一种基于云计算技术的分子动力学计算方法及其系统
文献发布时间:2023-06-19 10:11:51
技术领域
本发明涉及分子动力学计算技术,尤其涉及一种基于云计算技术的分子动力学计算方法及其系统。
背景技术
分子动力学是一种从原子、分子尺度研究材料性质的科学。在分子动力学的计算中,利用原子间的相互作用势以及每个原子的坐标,通过求解经典力学方程可以求出作用在每一个原子上的合力,计算每个原子的速度、位置,以及随时间的演化、模拟原子运动的轨迹,最终根据统计力学方法,得出体系的物化性质(热力学、动力学)。分子动力学的计算精度不如第一性原理计算方法,但由于牛顿(Newton)方程的数值求解相对容易,计算速度和计算规模远远大于第一性原理计算,因此分子动力学适合于较大空间尺度和较长时间尺度的分子动力学研究。
现有的大规模原子分子并行模拟器(Large-scale Atomic/Molecular MassivelyParallel Simulator,LAMMPS)是在材料计算领域广泛应用的一种分子动力学模拟软件,因其功能强大,支持包括气态、液态或固态相形态下、各种系统下、百万级的原子分子体系,并提供支持多种势函数,且具备良好的并行扩展性等原因,计算模拟软件LAMMPS拥有广大的使用者和支持者。但是,由于LAMMPS是一款非图形化界面的软件,需要安装该软件、经编译过程处理才能进行材料计算应用,并且参数设置较为复杂,对于使用者而言有较高的技术门槛要求。因此,限制了该LAMMPS软件的进一步应用。
发明内容
有鉴于此,本发明的主要目的在于提供一种基于云计算技术的分子动力学计算方法及其系统,无需安装和编译分子动力学模拟软件(如LAMMPS),通过利用云计算技术和力场参数的自动匹配技术,以降低使用者对开展分子动力学模拟软件的计算技术难度门槛要求,并简化其使用过程。
为达到上述目的,本发明的技术方案如下:
一种基于云计算技术的分子动力学计算系统,包括预处理子系统、交互与控制子系统、作业管控子系统和结果处理子系统;其中:
预处理子系统,用于定义和封装分子动力学模拟软件的核心组件模块,对所述分子动力学模拟软件的参数进行分类,并进行力场参数的自动匹配;
交互与控制子系统,利用工作流设计交互模块和结果交互处理模块,分别对分子动力学流程进行交互设计和结果交互处理;同时,通过MatCloud+工作流引擎,对整个工作流进行自动流程处理和过程控制;
作业管控子系统,用于分子动力学计算作业的创建,及将所述计算作业提交到本地的计算集群或超级计算机进行分子动力学计算;
结果处理子系统,用于将经分子动力学模拟软件计算完毕后,自动获取轨迹文件,根据预设的要求进行多种分析计算,并对分析完成后的数据进行关键物性数据的提取,然后存入物性数据库。
其中:所述的核心组件模块分为基本功能模块和高级功能模块;其中,基本功能模块包括:力场分配模块、几何优化模块、能量计算模块、动力学计算模块和轨迹文件分析模块;高级功能模块包括:热导分析计算、扩散分析计算、力学性能计算、粘度计算、交联计算、玻璃化转变计算等模块。
所述交互与控制子系统,还包括MatCloud+工作流引擎,用于对所述的工作流进行自动流程处理和过程控制。
所述作业管控子系统,还包括作业监控模块和负载均衡模块,分别用于对分子动力学计算作业过程和状态进行监控,以及对负载均衡进行调节和控制。
利用所述结果处理子系统自动获取轨迹文件后根据预设的要求进行多种分析计算,包括利用热力学分析模块、动力学分析模块分别进行热力学分析计算和动力学分析计算。
一种基于云计算技术的分子动力学计算方法,包括如下步骤:
A、利用预处理子系统定义和封装分子动力学模拟软件的核心组件模块,对所述分子动力学模拟软件的参数进行分类,并进行力场参数的自动匹配;
B、通过交互与控制子系统,利用工作流设计交互模块和结果交互处理模块,分别对分子动力学流程进行交互设计和结果交互处理;同时,通过MatCloud+工作流引擎,对整个工作流进行自动流程处理和过程控制;
C、通过作业管控子系统进行分子动力学计算作业的创建,及将所述计算作业提交到本地的计算集群或超级计算机进行分子动力学计算;
D、利用结果处理子系统,将经所述分子动力学模拟软件计算完毕后,自动获取轨迹文件,根据预设的要求进行多种分析计算,并对分析完成后的数据进行关键物性数据的提取,然后存入物性数据库。
其中,步骤A所述进行力场参数自动匹配的过程,具体为:在力场参数自动匹配模块中指定分子动力学模拟计算所需的力场文件,以及包含空间拓扑信息的构型文件,自动匹配合适的力场参数和指定体系原子间的相互作用力。
较佳地,所述步骤B还包括通过MatCloud+工作流引擎对所述的工作流进行自动流程处理和过程控制的过程。
较佳地,所述步骤C还包括通过作业监控模块和负载均衡模块,分别对分子动力学计算作业过程和状态进行监控,以及对负载均衡进行调节和控制。
其中,步骤D中所述根据预设的要求进行多种分析计算,包括利用热力学分析模块、动力学分析模块分别进行热力学分析计算和动力学分析计算。
本发明的基于云计算技术的分子动力学计算方法及其系统,具有如下有益效果:
应用本发明的实施例的分子动力学计算方法及其系统,只需要通过浏览器(如IE、Chrome等)通过计算机网络登录基于MatCloud+材料云计算平台/系统,不需要安装和编译分子动力学模拟软件,即可在图形化的用户界面下,进行力场参数的自动匹配、进行分子动力学模拟自动流程计算、进行物性数据的自动提取和入库,以及进行轨迹文件分析的分子动力学计算。
附图说明
图1为本发明实施例基于云计算技术的分子动力学计算系统的功能示意图;
图2为本发明实施例基于云计算技术的分子动力学计算系统的计算过程流程图;
图3为实施例一采用本发明的分子动力学计算系统对结晶聚乙烯醇体系的相变转变温度进行计算的过程示意图;
图4为实施例二采用本发明的分子动力学计算系统对聚丙烯类阻燃材料添加剂的模拟筛选进行计算的过程示意图;
图5(a)、图5(b)为实施例二中基于动力学模拟的最后10ps的轨迹文件分析产生的径向分布函数示意图。
具体实施方式
下面结合附图及本发明的实施例对本发明作进一步详细的说明。
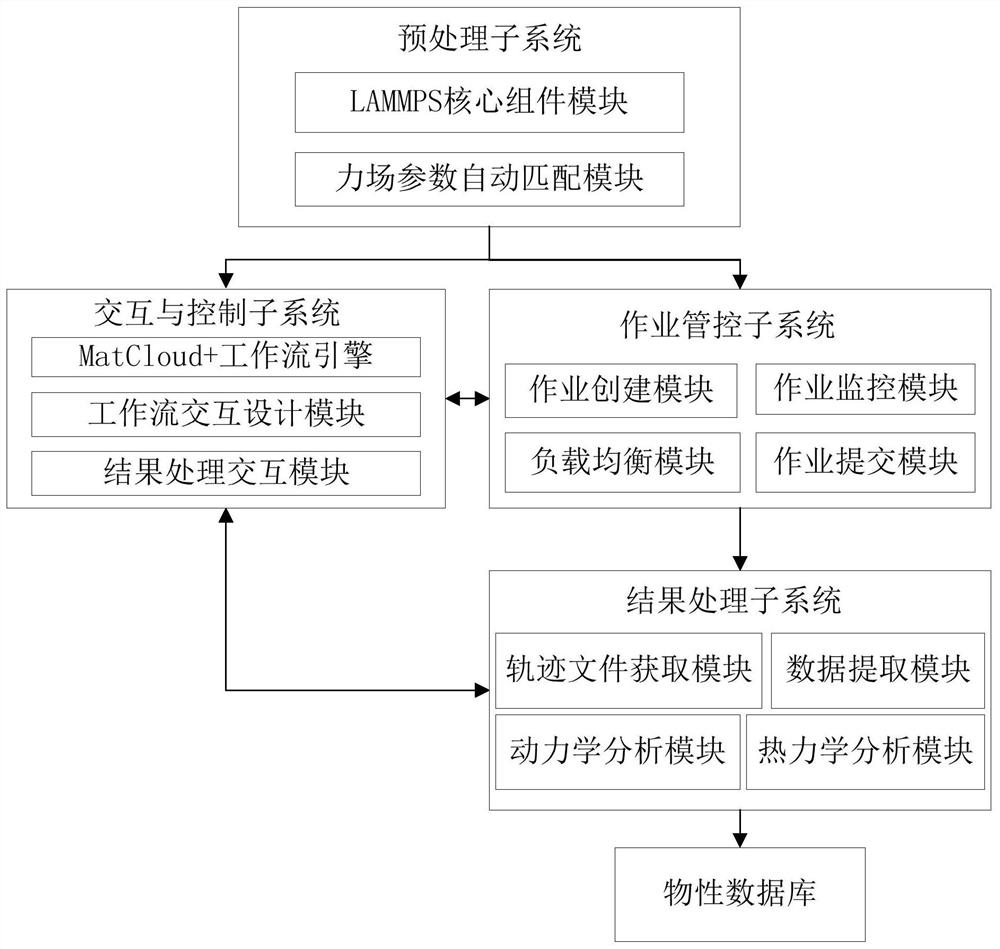
图1为本发明实施例基于云计算技术的分子动力学计算系统的功能示意图。
如图1所示,该分子动力学计算系统,包括预处理子系统、交互与控制子系统、作业管控子系统和结果处理子系统。其中:
所述预处理子系统,用于定义和封装分子动力学模拟软件(以LAMMPS软件为例)的核心组件模块,对LAMMPS软件的所能计算的物理化学性质按照分子动力学计算的难易程度、复杂程度进行分类和整理,并进行力场参数的自动匹配。本实施例中,预处理子系统主要包括:LAMMPS核心组件模块和力场参数自动匹配模块。所述LAMMPS核心组件模块,分为基本功能模块和高级功能模块。其中,基本功能模块包括5个核心组件,即力场参数自动匹配模块、几何优化模块、能量计算模块、动力学计算模块和轨迹文件分析模块;高级功能模块包括:热导分析计算、扩散分析计算、力学性能计算、粘度计算、交联计算、玻璃化转变计算等功能模块。这些组件分别用于特定的材料物理化学性质的计算,并可进行扩展。
由于轨迹文件存储了有序的时间序列上的所有体系原子位置、速度、受力等描述了体系原子的位置、速度、受力随着时间的变化过程。在某个时间点体系内所有原子都处于特定的位置(相当于影像文件的一帧图像),并随着时间的变化而变化(相当于整个影像文件)。轨迹文件分析模块,用于通过轨迹文件包含的信息(原子的位置、速度、受力)分析出和体系所处条件相关的性质(比如位置相对信息rdf等)。
所述的热导分析计算模块,用于计算扩散系数及分析扩散行为。所述的力学性能计算模块,用于计算材料分子间的力学性能。所述的粘度计算模块,用于计算粘度。所述的玻璃化转变计算模块,用于计算玻璃化转变温度值。所述的几何优化模块,用于获取优化后的几何构型。所述的能量计算模块,用于计算能量值。几何优化模块和能量计算模块不涉及时间尺度。
由于聚合物的不同高分子链间可以通过缩聚反应或者加聚反应形成共价键连接的网状高分子结构(化学反应)。LAMMPS软件的部分力场可以模拟化学反应过程,从而可以通过动力学计算模块来描述在特定条件下的交联过程(化学反应过程),并通过轨迹文件分析完成相关的性质计算。所述的交联计算模块,用于计算交联过程中与材料分子结构有关的性能数值。
所述交互与控制子系统,包括MatCloud+工作流引擎、工作流设计交互模块和结果处理交互模块。其中,工作流设计交互模块和结果交互处理模块,分别用于分子动力学流程的交互设计和结果交互处理。MatCloud+工作流引擎,用于对整个工作流进行自动流程处理和过程控制。
所述作业管控子系统,包括作业创建模块、作业提交模块、作业监控模块和负载均衡(控制/调整)模块。利用作业创建模块和作业提交模块,分别进行分子动力学计算作业的创建(包括LAMMPS软件的In文件和Data文件的生成),以及将所述作业提交到本地的计算集群或超级计算机;利用作业监控模块和负载均衡模块,分别实现对分子动力学计算作业过程和状态进行监控,以及对负载均衡进行调节和控制。
所述结果处理子系统,包括轨迹文件获取模块、数据提取模块、动力学分析模块和热力学分析模块;用于将经LAMMPS软件计算完毕后,自动将获取轨迹文件,根据预设的要求进行分析处理(包括热力学分析、动力学分析等),并对分析完成后的数据进行关键物性(即物理化学性质)数据的提取,然后存入物性数据库。
在本发明实施例中,动力学计算模块,用于模拟在特定条件下(如温度、压强等)体系内原子在一段时间内的运动过程。
图2为本发明实施例基于云计算技术的分子动力学计算系统的计算过程流程图。
如图2所示,该基于云计算技术的分子动力学计算过程,包括如下步骤:
步骤20:登录MatCloud+材料云计算平台/系统,进行必要的预处理的设置。包括:通过预处理子系统,设置分子动力学模拟软件(以LAMMPS为例)核心组件模块和力场参数自动匹配模块,进行定义和封装LAMMPS核心组件,并根据对LAMMPS所能计算的物理化学性质参数按照计算的难易程度、复杂程度进行分类和整理,并进行力场参数的自动匹配。
在本发明实施例中,进行分子动力学计算前只需在力场参数自动匹配模块中指定分子动力学模拟计算所需的力场文件,以及包含空间拓扑信息的构型文件,自动匹配合适的力场参数和指定体系原子间的相互作用力。
步骤21:通过交互与控制子系统和作业管控子系统,利用MatCloud+工作流引擎、工作流交互设计以及结果处理交互设计,实现图形化LAMMPS工作流(或调用预先设计的工作流模板)和力场参数自动匹配过程。
其中,实现图形化LAMMPS工作流的过程中,通过拖拽和点击图标的方式,即可实现图形化设计LAMMPS工作流(或调用预先设计的工作流模板)的目的,并可以通过不同的工作流连接方式组合来完成不同的分子动力学计算任务。图形化LAMMPS工作流的过程,由MatCloud+工作流引擎负责实现。
上述步骤中,实现力场参数的自动匹配过程的操作如下:用户只需在力场参数自动匹配模块中指定所需的力场文件,以及包含空间拓扑信息的构型文件,便可自动匹配合适的力场参数,指定体系原子间的相互作用力。
生成LAMMPS软件计算所需的Data文件和In文件的过程:用户在步骤20中的工作流组件的参数设置完成后,后台依据这些设置参数,即可自动完成LAMMPS软件计算所需的In、Data文件。
步骤22:通过作业管控子系统,创建并启动作业工作流。
这里,启动作业工作流后,利用底层MatCloud+工作流引擎,实现作业的自动提交、作业进度和状态及负载量的监控。并可通过作业管控子系统的负载均衡模块,调整分子动力学计算的负载量。
步骤23:获取轨迹文件,通过结果处理子系统进行包含热力学性质分析、动力学性质分析在内的轨迹文件处理过程,然后提取计算结果数据,将其存储在物性数据库。
该过程,可通过步骤20提供给用户的结果处理交互界面,通过用户页面参数的选择,在本地的计算集群或超级计算机的后台完成上述计算处理过程,将得到的处理结果展现给用户。
下面结合具体实施例对本发明的分子动力学计算方法及计算系统做进一步的详细说明。
【实施例一】
本实施例以采用该分子动力学计算系统对结晶聚乙烯醇体系的相变转变温度进行计算的过程为例进行说明,如图3所示。该实施例的具体步骤如下:
步骤31:导入结晶聚乙烯醇结构。所述聚乙烯醇结构,如图3(a)所示。
步骤32:设计工作流的步骤。工作流可通过图标拖拽与连接操作完成。
本实施例中,为了计算该聚乙烯醇结构相变转变温度,需要完成用户导入模型、分配力场、完成基本设置、结构优化、分子动力学缓慢升温过程等步骤。
基于上述步骤,首先完成工作流组件Topo structure data set,Model(力场分配,基本设置),Geometry optimization(结构优化),Molecular dynamic(动力学计算)的连接,该工作流设计过程,如图3(b)所示。
步骤33:各工作流部件的设置过程。
在连接工作流组件后,需要对各工作流组件进行详细的设置,从而能完成特定环境下的任务,具体的设置过程如下:
1)Model模块设置,首先完成原子间相互作用力即力场的设置,如图3(c11)所示。
在完成上述力场的设置后,还需要完成对体系的周期性情况以及是否扩胞等的通用设置,如图3(c12)所示。
2)Geometry optimization模块设置
设置好model模块后,进入结构优化模块设置页面,如图3(c21)所示。
结构优化模块可以提供有用的输出界面,输出界面,如图3(c22)所示。
3)Molecular dynamic设置
完成几何优化过程后,进行分子动力学连续缓慢升温过程,寻找相变点。设置页面如图3(c31)所示。
分子动力学过程输出有效信息,提供可供分析的输出文件,输出页面如图3(c32)所示。
生成LAMMPS分子动力学计算所需要的in文件以及data文件,通过MatCloud+材料云计算平台/系统,启动如图2所示的工作流。工作流负责完成结构优化、分子动力学等计算作业的提交、监控和计算结果数据采集。
步骤34:输出文件分析(热力学分析)
通过前端界面提供的热力学分析工具,可以有效提取分子动力学过程中的热力学信息。
其中,提取的能量、体积、熵值随时间的变化关系如图3(d1)~图3(d4)所示,通过分析可以确定该聚乙烯醇结晶体系的相转变温度。
所述总能量随时间变化关系曲线,如图3(d1)所示。
焓随时间变化的关系曲线,如图3(d2)所示。
密度随时间的变化关系曲线,如图3(d3)所示。
温度随时间的变化关系曲线,如图3(d4)所示。
从中不难看出,总能量、体系焓值、体系密度在时间步数约为7.8e8时候发生突变,对应的温度即为结晶聚乙烯醇的相变转变温度(~540K)。该工作过程完成了寻找结晶聚乙烯醇相变转变温度的任务。
【实施例二】
本实施例以采用该分子动力学计算系统对聚丙烯类阻燃材料添加剂的模拟筛选进行计算的过程为例进行说明,如图4所示。
聚磷酸铵(简称“APP”)填充的聚丙烯(简称“PP”)材料是无卤、低烟浓度、低腐蚀性的常用阻燃材料。想要达到满意的阻燃度需要较高的APP添加量,这时常发生无机的APP在有机的PP中的迁移析出,降低材料阻燃性能。研究人员致力于选取不同的增容剂增进阻燃材料PP/APP的相容性。
本实施例二在MatCloud+材料云计算平台/系统上,调用基于分子力学和动力学的LAMMPS软件,通过计算结合能、径向分布函数函数,分析不同增溶剂提高阻燃材料PP/APP相容性的效果,从而筛选出合适的增容剂,指导实验合成,以降低研发成本并提高研发效率。
该实施例的具体步骤如下:
步骤41:导入结构。
图4(a)为不加增容剂的PP/APP结构,图4(b)、图4(c)、图4(d)分别为添加了增容剂APES,NPCl6和APESP的结构,即图4(a)为PP/APP结构模型,图4(b)为PP/APP/APES的结构模型,图(c)为PP/APP/NPCl6的结构模型和图4(d)为PP/APP/APESP的结构模型。
步骤42:搭建结构平衡计算流程的步骤。
平衡结构的计算流程为:1)结构优化;2)从300K升温到500K再降温到300K的退火模拟,共经历5次循环,最终得到平衡结构。
为了完成以上流程,拖拽MatCloud+材料云计算平台/系统中的组件Lammps Model(力场分配,基本设置),Geometry optimization(结构优化),多个Molecular dynamic(动力学计算)组件,连接成如图4(e)所示的结构平衡计算流程图。
该计算流程中,每一步的分子动力学模拟所选的系综为NPT系综,步长为1fs,模拟时长为10ps。该分子动力学模拟参数设置界面及过程,如图4(f)所示。
步骤43:搭建动力学模拟计算流程。
动力学模拟计算流程为:1)500ps的NPT动力学模拟;2)500ps的NVT动力学模拟。其中第二步的NVT的动力学模拟设置每隔1ps保存一帧。并利用最后10ps的轨迹文件计算结合能和径向分布函数。
为了完成以上流程,拖拽组件Lammps Model(力场分配,基本设置),两个Molecular dynamic(动力学计算)组件,连接成如图4(g)所示的流程。
步骤44:增容剂的相容性评估
通过计算结合能、径向分布函数函数,分析不同增容剂提高阻燃材料PP/APP相容性的效果,从而筛选出合适的增容剂。
1)结合能
结合能(Binding energy)可以反映两组分的分子间相互作用,同时也反映了两组分的相容性。结合能的计算公式为:
E
E
E
其中:E
表1:PP/APP/增容剂的结合能计算数据(kJ/mol)
分析表1的结合能的计算结果可知:
1)未添加任何增容剂时,PP-APP结合能最低,二者结合不稳定;
2)三种增容剂均使得PP-APP的结合能增大,其中APESP的效果最明显,将PP-APP的相互作用能提高到161.46kJ/mol;
3)增容剂与PP/APP的相互作用由弱到强的顺序为NPCI6<APES<APESP,APESP与PP/APP有最强的相互作用;
2)径向分布函数
径向分布函数反应了与指定的原子在距离为r的时候,找到另外一个原子的可能性,对于相容性的评估非常重要。如果复合物不是均相的,每一种组分的g(r)将会比复合物的g(r)出现更强的第一峰。
基于动力学模拟的最后10ps的轨迹文件分析产生的径向分布函数,如图5(a)、图5(b)所示。
图5(a)为在PP/APP中径向分布函数示意图,图5(b)为在不同复合体系中,PP-APP的径向分布函数示意图。
从上述分析径向分布函数可知:
图5(a)中,PP-PP和APP-APP曲线远高于PP-APP,表明PP-APP是不相容的。
图5(b)图中,三种增容剂的加入均使得PP与APP的相互作用距离较未添加更近,说明增容剂提高了PP与APP的相容性。
以上所述,仅为本发明的较佳实施例,并非用于限定本发明的保护范围。尽管本发明提供的实施例是基于LAMMPS软件进行分子动力学模拟计算的,但是其方法和思路同样适用于GROMACS(Groningen Machine for Chemical Simulations)、NAMD(NAnoscaleMolecular Dynamics)等其他的分子动力学模拟软件,只要是借鉴本发明的思路基于云计算技术开展分子动力学模拟计算的,都在本发明的保护范围内。
- 一种基于云计算技术的分子动力学计算方法及其系统
- 一种基于云计算技术的电力系统潮流计算方法