplanMeier曲线。
图18示出了这样的箱线图:示出了TRACERx100组群(A)和TCGA组群(B)中血液样品中TCRA T细胞分数的性别差异,以及TRACERx100组群(C)和TCGA组群(D)中年龄与生殖系血液样品的TCRA T细胞分数之间的相关性。
图19示出了CPI1000+数据集的组群概述图。
图20是这样的小提琴图:示出了在CPI1000+meta组群中无应答者相对于应答者的计算的肿瘤TCRA T细胞分数之间的显著差异,0.1处的黑色虚线将肿瘤分类为免疫热性或免疫冷性。
图21是示出在CPI1000+数据中肿瘤TCRA T细胞分数相对于克隆TMB的图,虚线将组群分成具有高/低突变负荷和热/冷肿瘤的四个象限。插入饼图指示了对CPI治疗有响应的患者的百分比。
图22示出了包含至少10名个体癌症类型的患者以及DNA和RNA-seq数据二者的多个组群中免疫应答预测因子的单变量荟萃分析的结果。左图:示出了不同临床因素的OR值、以及就响应预测值而言相关的p值的森林图。右图:CPI1000+数据集中各个研究的OR值热图,集中于具有RNA-seq和TCRAT细胞分数二者的组群。
图23示出了用于预测CPI1000+数据集中CPI响应的GLM模型的ROC图(1:克隆TMB,3:克隆TMB+TCRAT细胞分数,2:克隆TMB+CD8A表达,4:克隆TMB+TCRAT细胞分数+CD8A表达)。
图24示出了A.CPI肺数据集的组群概述图,上图中的红线反映了对CPI有响应(0.10)或无响应(0.0070)的患者中的中位数TCRA T细胞分数,注意到肿瘤TCRAT细胞分数通常为零,以及B.跨三个具有DNA测序但无RNA测序数据的NSCLC CPI数据集的单变量荟萃分析的结果。
图25示出了TRACERx100组群中所有区域的TCRA T细胞分数,这取决于在读取深度归一化中使用的TCRA V区段的数量(参见图5中的步骤2)(上图),TRACERx100组群中非零区域的比例,这取决于使用的区段的数量(中图),以及与Danaher T细胞基因标记的相关性的对数(p值),这取决于用于归一化的V区段的数量(下图)。
图26示出了对TCGA数据中使用的外显子捕获过程中偏倚的影响的调查。A.示出了来自TCGA LUAD组群的单一样品中外显子的对数读取深度比的实例的图。某些区段,特别是J基因外显子,呈现显示覆盖偏倚的双峰分布。B.TRACERx100(蓝色)或TCGA LUAD(红色)组群中所选范围的外显子的中位数对数比值,其中TCGALUAD组群中的许多外显子具有低得多的覆盖,并因此具有低得多的对数比值。C至D.在GC校正之前(C)和之后(D),使用TCGA组群的减少的外显子组的TCRA T细胞分数。
图27示出了血液和恶性组织中TCRAT细胞分数的感染相关的决定因素的研究。A至B来自KRAKEN的微生物读取与血液和肿瘤样品中TCRAT细胞分数的相关性。C至D在LUAD(C)和LUSC(D)的肿瘤中,微生物菌种的归一化读取计数与TCRA T细胞分数显著相关。
图28示出了用多区域数据确定血液和恶性组织中的TCRAT细胞分数的研究。A.包含与正常血液样品配对的多区域显微切割组织的PNE组群的概要图。B至C.PNE组群中血液TCRAT细胞分数的预测因子。D至E.肿瘤样品位置对Messaoudene等人乳腺组群(D)和TRACERx100组群(E)中TCRAT细胞分数的影响。F.来自基因组因子的PNE样品中TCRAT细胞分数的线性模型。G.LUAD和LUSC中血液和肿瘤样品中测试的59种微生物与TCRAT细胞分数的相关性的Log10 p值。红线表示P=0.000423处的显著性阈值。H.PNE组群中平均TCRAT细胞分数的概括。
图29示出了多样品泛癌组群中亚克隆SCNA与免疫异质性的相关性的研究。A.跨多样品泛癌组群中免疫异质性概述图,红色虚线处于TCRA T细胞分数为0.1处B.三个类别之一中的患者的比例,均一免疫热性(在此限定为所有区域≥0.1TCRA T细胞分数)、均一免疫冷性(在此限定为所有区域<0.1TCRA T细胞分数)或具有热区域和冷区域二者的异质性。C.针对成对SCNA异质性绘制的具有异质性免疫浸润的多样品患者(n=58)的子集被限定为具有一对具有非常相似的TCRA T细胞分数(成对差异<0.1)的区域和一对具有非常不同的TCRA T细胞分数(成对差异≥0.1)的区域二者,所述成对SCNA异质性被限定为在比较中对任一区域独特的SCNA变化的基因组的组合比例。按肿瘤对数据进行平均以控制来自具有大量区域的肿瘤的任何偏差。D.下图:在整个基因组中具有亚克隆获得(暗红色-高于0水平线)或丢失(深蓝-低于0)的泛癌多样品组群中的肿瘤数目,水平线表示具有多于30个具有亚克隆获得或丢失的肿瘤(见方法)的区域。上图:测试TCRA T细胞分数与亚克隆获得(暗红色点)或丢失(暗蓝色点)之间相关的160个染色体区段区域的-log10(p值)。红色水平线标记显著性阈值,只有一个区域是显著的,即染色体12q24.31-32上的丢失事件。E.染色体12q24.31-32中有或没有亚克隆丢失事件的区域之间的TCRA T细胞分数变化。F.来自具有12q24.31-32亚克隆丢失的TRACERx100肿瘤的Limma-Voom分析的火山图,该分析是在有丢失的区域相对于无丢失的区域的情况下进行的。在多重假设调整之后,只有8个基因是显著的,其中三个(SPPL3,ABCB9和OGFOD2)位于丢失区并被标记。
图30示出了WGS数据的图5的方法的实现。示出了两个替代方案:使用基于bin的方法进行GC校正和质量控制的GAM模型、以及使用V和J区段的已知位置的区段模型。区段模型具有以下约束:1.将模型log读取深度比作为由与V(D)J区段位置比对的恒定区段组成的线性模型。2.模型在第一V区段之前的值为0。3.V区段按照它们在基因组中出现的顺序单调递减,例如TRVA2 图31示出了图30的GAM模型的示例性输出。 图32示出了使用TRACERx100组群的WGS T细胞ExTRECT模型的基准化分析的结果。A.WES和WES TCRA T细胞分数评分的比较B.WGS T细胞分数评分与T细胞的RNAseq Danaher评分的比较C.和D.在完整TRACERx100组群(C.)和在低区段模型对数似然样品移除的样品的组群(D.)的情况下,WGS B细胞分数评分与B细胞的RNAseq Danaher评分的比较E.来自WGS GAM模型的T细胞分数评分的比较F.来自WGS区段模型的T细胞分数评分的比较G.来自区段模型的TRDV1区段分数与来自Kallisto的TCRD基因区段的每百万转录本(transcriptsper million,TPM)RNA评分的比较。 图33示出了在测序深度降低的情况下,图30方法的稳健性的分析的结果。使用GAM或区段模型获得降采样bams的T细胞ExTRECT评分。 图34示出了使用图30中的区段模型来研究TCR和BCR多样性。A至D.患者CRUK0085(针对TCRA、TCRB和TCRG)和CRUK0045(针对IGH)的示例性V区段使用图。E.利用不同TCRA V区段的TRACERx100样品的比例热图。F至I.用MiXCR从匹配的RNAseq数据中调用的V区段数与从T细胞ExTRECT区段模型中调用的V区段数的比较。 具体实施方式 在描述本发明时,将采用以下术语,并且旨在如以下所指示进行定义。 本文中使用的“和/或”被认为具体公开了在具有或不具有另一个的情况下的两个指出的特征或组件中的每一个。例如“A和/或B”被认为具体公开了以下中的每一者:(i)A、(ii)B、以及(iii)A和B,就如同每一者在本文中单独陈述一样。 本文中所使用的“样品”可以是细胞或组织样品(例如活检物)、生物流体、提取物(例如从对象中获得的DNA提取物),从中可以获得基因组物质用于基因组分析,例如基因组测序(全基因组测序、全外显子组测序、靶向(也称为“小组”测序)或拷贝数阵列分析。样品可以是从对象中获得的细胞、组织或生物流体样品(例如活检物)。这样的样品可以被称为“对象样品”。特别地,样品可以是血液样品、或肿瘤样品、或来源于其的样品。样品可以是从对象中新鲜获得的样品,或者可以是在基因组分析之前已处理和/或储存(例如冷冻、固定或者经过一个或更多个纯化、富集或提取步骤)的样品。特别地,样品可以是细胞或组织培养样品。因此,如本文中所述的样品可以指包含来源于其的细胞或基因组物质的任何类型的样品,无论是来自从对象中获得的生物样品,还是来自从例如细胞系中获得的样品。样品优选来自有颌脊椎动物(例如如,有颌脊椎动物细胞样品或来自有颌脊椎动物对象的样品),合适地来自哺乳动物(例如如,哺乳动物细胞样品或来自哺乳动物对象,特别包括模型动物例如小鼠、大鼠等的样品),优选来自人(例如如,人细胞样品或来自人对象的样品)。在一些实施方案中,样品是从对象例如人对象中获得的样品。此外,样品可以被运输和/或储存,并且收集可在远离基因组序列数据采集(例如测序)位置的位置处进行,和/或计算机实施的方法步骤可以在远离样品收集位置和/或远离基因组数据采集(例如测序)位置的位置处进行(例如计算机实施的方法步骤可借助于联网计算机,例如借助于“云”提供者进行)。 “混合样品”是指假设包含多个细胞类型或来源于多个细胞类型的遗传物质的样品。从对象中获得的样品(例如如,肿瘤样品)通常是混合样品(除非它们经过一个或更多个纯化和/或分离步骤)。优选地,混合样品是包含淋巴细胞的样品,或者是被认为(预期)包含淋巴细胞的样品。合适地,样品包含淋巴细胞和至少一种其他细胞类型。例如,样品可以是肿瘤样品。“肿瘤样品”是指来源于肿瘤或从肿瘤获得的样品。这种样品可包含肿瘤细胞、免疫细胞(例如如,淋巴细胞)和其他正常(非肿瘤)细胞。在肿瘤样品的背景下,术语“纯度”是指样品中的细胞是肿瘤细胞的比例(有时也称为“癌细胞分数”或“肿瘤分数”),或者在样品包含来源于细胞的遗传物质的情况下,是指细胞的等效比例。在包含遗传物质的样品的背景下,可以使用尝试去卷积肿瘤和种系基因组的序列分析方法,例如ASCAT(Van Loo etal.,2010)、ABSOLUTE(Carter et al.,2012)、ichorCNA(Adalsteinsson et al.,2017)等来评估肿瘤分数。在肿瘤样品的背景下,样品中的淋巴细胞可被称为“肿瘤浸润淋巴细胞”(TIL)。肿瘤样品可以是原发性肿瘤样品、肿瘤相关的淋巴结样品或来自对象的转移部位的样品。包含肿瘤细胞或来源于肿瘤细胞的遗传物质的样品可以是体液样品。因此,来源于肿瘤细胞的遗传物质可以是循环肿瘤DNA或外排体中的肿瘤DNA。替代地或除此之外,样品可包含循环肿瘤细胞。混合样品可以是已被处理以提取遗传物质的细胞、组织或体液的样品。用于从生物样品中提取遗传物质的方法是本领域已知的。淋巴细胞可以是T淋巴细胞、B淋巴细胞或其混合物。特别地,T淋巴细胞可包含αβT淋巴细胞和/或γδ淋巴细胞。B淋巴细胞可包含具有IGL轻链的B细胞和/或具有IGK轻链的B细胞。 术语“淋巴细胞分数”是指混合样品中含有DNA的细胞是淋巴细胞的比例,或者是特定类型的淋巴细胞(例如如,T淋巴细胞、B淋巴细胞、αβT淋巴细胞、T淋巴细胞或B淋巴细胞,包括TCR或BCR基因中的任何所选择的基因区段等)的比例。术语“T淋巴细胞分数”是指混合样品中含有DNA的细胞是T淋巴细胞的比例。术语“B淋巴细胞分数”是指混合样品中含有DNA的细胞是B淋巴细胞的比例。在本公开内容的上下文中,基于来源于基因组区域的信号来评估淋巴细胞分数,所述基因组区域在被定量的特定淋巴细胞群体中经历VDJ重组。因此,淋巴细胞分数可更具体地是指混合样品中含有DNA的细胞是特征在于具有已经经历VDJ重组的目标的预定基因组区域的淋巴细胞的比例。目标的预定基因组区域可以是TCRA/TCRD、TCRB、TCRG、IGH、IGL或IGK基因的区域(本文也称为TCRA/TCRD、TCRB、TCRG、IGH、IGL和IGK基因座)。根据正在研究的物种,可以使用其他不太常见的基因座,例如存在于有袋类动物中的特定TCR的基因座(例如TCRμ)和存在于鲨鱼中的特定TCR的基因座。根据本方法中使用的目标的预定区域(基因组基因座)的选择,淋巴细胞分数可捕获不同的淋巴细胞的亚群。例如,使用TCRB基因座可允许量化αβT淋巴细胞分数。作为另一个实例,使用TCRA基因座可允许量化组合的αβ和γδT淋巴细胞分数,因为TCRD基因座被认为完全包含在TCRA基因座内。此外,使用对应于TCRA基因座内TCRD的一个或更多个区段可允许量化αβ和γδT淋巴细胞中是γδT淋巴细胞的分数。对应于TCRD的特定区段的选择可根据使用该区段所评估的分数与指示TCRD分数的正交度量之间的相关性来做出。作为另一个实例,使用TCRG或TCRD基因座可允许量化γδT淋巴细胞分数。对于γδT淋巴细胞分数的量化,使用TCRG基因座可以是优选的,因为TCRD基因座非常小,并因此预计不会包含同样多的信息信号。相反,使用TCRA基因座与TCRB基因座或TCRG基因座二者可使得分别定量αβ和γδT淋巴细胞分数。作为又一个实例,使用IGH基因座可使得定量B细胞(不管它们是否具有IGL或IGK轻链),而使用IGL或IGK基因座可使得定量具有IGL或IGK轻链的B细胞。此外,可以对多个淋巴细胞亚群定量单独的淋巴细胞分数。可以添加这些以获得代表多个或所有淋巴细胞亚群的淋巴细胞分数。另外,一些混合样品可能被认为主要或专门含有单个淋巴细胞群体。因此,使用目标的单一预定基因组区域评估的淋巴细胞分数可以被假定为代表样品的(全部)淋巴细胞分数。例如,可以假设样品主要含有T淋巴细胞(或者相反,可以假设与T淋巴细胞的比例相比,包含相对小比例的B淋巴细胞),其中大部分可以预期为αβT淋巴细胞。因此,在这样的情况下,使用对应于TCRA或TCRB基因座的目标的单一预定基因组区域来评估淋巴细胞分数。类似地,一些混合样品可被认为主要或专门含有单一T淋巴细胞群体。因此,使用目标的单一预定基因组区域评估的淋巴细胞分数可以被假定为代表样品的(全部)T淋巴细胞分数。例如,可以假定αβT淋巴细胞是样品中存在的主要类型的T淋巴细胞,使得可以使用对应于TCRA或TCRB基因座的目标单一预定基因组区域来评估T淋巴细胞分数。作为另一个实例,TCRD基因座被认为完全包含在TCRA基因座内(一起形成称为TCRA/TCRD的基因座),使得使用目标的该单一预定基因组区域评估的淋巴细胞分数可以被假定为代表样品(特别是包括αβT淋巴细胞和γδT淋巴细胞)的(全部)T淋巴细胞分数。 TCRA基因是指智人中具有基因ID 6955(HGNC符号:TRA)的T细胞受体α基因座,或另一种有颌脊椎动物中的任何同源区域。在智人中,该基因位于第14号染色体上的第NC_000014.9(21621904..22552132)(组装体GRCh38.p13)位置处,也称为第NC_000014.8(22090057..23021075)(组装体GRCh37.p13)位置处。同源基因座的一些实例包括小鼠tcra基因座(基因ID:21473,位于第14号染色体上,组装体GRCm39-NC_000080.7(52665424..54461655),组装体GRCm38.p6-NC_000080.6(52427967..54224198))。表7提供了可用作最大V(D)J重组的区域以及用作用于获得TCRA基因(和其中的任何基因,例如TCRD基因)的基线读取深度的区域的坐标的实例。 TCRB基因是指智人中具有基因ID 6957(HGNC符号:TRB)的T细胞受体β基因座,或另一种有颌脊椎动物中的任何同源区域。在智人中,该基因位于第7号染色体上的第NC_000007.14(142299011..142813287)(组装体GRCh38.p13)或NC_000007.13(141998851..142510972)(组装体GRCh37.p13)位置处。同源基因座的实例包括小鼠tcrb基因座(基因ID:21577),位于第6号染色体上,组装体GRCm39-NC_000072.7(40868230..41535305)或组装体GRCm38.p6-NC_000072.6(40891296..41558371)。表7提供了可以用作最大V(D)J重组的区域以及用作用于获得TCRB基因的基线读取深度的区域的坐标的实例。 TCRG基因是指智人中具有基因ID 6965(HGNC符号:TRG)的T细胞受体γ基因座,或另一种有颌脊椎动物中的任何同源区域。在智人中,该基因位于第7号染色体上的第NC_000007.14(38240024..38368055,补码)(GRCh38.p13)或NC_000007.13(38279625..38407656,补码)(GRCh37.p13)位置处。同源基因座的实例包括小鼠tcrg基因座(基因ID:110067,位于第13号染色体上,组装体GRCm39-NC_000079.7(19362212..19540646),组装体GRCm38.p6-NC_000079.6(19178042..19356476))。表7提供了可以用作最大V(D)J重组的区域以及用作用于获得TCRG基因的基线读取深度的区域的坐标的实例。 TCRD基因是指智人中具有基因ID 6964(HGNC符号:TRD)的T细胞受体δ基因座,或另一种有颌脊椎动物中的任何同源区域。在智人中,该基因位于第14号染色体上的第NC_000014.9(22422546..22466577)(GRCh38.p13)或NC_000014.8(22891537..22935569)(GRCh37.p13)位置处。同源基因座的实例包括小鼠tcrd基因座(基因ID:110066,位于第14号染色体上,组装体GRCm39-NC_000080.7(54183530..54396655),组装体GRCm38.p6-NC_000080.6(53946073..54159198))。表7(参考TCRA中使用的区域)和表8(参考TCRA基因座内的TCRD区段)中提供了可以用作最大V(D)J重组的区域以及用作用于获得TCRD基因的基线读取深度的区域的坐标的实例。 IGH基因是指智人中具有基因ID 3492(HGNC符号:IGH)的免疫球蛋白重基因座,或另一种有颌脊椎动物中的任何同源区域。在智人中,该基因位于第14号染色体上的第NC_000014.9(105586437..106879844,补码)(GRCh38.p13)或NC_000014.8(106032614..107288051,补码)(GRCh37.p13)位置处。同源基因座的实例包括小鼠Igh基因座(基因ID:11507,位于第12号染色体上,组装体GRCm39-NC_000078.7(113222388..115973574,补码),组装体GRCm38.p6-NC_000078.6(113258768..116009954,补码)。表7中提供了可以用作最大V(D)J重组的区域以及用作用于获得IGH基因的基线读取深度的区域的坐标的实例。 IGL基因是指智人中具有基因ID 3535(HGNC符号:IGL)的免疫球蛋白λ基因座,或另一种有颌脊椎动物中的任何同源区域。在智人中,该基因位于第22号染色体上的第NC_000022.11(22026076..22922913)(GRCh38.p13)或NC_000022.10(22380474..23265085)(GRCh37.p13)位置处。同源基因座的实例包括小鼠Igl基因座(基因ID:111519,位于第16号染色体上,组装体GRCm39-NC_000082.7(18845608..19079594,补码),组装体GRCm38.p6-NC_000082.6(19026858..19260844,补码)。 IGK基因是指智人中具有基因ID 50802(HGNC符号:IGK)的免疫球蛋白κ基因座,或另一种有颌脊椎动物中的任何同源区域。在智人中,该基因位于第2号染色体上的第NC_000002.12(88857361..90235368)(GRCh38.p13)或NC_000002.11(89890568..90274235),(89156874..89630436,补码)(GRCh37.p13)位置处。同源基因座的实例包括小鼠Igk基因座(基因ID:243469,位于第6号染色体上,组装体GRCm39-NC_000072.7(67532620..70703738),组装体GRCm38.p6-NC_000072.6(67555636..70726754))。 术语“序列数据”是指指示样品中具有特定序列的基因组物质的存在且优选地还指示样品中具有特定序列的基因组物质的量的信息。这样的信息可使用测序技术(例如如,下一代测序(NGS),例如如,全外显子组测序(WES)、全基因组测序(WGS)或捕获的基因组基因座的测序(靶向或小组测序))获得,或者使用阵列技术(例如,如拷贝数变异阵列,或其他分子计数确定)获得。当使用NGS技术时,序列数据可包括具有特定序列的测序读取的数量的计数。使用本领域已知的方法(例如如,Bowtie(Langmead et al,2009))将序列数据映射到参考序列(例如参考基因组)。因此,测序读取或等效非数字信号的计数可与特定的基因组位置相关(其中“基因组位置”是指序列数据映射到的参考基因组中的位置)。术语“读取深度”是指指示样品中映射到特定的基因组位置的基因组物质的量的信号。这种信号可使用测序技术例如如下一代测序(NGS,例如如WES、WGS、或捕获的基因组基因座的测序)来获得,或者使用阵列技术例如如拷贝数变异阵列来获得。当使用NGS技术时,读取深度可以是该词语常识内的读取深度,即映射到基因组位置的测序读取数的计数。当使用阵列技术时,读取深度可以是与特定基因组位置相关的强度值,其可以与对照进行比较以提供映射至特定位置的基因组物质的量的指示。术语“读取深度谱”是指与多个基因组位置相关的读取深度值的集合。例如,特定基因组第i位处的读取深度可指参考基因组中第i位处碱基处的读取深度,并且读取深度谱可指目标的一个或更多个区域内多个第i位的读取深度。 如本文所使用的“治疗”是指相对于治疗之前的症状,减少、减轻或消除正在治疗的疾病的一种或更多种症状。“预防”(或预防(prophylaxis))是指延迟或预防疾病症状的发作。预防可以是绝对的(使得不发生疾病)或者可仅在一些个体中或对有限的时间量是有效的。 如本文所使用的,术语“计算机系统”包括用于实现根据上述实施方案的系统或实施根据上述实施方案的方法的硬件、软件和数据存储装置。例如,计算机系统可包括处理单元(例如中央处理单元(central processing unit,CPU)和/或图形处理单元(graphicalprocessing unit,GPU))、输入装置、输出装置和数据存储器,其可作为一个或更多个连接的计算装置来实现。优选地,计算机系统具有显示器或者包括具有显示器的计算装置,以(例如在商务过程的设计中)提供视觉输出显示。数据存储器可包括RAM、磁盘驱动器或其他计算机可读介质。计算机系统可包括通过网络连接并且能够在该网络内相互通信的多个计算装置。明确设想了计算机系统可以由云计算机组成或包括云计算机。 如本文所使用的,术语“计算机可读介质”包括但不限于任何非暂时性介质或者可由计算机或计算机系统直接读取和访问的介质。所述介质可包括但不限于磁存储介质(例如软盘、硬盘存储介质和磁带)、光存储介质(例如光盘或CD-ROM)、电存储介质(例如存储器,包括RAM、ROM和闪存),以及上述的混合和组合(例如磁/光存储介质)。 确定混合样品中的淋巴细胞分数 本公开内容提供了使用来自样品的读取深度数据确定混合样品中的淋巴细胞分数的方法,所述读取深度数据包含目标的至少预定基因组区域的读取深度数据。将参考图1描述一种说明性方法。在任选的步骤10,可以从对象获得包含来自多种细胞类型的基因组物质的混合样品。在任选的步骤12,例如通过使用全外显子组测序、全基因组测序或小组测序之一对样品中的基因组物质进行测序可从混合样品中获得序列读取/读取深度数据。在步骤14,获得包含沿着目标的预定基因组区域的读取深度的样品的读取深度谱。这可包含获得目标的预定区域中的多个原始读取深度值的步骤14A和平滑和/或校正(例如GC校正)读取深度值的步骤14B。 步骤16,从目标区域的子集获得基线读取深度。这可包括将可用于获得基线读取深度的目标区域的子集鉴定为不太可能通过VDJ重组被缺失的目标区域的子集的任选的步骤16A。例如,不太可能通过VDJ重组而缺失的目标区域包含经历VDJ重组的基因组基因座的前n个V区段。可以使用适当的训练数据来确定参数n,例如,当与淋巴细胞分数的正交度量相比较时,通过选择在训练数据中产生最准确的淋巴细胞分数评估的n值。例如,可基于转录组数据和/或组织病理学数据获得淋巴细胞分数的正交度量。 在步骤18,通过参考在步骤16获得的基线读取深度来归一化沿着目标的预定区域的读取深度而获得多个读取深度比(r 在步骤22,混合样品中异常细胞(其中异常细胞是在目标的预定区域中的非整倍体的细胞)的分数(p)和目标的预定区域中异常细胞的拷贝数(ψ 在任选的步骤26,可将确定的淋巴细胞分数和/或从中推导出的任何值例如通过用户界面提供给用户。从中推导出的值可包括预后和/或诊断信息,如以下进一步所描述的。 应用 以上方法在多种临床环境中得到应用。例如,在癌症的背景下,确定肿瘤样品(或者优选地来自肿瘤的多个样品)中的淋巴细胞分数可以提供肿瘤免疫状态的指示。这已被证明对多种癌症具有预后价值,所述多种癌症包括例如非小细胞肺癌(如本文中所证明的)、卵巢癌(参见例如Zhang et al.,2003)、结直肠癌(参见例如Galon et al.,2006)、乳腺癌(参见例如Dieci et al.,2018)和黑素瘤。本发明人已经证明,血液和/或肿瘤样品中的T和/或B细胞分数指示至少一些癌症类型的预后(参见实施例6,图35)。 因此,本文中还描述了来自对象的肿瘤的免疫状态的方法,该方法包括确定来自对象的一个或更多个肿瘤样品中的淋巴细胞分数。 另外,本文中还描述了为已被诊断为患有癌症的对象提供预后的方法,该方法包括确定来自对象的一个或更多个肿瘤样品中的淋巴细胞分数。该方法可进一步包括根据样品中的淋巴细胞分数(例如如T淋巴细胞分数)将样品分类为免疫热性或免疫冷性。例如,淋巴细胞分数高于阈值(例如,约0.1,即约10%)的样品可被分类为免疫热性,而淋巴细胞分数处于或低于阈值的样品可被分类为免疫冷性。该方法可进一步包括根据来自被分类为免疫冷性的患者的样品数量,将对象在良好预后组与不良预后组之间分类。例如,当分析来自不同肿瘤区域的样品时,被分类为免疫冷性的样品的数量处于或高于阈值(例如如2)可以将对象分类在不良预后组中。与良好预后组相比,不良预后组可能与降低的无复发存活和/或降低的总存活有关。淋巴细胞分数和/或免疫冷性样品数量的阈值可以使用适当的训练组群,例如通过评估多种阈值在与显著不同预后相关的组之间对患者进行分类的能力来确定。 另外,仍然在癌症的背景下,已显示TIL的存在与对免疫治疗响应的可能性提高有关,所述免疫治疗尤其包括检查点抑制剂(CPI)(参见例如Litchfield et al.,2020),但也包括利用现有免疫功能的任何其他形式的免疫治疗,例如T细胞转移治疗(肿瘤浸润淋巴细胞(或TIL)治疗和CAR T细胞治疗)、治疗性抗体、癌症治疗疫苗和免疫系统调节剂(例如干扰素和白细胞介素)。因此,本文中还描述了确定已被诊断为患有癌症的对象是否可能受益于用免疫治疗进行治疗的方法,该方法包括确定来自对象的一个或更多个肿瘤样品中的淋巴细胞分数。免疫治疗可以是调节免疫系统的功能以治疗癌症的任何治疗。事实上,任何这样的治疗都被认为可能受到肿瘤内免疫细胞存在或不存在的影响。在一些实施方案中,免疫治疗是CPI治疗。CPI治疗包括例如用抗CTL4或抗PDL1药物的治疗。该方法还可包括将对象在可能对免疫治疗有响应的组与不太可能对免疫治疗有响应的组之间分类。例如,该方法可包括根据样品中的淋巴细胞分数(例如如T淋巴细胞分数)将样品分类为免疫热性或免疫冷性(如上所阐明的)。如果来自对象的一个或更多个样品是免疫冷性的,则对象可被分类在不太可能对免疫治疗(例如CPI治疗)有响应的组中。或者,如果来自对象的样品具有低于阈值的淋巴细胞分数,则对象可被分类在不太可能对免疫治疗(例如CPI治疗)有响应的组中,否则可被分类在可能对免疫治疗(例如CPI治疗)有响应的组中。或者,该方法还可包括基于来自对象的一个或更多个样品中的淋巴细胞分数和对象的评估肿瘤突变负荷,将对象在可能对CPI治疗有响应的组与不太可能对CPI治疗有响应的组之间分类。这种分类可以使用多变量模型来获得。可以使用适当的训练组群来确定分类模型的参数(例如,阈值、多变量模型的参数)。 如本文中所述的淋巴细胞分数也可用于确定对象是否患有严重的联合免疫缺陷(severe combined immune deficiency,SCID)和/或T细胞淋巴细胞减少症。这在新生儿护理的情况下特别有用。事实上,在本方法之前,通常使用全血细胞计数来鉴定SCID。这在理论上可使用TREC本身的测序和定量来证实,但这很少在真实的临床环境中进行。因此,使用来自WES的数据提供SCID诊断的能力是非常有价值的,这越来越多地作为新生儿基因组筛选的一部分来进行,以鉴定潜在的破坏性种系突变。因此,本公开内容还涉及鉴定对象,特别是新生儿对象的T细胞淋巴细胞减少症和/或SCID的方法,该方法包括确定来自对象的一个或更多个样品中的如本文所述的淋巴细胞分数。样品例如可以是血液样品。 本方法还可用于任何临床环境,其中样品中免疫细胞或特定类型免疫细胞的存在/不存在或丰度指示诊断或预后。例如,一些自身免疫性疾病与低淋巴细胞计数有关(例如狼疮、类风湿性关节炎),如一些骨髓障碍(例如再生障碍性贫血)和感染(例如HIV、败血症、流感、疟疾、病毒性肝炎、结核病、伤寒等)。因此,本文还描述了将对象诊断为患有与异常淋巴细胞计数相关的疾病、障碍或病症的方法,该方法包括确定来自对象的一个或更多个样品中的淋巴细胞分数。 如本文中所述的淋巴细胞分数也可用于确定一个或更多个种系突变例如单核苷酸多态性(single nucleotide polymorphism,SNP)是否与T/B细胞分数的提高或降低相关,例如在血液样品中进行,例如通过GWAS分析进行。这可有助于鉴定可能导致T细胞分数提高/降低的变体(种系突变)。 系统 图2示出了根据本公开内容用于确定混合样品中的淋巴细胞分数和/或用于至少部分基于淋巴细胞分数提供预后或治疗建议的系统的实施方案。该系统包括计算装置1,该计算装置1包括处理器101和计算机可读存储器102。在所示的实施方案中,计算装置1还包括用户界面103,其被示为屏幕,但是可包括例如如通过听觉或视觉信号向用户传达信息的任何其他手段。计算装置1例如如通过网络6可通信地连接至读取深度数据采集装置3,例如测序仪,和/或连接至存储读取深度数据的一个或更多个数据库2。一个或更多个数据库可另外存储可通过计算装置1使用的其他类型的信息,例如如参考序列、参数等。计算装置可以是智能手机、平板电脑、个人计算机或其他计算装置。如本文中所述的,计算装置被配置为实施用于确定混合样品中的淋巴细胞分数的方法。在替代实施方案中,计算装置1被配置为与远程计算装置(未示出)通信,远程计算装置本身被配置为实施如本文中所述的确定混合样品中的淋巴细胞分数的方法。在这样的情况下,远程计算装置还可被配置为将确定淋巴细胞分数的方法的结果发送至计算装置。计算装置1与远程计算装置之间的通信可通过有线或无线连接进行,并且可通过本地或公共网络(例如如通过公共互联网或通过WiFi)发生。读取深度数据采集装置可与计算装置1有线连接,或者可能能够通过无线连接例如如通过WiFi进行通信,如所示的。计算装置1与读取深度数据采集装置3之间的连接可以是直接的或间接的(例如如通过远程计算机)。读取深度数据采集装置3被配置为从核酸样品中获取读取深度数据,所述核酸样品例如是从细胞和/或组织样品提取的基因组DNA样品。在一些实施方案中,样品可以已经经受过一个或更多个预处理步骤,例如DNA纯化、区段化、文库制备、靶序列捕获(例如如,外显子捕获和/或小组序列捕获)。优选地,样品没有经受过扩增,或者当其已经经受过扩增时,这是在存在扩增偏倚(bias)控制装置(例如如使用独特的分子标识符)的情况下进行的。适合用于确定基因组拷贝数谱(无论是全基因组还是序列特异性)的任何样品制备方法都可在本公开内容的上下文中使用。读取深度数据采集装置优选地是下一代测序器。序列数据采集装置3可以与一个或更多个数据库2直接或间接连接,序列数据(原始或部分处理的)可以存储在数据库2上。 以下内容以实施例的方式呈现,并且不应被解释为对权利要求书的范围的限制。 实施例 免疫微环境影响肿瘤演变,并且可以既预后又预测对免疫治疗的响应。然而,测量肿瘤浸润淋巴细胞(TIL)受到缺乏合适的数据的限制。DNA的全外显子组测序(WES)经常用于计算肿瘤突变负荷并识别可操作的突变。在这些实施例中,发明人开发了使用在T细胞受体α(TCRA)基因的VDJ重组期间从T细胞受体切除环(TREC)丢失的信号从WES样品中评估T细胞分数的方法。该方法适用于多种类型的临床相关样品,包括癌症样品,例如适用于测量TIL。在这些实施例中,发明人表明该评分与正交TIL评估值显著相关,并且可以从新鲜冷冻或福尔马林固定的石蜡包埋样品中计算。血液TCRA T细胞分数与肿瘤中的免疫浸润、细菌测序读取的存在相关,并且在女性中较高。肿瘤TCRA T细胞分数在肺腺癌中具有预后意义,并且使用经免疫疗法治疗的肿瘤的荟萃分析显示,该评分可预测对免疫治疗的响应,提供了除突变负荷之外的价值。将该评分应用于多样品泛癌组群显示了肿瘤内免疫浸润的广泛多样性。涵盖SPPL3的12q24.31-32亚克隆丢失与降低的TCRA T细胞分数相关。本文中描述的方法,T细胞ExTRECT(T细胞外显子组TREC工具)阐述了WES样品的T细胞浸润。 实施例1–根据WES数据评估样品中的T细胞分数 介绍 确定肿瘤中TIL的水平对于理解肿瘤免疫微环境和预测对免疫治疗的响应二者都是至关重要的。然而,目前还没有通用的用于评估TIL的最先进的方法。微阵列特征例如CIBERSORT(Newman et al.,2015)以及RNA-seq特征例如RNA-seq的CIBERSORTx(Newman etal.,2019)、ESTIMATE(Yoshihara et al.,2013)和由Danaher等人汇编的那些,已经被用于量化肿瘤样品中TIL的转录组特征。或者,可以基于苏木精和伊红(H&E)组织病理学切片或者适当的细胞特异性染色(最常见的是免疫组织化学)来确定免疫浸润的存在。如果需要更多的T细胞表型知识,可以在编码TCR的高度多样化的VDJ重组CDR3区域的那些序列的富集之后,进行T细胞受体(TCR)测序(Bolotin et al.,2012)。 大多数现有的T细胞定量方法的显著缺点是它们需要除了WES之外的另外的物质、时间和专业知识。因此,虽然这些方法提供了可有助于预测免疫应答的另外的知识,但是这些方法在临床环境中的应用需要增加时间和支出二者的开销,从而增加了本已有限的免疫治疗的成本。 在该实施例中,发明人提出了直接根据WES数据评估样品中T细胞分数的方法。该方法利用了来自VDJ重组和T细胞受体切除环(TREC)切除的基于体细胞拷贝数的信号。 该方法具有从任何形式的NGS平台计算的潜力,包括全基因组测序和靶向的基于小组的方法二者,以及扩展至经历VDJ重组的所有基因。 结果 T细胞多样性是VDJ重组的产物,其中T细胞受体基因内的不同基因区段发生重组。这样的结果是从TCRA基因中切除大量未选择的基因区段作为TREC。这一过程导致T细胞与其他细胞之间的拷贝数差异,其中T细胞实际上经历了TCRA基因内的缺失事件。 标准的体细胞拷贝数改变评估工具(例如ASCAT(Van Loo et al.,2010)、Sequenza(Favero et al.,2015)、FACETS(Shen&Seshan,2015)或ABSOLUTE(Carter etal.,2012))主要依赖两个相关信号来获得癌症体细胞拷贝数改变谱;反映肿瘤样品中杂合SNP的相对频率的B等位基因频率,以及反映肿瘤样品与其匹配种系(通常是来自血液样品的血沉棕黄层)之间的读取比的对数的读取深度比。读取深度比的任何偏差都被假定为具体反映了肿瘤中拷贝数的变化(如图3A所示)。然而,在TCRA基因的背景下,该假设不成立;除了单独反映肿瘤中体细胞拷贝数变化,与TCRA基因重叠的读取深度比的偏差还将反映肿瘤样品中也存在的T细胞的TCRA基因内缺失事件的检测。这是丢失还是获得事件取决于与肿瘤样品相比,血液中存在的T细胞比例的差异(如图3B所示)。具体地,如果测序的肿瘤样品中的T细胞含量与血液样品相比较低,则将存在更多来自在肿瘤样品中鉴定的TCRA基因的读取,导致扩增信号(对数读取深度比>0)。相反,如果与血液相比,肿瘤中的T细胞含量更高,则TCRA读取将更少,并且体细胞拷贝数改变评估工具将推断出缺失或丢失事件(读取深度比<0)。 在检查了进行多区域测序的非小细胞肺癌(NSCLC)肿瘤的TRACERx100组群(Jamal-Hanjani et al.,2017)的ASCAT来源的体细胞拷贝数改变谱之后,发明人注意到,其中两个断点都在TCRA基因内的体细胞拷贝数改变区段出现在进行WES的肿瘤区域的165/327中(如图1B所示)。检查了TRACERx100组群中的两个这样的情况,发现读取深度比在D基因组区段的基因组位置(这是TREC中最常包含的位置)处形成峰,(如图4所示)。因此,这些数据表明可以从WES数据中识别TREC的信号。 为了明确地利用该信号来计算单独样品中的T细胞分数,发明人开发了:TILExTRECT(T细胞外显子组T细胞受体切除环工具)。简言之,TILExTRECT依赖于对TCRA基因内读取深度比的分析来直接测量WES样品中的T细胞浸润。首先,使用不受VDJ重组影响的TCRA起始和末端处的基因组区段作为对照,计算TCRA基因内每个位置处的读取深度,并在覆盖值的原始读取深度与对照基因组区段内的值的中位数之间计算修正的对数读取深度比。最后,只要已知测序样品中肿瘤细胞的分数(肿瘤纯度)和TCRA周围的局部体细胞拷贝数二者,就可以基于log(以2为底)读取深度比的偏差大小计算出T细胞分数的精确评估。如果无法获得肿瘤纯度和局部体细胞拷贝数的知识,也可以进行初始评估,我们证明其具有类似的预测值(如在如图5所示的方法部分详细解释的)。值得注意的是,与RNA-seq评分不同,TCRA评分代表了对T细胞比例的直接测量。这在此被称为“TCRA T细胞分数”。 TILExTRECT的完整描述及其参数的优化,例如用于所选外显子的归一化和质量控制的区段的选择在以下方法中给出。使用以下实施例2中描述的四种正交方法验证了TCRAT细胞分数的准确性。然后,在实施例3和4中,使用两种不同的方法证明了其临床效用。最后,在实施例5中研究了其在评价不同样品类型中T细胞免疫浸润的关键决定因素中的用途。 方法 肿瘤样品内不同细胞中TCRA基因座周围的局部拷贝数值的定义。 在已知为二倍体的T细胞和其他基质细胞内,有两个TCRA基因的拷贝,并且TCRA基因座周围的拷贝数可以说等于2。假设在T细胞内,存在TCRA基因座内的区域在VDJ重组期间总是丢失,这导致这个小基因组区段中的拷贝数为0(如图3B所示)。在肿瘤细胞中,第14号染色体(TCRA基因所在的位置)上可以存在拷贝数的增加和减少二者,导致TCRA基因的可不同于2的不同可能的拷贝数状态。本方法假设TCRA基因内不存在源自癌细胞中体细胞改变的断点(尽管该区域作为整体在肿瘤细胞中可以不是二倍体)。 如以下两个小节中更详细解释的,TCRA基因(chr14:22090057(hg19))起始处的平均拷贝数是使用ASCAT从样品中的所有癌细胞推断的,并被称为TCRA基因处的局部肿瘤拷贝数。这被用作由以下方程式(7)提供的SCNA感知TCRA T细胞分数中的术语。另外,当肿瘤体细胞拷贝数改变信息不可用时,SCNA-初始TCRA T细胞分数(方程式(3))可以被使用,并且被称为初始TCRA T细胞分数。 根据WES数据计算TCRA T细胞分数 Van Loo等人(2010)描述的ASCAT方法(肿瘤的等位基因特异性拷贝数分析)提供了由从包含肿瘤细胞和非异常细胞的混合物的样品中提取的DNA评估等位基因特异性拷贝数的方法。下式是ASCAT(Van Loo etal.,2010)用来评估基因组第i位处的肿瘤倍性的方程式,其与该位置处B等位基因频率的第二方程式组合(参见Van Loo等人): 其中r 在目前的工作中,目的是鉴定样品中细胞群中T细胞的分数。此外,该样品可含有在基因组基因座i处可具有未知倍性的异常细胞(例如肿瘤细胞)。通过与Van Loo等人研究的混合群体进行类比,r 其中f是样品的T细胞分数,n 或 在这一点上,可存在两种不同的情况:(a)可以假设Ψ 这被称为“初始TCRA T细胞分数”方程式。当(a)中的假设不成立(情况(b))时,可使用Ψ Ψ Ψ 其中ψ 将方程式(4)、(5)和(6)代入方程式(2)产生方程式(7): 方程式(7)表示方程式(3)的“更完整”版本(即,方程式(2)的解没有做出得出方程式(3)的所有假设)。因此,通过应用从方程式(7)开始的相同假设,可以恢复方程式(3)。特别是,在没有癌细胞的情况下(没有倍性不同于正常2的细胞;p=0),方程式(7)立即简化为方程式(3)。类似地,当任何存在的肿瘤细胞中TCRA周围的局部拷贝数与正常细胞中的相同时(ψ 换言之,样品的非癌症组分(1-p)可以通过将其分成T细胞和非T细胞亚群来重写: (1-p)=(1-p)f′+(1-p)(1-f′) 其中f’代表非肿瘤细胞中T细胞的分数,并与样品中T细胞的总分数T细胞分数(f)相关,如下: 现考虑T细胞中TCRA基因处的VDJ重组事件的基因组位置,假设在该位置n 将f’的方程式代入该方程式,并重新排列样品内T细胞分数的方程式,可以发现是由以上方程式(7)提供的。注意到,如果对数比r 从WES中单一样品中计算log读取深度比 为了由方程式(3)(对于T细胞含量的初始评估)或方程式(7)(如果肿瘤拷贝数和纯度是已知的)计算T细胞分数,需要从原始覆盖数据(即,从单一样品,而不是从分别包含和不包含待定量细胞群的匹配样品)来评估对数读取深度比r 总之,通过使用TCRA基因(或研究中的任何其他VDJ基因座,这取决于待定量的细胞类型)的最起始和最末端的基因组区域的中位数覆盖作为正常背景率来计算该对数R,该正常背景率被假定为具有不受任何VDJ重组影响的拷贝数值。如上所述,在任何实施方案中也可以使用研究中的VDJ基因座的起始或末端(而不是两者),或者可以假定具有不受VDJ重组影响的拷贝数值的其他(优选附近)区域。不希望被理论所束缚,认为使用较长的归一化区域(例如,使用起始和末端区域二者,和/或增加起始区域的长度)有利地补偿了读取深度数据中典型的噪声。例如,起始区域可以延伸至包括许多不太可能经常丢失的V区段,或者甚至基因外的区域,如果该数据是可获得的(例如如当使用全基因组测序数据时)。在本案例中,使用了下列区域:hg19的chr14:22090057至22298223和chr14:23016447至23221076。然后,将TCRA基因组区域(或研究中的任何其他VDJ基因座)的覆盖除以该中位数值,得到整个TCRA基因(在这种情况下,hg19的chr14:22090057至23221076)中r 更详细地说,从比对的BAM文件至hg19(基于GRCh37)或hg38(GRCh28)(取决于数据集,例如,TCGA数据是在与hg38预比对之前获得的,而TRACERx和其他数据是在内部与hg19比对的),使用samtools(第1.3.1版)深度函数(具有参数-q20-Q20)提取单独基准水平处的覆盖。完成此操作之后,只有TRACERx中使用的由SureSelect人全外显子探针(第5版)所限定的已知外显子内的碱基,然后用50个碱基对窗口中的滚动中位数对每个外显子中的读取进行归一化(即将每个50bp窗口中的中位数值作为新值)。注意到,窗口的大小不是固定的,并且在一些实施方案中,可以使用小于或大于50bp,例如20至200bP,或50至150bP的窗口。通过将使用本方法利用多种候选窗口大小获得的淋巴细胞分数评估值与淋巴细胞分数的正交度量(例如Danaher评分)进行比较,可以针对特定的数据集、数据类型或背景凭经验确定合适的窗口尺寸。 在假定不受VDJ重组影响的TCRA区域中取基因的起始和末端区域(hg19的chr14:22090057至22298223和chr14:23016447至23221076-对应于TCRA的前12个V区段和C区段),计算中位数基线覆盖值。然后将所有覆盖值除以该值,然后取对数来计算“单一样品”对数比。为了完成r 总之,对于许多肿瘤样品,可以使用方程式(7),这需要已知肿瘤纯度和TCRA基因的肿瘤拷贝数二者。在特殊情况下,当TCRA的拷贝数正好是2时,如在所有血液来源的样品和许多肿瘤中,方程式(3)可用于计算T细胞分数,并且是精确的。另外,如果肿瘤纯度和TCRA的肿瘤拷贝数未知,方程式(3)可以作为T细胞分数的初始评估(尽管取决于上述假设是否成立,该初始评估可能不精确)。这将在下一节中进行研究。 初始TCRA T细胞分数相对于精确评估的T细胞分数 图6A示出了基于从方程式(2)、(3)和(7)推导出的理论值,对于一系列局部拷贝数值和肿瘤纯度,T细胞的评估初始值与不同真实T细胞分数值之间的(理论)差异。在低T细胞分数和接近于2的局部拷贝数值下,这种评估是非常准确的。图6B至C示出了使用来自TRACERx100组群的真实数据(在实施例2中提供了关于该数据的更多信息),局部TCRA拷贝数的分布的模式为2,而肿瘤纯度值通常较低。对于局部TCRA拷贝数不为2的情况,图6D示出了初始评估值与精确计算的T细胞分数之间的相关性。 用于评估对数比(rVDJ)的区段的优化 rVDJ的计算需要选择(i)最大VDJ重组的区段,和(ii)用于计算“正常基线”的区段,TCRA基因组区域中的覆盖与该“正常基线”进行比较(通过计算比率,如上所解释的)。 对于代表最大VDJ重组位置的焦点区段,使用了最终V基因区段与第一区段之间的间隙(hg19,chr14:22800000至22880000),所述第一区段编码TCRδ链的一部分。基因组的这个区域理论上可能在T细胞受体切除环(TREC)内,并且与先前用于测量TREC的PCR引物的区域重叠(Kuss et al.,2005)。如上所解释的,可以使用目标VDJ基因座的最终V区段与第一J区段之间的任何区域。 由于测序噪声,在使用在VDJ重组中通常不太可能丢失的V基因区段计算正常基线时,希望具有尽可能宽的区域(参见WES中单一样品的对数比的计算)。然而,选择的V基因区段越多,T细胞克隆型存在的可能性就越大,其中这些基因区段已经被TREC切除(使得该区域中的覆盖将受到VDJ重组的影响)。 对于局部区段,使用以下概述的优化方案来选择第一n-V基因区段以及最终C区段。依次取第一n-V区段,计算整个TRACERx100组群的评分(TCRA T细胞分数,f,非GC校正)(参见实施例2中关于该组群的更多细节)。基于哪个n具有最大的非零f值并最大化计算的T细胞分数与T细胞浸润的Danaher转录组评分(T细胞浸润的比较指标,参见实施例2)之间的相关性来选择V区段的数目。发现所用区段的长度可用于降低噪声水平(即,从噪声角度来看,使用的区段越多,归一化越好),但如果在某些情况下太长,则将与从某些T细胞克隆型中丢失的区段重叠(即,较长的区段更有可能包括在一个或更多个T细胞克隆型中丢失的一个或更多个区段)。因此,为了保守起见,选择前12个V区段用作与Danaher评分的高度相关与尽可能短之间的折衷(如图25所举例说明的,其中顶部图示出了TCRA T细胞分数作为用于归一化的V区段数目的函数,在TRACERx100数据中,中间图示出了非零样品的相应分数,并且底部图示出了使用Spearman方法所评估的与Danaher T细胞特征相关的显著性)。注意到,在不对结果产生显著影响的情况下,可以使用其他数目的区段,例如,如前11个V区段。 TIL ExTRECT内使用的GC归一化方法 已知GC含量会使序列覆盖值产生偏差。因此,在计算T细胞分数之前,TILExTRECT可以通过GC含量将覆盖值归一化。为了做到这一点,GC含量在两个尺度上计算,即计算每个TCRA外显子的GC含量的局部外显子水平(GC 为了使GC含量归一化,将所有TCRA外显子内每个碱基对的覆盖值放入到以下线性模型中: 然后将该模型的残差作为新的GC归一化覆盖值。除非另有说明,否则本文中使用的所有TCRA T细胞分数都经过了GC校正。 在GC校正后,通过取TCRA基因(hg19的chr14:22090057至22298223和chr14:23016447至23221076)起始和末端处的中位数值、以及最大VDJ重组位置(chr14:22800000至22880000)处拟合的广义加性模型(generalised additive model,GAM)的中位数值来重新调整评分的基线。由于理论上T细胞分数不可能为负,并且必然如此,所以chr14:22800000至22880000处的对数读取深度比必须为≤0,两个中位数值的最大值被当作新的基线并设定为0。 TIL ExTRECT内置信区间的计算 计算TCRAT细胞分数的95%置信区间时考虑了两个因素,1)GC校正后最终基线调整中的噪声,2)拟合TCRA T细胞分数最终计算中使用的GAM模型的不确定性。 为了考虑基线校正中的噪声,调整值的95%置信区间被计算为用于归一化的区域中覆盖值标准偏差的1.96倍。为了拟合GAM模型,使用R包“gratia”(v0.5.1)来计算同时置信区间(使用confint函数)。最后,将这两个不确定性来源组合以得出95%的置信区间。 由于高估肿瘤TCRA拷贝数而导致TCRA T细胞分数偏差 TCRA肿瘤拷贝数的过高评估值将导致TCRA T细胞分数的夸大值。这可能是由于从拷贝数分割算法(例如ASCAT)中选择的差质量的解而造成的。这种情况的一个指示是,从TIL ExTRECT计算的TCRA T细胞分数是否超过1-肿瘤纯度的值。在这些情况下,给定的拷贝数解被认为是不可靠的,并且相反假设TCRA肿瘤拷贝数为2的T细胞分数的简单评估被用作替代解。 实施例2-WES来源的TCRAT细胞分数的验证 介绍 为了评价实施例1中描述的TCRA T细胞分数度量的准确性,发明人使用了四种正交方法。 首先,作为T细胞含量存在的阳性和阴性对照,使用了来自细胞系的WES数据。第二,使用了具有一系列T细胞分数值的模拟的下一代测序(NGS)数据。模拟数据还用于研究局部肿瘤TCRA拷贝数和肿瘤纯度对评分准确性的影响。第三,发明人检查了该评分如何与多个NSCLC组群内的正交免疫相关数据进行比较。第四,发明人计算了它们的评分与用于推断免疫含量的替代的基于DNA的方法的相似程度。 结果 作为对实施例1中所述方法的第一次验证,发明人利用了来自细胞系的WES数据,所述细胞系由以下构成:来源于HCT116细胞系的14个独立样品,包括四倍体和二倍体克隆,其具有不同程度的基因组复杂性(López et al.,2020);以及源自癌细胞系百科全书(encyclopaedia)的起源于T细胞淋巴瘤(JURKAT,PEER和HPB-ALL)的细胞系的三个样品(Ghandi et al.,2019)。理论上,来自在HCT116细胞系的NGS中TCRA T细胞分数应为零,而T细胞淋巴瘤来源的细胞系(应该都经历了VDJ重组)的TCRAT细胞分数应为1(反映了100%的细胞为T细胞)。令人欣慰的是,HCT116细胞系的计算分数都为0,无论它们是二倍体还是四倍体状态(如图7所举例说明的)。相反,三种T细胞来源的细胞系的用初始评估计算的评分接近1(约0.95至约0.96)(参见图7)。这种与精确100%的评估分数的微小差异可由技术(例如,错位读取)或生物因素所解释。 作为第二种验证方法,为了进一步评价除二元存在或不存在之外的TCRA T细胞分数的准确性,并且对于一系列T细胞分数,获得了具有一系列T细胞分数值的模拟NGS数据(参见方法)。如图8A所示,在背景TCRA拷贝数为2的样品中,模拟的T细胞分数与计算的T细胞分数之间存在高度显著、近乎完美的关系(ρ=1,p<2.2e-16)。用模拟数据研究了局部肿瘤TCRA拷贝数(参见方法)和肿瘤纯度对评分准确性的影响的进一步研究,并且还证实了模拟的TCRA T细胞分数与推断的TCRA T细胞分数之间的显著关系(如图8C至D所示)。 作为第三种验证方法,本发明人检查了该评分如何与TRACERx100组群内以及肺腺癌(lung adenocarcinoma,LUAD)TCGA(癌症基因组图谱研究网络,2014)和肺鳞状细胞癌(lung squamous cell carcinoma,LUSC)(癌症基因组图谱研究网络,2012)组群内的正交免疫相关数据进行比较。这些NSCLC组群已经根据一系列不同的数据类型(包括RNA-seq、组织病理学切片和甲基化数据)对T细胞含量进行了评估。 TRACERx100组群由经受WES的327个肿瘤区域构成,所述肿瘤区域来源自100个患者的100个肿瘤,每个患者具有也经受WES的匹配种系血液样品(参见表1)。这些肿瘤区域中的189个也具有匹配的RNA-seq数据,由此可以计算出基于转录组的免疫评分。 表1:TRACERx100组群中样品的概述。 特别地,发明人检查了该评分如何与TRACERx100组群内的正交非WES免疫相关数据进行比较。对于具有RNA-seq数据的189个TRACERx100区域,计算了来自Danaher等人、Davoli等人、xCell、TIMER、EPIC和CIBERSORT的多种细胞类型的免疫相关特征评分(Rosenthal et al.,2019)。发现来自WES的TCRA T细胞分数与来自不同方法的多个免疫评分具有显著的正相关。相关性最强的前三个项都是T细胞相关的(Danaher Th1:ρ=0.68,P=9.0e-23,xCell CD8 T细胞记忆ρ=0.67,P=2.3e-22和Danaher T细胞:ρ=0.67,P=2.94e-22)。其他非T细胞评分也得分非常高,例如NK细胞的Davoli评分(ρ=0.67,P=3.92e-22)和xCell鉴定的活化树突细胞(ρ=0.61,P=1.26e-17),这表明肿瘤免疫微环境的复杂性,并且表明T细胞浸润与来自其他免疫细胞的浸润密切相关。发现来自WES的TCRAT细胞分数与对应于CD8+细胞(ρ=0.62,P=9.6e-20)、T细胞(ρ=0.63,P=2.3e-20)和总TIL(ρ=0.59,P=1.4e-17)的Danaher转录组特征具有显著的正相关关系(如图9A、9C所示)。 为了进一步证实TCRA T细胞分数,发明人基于病理学家对88名患者进行手动检查和评分的组织病理学H&E切片,评估了其与TIL分数的相关性。令人欣慰的是,TCRAT细胞分数与病理TIL分数评估显著相关。值得注意的是,病理学TIL分数评估不会专门包括CD8+和CD4+T细胞,因此预计其与TCRAT细胞分数之间不会有完美的相关性。 作为第四种方法,本发明人计算了它们的评分与推断免疫含量的替代的基于DNA的方法的相似程度{Levy et al},该方法基于在VDJ重组后与CDR3区域比对的每个WES中的读取相对于总覆盖的数量(CDR3·VDJ评分,详情参见方法)。在TRACERx100组群中,发明人观察到TCRA T细胞分数与CRD3 VDJ评分之间的显著正相关(图9D,ρ=0.36,P=1.4e-13)。然而,我们注意到,尽管TRACEER×100组群中的覆盖很高,但CDR3·VDJ评分受到测序深度的显著限制;与CDR3区域比对的读取数通常非常低(Q1=0,中值=2,平均值=2.335,Q3=3,最大值=14)。为了明确量化T细胞ExTRECT来源的TCRA T细胞分数以及CDR3·VDJ评分对测序覆盖差异稳健的程度,发明人采用了两种互补的方法(如下所述)。 接下来,选择具有RNA-seq数据和病理学来源的TIL评分二者的肿瘤区域的子集(147个区域),发明人还能够评价两种TCRA T细胞分数、CDR3 VDJ评分和CD8+细胞的六种基于RNA-seq的免疫测量(Danaher、Davoli、xCell、TIMER、CIBERSORT和EPIC)与TIL评分相比如何(如图9B所示)。Danaher CD8+评分具有与病理TIL最强的相关性(ρ=0.49),随后是我们的TCRA T细胞分数(ρ=0.41)、Davoli(ρ=0.4)、xCell(ρ=0.36)、CIBERSORT(ρ=0.23)、TIMER(ρ=0.2)、CDR3 VDJ评分(ρ=0.2)和EPIC(ρ=0.082),其中除EPIC外,其他均显著相关。因此,TCRA T细胞分数评估是对替代的基于DNA的测量的巨大改进,并且在揭示免疫细胞肿瘤浸润含量方面与最先进的基于RNA-seq的免疫特征相竞争,但是与许多现有方法不同,提供了T细胞分数的精确点评估。 另外,发明人进一步评价了来自TCGA的具有较低覆盖(与TRACERx中的中位数深度为426X相比,中位数覆盖:LUAD=84,LUSC=88.2)的独立NSCLC数据集中的TIL ExTRECT。构成LUAD和LUSC组群的样品如表2中所述。发明人将TCRA T细胞分数与来自Thorsson等人的免疫相关数据(Thorsson et al.,2018)相关联。Thorsson等人由DNA甲基化计算了称为“白细胞分数”的样品中总TIL的测量值,以及基于RNA-seq CIBERSORT的T细胞CD8分数。 表2:TCGA LUAD和LUSC组群中样品的概述。 与基于CIBERSORT的CD8+T细胞评估值相比,TCRA T细胞分数与由Thorrson等人使用的甲基化来源的白细胞分数具有更强的相关性(如图10所示,ρ=0.29,P=1.2e-18相对于ρ=0.1,P=0.00097),这既表明TCRA T细胞分数与基于甲基化的总TIL测量值的相关性良好,并且又表明可能优于T细胞含量的某些RNA测量值。 另外,本发明人试图测试TIL ExTRECT来源的TCRA T细胞分数对测序覆盖差异稳健的程度。具体地,为了确定产生准确的T细胞分数评估所需的最小测序深度,他们采用了两种互补的方法。首先,选择来自TRACERx100的五个NSCLC肿瘤区域,其中来自TIL ExTRECT的TCRA T细胞分数范围为0至0.35。然后将这些区域独立地降采样至5、10、20、30、40、50、75、100和200X覆盖10次。本发明人发现,T细胞分数评估在高于并包括30X的覆盖下保持一致(ρ=0.84,P=1.4e-14)(如图11A所示)。与此一致的是,模拟数据显示了30X覆盖与已知T细胞分数之间的可靠对应(如图11B所示,ρ=0.96,P=1.9e-14)。在20X的覆盖及以下时,发明人观察到TCRA T细胞分数开始失去保真度,尽管即使在10X的覆盖下也可获得有用的信息(如图11A所示);这可能是由于噪声提高,特别是对于开始低于检测限度的较低T细胞分数。因此,低于30x(包括10X和20X)的覆盖可仍然有助于区分高的T细胞分数与低的T细胞分数,但可能不足以获得非常低的T细胞分数的可靠评估。由于低于0.1的T细胞分数是常见的,使用30X及以上的覆盖可有利于提供另外的确定性和广泛的适用性。相比之下,本发明人发现用于确定T细胞浸润的CDR3方法由于低测序覆盖而严重偏斜,选择具有最高CDR3读取的五个样品并降采样至50X覆盖,有一个样品检测到3个读取,并且其余的仅检测到单个CDR3读取(图11D)。与此相一致,在由Levy等人的TCGABRCA分析中值得注意的是,在56%的肿瘤中没有鉴定到CDR3读取。 最后,从对多个不同数据集的测试中,本发明人鉴定出根据样品是新鲜冷冻的还是福尔马林固定的石蜡包埋(formalin-fixed paraffin-embedded,FFPE)的T细胞分数没有系统性的显著差异(参见方法和图11C),这意味着该方法对任何FFPE特异性DNA改变不敏感。 因此,数据表明,本文中所述的TCRAT细胞分数评估方法是对替代的基于DNA的测量的改进,并且在揭示免疫细胞肿瘤浸润含量方面与最先进的基于RNA-seq的免疫特征相竞争,但与许多现有方法不同,其提供了T细胞分数的精确点评估。此外,T细胞ExTRECT可以用于任何经受WES的样品,从而允许分析肿瘤和血液样品二者中的T细胞分数。 方法 统计数 本实施例和所有其他实施例中的所有统计检验都在R 3.6.1中进行。没有使用统计方法来预先确定样品量。使用来自R包ggpubr(v0.4.0)中的“stat_cor”与Spearman方法一起进行涉及相关性的检验。除非另有说明,否则涉及分布比较的检验是使用‘stat_compare_means’或使用‘wilcox.test’使用未配对选项完成的。使用来自rstatix包(v0.6.0)中的“wilcox_effsize”函数测量相应Wilcoxon检验的效应大小。使用KaplanMeier曲线和Cox比例风险模型二者的“存活”包(v3.2-3)计算风险比和p值。对于所有的统计检验,所包括的数据点的数量在相应的图中被标绘或注释。 免疫热性和冷性肿瘤区域的限定 在所有这些实施例中,免疫热性区域被限定为通过TIL ExTRECT测量的含有大于10% T细胞的区域,以及免疫冷性区域被限定为含有小于10% T细胞的区域。该阈值是基于专业知识限定的,并且可以根据分析的背景和目的使用其他阈值。 新鲜冷冻样品相对于FFPE样品 为了测试TCRAT细胞分数对于新鲜冷冻样品和FFPE样品二者是可靠的且一致的,针对CPI响应组群中六项不同研究,计算了非GC校正的TCRA T细胞分数。其中三项研究使用了来源于FFPE组织的WES(n=460),而另外三项使用了来源于新鲜冷冻组织的WES样品(n=357)。通过组织学和FFPE状态拟合线性模型来预测TCRA T细胞分数(表6)揭示了癌症类型是这一显著性的主要驱动因素,而FFPE状态并不显著。另外,对于具有FFPE和新鲜冷冻WES样品的黑素瘤和膀胱肿瘤,没有发现显著差异(图11C)。因此可以得出结论,无论WES样品是来源于新鲜冷冻组织还是FFPE组织,都不会显著影响通过TIL ExTRECT计算的TCRA T细胞分数的值。 表6:CPI组群内从组织学和FFPE样品状态预测非GC校正的TCRA T细胞分数的线性模型汇总。 模拟数据 使用与MASCoTE(Zaccaria&Raphael,2020)肿瘤模拟方法的工具组合的ARTIllumina(第2.5.8版)(Huang,Li,Myers,&Marth,2012),创建了测试非GC校正的TCRA T细胞评分结果的模拟数据。总之,ART用于为人第14号染色体生成模拟的成对末端FASTQ文件,该文件来自HiSeq2500DNA测序仪,其中覆盖为30,并且读取长度为150(参数-p-ss HS25-f30-na-l 150-m 200-s 10,其中-ss是指测序平台,-f是指覆盖,-l是指读取长度,-m是指DNA区段的平均大小,-s是指区段大小的标准偏差,以及-na表示不提供输出比对文件-详情参见https://manpages.debian.org/stretch/art-nextgen-simulation-tools/artillumina.l)。然后将这些FASTQ文件用bwa-mem(v0.7.15)与人基因组hg19进行比对。然后使用Picard工具(第1.107版)对生成的BAM文件进行清理、排序和索引。因为只有TCRA基因处的覆盖与模拟数据的测试相关,所以用samtools(第1.3.1版)创建了BAM文件,所有读取的视图映射到chr14:20000000至24000000。该BAM文件用作正常细胞和肿瘤细胞的模板。通过仅提取映射至chr14:20000000至22500000或者chr14:23000000至24000000的读取,并用samtools合并将结果合并到单个BAM文件中来从该BAM文件创建T细胞模板。因此,该T细胞BAM文件在chr14:22500000至23000000之间具有手动创建的100%缺失。包括部分映射至该间隙任一侧的任一区域的任何读取(即不缺失)。由于读取大小为150bp,因此这些读取的存在或不存在被认为与缺失的大小相比是无关紧要的。使用该T细胞和正常/癌症BAM文件,使用MASCoTE方法中使用的Mi×Bam.py模块产生不同的混合BAM文件。这通过根据混合物的基因组长度和拷贝数二者对正常/癌症和T细胞BAM进行采样来产生混合BAM。通过这种方式,可以创建包含具有不同癌症体细胞拷贝数改变值的正常细胞、T细胞和癌症细胞群的不同比例的模拟数据。 对于肿瘤纯度=[0.25,0.5,0.75]、肿瘤拷贝数=[1,3,4,5]和T细胞分数=[0.01至0.25,间隔为0.01]的所有可能组合,以及对于其中T细胞分数范围为0.01至0.99、间隔为0.01的肿瘤拷贝数2,创建了使用该方法的模拟数据。在创建该模拟数据后,仅使用TCRA基因的外显子内位置的读取计算T细胞分数,如应用于WES的标准TIL ExTRECT方法中所进行的(如实施例1中所述)。图8B给出了TCRA周围的局部拷贝数为1的24%T细胞和75%肿瘤的单次模拟运行的输出的一个实例,而图8C和8D示出了不同局部肿瘤拷贝数和纯度值的模拟值与计算值之间的差异。当拷贝数低于2时,初始评分高估了T细胞分数,并且当拷贝数高于2时,初始评分低估了T细胞分数。然而,精确的评分遵循线y=x,这表明其确实是精确的。 研究测序深度对TCRA T细胞分数评估的影响 为了评估较低覆盖的影响,基于它们所代表的TCRA T细胞分数的范围选择了五个TRACERx100区域(高=0.35,中=0.156,低=0.053,非常低=0.010,无=0)。为简单起见,所有区域的局部TCRA肿瘤拷贝数为2。作为一项研究,TRACERx100的中位数覆盖为430.91X。尽管这些样品具有一系列深度,但考虑到该区域的原始深度,使用samtools视图和-s选项将比对的BAM文件降采样至特定深度。以这种方式,创建了从不同随机种子创建的10个降采样深度,以制作深度为200、100、75、50、40、30、20、10和5X覆盖的BAM文件。在降采样以产生这些BAM后,使用上述方法评估非GC校正的TCRA T细胞分数。 捕获试剂盒外显子偏倚检测和用于计算TCRA T细胞分数的外显子的质量控制 作为一般性质量控制步骤,在用于计算TCRA T细胞分数之前,检查每个外显子是否充分覆盖。去除中位数值小于15X的外显子,并且如果超过30个外显子低于该阈值,则样品由于低覆盖而被标记为失败。 在分析TCGA数据集的过程中,观察到由于使用的捕获试剂盒(基于AgilentSureSelect v2的广泛自定义集)而产生的偏差。先前由Wang等人(Wang,Kim,&Chuang,2018)已描述了这种偏差,影响包括TCRA的4833个基因。这种偏差导致某些外显子的覆盖远低于所预期的,从而干扰了评分的计算。这种偏差的一个实例可以在图26A中看到。为了解决该问题,本发明人计算了TRACERx100和TCGALUAD组群内每个单独外显子的平均logR比(即外显子中读取覆盖与前12V区段和C区段中的中位数读取覆盖的log比的平均值-如上所解释的)(参见图26B)。TCGA·LUSC组群未用于此分析,尽管发明人预计来自该组群的结果不会与来自TCGALUAD组群的那些结果有显著差异。将中位数差异>0.5的外显子(其中对于每个外显子,在每个组群的所有样品中计算中位数,然后检查组群之间的中位数差异)标记并从TCGA分析中去除。这导致99/192外显子被排除。注意到,在以下分析中,在TRACERx100数据中去除了这些外显子(尽管预期该数据不经受与TCGA数据相同的偏差,因为该偏差被认为与所使用的捕获试剂盒相关)以表明,当没有偏差时,在使用该降低的组中存在最小的差异(与当偏差存在时可观察到的大影响相比)。也观察到使用Agilent SureSelect v2捕获试剂盒的其他组群,尤其是CPI数据集内的组群(Rizvi et al.,2015;Shim et al.,2020)具有相同的偏差(即偏差影响相同的外显子-如上文所述计算所有外显子的中位数值,并检查这些中位数值与受偏差影响的数据集(即本例中的TCGALUAD组群)中外显子的中位数值之间的相关性所示),并用该降低的外显子集计算评分。另外,GC校正后,在TCRδ基因区段中的一者内使用Agilent SureSelect v2捕获试剂盒时,在单个外显子中观察到偏差。由于该区段位于用于计算TCRA分数的焦点区域内,并且由于缩减集中的外显子数量较少,因此该外显子的任何微小变化都会对计算的评分产生大影响。GC校正后发现这一点更加明显,因此在组群例如TCGA中,这一效应很大,该外显子也从分析中被去除。 当本发明人将降低的外显子集应用于也去除了另外的TCRδ基因区段的TCGA组群,并比较TCRA T细胞分数时,他们观察到TCGA与TRACERx组群之间对于来自LUAD和LUSC二者的肿瘤样品的大差异(图26C:Wilcoxon检验,LUAD:P<2.2e-16LUSC:P<2.2e-16)。然而,这种差异完全是由于该降低的外显子集中的GC偏差。当使用GC校正方法时,LUAD患者的TCRA T细胞分数仅存在显著差异,并且这可能是由于组群人群的差异而仅略有显著性(图26D,Wilcoxon检验,LUAD:P=0.028,LUSC:P=0.97)。 除了在本文的任何组群中使用的任何其他捕获试剂盒中检测到的通过GC校正可以修复的偏差之外,没有其他偏差。然而,使用Nimblegen捕获试剂盒的组群被发现与由Agilent捕获试剂盒限定的外显子的那些有足够的差异,其中来自该试剂盒的所有样品都是使用直接由Nimblegen外显子捕获区限定的外显子计算的。还存在一些鉴定的捕获试剂盒,例如Nextera快速捕获和IDT xGen外显子研究小组在TCRA基因中的覆盖极低。手动检查这些试剂盒的外显子组捕获区域显示,TCRA基因中没有区域被包括在其设计中。对于使用这些试剂盒的任何数据集,无法使用TIL ExTRECT来计算TCRA T细胞分数。 CDR3·VDJ评分的计算 按照Levy等人概述的程序计算CDR3·VDJ评分。首先用samtools提取与TCRB(hg19:chr7:142000817至142510993)比对的读取和未比对的读取,使用bedtools将所得bam转化为fastq,随后在所得输出中使用工具IMSEQ(v1.1.0)来识别与CDR3区域比对的VDJ重组读取,然后将比对的读取数通过原始bam文件中的总读取数(如由samtools flagstat所测量的)进行归一化以产生CDR3 VDJ评分。 TRACERx100患者 在本研究中使用了由NSCLC TRACERx研究(https://clinicaltrials.gov/ct2/show/NCT01888601,经独立研究伦理委员会批准)进行前瞻性分析的前100名患者。这与Jamal-Hanjani等人(Jamal-Hanjani et al.,2017)最初描述的100名患者组群相同。简要描述该组群,知情同意是进入TRACERx研究的强制性要求。该NSCLC组群由68名男性和32名女性患者组成,其中中位年龄为68岁。最后,该组群主要由早期肿瘤(Ia(26)、Ib(36)、IIa(13)、IIb(11)、IIa(13)和IIIb(1))构成,并且28名患者还接受了辅助治疗。WES(与hg19比对的)和RNA-seq样品二者均获自针对前100名患者的TRACERx研究,处理这些样品的方法如前所述(Jama1-Hanjani et al.,2017)。值得注意的是,对于WES样品,外显子组捕获是按照制造商说明使用定制版的Agilent人全外显子V5试剂盒(Agilent Human All Exome V5kit)进行的。 TCGA LUAD和LUSC组群 从基因组数据共享(数据集ID:phs000178.v10.p8)下载TCGALUAD和LUSC组群的比对BAM文件(hg38)。样品纯度和倍性调用使用ASCAT(v2.4.2)生成,并从TCGA数据的先前分析中获得(Middleton et al.,2020)。总之,来自成对的肿瘤-正常样品(数据集ID:hs000178.v10.p8)的Affymetrix SNP6谱通过PennCNV文库(K.Wang et al.,2007)进行处理以获得BAF和log比(在用ASCAT处理之前对其进行GC校正)。免疫相关数据(包括白细胞分数和CD8+分数)从Thorsson et al.,2018获得。 癌细胞系数据 用Illumina HiSeq 2500对非T细胞来源的细胞系HCT116进行测序,并使用hg19与bwa mem进行比对,如López et al.,2020所述的。T细胞来源的细胞系来自Ghandi et al.,2019中描述的数据集,其从序列读取档案(Sequence Read Archive,SRA)下载,登记号为PRJNA523380。选择来源于T细胞的细胞系,确保排除来源于前体T细胞急性淋巴细胞白血病的任何细胞系,因为这些细胞系没有经历VDJ重组。这一过程导致从三种细胞系:JURKAT、HPB-ALL和PEER中选择WES数据。 由于在没有匹配的种系样品的情况下运行ASCAT很困难,因此对于所有细胞系工作,都使用了初始TCRAT细胞分数。 正交免疫测量:1.RNA-seq特征 Danaher等人的方法(Danaher et al.,2018)被用作从RNA-seq测量中评估T细胞含量的主要方法,因为之前已经证明这与TRACERx中计算的TIL评分最强相关(Rosenthalet al.,2019)。针对TCRA T细胞分数测试的其他RNA-seq特征为Davoli方法(Davoli,Uno,Wooten,&Elledge,2017)、xCell(Aran,Hu,&Butte,2017)、TIMER(Li et al.,2017)和EPIC(Racle,de Jonge,Baumgaertner,Speiser,&Gfeller,2017)。 正交免疫测量:2.病理学TIL评分 如Rosenthal等人(Rosenthal et al.,2019)先前所述的,使用由国际免疫肿瘤生物标志物工作组(Hendry et al.,2017)开发的国际既定指南,从组织病理学切片评估TIL。简言之,由给定肿瘤区域的病理切片确定基质面积与肿瘤面积的相对比例。报告了基质隔室的TIL(=基质TIL的百分比)。用于确定基质TIL百分比的分母是基质组织的面积(即由单核炎性细胞占总肿瘤内基质面积的面积),而不是基质细胞的数量(即代表单核炎性细胞核的总基质细胞核的分数)。该方法已被证明可在训练有素的病理学家中重现(Denkert etal.,2016)。进行了人与人之间的一致性,并且这证明了高的可重复性。国际免疫肿瘤生物标志物工作组已开发了免费可获得的培训工具,用于培训病理学家在苏木精-伊红切片上进行最佳TIL评估(www.tilsincancer.org)。 实施例3-TRACERx100和TCGA NSCLC组群中WES来源的TCRA T细胞分数的预后值的 介绍 为了探究TILExTRECT的潜在临床效用,发明人首先考虑了在TRACERx100 NSCLC组群中TCRAT细胞分数是否具有预后性。从TRACERx100组群中的样品的组织病理学H&E切片推断的TIL水平先前与无疾病存活相关(AbdulJabbar et al.,2020)。因此,本发明人探究了他们是否可以使用本文所述的新的TCRAT细胞分数来鉴定类似的相关性。使用来自TCGA的数据也进行了类似的研究。 最后,发明人使用他们的方法来定量在这些组群中匹配血液样品中的TCRA T细胞分数。 结果 根据患者肿瘤内免疫冷性区域的数量将TRACERx100 NSCLC组群中的患者分为两组(≥2个免疫冷性区域,其中免疫冷性区域被限定为TCRAT细胞分数≤0.1的免疫冷性区域),显示出显著差异:发现观察到的两个或更多免疫冷性区域的存在与降低的无复发存活相关(参见图12A,对数秩检验P=0.0068,HR=2.3)。基于专业知识选择≥2个免疫冷性区域的阈值(例如,参见AbdulJabbar et al.,2020)。通过组织学来区分组群而显示出,这种显著性主要来源于LUAD患者,而在LUSC患者的无复发存活方面没有显著性(参见图12B,LUAD:P=0.0037,HR=4.1;图12C,LUSC:P=ns,HR=1.12)。 TRACERx100组群的结果表明,TCRA T细胞分数与LUAD患者的无复发存活具有显著相关性。然而,当本发明人评估TCGALUAD数据集(表2)中的总存活是否也与TCRA T细胞分数相关时,当基于TCRAT细胞分数<0.1的免疫冷性区域的定义将患者分为两类时,他们观察到没有显著的关系(参见图13,对数秩检验,P=ns)。TCGA数据集是单区域的,并且经受采样偏差,并且不能区分具有均匀冷性相对于仅具有单个冷性区域的肿瘤的患者。与TRACERx100相比,TCGA组群内缺乏显著性,这表明多区域免疫数据在预测存活时的重要性。 除了评估肿瘤样品中的TCRA T细胞浸润,TIL ExTRECT还可以应用于来自TRACERx100组群的匹配血液样品。发明人观察到匹配的血液样品比它们相应的配对原发性肿瘤样品具有显著更高的TCRA T细胞分数(参见图14,wilcoxon检验P=0.0012,效应大小=0.16)。然而,值得注意的是,这一比例反映了具有DNA的细胞的比例,因此,如果计入红细胞,则血液中T细胞的这一比例可能会低得多。值得注意的是,与匹配的血液相比,肿瘤区域中的肿瘤TCRA T细胞分数并不是始终较低。事实上,相当一部分(139/339)的肿瘤区域在肿瘤区域内比匹配血液中含有更高的TCRA T细胞分数。在患者而不是区域层面总结这一点,44名患者的所有区域的评分都低于匹配的血液样品,24名患者的所有区域的评分都高于匹配的血液样品,其余32名患者是异质的。发明人还观察到(使用TRACERx100数据,其中由于数据的多区域性质,一个血液样品与多个肿瘤区域相匹配)在LUAD和LUSC二者中肿瘤TCRAT细胞分数与血液TCRA T细胞分数显著相关(LUAD:ρ=0.39,P=1.2e-07,LUSC:ρ=0.45,P=7.8e-07,参见图15),这表明血液中的TCRA T细胞分数和来自同一患者的肿瘤相关。 通过组织学划分TRACERx100组群,发明人发现与LUSC相比,血液TCRA T细胞分数在LUAD患者中显著更高(参见图16A,左图,Wilcoxon检验P=0.0053,效应大小=0.29),与LUSC肿瘤相比,原发性肿瘤样品在LUAD中也具有鉴定更高的TCRA T细胞分数(参见图16A,右图,Wilcoxon检验,P=2.2e-12,效应大小=0.42)。在TCGA肿瘤中观察到类似的结果(参见图17A至D),其中发明人还发现根据样品类型的显著差异,其中在血液中存在的TCRA T细胞分数高于原发性肿瘤(参见图17A和B,Wilcoxon检验LUAD:P<2.2e-16,效应大小=0.28,LUSC:P<2.2e-16,效应大小=0.38)。这些结果表明,刺激强免疫应答的肿瘤也可导致血液中循环的T细胞水平更高,或者血液中更高的T细胞水平可能允许在肿瘤中发生更强的免疫应答。 为了进一步研究可以解释血液样品中TCRA T细胞分数变化的因素,发明人考虑了患者的临床特征,包括他们的性别和年龄。事实上,已经注意到,在衰老期间,人免疫系统中存在相当大的性别二态性(Márquez et al.,2020),其中CD8+T细胞含量的减少与年龄增长和男性性别相关。对于TRACERx100和TCGA组群二者,男性与女性患者在血液样品中的TCRAT细胞分数方面存在显著差异,其中女性中存在的水平提高(TRACERx100:图18A,Wilcoxon检验P=0.027,效应大小=0.22,TCGA:图18B,Wilcoxon检验,P=1.2e-5,效应大小=0.12)。年龄的影响较弱;TRACERx100组群显示出轻微的负趋势,其不显著(参见图18C:ρ=-0.091,P=0.37),而TCGA组群显示出微弱但显著的相关性(参见图18D:ρ=-0.095,P=0.013)。 在进一步的分析中,发明人分别分析了LUAD和LUSC,并基于肿瘤中存在的冷性区域的数量将患者分成免疫热性组和免疫冷性组。在此,他们使用了基于所有肿瘤区域的平均TCRA T细胞分数(0.081)的简单阈值,并将他们的分析限于每个阈值中使用的多于冷性区域数量的肿瘤。图16C中的结果显示,对于LUAD,随着阈值中使用的冷性区域的数量增加,免疫热性组与冷性组之间的存活率更显著(LUAD:>=2个冷性区域,HR=3.1,P=0.0063对数秩检验,LUAD:>=3个冷性区域,HR=7.3,P=0.00024对数秩检验)。相比之下,对于LUSC,任何不同的阈值的免疫冷性组或热性组之间的存活率没有显著差异(图16C)。使用中位数(0.074)代替平均值作为免疫热性区域或冷性区域的阈值而产生了类似的结果(图16F)。 在单区域TCGALUAD组群中,发明人还发现使用平均值作为热肿瘤与冷肿瘤之间的阈值(0.11),热肿瘤与冷肿瘤之间的存活具有显著性(总存活(OS):HR=0.61,P=0.0043,无进展存活(PFS):HR=0.67,P=0.016-参见图17F)。对于OS和PFS二者,存在一系列将产生类似结果的可能阈值(图17H)。使用相同的阈值来限定免疫热性肿瘤和免疫冷性肿瘤,在TCGALUSC组群中,OS或PFS的存活没有显著相关性(图17I)。 然后,本发明人使用Cox回归模型来确定使用与TCRA T细胞分数相关的不同多区域评分的连续分析是否对存活具有同样的预后性。发明人选择研究每个患者的以下四个评分:1)所有区域中的平均TCRA T细胞分数(肿瘤中总T细胞浸润的指标),2)所有区域的最小TCRA T细胞分数,3)所有区域的最大TCRA T细胞分数,和4)TCRA T细胞分数差异,其被限定为最大区域TCRA T细胞分数除以最小区域TCRA T细胞分数的上95%置信区间(以避免除以0或非常小的数)。后者评分代表具有最大与最小TCRA T细胞分数的区域之间的倍数差异,并且当高时,可指示T细胞差异/异质性,可能指示已经经历免疫逃逸的亚克隆。由于相对区域免疫逃逸评分由最小和最大评分决定,本发明人还建立了包括这两个评分的Cox模型。图16D和17G中汇总了该分析的结果。与具有最低TCRA评分32的肿瘤区域的重要性一致,在整个TRACERx组群中,肿瘤区域中的最小TCRA分数具有预后性(HR=0.52,P=0.048)。然而,平均和最大TCRAT细胞分数二者在任何组中都不显著。TCRA T细胞分数差异评分在LUAD中显著(HR=2.2,P=0.023对数秩检验)。包含最小和最大TCRA评分二者的模型在整个组群(最小值:HR=0.5,P=0.005,最大值:HR=1.5,P=0.061)和LUAD亚组(最小值:HR=0.36,P=0.016,最大值:HR=2.52,P=0.029)二者中具有显著性,参见图16D,这表明当考虑到TCRAT细胞分数的异质性时,有增加的预测潜力。在LUSC中,没有TCRA相关的评分是显著的,并且最大TCRA T细胞分数仅与最小值结合时是显著的并且仅在LUAD中是显著的。 当将最小和最大TCRA T细胞分数与其他临床表型(例如肿瘤分期、性别和年龄)相结合时,最小TCRA T细胞分数仍然是显著的(HR=0.52,P=0.022,参见图16E)。检查单区域TCGA的总存活(OS)和无进展存活(PFS)的存活数据,在任何比较的单变量Cox模型中,TCRAT细胞分数都不显著,最密切的相关性是OS模型中的TCGALUAD(HR=0.85,P=0.069)。 综上所述,本发明人认为该数据暗示TCRA T细胞对LUAD患者的存活具有预测性。特别地,本发明人注意到,尽管TRACERx100组群规模较小,但在TRACERx100组群中观察到了更强的整体信号,这表明了通过多区域数据揭示的肿瘤内免疫异质性的重要性。 方法 参见实施例1-2。 多样品肿瘤患者组群 多样品泛癌组群(参见表5)是通过将TRACERx组群与由Watkins等人最近提出的组群的子集相结合而创建的。如果肿瘤在原发性肿瘤中至少有两个测序区域,可以使用TILExTRECT计算TCRA T细胞分数,则肿瘤被包括在内。因此,最终组群由多区域原发性肿瘤数据集组成,其中添加了也对这些患者进行测序的任何转移样品。 表5:多样品泛癌组群中患者的概述 除TRACERx100外,以下数据集被合并到最终的多样品泛癌组群中: 1.Brastianos等人-集中研究来源于不同组织学的脑转移瘤的组群,仅包括来自该组群的具有多区域原始样品的肿瘤。 2.Gerlinger等人-肾脏肾透明细胞癌(kidney renal clear cell carcinoma,KIRC)患者的多样品主要组群。 3.Harbst等人-皮肤黑素瘤(skin cutaneous melanoma,SKCM)患者的多区域主要组群。 4.Lamy等人-膀胱癌(bladder cancer,BLCA)患者的多区域主要组群 5.Savas等人-ER+和三阴性乳腺癌(BRCAER+和TNBC)患者的多样品组群 6.Suzuki等人-胶质瘤的多区域主要组群。 7.Turajlic等人-肾透明细胞肾细胞癌(KIRC)、肾乳头状细胞癌(Kidney renalpapillary cell carcinoma,KIRP)和肾嫌色细胞癌(Kidney Chromophobe,KICH)患者的多区域主要组群。 8.Messaoudene等人-HER2+和ER+乳腺癌患者的多区域主要组群。不同数据集中多区域测序的亚区域的选择 在所有的多区域组群中,通过不同的方法(参见相关出版物)选择考虑两个主要标准的区域,第一,以基质为代价使肿瘤含量最大化,以确保用于基因组分析的主要目标的高质量的突变和拷贝数分析,以及第二,每个区域代表肿瘤的物理上分离和不同的部分。在不在单独位点的情况下,使用了不同的测量。例如,在TRACERx100组群中,测序区域间隔至少为3mm。 实施例4-TCRAT细胞分数可预测对免疫检查点阻断的响应 介绍 为了进一步探究TCRA T细胞分数可能具有临床效用的地方,本发明人使用来自CPI1000+组群(Litchfield et al,2020)的数据评价了其是否可用于预测对免疫检查点阻断的响应。 结果 CPI1000+组群(Litchfield et al,2020)多项研究数据集由8种主要癌症类型中的接受抗CTL4或抗PDL1治疗的1070个CPI(检查点抑制剂)治疗的肿瘤组成(参见图19和表3)。根据RECIST标准,响应被限定为具有完全响应(complete response,CR)或部分响应(partial response,PR)的放射响应的患者,而无响应被限定为疾病稳定(stabledisease,SD)或疾病进展(progressive disease,PD)。表3详细说明了具有WES和RNA-seq数据二者的肿瘤的百分比。 表3:CPI1000+组群(不包括Snyder et al.,2017)中样品的概述 与肿瘤TCRA T细胞分数在预测对CPI的响应中的重要性一致,发明人观察到在整个组群中响应者与无响应者之间的TCRA T细胞分数有显著差异(参见图20,P=2.3e-7,效应大小=0.17)。响应者的肿瘤DNA样品中TCRA T细胞分数的中位数为0.053(Q1=0,Q3=0.17),相比之下,在无响应者中为0.000268(Q1=0,Q3=0.084)。关于免疫冷性肿瘤(在此限定为TCRA T细胞分数<0.1的肿瘤),在无响应者中有非常显著的富集(图21,免疫冷性肿瘤是虚线以下的那些,78%相对于63%,Fisher精确检验,优势比(odd ratio,OR)=0.47,P=2.25e-06)。 通过突变负荷(高:总克隆TMB≥68,低:克隆TMB<68)和免疫微环境(热性:TCRA T细胞分数≥0.016,冷性:TCRAT细胞分数<0.016)二者来区分组群,揭示了TCRA T细胞分数与响应之间的相关性独立于突变负荷(参见图21)。在低突变负荷患者中,免疫热性肿瘤的响应率为21%,相比之下在免疫冷性肿瘤中为8%。在高突变负荷肿瘤中,当肿瘤为免疫热性时有45%的响应率,并且当肿瘤免疫为冷性时有30%的响应率。 为了进一步评价TCRA T细胞分数与基于RNA-seq的测量相比的效用,选择了来源于具有RNA-seq和TCRA T细胞分数二者的个体癌症类型的超过10个样品的所有研究进行单变量荟萃分析(参见图22:7项研究和5种癌症类型中的557名患者)。如所预期的,发现TCRAT细胞分数(优势比(OR)=1.39,P=0.00858)、克隆TMB(OR=1.59,P=6.021e-05)和CD8A表达(OR=1.45,P=0.0004479)都与响应显著相关。未发现来自血液中的TCRA T细胞分数和肿瘤纯度与响应显著相关,而肿瘤与血液TCRA T细胞分数之间的差异是略微显著的(OR=1.27,P=0.016)。 然后,本发明人评估了肿瘤TCRA T细胞分数是否提供了超过TMB(肿瘤突变负荷)的另外临床用途,此外,它是否比RNA-seq测量(例如CD8A表达)更大程度地改善了响应的预测。他们创建了四个一般线性模型(GLM)来预测患者是响应者还是无响应者。第一模型仅由克隆TMB组成,而第二和第三模型使用了与CD8A RNA-seq表达或TCRA T细胞分数组合的克隆TMB。第四模型使用了与CD8A表达和TCRA T细胞分数二者组合的克隆TMB。使用CD8A表达或TCRA均提高了模型的预测值(参见图23,与单独的克隆TMB为0.62相比,CD8A表达的AUC=0.66,TCRA T细胞分数的AUC=0.70)。然而,在这些模型中,与单独的克隆TMB相比,只有克隆TMB+TCRA T细胞分数的模型(ROC检验,P=0.0028,GLM:克隆TMB+TCRA,AUC=0.68,GLM:克隆TMB,AUC=0.62)是显著的。将TCRA T细胞分数与CD8A表达相组合未能显著提高模型的预测值(参见图23,AUC=0.68,ROC检验相对于克隆TMB+TCRA模型P=0.72)。然而,检查所有4个模型中变量的显著性,TCRA T细胞分数比CD8A更显著(GLM:克隆TMB+TCRA,P=4.62e-05;GLM:克隆TMB+CD8A,P=0.000431),并且当组合成单一模型时,TCRA T细胞分数仍然显著,但CD8A表达不显著(TCRA,P=0.00601,CD8A,P=0.06246)。 综上所述,这些结果表明肿瘤TCRA T细胞分数可以用作CD8+浸润的RNA-seq测量的替代物,此外,基于WES的TCRA T细胞分数评估增加了TMB评估的预后值。 鉴于在许多情况下无法获得RNA-seq,发明人接下来评估了组合的NSCLC CPI组群中TCRA T细胞分数的预测潜力(参见表4)。图24A提供了该组群的概况,该组群包含266名经WES测定的患者,并且关键是缺乏任何RNA-seq或其他正交免疫测量。对肺CPI组群进行单变量分析的分析(参见图24B),发现TCRA T细胞分数(OR=1.44,P=0.005)和血液TCRA T细胞分数(OR=1.39,P=0.0015)与对CPI的响应显著相关。在单个研究的基础上,TCRA T细胞分数在Hellman和Shim组群二者中都具有OR>1,但在Rizvi组群中没有,相反,血液TCRA在所有三个独立的研究中都具有OR>1。这些结果表明,TCRA T细胞分数可以根据单独的WES数据来计算,并且来自NSCLC以及来自匹配的血液中的这样的评估可预测对免疫治疗的响应。 表4:CPI肺组群中患者的概况。 方法 参见实施例1-2。 CPI1000+组群的荟萃分析 CPI1000+组群在Litchfield等人(2020)中充分描述并且包含以下数据集:1.Snyder等人(Snyder et al.,2014),晚期黑素瘤抗CTLA-4治疗的组群。2.Van Allen等人(Van Allen et al.,2016),晚期黑素瘤抗CTLA-4治疗的组群。3.Hugo等人(Hugo et al.,2016),晚期黑素瘤抗PD-1治疗的组群。4.Riaz等人(Riaz et al.,2017),晚期黑素瘤抗PD-1治疗的组群。5.Cristescu等人(Cristescu et al.,2018)晚期黑素瘤抗PD-1治疗的组群。6.Cristescu等人(Cristescu et al.,2018)晚期头颈癌抗PD-1治疗的组群。7.Cristescu等人(Cristescu et al.,2018)用抗PD-1治疗的“所有其他肿瘤类型”组群(来自KEYNOTE-028和KEYNOTE-012研究)。8.Snyder等人(Snyder et al.,2017),转移性尿路上皮癌症抗PD-L1治疗的组群。9.Mariathasan等人(Mariathasan et al.,2018),转移性尿路上皮癌症抗PD-L1治疗的组群。10.McDermot等人(McDermott et al.,2018),转移肾细胞癌抗PD-L1治疗的组群。11.Rizvi等人(Rizvi et al.,2015),非小细胞肺癌抗PD-1治疗的组群。12.Hellman等人,由Litchfield et al.,2020使用的经抗PD-1治疗的非小细胞肺癌样品的组群。13.Le等人(Le et al.,2015),用抗PD-1治疗治疗的结直肠癌组群。 在这些研究中,Snyder等人(Snyder et al.,2017)被排除在分析之外,因为其在TCRA基因中的覆盖极低。使用bwa mem(v0.7.15)将所有样品与hg19进行比对,其中纯度和拷贝数数据由如Litchfield et al.(2020)所述的ASCAT进行计算。值得注意的是,1008/1125(90%)的样品具有WES数据,941/1125(83%)具有足够的纯度和覆盖以能够计算拷贝数,从而能够计算TCRA T细胞分数。这些样品中的833/1125(74%)具有匹配的RNA-seq数据,允许T细胞评估的正交评估。为了扩展至该数据集,Shim等人(Shim et al.,2020)添加了NSCLC抗PD-1治疗的组群用于特定的NSCLC分析。 CPI响应的单变量和多变量模型 对于单变量模型,遵循Litchfield等人(2020)的改编程序,其中主要区别在于仅包括具有完整数据(CD8A的RNA-seq、克隆TMB和TCRA T细胞分数)的样品。使用R包“meta”(第4.13-0版)进行单变量模型荟萃分析。多变量模型是使用来自“stats”R包中的函数“glm”使用默认值用广义线性模型创建的。R包“ROCR”(第1.0-11版)用于ROC曲线分析。 实施例5-血液、正常和恶性组织中TCRA T细胞分数的决定因素 虽然先前的分析必然集中于肿瘤组织中的T细胞浸润,但是本文中所述的TILExTRECT方法提供了研究任何经受WES的样品中T细胞浸润的机会。因此,在本实施例中,发明人进行了分析以评价不同样品类型中T细胞免疫浸润的关键决定因素。 结果 发明人首先探究了作为用于肿瘤分析的正常对照的血液中T细胞浸润的程度。在TRACERx100组群中,血液中的TCRA T细胞分数在女性中相比于男性明显更高,并且我们观察到肿瘤样品TCRA T细胞分数与血液TCRAT细胞分数之间有显著的正相关(图16B,组织学:P=0.066,ES=0.19;性别:P=0.0057,ES=0.28;平均肿瘤浸润:ρ=0.42,P=1.7e-05)。这些数据表明肿瘤免疫浸润可影响循环中的血液中的T细胞水平。性别和平均肿瘤浸润是TCRAT细胞分数的显著单变量预测因子,并且在控制吸烟状态、年龄和组织学的预测血液TCRAT细胞分数的线性模型中仍然显著。相比之下,在多变量模型中,在LUAD与LUSC患者之间没有观察到血液中T细胞浸润的显著差异。发明人还分析了取自TCGA LUAD和LUSC组群的匹配血液样品,并发现了类似的结果,其中在预测血液中TCRA T细胞分数的线性模型中,性别和肿瘤组织学密切相关(图17E)。 然后,发明人在具有较低的TCGA覆盖的独立的NSCLC数据集中评价了T细胞ExTRECT,如以上和在表2中所述的。他们在来自LUAD和LUSC·TCGA患者的血液样品中观察到大体上一致的结果(图17E)。 另一个可能影响血液中T细胞浸润的因素是身体其他部位存在病毒或细菌感染。为了探究这一假设,发明人使用了Poore等人提出的使用生物信息学工具KRAKEN的数据来量化来自LUAD和LUSC·TCGA组群的WGS血液样品和RNA-seq肿瘤样品的微生物组读取数量。发明人观察到,具有升高的微生物读取数量(大于中位数,6.81)的血液样品具有比低组显著更高的血液TCRAT细胞分数(图27A P=0.00092,ES=0.31,Wilcoxon检验)。相比之下,没有观察到总微生物读取数量与肿瘤TCRA T细胞分数之间的相关性,这表明病毒或细菌的存在并不驱动我们在癌症中所看到的T细胞分数的水平(图27B P=n.s)。然后,发明人检查了在单一物种水平上是否存在任何与TCRA T细胞分数相关的病毒或细菌(参见方法)。在对血液样品进行多重假设校正之后,没有显著的相关性。对于肿瘤样品,LUAD和LUSC肿瘤各有一个命中,分别为威廉姆氏菌属(Williamsia)和拟孢菌属(Paeniclostridium)细菌(图27C和28G,LUAD的威廉姆氏菌:ρ=-0.17,P=0.00011,FDR P=0.013,图27D和28G,LUSC的拟孢菌:ρ=-0.2,P=0.00013,FDR P=0.015)。然而,当TCRAT细胞分数较低时,这两种都具有较高数量的归一化log-cpm值,这表明这些细菌物种的存在可能不会导致T细胞浸润增加,相反,它们可以是利用免疫冷性肿瘤微环境的机会性物种。 为了进一步理解血液TCRA T细胞分数的关键决定因素,发明人探究了来源于血液和生理正常食管上皮(PNE)组织的WES测序样品。特别地,本发明人检查了这样的数据集:包含来源于生理正常食管上皮(PNE)组织的显微解剖的WES测序样品,如由Yokyama等人所述的。在此,在血液样品中计算了宽范围的TCRAT细胞分数,但是大多数PNE组织没有检测到T细胞浸润(图28A,图28H)。将参与者分成PNE样品中有或没有T细胞浸润的参与者,显示出与血液TCRAT细胞分数显著相关(图28B,P=0.021,ES=0.29),再次表明与肿瘤样品相似,正常组织中高水平的T细胞浸润影响血液中可见的TCRAT细胞分数。在预测血液中T细胞分数的线性模型中,只有正常组织中的浸润水平具有显著的独立预测性(图28C)。在单独的线性模型中,没有发现基因组因素(例如正常组织突变负荷或癌症驱动突变状态)预测PNE组织中的T细胞浸润(图28F)。这表明检测到的T细胞浸润不是由于持续存在的免疫监测,而可能是由于微生物感染的存在。 为了进一步评价影响肿瘤组织中T细胞浸润的因素,发明人利用了最近发表的多样品数据的泛癌组群(Watkins et al.),允许研究同一肿瘤的不同区域可表现出不同水平的免疫浸润的程度,以及其是否有可能鉴定这种差异的基因组基础。总之,本发明人能够评价来自14种癌症类型的182个肿瘤的739个肿瘤区域中的T细胞浸润(参见表5)。 在组群中观察到一系列T细胞浸润(图29A,范围0%至58%)。有趣的是,发现具有51%测序细胞的第三高评分代表来自患者RMH002的KIRC肿瘤的单一区域中的T细胞。值得注意的是,在肾切除术之前,患者RMH002已经用抗血管生成药物舒尼替尼(sunitinib)治疗了14周,并且舒尼替尼治疗已经显示增加KIRC肿瘤中的T细胞浸润(Haywood etal.)。为了量化肿瘤内T细胞浸润多样性的程度,发明人基于计算出的TCRAT细胞分数是否在所有区域中都是均匀热性的(在此限定为所有区域≥0.11,组群中的平均TCRAT细胞分数),其是否是均匀冷性的(限定为所有区域<0.11)或者T细胞分数是否是异质的,将各肿瘤分为三类中的一类。由癌症类型评估的均匀热性、均匀冷性以及具有异质性免疫浸润的肿瘤的比例存在显著差异(图29B卡方检验:P=1.62e-07),其中BRCAER+肿瘤的异质性最高(83%),而LUSC肿瘤最低(22%)。在免疫浸润的程度和在癌症类型中不同免疫组类别(均匀热性的、冷性的或异质的)的患病率方面也有明显的差异。例如,虽然BLCA和LUSC有相似数量的异质性肿瘤(36%相对于37%),但约64%的BLCA肿瘤是均匀热性的,0%是均匀冷性的,而在LUAD中,37%的肿瘤是均匀冷性的,25%是均匀热性的。这表明,对于某些癌症类型,最显著的是BRCAER+、BRCAHER+、LUAD和KIRC,存在高度局部化的免疫浸润,这可能受到相当大的取样偏差的影响。 接下来,发明人检查了基因组多样性与免疫多样性之间是否存在任何关系,研究了亚克隆SCNA异质性作为可导致单个肿瘤内异质性免疫应答的潜在机制。发明人首先将分析限于具有至少三个样品和T细胞分数的异质混合物的肿瘤(参见方法)。将任意两个区域之间的成对SCNA异质性计算为任一区域中具有独特SCNA的基因组比例的之和。图29C通过成对分析显示,在肿瘤中,具有较大TCRA T细胞分数差异(>=所有成对距离的平均值,0.065)的区域对更可能具有较高水平的成对SCNA异质性(所有事件:P=0.00021,效应大小=0.352;获得事件:P=0.0045,效应大小=0.324;丢失或LOH事件:P=0.024,效应大小=0.257,n=77)。 接下来,为了探究任何特定的亚克隆SCNA事件是否与免疫耗竭或激活相关,发明人鉴定了在全泛癌多样品组群中超过30个肿瘤中亚克隆丢失或获得的染色体区段(cytoband)区域(图29D),并测试了这些获得或丢失事件是否与TCRA T细胞分数的变化相关。只有一个染色体区段水平的事件,即12q24.31-32的亚克隆丢失被发现与TCRA T细胞分数的降低显著相关(图29E:P=1.3e-05,效应大小=0.735)。 为了确定这种效应是否是由于任何单个基因引起的,发明人在TRACERx100组群内选择了具有12q24.31-32亚克隆丢失的肿瘤,并对相关的RNA-seq数据进行了差异基因表达分析。在测试了16,168个基因之后,在多次测试校正之后,只有8个保持显著性:SPPL3、C12orf76、LYRM9、CIT、UBE3B、ABCB9、OGFOD2和USP30(图29F)。值得注意的是,SPPL3是最重要的基因,并且与ABCB9和OGFOD2一起位于12q24.31上。最近发现SPPL3可增强B3GNT5酶活性,从而上调细胞表面糖脂,进而抑制I类HLA功能并降低CD8+T细胞活化(Jongsma etal.)。因此,这些数据表明,12q24.31的亚克隆丢失可在跨癌症类型中的肿瘤演变中被选择(发生在组群中18.7%或34/182的肿瘤中)作为免疫逃避的机制。 方法 参见实施例1至4。 KRAKEN TCGA分析 由Poore等人进行的来自KRAKEN(Wood&Salzberg)分析的预处理微生物组数据输出从ftp://ftp.microbio.me/pub/cancer_micrcbiome_analysis/下载。 为了创建血液和肿瘤样品二者的高和低KRAKEN微生物组,下载了包含归一化log-cpm值的文件Kraken-TCGA-Voom-SNM-Most-Stringent-Filtering-Data.csv,对每个样品的行求和,给出总体“微生物组”评分。然后基于该评分的中位数将样品分成高分组和低分组。 为了研究任何单个微生物物种在影响TCRA T细胞分数方面的作用,通过从原始数据文件Kraken-TCGA-Raw-Data-17625-Samples.csv中去除了TCGA LUAD和LUSC组群中小于1000总原始读取的所有物种,从Kraken-TCGA-Voom-SNM-Most-Stringent-Filtering-Data.csv文件中选择了物种的简化列表。这留下了总共59种微生物物种,这些微生物物种使用LUAD和LUSC血液和肿瘤样品二者的Spearman相关性单独测试与TCRA T细胞分数的相关性。 泛癌多样品组群中获得、丢失和LOH事件的鉴定 如Jamal-Hanjani等人先前所述的进行全外显子组测序分析。如先前所述的(Jamal-Hanjani et al.)使用ASCAT(Van Loo et al.)评价每个样品的拷贝数分割、肿瘤纯度和倍性。这些数据被用作多样品SCNA评估方法的输入,以产生杂合性丢失以及与样品倍性相关的丢失、中性、获得和扩增拷贝数状态的存在的全基因组评估。使用P<0.01阈值的单尾t检验,相对于三个样品倍性调整的对数比阈值,检查了肿瘤的所有样品中对数比值≥5的每个拷贝数区段中存在的对数比值。在二倍体肿瘤中,这些对数比阈值相当于丢失为<log2[1.5/2],获得为>log2[2.5/2]。任何未分类为丢失或获得的区段均被分类为中性。对于每个区段,这些相对于倍性的定义与来自单一肿瘤的所有样品中的杂合性丢失检测组合。 成对亚克隆SCNA评分 为了计算成对亚克隆SCNA测量值,使用在先前方法部分中概述的分类来创建三组成对亚克隆SCNA评分。首先,本发明人认为受任何与倍性或LOH相关的获得或丢失影响的任何区段都是异常的,并比较了来自单个患者疾病的每对区域,如果在两个样品中均异常,则将异常区域分类为克隆的,或者如果仅在一个样品中异常,则分类为亚克隆性的。对单独相对于倍性的获得重复同样的过程,然后同时考虑相对于倍性和LOH的丢失。 染色体区段水平SCNA分析 为了能够跨肿瘤进行比较,将区段映射到hg19染色体区段。如果多个区段映射到染色体区段,选择与染色体区段重叠最大的区段的SCNA状态(相对于倍性的获得或丢失)。对于SCNA获得或丢失分析,如果染色体区段水平事件在整个组群中亚克隆发生超过30次,则选择染色体区段水平事件。然后将同一区域内超过该阈值的条带(例如1p36上的所有染色体区段)分组在一起。使用Wilcoxon配对检验来评估具有亚克隆SCNA事件的单个患者内的肿瘤区域与在没有该事件的那些区域的TCRA T细胞分数方面是否有显著差异。 具有异质免疫浸润的多样品肿瘤的选择 要被包括在内,肿瘤必须具有至少3个测序的区域并且满足以下两个要求,1)具有免疫浸润变化大(限定为TCRA T细胞分数差异≥0.1)的区域对,以及2)具有免疫浸润变化小或无变化(限定为TCRA T细胞分数差异<0.1)的区域对。符合这一要求的肿瘤的一个实例是具有其中TCRA T细胞分数分别为0.01、0.01和0.2的三个区域R1、R2和R3的肿瘤。R1-R2对的TCRA T细胞分数的差异为0,而R1-R3和R2-R3对均具有为0.19的大的差异。在多样品肿瘤组群中,58名患者符合这些标准。 具有亚克隆12q24.31-32丢失的患者的RNA-seq差异基因表达分析 对具有亚克隆12q24.31-32丢失的TRACERx100 RNA-seq患者进行差异基因表达分析。使用R4.0.0,首先将edgeR R包(版本3.32.1)用于样品特异性TMM(M值的修整平均值)归一化,然后使用标准edgeR过滤方法过滤掉任何低表达的基因,然后使用来自limma R包(版本3.46.0)的Limma-Voom方法计算Voom拟合并获得基因表达差异的p值。将患者和组织学作为阻断因素进行对照比较,并对多重测试的p值进行FDR校正。然后用REnhancedVolcano软件包(版本1.8.0)将结果可视化。 实施例6-来自WGS数据的样品中T细胞分数的评估、B细胞分数的评估和免疫克隆 介绍 实施例1中描述的和实施例2至5中例示的方法可用于研究进一步的情况,例如全基因组测序数据可用的情况,除αβT细胞之外的其他免疫细胞区室是令人感兴趣的情况,和/或特定V和J区段的使用令人感兴趣的情况,例如如用于研究克隆型。在该实施例中,发明人示出了实施例1中描述的用于WGS数据的通用方法的实现。通过利用具有WES和WGS数据二者的TRACERx100组群的子集,他们验证了该方法的WGS版本,显示了使用WES数据与使用WGS数据之间的高度一致性。它们进一步提供了用于读取深度比建模的替代模型(区段模型),该模型考虑了利用WGS样品内的更大覆盖的V(D)J重组的性质。他们使用正交RNA-seq数据表明评分准确地描述了免疫微环境并且使用降采样表明该方法保持精确低至2X的深度。然后他们还表明,该方法可以用于研究γδT细胞的分数以及TCR和BCR多样性。最后,他们还示出了使用该方法来研究大的泛癌组群中的存活(数据未显示)。 结果 与全外显子组测序(WES)不同,WGS包含整个基因组中的覆盖。在评估拷贝数改变的背景下,这提供了许多重要的进步,1)均匀的覆盖允许精确鉴定拷贝数断点的位置,以及2)存在更少的偏差,例如由于GC含量或参考等位基因偏差而引起的那些偏差。因此,本发明人推断,实施例1中描述的应用于WGS的T细胞ExTRECT 通过将GC校正和质量控制应用于1000bp窗口而不是来自WES捕获试剂盒的外显子区段,并应用另外的质量控制来鉴定任何100bp基因组窗口(该窗口具有表明在比附近区域高得多或更低的读取深度方面的异常值的证据),使上述方法适用于WGS(全部细节参见方法)。图30中给出了这种调整的方法(称为“调整的GAM”)的概述。另外,通过为聚焦区域和归一化区域选择不同的基因组基因座,本发明人能够将相同的WGS方法应用于TCRB、TCRG和IGH(参见表7中使用的基因组位置和图31中作为实例的CRUK0085 T1-R3的图)。由于这些基因座内序列比对质量的严重问题,这种特定的数据集不适于IGL和IGK方法的例证,可能是由于它们的高度复杂性和参考序列的质量导致难以以一致和无偏差的方式映射短读取序列。然而,如果可以获得更高质量的比对,例如如通过使用更高质量的参考序列,同样的方法应该适用于这些基因座。同样,由于在TCRA内的其基因座的尺寸较小,该方法并未直接应用于TCRD。然而,如下所述的基于片段使用的方法能够评估包括TCRD区段的T细胞的分数。 WGS数据应用的基准 为了利用WGS数据的均匀覆盖,设计了平滑广义线性模型(GAM)的替代模型。这是分段约束的线性模型,其中断点被强制定位在V(D)J基因座内V和J段的已知位置处。该“区段模型”在方法和图30中进行了描述,并与GAM模型相比具有以下理论优势:1)其基于真实的V(D)J重组生物学;2)可以计算单独的V和J基因区段的单独分数,从而了解区段的使用和T或B细胞受体多样性;以及3)TCRD特异性区段的分数可用作γδT细胞的指标。该模型可以用于在整个目标区域具有足够覆盖的任何测序数据。对于特定的WES数据,这种方法并不具有优势(尽管理论上是可能的),因为所用的特定外显子捕获试剂盒在目标区域的覆盖上具有大间隙。然而,在其他捕获数据集中可以不是这种情况。 应用于WGS数据的GAM模型和区段模型二者的准确性通过确定一组322个TRACERx100样品(可获得匹配的WES和WGS)和126个WGS样品(具有正交RNAseq数据)的T和B细胞分数来测试。使用GAM模型比较WES和WGS·TCRA T细胞分数,发明人发现在血液(ρ=0.71,P=4.7e-16,图32A)和肿瘤(ρ=0.72,P<2.2e-16,图32A)样品二者中均有显著相关性。使用区段模型,本发明人发现了与基于GAM的WES评分的类似显著相关性(血液:ρ=0.69,P=6.7e-15;肿瘤:ρ=0.7,P<2.2e-16,图32A)。然而,在这两个模型中,发现WGS值在较低的T细胞分数下较高,这可能是由于WGS数据的可能提高的准确性和敏感性。支持这一点的是:当检查正交TRACERx100 RNAseq数据时,发现与匹配的WES评分相比,WGS评分与Danaher T细胞评分的相关性更强(WES:ρ=0.64,P=1.6e-15,(Danaher T细胞评分)=1.9+8.3(ExTRECT T细胞分数(WES)),WGS(GAM):ρ=0.82,P<2.2e-16,WGS(区段模型):ρ=0.81,P<2.2e-16,图32B)。 其他基因座的方法验证 根据匹配的RNAseq数据计算的Danaher评分用于对从TCRB和TCRG计算的T细胞分数和从IGH计算的B细胞分数的准确度进行正交验证。发现TCRB(GAM:ρ=0.66,P<2.2e-16;区段模型:ρ=0.81,P<2.2e-16,图32B)和TCRG(GAM:rho=0.64,P=3.1e-16;区段模型:ρ=0.8,P<2.2e-16,图32C)与T细胞Danaher评分显著相关,并且IGH与B细胞Danaher评分显著相关(GAM:ρ=0.47,P=2.3e-8;区段模型:ρ=0.44,P=2.1e-07,图32C)。对于来自区段模型的B细胞分数,存在一些具有高B细胞分数但低B细胞Danaher评分的明显的异常值点。这些评分倾向于具有来自区段模型的非常低的对数似然值,这表明非常嘈杂的样品和不良拟合。去除所有对数似然值<0的样品,导致与B细胞Danaher评分的相关性提高(GAM:ρ=0.41,P=4.3e-06;区段模型:ρ=0.6,P=5.1e-13)。总的来说,这些结果表明,与来自GAM模型的相应值相比,区段模型产生与Danaher评分具有更强的相关性的评分。 比较来自TCRA、TCRB和TCRG的不同T细胞分数,TCRB T细胞分数与TCRA T细胞分数高度相关,但在GAM模型中大约是其值的一半(ρ=0.69,P<2.2e-16,y=0.00059+0.49x,图32E),并在segment模型中略高于一半(ρ=0.91,P<2.2e-16,y=0.033+0.57x,图32F)。这大概是由于其中经常只有一个TCRB等位基因经历V(D)J重组的等位基因排斥。相比之下,TCRGT细胞分数出乎意料地高于1%至5%的CD3+T细胞,所述CD3+T细胞通常被表征为γδT细胞,并且还被发现与TCRA T细胞分数高度相关(GAM:ρ=0.71,P<2.2e-16,y=-0.028+0.71,图32E和区段模型:ρ=0.94,P<2.2e-16,y=0.005+x)。尽管与GAM模型中的TCRA相比,TCRG分数接近0的样品数量明显更多(32.8%的TCRG T细胞分数<1e-5相对于1.7% TCRA T细胞分数<1e-5),但如果该信号完全是由γδT细胞引起的,则大多数样品中的高分数和区段模型中两者之间近乎精确的相关性将被广泛认为是生物学上不可信的。相反,已知αβT细胞在分化为它们的最终谱系之前首先经历TCRG基因座的重排,来自区段模型的结果表明,具有重排的TCRG基因座是αβT细胞的一个几乎普遍的特征,并且TCRG基因座可以提供T细胞分数的精确测量。 TCRD基因座完全位于TCRA基因座内,并且由于大量的噪声,尺寸太小以致于不能使用具有可用数据的GAM模型来精确地产生γδT细胞分数的评分。然而,使用区段模型,可以计算具有来自TCRD基因区段的区段的T细胞的分数,从而计算起源于γδT细胞的T细胞分数。在TRACERx100组群中,发现TRDV1的基于区段的评分与以下的TPM RNAseq值显著相关:TCRD区段TRDV1(ρ=0.25,P=0.0011,图32G)、TRDJ1+TRDJ2+TRDJ3+TRDJ4(ρ=0.15,P=0.047,图32G)、TRDC(ρ=0.23,P=0.0021,图32G)。 深度要求的研究 选择代表来自TCRA、TCRB、TCRG或IGH基因座的一系列T和B细胞分数的TRACERx100样品的子集(n=19)用于降采样分析,以确定GAM或区段模型能够概括全部深度评分的最低深度。使用samtools对每个样品进行随机降采样四次,深度分别为60X、30X、10X、5X、2X、1X、0.5X或0.1X。图33给出了该分析的结果,并示出了所有情况下对于GAM和区段模型二者在2X下对全深度值的高保真度(TCRA GAM:ρ=0.83,P<2.2e-16,TCRA seg:ρ=0.75,P=7.5e-15,TCRB GAM:ρ=0.46,P=2.7e-05,TCRB seg:ρ=0.53,P=8.4e-07,TCRG GAM:ρ=0.5,P=4.2e-06,TCRG seg:ρ=0.61P=5e-09,T细胞平均GAM:ρ=0.74,P=1.9e-14,T细胞平均seg:ρ=0.76P=1.4e-15,IGH GAM:ρ=0.37,P=0.0011,IGH seg:ρ=0.56,P=1.3e-07,图33A至33J),尽管在这种低深度下,在许多情况下,这些值变为紧缩的(例如,当与全深度值相比时,1X下的T细胞平均区段模型具有rho=0.59,但是点位于线y=-0.028+1.6x上)。由于在这些低深度下降采样文件中观察到的评分的高保真度,很可能通过计算信息仓中的覆盖(例如如约100bp至约10,000bp,特别是100bp至1000bp的值的信息仓可能特别有用-可以使用如上所述的基准方法选择合适的值),T细胞ExTRECT方法可以被设计为用GAM或区段模型在低通WGS样品(例如如0.1x及以上、0.1x至0.5x、0.1x至2x)上非常好地工作。 该ExTRECT方法可用于研究免疫细胞库多样性 研究了使用上述方法来深入了解更一般的TCR和BCR多样性。这是通过检查由区段模型的最佳拟合预测的V和J区段使用在样品内完成的。图34A至34C示出了TCRA、TCRB和TCRG的TRACERx患者CRUK0085在不同肿瘤区域和种系血液样品中使用的V区段的比例,并且图34D示出了IGH在TRACERx患者CRUK0045中的V区段使用。这些可以基于最大重组的位置,使用如上所阐述的f的相同方程式,而不是使用相关区段的模型评估作为r 泛癌组群中癌症免疫环境与生存率、临床决定因素和癌症类型之间的相关性 将T细胞ExTRECT区段模型应用于大的泛癌WGS组群,所述大的泛癌WGS组群由20种不同癌症类型的>15000名参与者组成。该分析提供了来自TCRA、TCRB和TCRG的T细胞分数,和来自IGH的B细胞分数,以及单个V和J基因区段的分数。这些参与者中的大多数具有匹配的血液种系WGS样品,其中剩余的大多数血液癌症患者具有来自唾液或其他正常组织的匹配种系样品。这使得在大的泛癌组群中首次有机会来研究取样时患者血液中免疫分数的差异。 该分析揭示了在血液和肿瘤样品中在癌症类型内和癌症类型之间免疫景观范围的广泛多样性(TCRAT细胞分数,TCR多样性,如由分数超过0.01的模型调用的TRAV区段的数量所测量的)。对于区段模型中的调用的TRAV区段数和TCRA T细胞分数二者,也可以拟合线性模型来确定这些值对性别、年龄和血统的依赖性。这些模型可以适用于血液TCRA T细胞分数和预测的TRAV区段数,以研究例如年龄与泛癌组群中T细胞分数和多样性的降低相关,以及这是否在不同的癌症亚型中观察到不同的程度。 还可以对来自T细胞ExTRECT的IGH B细胞分数评分进行这样的分析,研究不同癌症类型的血液和肿瘤中的IGH B细胞分数,以及IGH B细胞分数和多样性的临床决定因素(例如性别、年龄、血统)。 为了确定种系和血统对确定血液中T细胞分数的贡献,可以使用PLINK和癌症或泛癌组群中的一组不相关患者进行GWAS分析(参见方法)。为了提高效力,可以分别对血液TCRA、TCRB和TCRG T细胞分数值运行GWAS。这些值是从基因组的不同区域独立计算的,并且通过查看所有三个区域中高于建议显著性水平(P<1e-5)的命中,我们可以确信它们不是由与映射质量和覆盖值(而不是真实的T细胞分数信号)降低相关的V(D)J基因内的SNP引起的数据中的噪声或伪影的结果。然后,对于在这些GWAS的一个或所有三个中鉴定的任何SNP,可以看到这些SNP在不同癌症类型中的富集。 最后,可以分析存活数据,例如如使用来自医院事件统计中最近记录的癌症诊断日期、死亡记录和最近随访时间的数据。然后可以获得例如血液和肿瘤样品中TCRA T细胞和IGH B细胞分数的Kaplan Meier曲线,在每种情况下按照中位数值将组群分成高分数或低分数。该分析可用于研究血液/肿瘤中的TCRA T细胞分数或血液/肿瘤中的IGH B细胞分数是否与存活显著相关。 最后,为了更好地理解免疫细胞分数和其他临床特征之间的关系,以及其在不同癌症类型中的异质性,可以拟合由血液和肿瘤中的TCRA T细胞分数、血液和肿瘤中的IGH B细胞分数以及作为血液和肿瘤中γδT细胞替代物的TRDV1 T细胞分数构成的Cox比例模型。Cox模型还可以包括年龄和性别,以控制与T或B细胞分数的可能混杂相关性,以及患者在手术前是否接受了化学治疗。这最终因素可以作为癌症在临床上的侵袭性或晚期程度如何的替代因素。可以使用替代指标例如癌症分期。这种分析的结果可以通过查看在任何特定组群上运行的Cox模型的z评分的热图来检查。这可以揭示血液/肿瘤中的TCRA T细胞分数或血液/肿瘤中的IGHB细胞分数是否是泛癌组群以及任何个体癌症类型中的显著风险。 方法 计算来自WGS的T细胞分数的整个过程严格遵循实施例1中的方法。这包括使用先前所限定的TCRA基因座在起始和末端处的区域来归一化覆盖并计算单一样品读取深度比,以及使用方程式(3)或(7)中的式用于T细胞分数,对于Illumina HiSeq使用γ=1。唯一的区别在于计算r 这与实施例1中描述的方法相同,除了局部GC含量是在1000bp窗口的水平(GC 作为最后一步,稳健的质量控制过程鉴定了任何V(D)基因内的任何100bp区段,这些区段就以下的覆盖而言是异常值:1)在整个组群中,或者2)与组群的子集相关并与已知种系基因组变异体相连。对于第一步,计算所有GEL肺组群集的所有V(D)J基因的平均GC校正比。这显示了与区段周围区域相比覆盖极低或者覆盖极高的区段。通过将GAM模型拟合至平均GC校正比,可以从拟合线(+/-0.25)中排除高于和低于特定阈值的那些。这对TCRA来说很有效,但对其他基因例如TCRB来说,大区段聚集在一起干扰了GAM模型的拟合。对于这些,一些明显的异常区域最初被手动去除,例如TCRB的平均GC比高于0.25的所有区域。在去除以这种方式鉴定的区段后,在使用PLINK软件进行GWAS分析后,鉴定出另外的有偏差的区段(参见Renteria et al.,2013)。特别是,进行了GWAS分析以鉴定任何与T或B细胞分数相关的SNP。鉴定并标记在某些外显子中的覆盖中有偏差的基因区域以便去除。重新计算评分并重复GWAS过程以检查偏差的去除。被去除的区段相对于与特定种系基因型相关的周围区域,覆盖不足或覆盖过度,并导致基因内的某些变体纯粹由于这种伪影而与T或B细胞分数密切相关。在这些情况下,选择具有相关基因型的特定样品,并计算平均GC校正比。然后,通过如上所述的相同程序来鉴定和标记另外的异常区段以便移除,并且重新运行PLINK/GWAS分析以确保没有进一步的伪影区段。 使用上述方法将该方法扩展至经历V(D)J重组的其他T细胞受体基因、TCRB和TCRG以及B细胞受体IGH,但是重新定义了用于读取深度比归一化的位置以及预期最大偏差的位置。一般而言,预计最大偏差位于最终V段与第一个J段之间,并相应地选择区域并在表7中给出。 表7.用于单一样品归一化和r 类似地,该方法还应用于研究区段使用,使用上述方法,但对于r 表8.用于r /> /> /> /> 新的T细胞ExTRECT V(D)J区段模型的创建 实施例1中描述的方法使用GAM模型来计算r 为了适合该模型,我们将每个可能的区段(具有对应于V和J基因的已知断点,参见表8)转换成1s和0s的向量(在它们的区域内等于1,在区域外等于0)。然后,我们使用约束线性模型(使用来自R包restriktor v0.3中的函数)将这些向量拟合至归一化的读取率数据,其中对于n个V段和m个J区段中的每一个,如下选择不等式约束:V 用PLINK的GWAS分析 在一群不相关的参与者(这些参与者具有来自血液的WGS种系样品)中运行PLINK,并且使用前20个遗传PC、性别、年龄和疾病类型的协变量进行控制。PLINK的输入是已经预处理的变体,使得它们在整个组群中具有>0.001的MAF,通过包括缺失和足够深度的QC过滤器,并且经历了变体归一化,例如使得所有多等位基因变体都是双等位基因,以及确保所有变体都是左对齐的和简约的。除了这些预设的过滤器和在运行PLINK之前的QC之外,我们在500kb窗口中进行了LD修剪,其中R2阈值为0.2,以及确保我们组群中所有变体具有>0.001的MAF,并且基因型缺失不超过0.2。最后,在运行PLINK之前,用0.000001的阈值进行Hardy-Weinberg测试。 实施例7–讨论 免疫系统被广泛认为是影响肿瘤发生和随后癌症演变二者的关键因素之一。T细胞通过清除含有新抗原的癌细胞而在癌症免疫微环境中发挥重要作用,因此肿瘤样品中T细胞的数量是可决定疾病进程的重要临床因素。在癌症的背景下,肿瘤组织的大量DNA测序主要用于表征驱动肿瘤发生的体细胞变化。然而,通过本文中给出的方法,我们证明了DNA测序也可以用于研究样品的免疫微环境。 先前曾在新生儿严重联合免疫缺陷(severe combined immune deficiency,SCID)筛查的背景下对TREC进行过临床检查(van der Spek,Groenwold,van der Burg,&van Montfrans,2015),其中它们的缺乏被用于推断T细胞淋巴细胞减少症。然而,这些筛查方法是基于TREC本身的测序和定量,而不是基于WES数据。最近的腺癌单细胞基因组测序研究鉴定了TCRA基因内T细胞中的缺失事件(Baslan et al.,2020)。在此,本发明人在这项工作的基础上明确使用TREC来评估WES样品中的T细胞分数,如实施例1中所详细描述的。 本文中所描述的方法(TILExTRECT)提供了免疫浸润的准确评估,并且其评估显示了临床实用性。发明人发现TCRA T细胞分数评估与正交免疫测量密切相关,并且癌细胞系和模拟WES数据证实了它们的可靠性(实施例2)。此外,本发明人证明了推断的TCRA T细胞分数在LUAD中是预后性的,并在TCGA LUAD组群中验证了这一发现(实施例3)。相关地,本发明人表明,在泛癌组群中,TCRA T细胞分数与对CPI的响应相关,并且提高了单独的克隆TMB的预测值(实施例4)。本发明人进一步证明,血液和肿瘤样品中的TCRA T细胞分数可预测泛癌组群以及许多特定癌症类型中的存活,IGH B细胞分数也是如此(实施例6)。发现基于TRDV1 T细胞分数评估的γδT细胞的分数可预测特定癌症类型的存活。 另外,TIL ExTRECT使T细胞(或B细胞)分数能够在先前不可能的数据集中计算出来。利用这一点,本发明人证明了T细胞分数在血液中是异质的,并且在女性中显著高于男性,这与当前的发现一致,并且也如所预期的与微生物感染相关(实施例5)。除了概述已知的免疫相关性,我们还表明T细胞ExTRECT可用于没有先前免疫注释的数据集。在缺乏RNA-seq的泛癌多样品组群中,我们观察到不同癌症类型中T细胞浸润的异质性程度的显著变化(实施例5)。这种异质性中大部分看起来是由亚克隆SCNA驱动的,并且他们确定了12q24.31-32的亚克隆丢失与T细胞浸润的显著减少相关。这可能与SPPL3的表达减少有关,导致细胞表面鞘糖脂上调,从而阻碍I类HLA分子的功能。这种丢失可能是多种癌症类型中的免疫逃逸机制,通常是亚克隆发生,因此处于肿瘤的进化轨迹的晚期。 在实施例2中,本发明人已经证明了TCRA T细胞分数与来自不同平台和多个不同数据集中的正交免疫测量密切相关。癌细胞系和模拟的WES数据证实了该评分为测序样品中T细胞的分数提供了可靠的评估。高深度TRACERx100样品的模拟和降采样表明,30X覆盖提供了足够的信号来计算可靠的T细胞分数(尽管较低的覆盖可用于区分高和低的T细胞含量,这可用作更原始的生物标志物)。这种相对低的覆盖意味着其应该适用于大多数DNA测序数据集。在实施例6中,发明人证明了该方法可以应用于WGS数据,其中可以在TCRA区域之外并且甚至在更低的深度处获得具有比WES更高分辨率的信号。他们使用正交RNA-seq数据表明了评分准确地描述了免疫微环境,并使用降采样表明了该方法保持精确低至2X的深度。在实施例6中,他们进一步证明了该方法研究以下二者的潜力:γδT细胞分数以及TCR和BCR多样性二者。除了WES和WGS,任何靶向TCRA基因(或研究中的进行V(D)J重组的任何基因)的NGS方法都可能与这种方法兼容。不希望被理论所束缚,认为测序数据优于例如SNP阵列数据,因为后者通常在目标VDJ基因座的关键区域中具有明显较低的分辨率。另外,可以使用靶向的基于组的测序方法,该方法将对TCRA基因(或研究中的进行V(D)J重组的任何基因)进行特异性靶向并提供TCRA T细胞分数(或TCRD、TCRG、TCRB、IGH、IGK或IGL细胞分数)。事实上,很容易设想将这种方法与目前评估TMB的基于组的测序方法组合,以便促进其作为诊断工具的用途。测序平台特定常数(方程式(2)、(3)和(7)中的γ-其中值为1适合于WES或WGS数据)可以根据使用的测序平台进行调整。此外,可以评估和考虑平台特定偏差,例如GC含量偏差。 由于这些原因,TIL ExTRECT在重新分析先前发表的原本缺乏对T细胞含量的无偏评估的NGS癌症数据集的背景下具有潜在的价值。TCRA T细胞分数也具有在临床上作为简单的生物标志物的潜在价值,因为其与对免疫治疗的响应相关,如实施例4所示,并且其与存活相关,如实施例6所示。 虽然存在提供来自样品的免疫相关数据的其他方法,但这些方法通常是不可用的或者在逻辑上复杂的,例如成像切片需要在手动或数字评价之前取回、切割、染色FFPE块。同时,T细胞ExTRECT可以与通常用于研究和提高临床的DNA测序平台组合使用。 虽然这种方法是在癌症研究框架内开发的,但值得考虑将其应用于更广泛的临床环境。这种测量可以从任何NGS分析中计算,从健康或疾病的任何组织或血液中提取。对癌症组群中匹配血液样品的分析证明了血液TCRA T细胞分数与患者的癌症类型以及性别之间的显著关系,这显示了这种方法的潜力(参见实施例3以及实施例6)。此外,仅分析肺CPI组群中的血液样品发现,推断的TCRA T细胞分数可预测对免疫治疗的反应,而无需对肿瘤样品本身进行任何分析(图24B)。此外,血液样品中TREC水平的非NGS聚合酶链式反应(polymerase chain reaction,PCR)定量已经用于在乳腺癌(Page et al.)和前列腺癌(Page et al.)的免疫治疗试验中监测T细胞的胸腺生成。另外,T细胞ExTRECT可具有在肿瘤学之外的应用。血液TCRA T细胞分数的一个未来临床应用可以是在SCID(通常导致严重的T细胞耗竭的一组罕见的先天性综合征,其的治疗需要快速造血干细胞移植)的情况下,其中TIL ExTRECT可以与新生儿的基因组筛查组合,以同时鉴定潜在的破坏性种系突变和降低的T细胞含量。遗憾的是,在撰写时,SCID虽然影响到50,000例活产儿中的1例,但在大多数国家并不是常规新生儿筛查的一部分,并且大多数病例仅在严重的机会性感染之后才被诊断出来,而且往往为时已晚而无法进行治疗(Chan etal.)。TILExTRECT可通过将TCRA包含在新生儿遗传病筛查基因组中来鉴定SCID。 由于T细胞和VDJ重组是有颌脊椎动物适应性免疫系统的决定性特征,本文中提出的一般性方法是物种不可知的。虽然上述实施例中描述的TIL ExTRECT方法已经针对人基因组进行了优化,但是该方法可以扩展至其他物种(主要是通过限定要使用的相应的特定基因组区域并评估和解决任何区域特异性偏差),包括大量研究的模式生物(包括例如小鼠、鸡、雪貂等)。还值得注意的是,VDJ重组虽然支持这种新的TCRA T细胞分数评估的方法,但并不是编码TCR-α受体的基因所独有的。其他TCR基因,β、γ和δ,也经历VDJ重组,BCR免疫球蛋白基因IGH、IGL和IGK也是如此。因此,该方法也可用于计算具有IGL或IGK轻链的α-β、γ-δT细胞以及B细胞的分数,如实施例6所示。这些可以使用合适的染色体区域,例如如 (hg19):TCRB chr7:141998851-142510972;TCRG chr7:38279625-38407656;IGHchr14:106032614-107288051;IGL chr22:22380474-23265085;IGK chr2:89156674-90274235 或表7和表8中的区域;并评估和解决任何区域特异性偏差。根据数据中的噪声和样品中目标细胞的分数,这些中的一些可能表现不佳,因为这些其他免疫细胞类型通常不太普遍,但是这种扩展的方法具有仅利用DNA测序数据详细说明癌症样品的免疫微环境的潜力(如实施例6所示)。 总之,本文中所述的方法,TIL ExTRECT,可通过提供经济有效的技术在不需要RNA测序的情况下来表征人和模型系统数据集二者中的免疫浸润和体细胞变化,而在基础和转化研究二者中具有重要的应用。 参考文献 AbdulJabbar et al.(2020).Geospatial immune variability illuminatesdifferential evolution of lung adenocarcinoma.Nature Medicine,26(7),1054-1062. Aran,D.,Hu,Z.,&Butte,A.J.(2017).xCell:Digitally portraying the tissuecellular heterogeneity landscape.Genome BiologY,18(1),1-14. Aversa I,Malanga D,Fiume G,Palmieri C.Molecular T-Cell RepertoireAnalysis as Source of Proqnostic and Predictive Biomarkers for CheckpointBlockade Immunotherapy.Int J Mol Sci.2020Mar30;21(7):2378. Baslan et al.(2020).Novel insights into breast cancer copy numbergenetic heterogeneity revealed by single-cell genome sequencing.ELife,9,1-21. Bolotin et al.(2012).Next generation sequencing for TCR repertoireprofiling:Platform-specific features and correction algorithms.EuropeanJournal of Immunology,42(11),3073-3083. Bolotin,D.A.et al.MiXCR:software for comprehensive adaptive immunityprofiling.Nat.Methods 12,380-381(2015). Brastianos,P.K.et al.Genomic characterization of brain metastasesreveals branched evolution and potential therapeutic targets.Cancer Discov.5,1164-1177(2015). Cancer Genome Atlas Research Network.(2012).Comprehensive genomiccharacterization of squamous cell lung cancers.Nature,489(7417),519-525. Cancer Genome Atlas Research Network.(2014).Comprehensive molecularprofiling of lung adenocarcinoma.Nature,511(7511),543-550. Carter et al.(2012).Absolute quantification of somatic DNAalterations in human cancer.Nature Biotechnology,30(5),413-421. Chan,K.&Puck,J.M.Development of population-based newborn screeningfor severe combined immunodeficiency.J.Allergy Clin.Immunol.115,391-398(2005). Cristescu et al.(2018).Pan-tumor genomic biomarkers for PD-1checkpoint blockade-based immunotherapy.Science,362(6411). Danaher et al.(2018).Pan-cancer adaptive immune resistance as definedby the TumorInflammation Signature(TIS):Results from The Cancer Genome Atlas(TCGA).Journal for ImmunoTherapy of Cancer,6(1),1-17. Davoli,T.,Uno,H.,Wooten,E.C.,&Elledge,S.J.(2017).Tumor aneuploidycorrelates with markers of immune evasion and with reduced response toimmunotherapy.Science,355(6322). Denkert et al.(2016).Standardized evaluation of tumor-infiltratinglymphocytes in breast cancer:Results of the ring studies of the internationalimmuno-oncology biomarker working group.Modern Pathology,29(10),1155-1164. Dieci et al.(2018)Update on tumor-infiltrating lymphocytes(TILs)inbreast cancer,including recommendations to assess TILs in residual diseaseafter neoadj uvant therapy and in carcinoma in situ:A report of theInternational Immuno-Oncology Biomarker Working Group on BreastCancer.Seminars in Cancer Biology,Volume 52,Part 2,2018,Pages16-25. Favero et al.(2015).Sequenza:Allele-specific copy number and mutationprofiles from tumor sequencing data.Annals of Oncology,26(1),64-70. Galon et al.(2006).Type,density,and location of immune cells withinhuman colorectal tumors predict clinical outcome.Science,313(5795),1960-1964. Gerlinger,M.et al.Intratumor Heterogeneity and Branched EvolutionRevealed by Multiregion Sequencing.N.Engl.J.Med.366,883-892(2012). Gerlinger,M.et al.Genomic architecture and evolution of clear cellrenal cell carcinomas defined by multiregion sequencing.Nat.Genet.46,225-233(2014). Ghandi et al.(2019).Next-generation characterization of the CancerCell Line Encyclopedia.Nature,569(7757),503-508. Goodman et al.(2017).Tumor mutational burden as an independentpredictor of response to immunotherapy in diverse cancers.Molecular CancerTherapeutics,16(11),2598-2608. Harbst,K.et al.Multiregion whole-exome sequencing uncovers thegenetic evolution and mutational heterogeneity of early-stage metastaticmelanoma.CancerRes.76,4765-4774(2016). Haywood et al.Sunitinib′s effect on tumor infiltration of CD8 T cellsin renal cell carcinoma(RCC)and modulation of their function by alteringVEGF-induced upregulation of PD1 expression.Journal of Clinical Oncology,34(2),591(2016). Hendry,S.,Salgado,R.,Gevaert,T.,Russell,P.A.,John,T.,Thapa,B.,...Fox,S.B.(2017).Assessing rumor-infiltrating Lymphocytes in Solid Tumors.AdvancesIn Anatomic Pathology(Vol.24). Hopkins et al.T cell receptor repertoire features associated withsurvival in immunotherapy-treated pancreatic ductal adenocarcinoma.JCIInsight.2018 Jul 12;3(13):e122092. Huang,W.,Li,L.,Myers,J.R.,&Marth,G.T.(2012).ART:A next-generationsequencing read simulator.Bioinformatics,28(4),593-594.Hugo et al.(2016).Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy inMetastatic Melanoma.Cell,165(1),35-44.Iglesia et al.Prognostic B-cellsignatures using mRNA-seq in patients with subtype-specific breast andovarian cancer.Clin Cancer Res.2014Jul 15;20(14):3818-29. Jamal-Hanjani et al.(2017).Tracking the Evolution of Non-Small-CellLung cancer.New Engiand Journal of Medicine,376(22),2109-2121.Jongsma etal.The SPPL3-Defined Glycosphingolipid Repertoire Orchestrates HLA Class I-Mediated Immune Responses.Immunity.2021Jan 12;54(1):132-150.e9.doi:10.1016/j.immuni.2020.11.003.Epub 2020Dec 2.Erratum in:Immunity.2021Feb 9;54(2):387.PMID:33271119.Lamy,P.et al.Paired exome analysis reveals clonal evolutionand potential therapeutic targets in urothelial carcinoma.Cancer Res.76,5894-5906(2016). Le,D.T.,Uram,J.N.,Wang,H.,Bartlett,B.R.,Kemberling,H.,Eyring,A.D.,...Diaz,L.A.(2015).PD-1 Blockade in Tumors with Mismatch-RepairDeficiency.New England Journal of Medicine,372(26),2509-2520. Levy,E.,Marty,R.,Gárate Calderón,V.et al.Immune DNA signature of T-cell infiltration in breast tumor exomes.Sci Rep 6,30064(2016). Li et al.(2017).TIMER:A web server for comprehensive analysis oftumor-infiltrating immune cells.Cancer Research,77(21),e108-e110.Litchfieldet al.Meta-analysis of tumor and T cell-intrinsic mechanisms of sensitizationto checkpoint inhibition.Cell 184.3(2021):596-614. López et al.(2020).Interplay between whole-genome doubling and theaccumulation of deleterious alterations in cancer evolution.Nature Genetics,52(3),283-293. Madan,R.A.et al.Clinical and immunologic impact of short-courseenzalutamide alone and with immunotherapy in non-metastatic castrationsensitive prostate cancer.J.Immunother.Cancer 9,(2021). Mariathasan et al.(2018).TGFβ attenuates tumour response to PD-L1blockade by contributing to exclusion of T cells.Nature,554(7693),544-548. Márquez et al.(2020).Sexual-dimorphismin human immune systemaging.Nature Communications,11(1). McDermott et al.(2018).Clinical activity and molecular correlates ofresponse to atezolizumab alone or in combination with bevacizumab versussunitinib in renal cell carcinoma.Nature Medicine,24(6),749-757. McGrath et al.Detecting T cell receptor rearrangements in silico fromnon-targeted DNA-sequencing(WGS/WES).bioRxiv preprint doi.org/10.1101/201947;October 13,2017. Messaoudene,M.et al.T-cell bispecific antibodies in node-positivebreast cancer:Novel therapeutic avenue for MHC class i lossvariants.Ann.Oncol.30,934-944(2019). Middleton et al.(2020).The National Lung Matrix Trial of personalizedtherapy in lung cancer.Nature,583(7818),807-812.Newman et al.(2015).Robustenumeration of cell subsets from tissue expression profiles.Nature Methods,12(5),453-457. Newman et al.(2019).Determining cell type abundance and expressionfrom bulk tissues with digital cytometry.Nature Biotechnology,37(7),773-782. Page,D.B.et al.A phase II study of dual immune checkpoint blockade(ICB)plus androgen receptor(AR)blockade to enhance thymic T-cell productionand immunotherapy response in metastatic breast cancer(MBC).J.Clin.Oncol.37,TPS1106-TPS1106(2019). Poore GD,Kopylova E,Zhu Q,et al.Microbiome analyses of blood andtissues suggest cancer diagnostic approach.Nature.2020;579(7800):567-574. Racle et al.(2017).Simultaneous enumeration of cancer and immune celltypes from bulk tumor gene expression data.ELife,6,1-25.Riaz et al.(2017).Tumor and Microenvironment Evolution during Immunotherapy withNivolumab.Cell,171(4),934-949.e15. Rentería ME,Cortes A,Medland SE.Using PLINK for Genome-WideAssociation Studies(GWAS)and data analysis.Methods Mol Biol.2013;1019:193-213. Riester,M.,Singh,A.P.,Brannon,A.R.et al.PureCN:copy number callingand SNV classification using targeted short read sequencing.Source Code BiolMed 11,13(2016). Rizvi et al.(2015).Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer.Science,348(6230),124-128. Robert et al.(2011).Ipilimumab plus Dacarbazine for PreviouslyUntreated Metastatic Melanoma.New England Journal of Medicine,364(26),2517-2526. Rosenthal et al.(2019).Neoantigen-directed immune escape in lungcancer evolution.Nature,567(7749),479-485. Samstein et al.(2019).Tumor mutational load predicts survival afterimmunotherapy across multiple cancer types.Nature Genetics,51(2),202-206. Savas,P.et al.The Subclonal Architecture of Metastatic Breast Cancer:Results from a Prospective Community-Based Rapid Autopsy Program″CASCADE″.PILoS Med.13,1-25(2016). Schadendorf et al.(2015).Pooled analysis of long-term survival datafrom phase II and phase III trials of ipilimumab in unresectable ormetastatic melanoma.Journal of Clinical Oncology,33(17),1889-1894. Shen,R.,&Seshan,V.(2015).FACETS:Fraction and Allele-Specific CopyNumber Estimates from Tumor Sequencing.Memorial Sloan-Kettering CancerCenter,Dept.of Epidemiology&Biostatistics Working Paper Series.,1,50.Retrieved from http://biostats.bepress.com/mskccbiostat/paper29 Shim et al.(2020).HLA-corrected tumor mutation burden and homologousrecombination deficiency for the prediction of response to PD-(L)1blockade inadvanced non-small-cell lung cancer patients.Annals of Oncology,31(7),902-911. Snyder et al.(2014).Genetic Basis for Clinical Response to CTLA-4Blockade in Melanoma.New England Journal of Medicine,371(23),2189-2199. Snyder et al.(2017).Contribution of systemic and somatic factors toclinical response and resistance to PD-L1 blockade in urothelial cancer:Anexploratory multi-omic analysis.PLoS Medicine,14(5),1-24. Suzuki,H.et al.Mutational landscape and clonal architecture in gradeII and III gliomas.Nat.Genet.47,458-468(2015). Thorsson et al.(2018).The Immune Landscape of Cancer.Immunity,48(4),812-830.e14. Topalian et al.(2012).Safety,Activity,and Immune Correlates of Anti-PD-1 Antibody in Cancer.New England Journal of Medicine,366(26),2443-2454. Turajlic,s.et al.Deterministic Evolutionary Trajectories InfluencePrimary Tumor Growth:TRACERx Renal.Cell 173,595-610.ell(2018). Valpione,S.,Mundra,P.A.,Galvani,E.et al.The T cell receptorrepertoire of tumor infiltrating T cells is predictive and prognostic forcancer survival.Nat Commun12,4098(2021). Van Allen et al.(2016).Erratum for the report″genomic correlates ofresponse to CTLA-4 blockade in metastatic melanoma.″Science,352(6283),207-212. van der Spek,J.,Groenwold,R.H.H.,van der Burg,M.,&van Montfrans,J.M.(2015).TREC Based Newborn Screening for Severe Combined ImmunodeficiencyDisease:A Systematic Review.Journal of Clinical Immunology,35(4),416-430. Van Loo et al.(2010).Allele-specific copy number analysis oftumors.Proceedings of the National Academy of Sciences of the Uhited Statesof America,107(39),16910-16915. Wang et al.(2007).PennCNV:An integrated hidden Markov model designedfor high-resolution copy number variation detection in whole-genome sNPgenotyping data.Genome Research,17(11),1665-1674. Wang,V.G.,Kim,H.,&Chuang,J.H.(2018).Whole-exome sequencing capturekit biases yield false negative mutation calls in TCGA cohorts.PLoS ONE,13(10),1-14. Watkins,T.B.K.et al.Pervasive chromosomal instability and karyotypeorder in tumour evolution.Nature 587,126-132(2020). Yokoyana,A.,Kakiuchi,N.,Yoshizato,T.et al.Age-related remodelling ofoesophageal epithelia by mutated cancer drivers.Nature565,312-317(2019). Yoshihara et al.(2013).Inferring tumour purity and stromal and immunecell admixture from expression data.Nature Communications,4.Zaccaria,s.,&Raphael,B.J.(2020).Accurate quantification of copy-number aberrations andwhole-genome duplications in multi-sample tumor sequencing data.NatureCommunications,11(1). Zhang et al.(2003).Intratumoral T Cells,Recurrence,and Survival inEpithelial ovarian Cancer.New England Journal of Medicine,348(3),203-213. Wood,D.E.&Salzberg,S.L.Kraken:Ultrafast metagenomic sequenceclassification using exact alignments.Genome Biol.15,(2014). 本文中引用的所有参考文献均通过引用整体并入本文并且用于所有目的,其程度如同每个单独的出版物或专利或专利申请被具体地和单独地指示为通过引用整体并入。 本申请要求2021年7月22日提交的GB申请号2110555.6和2022年3月11日提交的GB申请2203451.6的优先权。出于所有目的,这两个优先权申请的全部内容通过引用在此并入。 本文中描述的具体实施方案是通过示例而非通过限制来提供的。在不脱离所述技术的范围和精神的情况下,所描述的技术的组合物、方法和用途的各种修改和变化对于本领域技术人员将是显而易见的。本文中包括的任何小标题仅是为了方便起见,而不应被解释为以任何方式限制本公开内容。 本文中描述的任何实施方案的方法均可作为计算机程序或作为计算机程序产品或携带计算机程序的计算机可读介质来提供,所述计算机程序在计算机上运行时被安排以执行上述方法。 除非上下文中另外指示,否则对上述特征的描述和限定不限于本发明的任何特定方面或实施方案,并且同等地适用于所描述的所有方面和实施方案。 在整个说明书和权利要求书中,除非上下文另外明确指出,否则以下术语采用与本文明确相关的含义。如本文所用的短语“在一个实施方案中”不一定是指相同的实施方案,但是其可以指相同的实施方案。此外,如本文所用的短语“在另一实施方案中”不一定是指不同的实施方案,但是其可以指不同的实施方案。因此,如下所述,在不脱离本发明的范围或精神的情况下,可以容易地组合本发明的各种实施方案。 必须注意的是,除非上下文另外明确指出,否则说明书和所附权利要求书中使用的没有数量词修饰的名词表示一个/种或更多个/种。范围可在本文中表示为从“约”一个特定值和/或至“约”另一特定值。当表示这样的范围时,另一个实施方案包括从一个特定值和/或至另一个特定值。类似地,当通过使用先行词“约”将值表示为近似值时,将理解的是,特定值形成另一个实施方案。与数值相关的术语“约”是任选的,并且意指例如+/-10%。 在整个本说明书中(包括所附的权利要求书),除非上下文另有要求,否则词语“包含”和“包括”以及变化形式将被理解为暗示包含所指出的整体或步骤或者整体或步骤的组,但是不排除任何其他整体或步骤或者整体或步骤的组。 除非上下文另外说明,否则本发明的另一些方面和实施方案提供了由术语“由......组成”或“基本上由......组成”替换术语“包含/包括/含有”的上述方面和实施方案。 在前述说明书中、或在所附权利要求书中、或在附图中公开的以它们的特定形式或者在用于进行所公开的功能的方式、或者用于获得所公开的结果的方法或过程方面表示的特征(视情况而定)可单独地或以这样的特征的任何组合用于以其多种形式实现本发明。
- 一种确定样品中靶分子丰度的方法
- 一种确定样品中靶分子丰度的方法