刺五加对照提取物的制备工艺及其质量控制方法
文献发布时间:2023-06-19 10:54:12
技术领域
本发明属于中药制剂及检测领域,具体涉及刺五加对照提取物的制备工艺及其质量控制方法。
背景技术
随着现代中药制剂的研究和发展,传统的煎煮方式存在的不便性和疗效上的不确定性越显突出,并在很大程度上影响并制约了中药制剂在市场上的应用和推广,为最大限度的提高药效,降低毒副作用,满足中医临床要求,改善中药制剂的提取工艺并建立专属的质量控制是关键。
刺五加为五加科(Acanthopanax)灌木植物刺五加(Acanthopanax senticosus(Rupr.et Maxim.)Harms)的干燥根和根茎或茎。其茎、叶也可入药。主要分布于黑龙江、吉林、辽宁及河北等地区。刺五加性温,味辛,微苦,无毒,入脾,肾、心经,有扶正固本、补中益气、补肾健脾及安神益智之功效。临床用于脾肾阳虚,腰膝酸软,体虚乏力,失眠多梦,食欲不振等。刺五加对照提取物是由刺五加经煎煮提取有效成分后真空冷冻干燥而成,其中主要含有各种苷类如紫丁香苷(苷B)、紫丁香树脂苷(苷E)及多种木脂素类、异秦皮定和黄酮类等多种药理成分,化学成分复杂。
公开号CN109745349A,名称为一种刺五加对照提取物及其制剂的工艺制备的发明专利申请中,记载了采用刺五加饮片,加3-8倍量水,加热煎煮2次,每次30-70分钟,合并煎煮液,浓缩至相对密度1.0-1.2的浓缩液,待温度降低到40度以下时,加入乙醇溶液,搅拌均匀后,静沉2-3次,静沉7-20小时,过滤,合并过滤液得浸膏,干燥,粉碎,得浸膏粉,即得刺五加对照提取物,可用于制备刺五加片制剂。但该专利并未针对该中药制剂的药效成分及制备过程建立专属的质量控制方法,该提取物的药效成分含量难以控制。
公开号CN102048777A,名称为一种刺五加对照提取物的检测方法的发明专利申请中,记载了采用高效液相色谱法对刺五加对照提取物中的药物成分紫丁香苷的含量进行测定以及采用红外一维光谱法对刺五加对照提取物的相似度进行的比较,通过对刺五加提取的成分进行检测,可以更加准确的把握有关药品的质量,降低用药风险,提高产品质量,同时,通过与刺五加对照提取物标准对照品的一维光谱图进行比较并判断相似度,可以进一步的保证产品的质量。但该专利仅针对其药物成分紫丁香苷进行的含量测定,由于刺五加对照提取物的药效成分复杂,无法有效的实现其质量控制,另外,采用刺五加对照提取物对照品作为对照标准,无法建立对刺五加对照提取物制备工艺的全面控制。
鉴于现有技术中对刺五加对照提取物制备方法及其专属性的质量控制方法的缺陷,本发明应运而生。
发明内容
本发明的目的在于提供刺五加对照提取物的制备工艺,采用刺五加饮片为原料经煎煮提取而制得,制备过程中,通过合理控制煎煮次数、加水量、煎煮时间等,能有效控制收粉率及其有效物质紫丁香苷的含量,保证了刺五加对照提取物成分的稳定性,且工艺运行稳定。
为实现刺五加对照提取物制备工艺的稳定性和可靠性,本发明还提供了一种刺五加对照提取物的质量控制方法。
本发明通过下述技术方案实现:刺五加对照提取物的制备工艺,取刺五加饮片,加水煎煮两次,一煎加10倍水,浸泡30分钟,煮沸,保持微沸煎煮60分钟,过滤,冷却至室温;二煎加8倍水,煮沸,保持微沸煎煮40分钟,过滤,合并水煎液,冷却至室温,真空冷冻干燥,即得刺五加对照提取物。
所述刺五加对照提取物中紫丁香苷的含量为0.4~1.40%,水分含量≤9.0%。
刺五加对照提取物的质量控制方法,包括构建所述刺五加对照提取物的特征图谱,步骤如下:
A.分别制备参照物溶液和刺五加对照提取物的供试品溶液;
B.分别取参照物溶液和供试品溶液,按高效液相色谱法进行测定,得到对应的特征图谱;
C.以参照物溶液的特征图谱为参照图谱,从供试品溶液的特征图谱中选择共有峰,构建刺五加对照提取物的特征图谱。
所述步骤A中,参照物溶液的制备包括:
刺五加对照药材的参照物溶液:取刺五加药材,加70%甲醇25ml,超声处理30分钟,放冷,摇匀,滤过,取续滤液,即得;
紫丁香对照品的参照物溶液:取紫丁香苷对照品,加甲醇制成每1ml含80μg的溶液,即得。
所述步骤A中,供试品溶液的制备包括:取刺五加对照提取物,加入70%甲醇25ml,称定重量,超声处理20分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
所述步骤B中,按高效液相色谱法,以十八烷基硅烷键合硅胶为填充剂,乙腈为流动相A,0.1%磷酸溶液为流动相B,进行梯度洗脱,在柱温为25℃,流速为1.2ml/min,检测波长为220nm下进行测定。
所述梯度脱洗满足以下条件:
0~50min,流动相A:4%→20%,流动相B:96%→80%;
50~60min,流动相A:20%,流动相B:80%。
所述共有峰包括10个特征峰,与紫丁香苷参照物峰相对应的峰为S峰,计算各特征峰与S峰的相对保留时间,各特征峰相对保留时间应在规定值的±10%范围之内,规定值为:峰1:0.569,峰2:0.711,峰4:1.059,峰5:1.107,峰6:1.168,峰7:1.187,峰8:1.821,峰9:1.930,峰10:2.034。
还包括对所述刺五加对照提取物中紫丁香苷的含量进行测定,步骤如下:
A.分别制备紫丁香苷对照品溶液和刺五加对照提取物的供试品溶液,
紫丁香苷对照品溶液的制备包括:取紫丁香苷对照品,加甲醇制成每1ml含80μg的溶液,即得。
供试品溶液的制备包括:取刺五加对照提取物,加入70%甲醇25ml,称定重量,超声处理20分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
B.分别取紫丁香苷对照品溶液和供试品溶液各10μl,注入高效液相色谱仪,以十八烷基硅烷键合硅胶为填充剂,按20:80的甲醇-水为流动相,进行等度洗脱,在检测波长为265nm下进行测定,测定得到刺五加对照提取物中紫丁香苷的含量限度为0.4~1.40%。
本发明与现有技术相比,具有以下优点及有益效果:
(1)本发明方法提供了基于煎煮工艺实现的刺五加对照提取物的制备方法,通过合理控制煎煮过程的工艺参数条件,如加水量及煎煮时间,能合理控制收粉率及其有效物质紫丁香苷的含量,保证了刺五加对照提取物成分的稳定性,为后续刺五加制剂在中药自动配药机中的应用提供更加稳定和精确的数据。
(2)本发明为刺五加对照提取物的制备工艺建立了专属的质量控制方法,以刺五加对照药材、紫丁香苷对照品为参照物,在特定的检测条件下,构建得到刺五加对照提取物的特征图谱,可快速并高效地实现刺五加对照提取物的质量控制。
(3)本发明同时还建立了刺五加对照提取物中紫丁香苷含量的测定方法,用于实现刺五加对照提取物中有效物质紫丁香苷含量的质量控制。
综上所述,本发明提出了刺五加对照提取物的制备工艺并为此建立了专属的质量控制方法,实现了从药材、制备工艺过程及有效物质含量的全面反映,更好的符合中药制剂的规范要求,保证了中药制剂含量的合理性。
附图说明
图1为刺五加吸水率曲线。
图2为刺五加对照提取物冻干工艺。
图3为18批刺五加对照提取物收粉率分布图。
图4为18批刺五加对照提取物水分分布图。
图5为刺五加对照提取物对照特征图谱。
图6为原儿茶酸紫外吸收光谱图。
图7为紫丁香苷紫外吸收光谱图。
图8为绿原酸紫外吸收光谱图。
图9为香草酸紫外吸收光谱图。
图10为刺五加苷E紫外吸收光谱图。
图11为异嗪皮啶紫外吸收光谱图。
图12为刺五加对照提取物3D色谱图。
图13为刺五加对照提取物不同波长色谱图。
图14为柱温考察图谱。
图15为流速考察图谱
图16为延迟性实验图谱。
图17为提取方法考察图谱。
图18为提取溶剂考察图谱。
图19为提取时间考察图谱。
图20为刺五加对照提取物特征图谱色谱峰指认图谱。
图21为刺五加对照药材特征图谱。
图22为不同仪器考察图谱。
图23为刺五加对照提取物色谱柱耐用性考察图谱。
图24为刺五加对照提取物特征图谱(a)。
图25为刺五加对照提取物特征图谱(b)。
图26为紫丁香苷紫外吸收光谱图(265nm)。
图27为刺五加对照提取物色谱图。
图28为不同流速色谱图(紫丁香苷)。
图29为不同柱温色谱图(紫丁香苷)。
图30为刺五加对照提取物专属性叠加图。
图31为紫丁香苷标准曲线图。
图32为刺五加对照提取物紫丁香苷含量分布图。
具体实施方式
下面结合实施例对本发明作进一步地详细说明,但本发明的实施方式不限于此。
实施例1:
本实施例提出了刺五加对照提取物的制备工艺。
取刺五加饮片100g,加水煎煮二次,一煎加10倍水,浸泡30分钟,煮沸,保持微沸煎煮60分钟,200目筛网过滤,立即冷却至室温;二煎加8倍水,煮沸,保持微沸煎煮40分钟,200目筛网过滤,合并水煎液,立即冷却至室温,真空冷冻干燥,分装,即得。
以下对刺五加对照提取物制备工艺的研究。
1.实验仪器与设备
电子天平:上海方瑞仪器有限公司JY系列百分之一天平;
煎药壶:美味世家全自动分体式陶瓷煎药壶4升;
筛网:九峰筛网200目;
真空冷冻干燥机:北京松源华兴科技发展有限公司LGJ-100F;
2000ml不锈钢量杯若干;西林瓶:10ml,30ml,50ml若干。
2.刺五加饮片的选择
参考《中国药典》2015年版一部刺五加【炮制】项下相关规定及第四部通则0213炮制通则相关规定进行炮制,具体方法为:除去杂质,洗净,稍泡,润透,切厚片,干燥;产地已切片者,筛去灰屑。
3.提取工艺
3.1提取溶媒
依据《中药配方颗粒质量控制与标准制定技术要求(征求意见稿)》、《医疗机构中药煎药室管理规范》中相关规定,确定刺五加对照提取物的制备提取溶媒为水。
3.2取样量
用于制备刺五加对照提取物的饮片取样量定为100g。
3.3浸泡
依据《中药配方颗粒质量控制与标准制定技术要求(征求意见稿)》、《医疗机构中药煎药室管理规范》中相关规定,待煎饮片应当先行浸泡,浸泡时间应根据饮片的质地确定,一般不少于30min。确定浸泡时间为30min。
3.4煎煮次数
依据《中药配方颗粒质量控制与标准制定技术要求(征求意见稿)》、《医疗机构中药煎药室管理规范》中相关规定,确定煎煮次数定为2次。
3.5加水量
取同一批号刺五加50g(批号:XLS201808511),两份,分别加入10倍量的水(500g),分别在浸泡10min、20min、30min、50min、1h、1.5h、2h、3h、4h、7h、22h测定,计算吸水率,结果见下表1及图1。
表1刺五加吸水率变化
由图1可知,其在前30分钟急剧吸水,30分钟时的平均吸水率为72.1%,30分钟后增长率逐渐减小,7~22h吸水接近饱和,22h时的平均吸水率为179.7%。
取饮片100g(批号:CWJ180927)于煎药壶,分别加入0倍、4倍水、8倍水、10倍水、12倍水、14倍水,观察水面没过饮片的高度,结果见下表2。
表2不同加水量液面高度
结果可知当加水量为8~14倍时,浸过药面高度均在2~5cm范围,参考其吸水率,同时保证不同批次饮片均能在2~5cm范围,确定一煎加水倍数为10倍,二煎加水量为8倍。
3.6煎煮时间
刺五加属于滋补类药,按照《中药配方颗粒质量控制与标准制定技术要求(征求意见稿)》、《医疗机构中药煎药室管理规范》中对照提取物制备的规定:煎煮时间应当根据方剂的功能主治和药物的功效确定。一般药物煮沸后再煎煮20-30分钟;解表类、清热类、芳香类药物不宜久煎,煮沸后再煎煮15-20分钟;滋补药物先用武火煮沸后,改用文火慢煎约40-60分钟。药剂第二煎的煎煮时间应当比第一煎的时间略缩短。确定刺五加对照提取物第一煎先用武火煮沸后,改用文火慢煎60min,第二煎先用武火煮沸后,改用文火慢煎40min。
3.7固液分离
根据《中药配方颗粒质量控制与标准制定技术要求(征求意见稿)》、《医疗机构中药煎药室管理规范》,并结合大生产,采用200目筛网趁热过滤。
3.8真空冷冻干燥
根据《中药配方颗粒质量控制与标准制定技术要求(征求意见稿)》、《医疗机构中药煎药室管理规范》的要求,对照提取物干燥过程推荐采用真空冷冻干燥方法制备为宜,可保证其质量的稳定、易于保存。真空冷冻干燥工艺参数见下表3及图2。
表3冻干工艺
实施例2:
本实施例涉及18批次刺五加对照提取物的制备。
1.来源
按照《中药配方颗粒质量控制与标准制定技术要求(征求意见稿)》的要求共收集18批来自黑龙江鸡西市、黑龙江林口县、吉林白山等产区的刺五加药材,见下表4,均符合《中国药典》2015版的要求。
表4检验结果汇总表
2.炮制
按照《中药饮片质量标准通则(试行)》以及《中国药典》2015年版一部刺五加【炮制】项下相关规定对上述18批刺五加药材进行炮制。
具体方法为:除去杂质,洗净,稍泡,润透,切厚片,干燥。产地已切片者,筛去灰屑。
因其药材已经在产地加工时切片,药材中几乎无杂质存在,可在此将刺五加药材同时视为产地饮片,作为制备对照提取物的原料药。
18批刺五加饮片与药材批号对应情况见下表5。
表5 18批刺五加饮片与药材批号对应表
综上所述,刺五加18批次饮片均检验合格。
3.制备
按实施例1所述方法制备刺五加对照物提取物。
4.性状
用18批刺五加饮片进行对照提取物的制备,每批刺五加饮片制1批对照提取物,共制得18批冻干粉,各对照提取物批号所对应饮片及药材批号及各批对照提取物性状见下表6。
表6 18批次刺五加对照提取物性状
5.出膏率
对18批刺五加对照提取物进行测定,所得各批次对照提取物出膏率结果见下表7和图3。
表7 18批刺五加对照提取物制备数据汇总
结果表明,18批刺五加对照提取物出膏率范围为6.0~7.8%,平均值为6.7%,SD为0.6%。平均值的70~130%的范围为4.7~8.7%,平均值加减3倍SD范围为4.9~8.5%。
根据以上结果可以知道,刺五加对照提取物收粉率应为4.0~9.0%。
6.水分
按照《中国药典》2015年版四部通则0832第二法进行测定,18批刺五加对照提取物水分测定结果,见下表8和图4。
表8 18批刺五加对照提取物水分测定结果
结果表明,18批刺五加对照提取物水分在5.6~7.7%范围之内,平均值为6.7%。平均值的±30%范围为4.7%~8.7%。
根据以上结果可以知道,刺五加对照提取物水分应为不大于9.0%。
实施例3:
本实施例是对刺五加对照提取物的质量控制方法,当然,该质量控制方法也可适用于刺五加配方颗粒或其单方中药制剂的质量控制。
1.实验仪器与材料
高效液相色谱仪:安捷伦1260型高效液相色谱仪、Waters e2695型高效液相色谱仪、岛津LC-20AD型高效液相色谱仪;
电子天平:ME204E/02、MS205DU、XP26(梅特勒-托利多仪器有限公司);
超纯水机:细胞型1810A(上海摩勒科学仪器有限公司);
超声波清洗器:KQ-600DB型(600W,40KHz;昆山市超声仪器有限公司);
色谱柱:ZORBAX Eclipse Plus C18 Analytical 4.6×250mm 5-Micron、ZORBAXEclipse XDB C18 Analytical 4.6×250mm 5-Micron、Kromasil 100-5-C18 4.6×250mm。
2.试剂及试药
乙腈、磷酸为色谱纯,水为超纯水,其余试剂均为分析纯。
紫丁香苷(中国食品药品检定研究院,批号:111574-201605,含量以95.2%计);
原儿茶酸(中国食品药品检定研究院,批号:110809-201205,含量以99.9%计);
绿原酸(中国食品药品检定研究院,批号:110753-201415,含量以96.2%计);
刺五加苷E(北京世纪奥科生物技术有限公司,批号:39432-56-9,含量以98%计);
异嗪皮啶(中国食品药品检定研究院,批号:110837-201608,含量以100.0%计);
香草酸(中国食品药品检定研究院,批号:110776-201503,含量以99.8%计);
刺五加对照药材(中国食品药品检定研究院,批号:120991-201610);
刺五加对照提取物(批号:CWJBT180901、CWJBT180902、CWJBT180903、CWJBT180904、CWJBT180905、CWJBT180906、CWJBT180907、CWJBT180908、CWJBT180909、CWJBT180910、CWJBT180911、CWJBT180912、CWJBT180913、CWJBT180916、CWJBT180917、CWJBT180918、CWJBT180919、CWJBT180920)。
3.特征图谱
参照物溶液的制备包括:取刺五加对照药材3g,置具塞锥形瓶中,加70%甲醇25ml,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,即得。另取紫丁香苷对照品适量,加甲醇制成每1ml含80μg的溶液,作为对照品参照物溶液。
供试品溶液的制备包括:取实施例1制得的刺五加对照提取物0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,称定重量,超声处理(功率600W,频率40kHz)20分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
精密吸取参照物溶液和供试品溶液各10μl,注入高效液相色谱仪,以十八烷基硅烷键合硅胶为填充剂(柱长为250mm,内径为4.6mm,粒径为5μm),乙腈为流动相A,0.1%磷酸溶液为流动相B,进行梯度洗脱(见下表10),在柱温为25℃,流速为1.2ml/min,检测波长为220nm下进行测定。
表10梯度脱洗
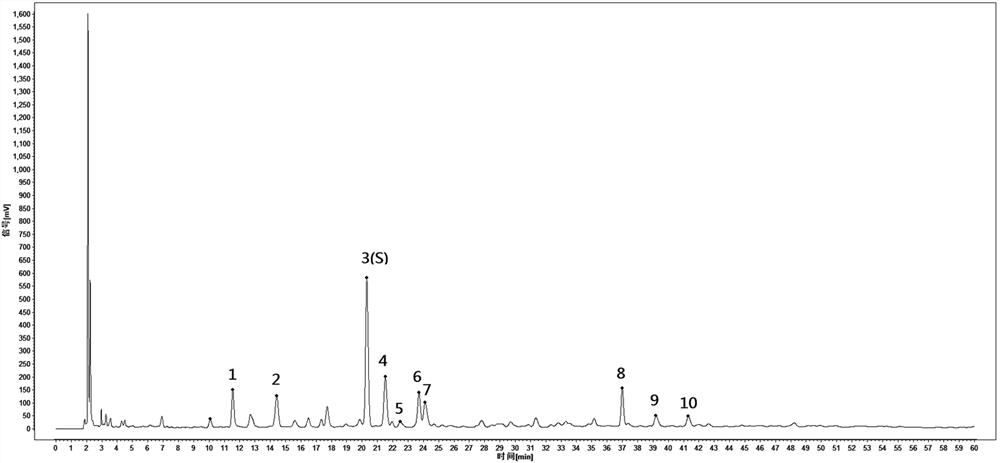
以参照物溶液的特征图谱为参照图谱,从供试品溶液的特征图谱中选择共有峰,构建刺五加对照提取物的特征图谱,见图5(峰1:原儿茶酸;峰3S:紫丁香苷;峰4:绿原酸;峰5:香草酸;峰8:刺五加苷E;峰9:异嗪皮啶)。
供试品色谱中应呈现10个特征峰,并应与对照药材参照物色谱中的10个特征峰保留时间相对应,与紫丁香苷参照物峰相对应的峰为S峰,计算各特征峰与S峰的相对保留时间,各特征峰相对保留时间应在规定值的±10%范围之内。规定值为:0.569(峰1)、0.711(峰2)、1.059(峰4)、1.107(峰5)、1.168(峰6)、1.187(峰7)、1.821(峰8)、1.930(峰9)、2.034(峰10)。
4.0色谱条件与系统适用性试验
4.1波长选择
在以上拟定的实验条件基础上,利用二极管阵列检测器分别对原儿茶酸对照品溶液、紫丁香苷对照品溶液、绿原酸对照品溶液、香草酸对照品溶液、刺五加苷E对照品溶液、异嗪皮啶对照品溶液、供试品溶液进行全波段扫描,并提取供试品溶液3D图以及在200nm、210nm、220nm、230nm、240nm、250nm、260nm、270nm波长下的色谱图,见图6-13。
结果表明,在检测波长为220nm波长下各色谱峰峰形和对称性更好,整个色谱图信息量较大,故检测波长为220nm。
4.2柱温考察
在以上拟定的实验条件基础上,分别对柱温为20℃、25℃、30℃、35℃时进行考察。见图14。
结果表明,在柱温为25℃时,各色谱峰分离度更好,故暂定柱温为25℃进行后续考察。
4.3.流速考察
在以上拟定的实验条件基础上,分别对流速为0.8ml/min、1.0ml/min、1.2ml/min时进行了考察。见图15。
结果表明,在流速为1.2ml/min时,各色谱峰分离度最好,故选择流速为1.2ml/min进行后续考察。
4.4.延迟性实验
在以上拟定的实验条件基础上,将色谱图采集时间延长至120min。见图16。
结果表明,在色谱图采集到60分钟时,已将色谱峰采集完全。故将色谱图采集时间确定为60分钟。
综上所述,刺五加对照提取物特征图谱色谱条件与系统适应性试验确定为:
以十八烷基硅烷键合硅胶为填充剂(柱长为250mm,内径为4.6mm,粒径为5μm);以乙腈为流动相A,以0.1%磷酸溶液为流动相B,按规定进行梯度洗脱(见表10);流速为每分钟1.2ml;柱温为25℃;检测波长为220nm。理论板数按紫丁香苷峰计算应不低于2000。
5.供试品溶液的制备考察。
5.1.提取方法考察
取刺五加对照提取物(批号CWJBT180908)0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,分别对供试品提取方法为回流、超声时进行考察,提取时间30min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。见图17。
结果表明,对供试品分别进行超声提取与回流提取时效果无显著差异。因超声提取操作更为简便,故供试品提取方法确定为超声提取。
5.2.提取溶剂考察
取刺五加对照提取物(批号CWJBT180908)0.5g,精密称定,置具塞锥形瓶中,分别对供试品提取溶剂为甲醇、70%甲醇、水时进行考察,溶剂加入量为25ml,密塞,称定重量,超声处理(功率600W,频率40kHz)30分钟,放冷,再称定重量,用提取溶剂补足减失的重量,摇匀,滤过,取续滤液,即得。见图18。
结果表明,当提取溶剂为水和70%甲醇时,色谱峰信息量较大,峰型最好,且差异不大。故选择更易操作的70%甲醇作为提取溶剂。
5.3.提取时间考察
取刺五加对照提取物(批号CWJBT180908)0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理(功率600W,频率40kHz),分别对供试品提取时间为20分钟、30分钟、40分钟时进行考察,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。见图19。
结果表明,在提取时间为20分钟,即可充分提取。故供试品提取时间确定为20分钟。
综上所述,刺五加对照提取物特征图谱供试品溶液的制备方法确定为:
取刺五加对照提取物0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,密塞,称定重量,超声处理(功率600W,频率40kHz)20分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
6.方法学考察。
6.1.色谱峰指认
供试品溶液的制备:按以上拟定的实验条件,制备刺五加对照提取物供试品溶液。
紫丁香苷对照品溶液:取紫丁香苷对照品适量,精密称定,加甲醇制成每1ml含80μg的溶液,即得。
原儿茶酸对照品溶液:取原儿茶酸对照品适量,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,即得。
绿原酸对照品溶液:取绿原酸对照品适量,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,即得。
刺五加苷E对照品溶液:取刺五加苷E对照品适量,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,即得。
异嗪皮啶对照品溶液:取异嗪皮啶对照品适量,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,即得。
香草酸对照品溶液:取香草酸对照品适量,精密称定,加70%甲醇制成每1ml含0.1mg的溶液,即得。
刺五加对照药材溶液:取刺五加对照药材3g,置具塞锥形瓶中,加入70%甲醇25ml,密塞,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,即得。
阴性对照溶液的制备:按以上拟定的实验条件,制备缺刺五加对照提取物的阴性对照溶液。
对刺五加对照提取物特征图谱峰进行定位。见图20和图21。
结果表明,峰2为原儿茶酸,峰4为紫丁香苷,峰5为绿原酸,峰6为香草酸,峰9为刺五加苷E,峰10为异嗪皮啶。对照提取物中的11个特征峰在刺五加对照药材图谱中能够一一对应。在以下方法学考察中,暂将峰4设为S峰,对样品中的11个峰进行考察。
6.2.仪器精密度试验
精密称取刺五加对照提取物(批号:CWJBT180908)1份,按拟定实验方法进行制备,连续进样5次,每次10μl。见下表11和表12。
表11精密度考察-特征峰相对保留时间
表12精密度考察-特征峰相对峰面积
结果表明,仪器精密度考察中,各特征峰相对保留时间的RSD在0.04~0.23%,各特征峰相对峰面积的RSD在0.19~6.62%。
6.3.重复性考察
精密称取刺五加对照提取物(批号:CWJBT180908)6份,按拟定实验方法进行制备及测定。见下表13和表14。
表13重复性考察—特征峰相对保留时间比值
表14重复性考察—特征峰相对峰面积比值
结果表明,各特征峰相对保留时间的RSD为0.04~0.31%,各特征峰相对峰面积的RSD在0.91~3.45%。
6.4.中间精密度考察
6.4.1不同仪器考察
在以上拟定的实验条件基础上,分别精密称取刺五加对照提取物(批号:CWJBT180908)一份,制备供试品溶液,分别在安捷伦1260、Waters e2695、岛津LC-20AD型高效液相色谱仪上进行测定(色谱柱均为ZORBAX Eclipse Plus C18 Analytical 4.6×250mm 5-Micron)。见下表15、表16和图22。
表15不同仪器考察—特征峰相对保留时间比值
表16不同仪器考察—特征峰相对峰面积比值
结果表明,用上述3种仪器对供试品进行检测时,各特征峰相对保留时间的RSD在0.24~2.01%;各特征峰相对峰面积的RSD在1.70~40.88%。
6.4.2不同人员和时间考察
在以上拟定的实验条件基础上,由不同人员(A、B)在不同时间(T1、T2)分别精密称取刺五加对照提取物(批号:CWJBT180908)各两份,制备供试品,进行测定。见下表17、表18。
表17不同人员与时间考察—特征峰相对保留时间比值
表18不同人员与时间考察—特征峰相对峰面积比值
结果表明,不同样品制备人员和不同样品制备时间条件下,各特征峰相对保留时间的RSD在0.00~0.10%;各特征峰相对峰面积的RSD在0.00~0.88%。
6.5.耐用性考察
6.5.1色谱柱耐用性考察
在以上拟定的实验条件基础上,分别对ZORBAX Eclipse Plus C18 Analytical4.6×250mm 5-Micron(色谱柱1)、ZORBAX Eclipse XDB-C18 Analytical 4.6×250mm 5-Micron(色谱柱2)、Kromasil 100-5-C18 4.6×250mm(色谱柱3)色谱柱进行考察。见下表19、表20和图23。
表19不同色谱柱考察—特征峰相对保留时间比值
表20不同色谱柱考察—特征峰相对峰面积比值
结果表明,用上述3种色谱柱对样品进行检测,色谱柱3的峰6和峰8分离度差,其余各特征峰相对保留时间的RSD在0.14~5.30%;各特征峰相对峰面积的RSD在2.59~34.05%。
6.5.2稳定性考察
在以上拟定的实验条件基础上,取同一供试品溶液,分别于0h,3h,6h,12h,18h,24h时测定。见下表21和表22。
表21稳定性考察-特征峰相对保留时间
表22稳定性考察-特征峰相对峰面积
结果表明,各特征峰相对保留时间的RSD在0.05~0.27%,各特征峰相对峰面积的RSD在0.39~3.23%。该方法供试品在24小时内稳定性良好。
综上所述,在色谱柱耐用性考察中,峰6、峰8无法保证完全分离,峰2的相对保留时间的RSD为5.30%,峰1的峰面积占比较小,故暂定将特征峰峰1删除,并将其他特征峰相对保留时间的限度范围放宽至±10%,同时将该检测方法色谱柱规定为ZORBAX EclipsePlus C18 Analytical 4.6×250mm 5-Micron。以紫丁香苷峰作为S峰,将剩余的10个特征峰纳入后续考察。
6.6特征峰的确定及对照图谱的建立
采用拟定的方法对刺五加对照提取物18批样品进行特征图谱的测定,计算相对保留时间、相对峰面积。见图24、图25以及下表23、表24。(图24,S1:CWJBT180901;S2:CWJBT180902;S3:CWJBT180903;S4:CWJBT180904;S5:CWJBT180905;S6:CWJBT180906;S7:CWJBT180907;S8:CWJBT180908;S9:CWJBT180909。图25中,S1:CWJBT180910;S2:CWJBT180911;S3:CWJBT180912;S4:CWJBT180913;S5:CWJBT180916;S6:CWJBT180917;S7:CWJBT180918;S8:CWJBT180919;S9:CWJBT180920)
表23 18批刺五加对照提取物相对保留时间
表24 18批刺五加对照提取物相对峰面积
根据相对保留时间稳定及各批次样品均能检出且峰相对较高的原则,共选择了10个重复性较好的峰作为特征峰。结果表明,18批次刺五加对照提取物10个特征峰相对保留时间RSD均小于2.0%。最终规定:供试品色谱中应呈现10个特征峰,并应于对照药材参照物色谱中的10个特征峰保留时间相对应,与紫丁香苷参照物峰相对应的峰为S峰,计算各特征峰与S峰的相对保留时间,各特征峰相对保留时间应在规定值的±10%范围之内。规定值为:0.569(峰1)、0.711(峰2)、1.059(峰4)、1.107(峰5)、1.168(峰6)、1.187(峰7)、1.821(峰8)、1.930(峰9)、2.034(峰10)。
采用中药色谱指纹图谱相似度评价系统(2012版)对18批次刺五加对照提取物特征图谱进行合成,建立了刺五加对照提取物特征图谱的对照特征图谱。见图5。
实施例4:
本实施例是在实施例3的基础上,对刺五加对照提取物的质量控制,当然,该质量控制方法也可适用于刺五加配方颗粒或其单方中药制剂的质量控制。
1.试验仪器与材料
1.1仪器
高效液相色谱仪:安捷伦1260型高效液相色谱仪、岛津LC-20AD型高效液相色谱仪、Waters e2695型高效液相色谱仪;
电子天平:ME204E/02、MS205DU、XP26(梅特勒-托利多仪器有限公司);
超纯水机:细胞型1810A(上海摩勒科学仪器有限公司);
超声波清洗器:KQ-600DB型(600W,40KHz;昆山市超声仪器有限公司);
色谱柱:Agilent 5TC-C18(2)250*4.6mm、Shim-pack GIST 4.6*250mm、Diamosil5um C18(2)250*4.6mm。
1.2材料
紫丁香苷对照品:(中国食品药品检定研究院,批号:111574-201605,纯度95.2%);
甲醇为色谱纯,水为超纯水,磷酸为色谱纯,其它试剂均为分析纯;
刺五加对照提取物(同上)。
2.含量测定
紫丁香苷对照品溶液的制备:取紫丁香苷对照品适量,精密称定,加甲醇制成每1ml含80μg的溶液,即得。
刺五加对照提取物的供试品溶液:取实施例1制得的刺五加对照提取物0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,称定重量,超声处理(功率600W,频率40kHz)20分钟,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
精密吸取紫丁香苷对照品溶液和供试品溶液各10μl,注入高效液相色谱仪,以十八烷基硅烷键合硅胶为填充剂,甲醇-水(20:80)为流动相,进行等度洗脱,在检测波长为265nm下进行测定。
3.色谱条件选择与系统适用性考察
3.1检测波长确定
对紫丁香苷对照品溶液进行全波长光谱采集,通过对光谱图的分析,并且参考《中国药典》2015年版一部刺五加【含量测定】项下的检测波长,确定刺五加对照提取物中紫丁香苷的含量测定的最佳检测波长为265nm。见图26。
3.2流动相选择
参考刺五加对照提取物项下含量测定方法及《中国药典》2015年版刺五加药材【含量测定】项下流动相,以甲醇-水(20:80)为流动相进行等度洗脱。供试品色谱图、色谱指数,见图27及下表25:
表25色谱指数
结果表明,按上述流动相进行等度洗脱时,紫丁香苷与相邻的色谱峰分离度较佳,且达到较高的理论塔板数,故选择甲醇-水(20:80)为流动相进行等度洗脱。
3.3流速考察
在以上拟定的实验条件下,分别考察在0.8ml/min、1.0ml/min、1.2ml/min流速下,以紫丁香苷色谱峰的分离度、理论塔板数、拖尾因子等作为评价指标。结果见图28及下表26。
表26不同流速分析结果
结果表明,三种流速均能对紫丁香苷进行分离和检测,随着流速增加,理论塔板数和分离度逐渐减小,拖尾因子逐渐增大,选择流速为1.0ml/min进行后续考察。
3.4柱温考察
在以上拟定的实验条件下,考察供试品溶液在20℃、25℃、30℃、35℃柱温下的分析情况,以紫丁香苷分离度、理论塔板数、拖尾因子等作为评价指标。见图29及下表27。
表27不同柱温的分析结果
结果表明,在20℃、25℃、30℃、35℃柱温下,紫丁香苷色谱峰的理论塔板数和分离度逐渐升高,拖尾逐渐减小,选择柱温为25℃进行后续考察。
综上所述,色谱条件和系统适应性试验结果如下:
以十八烷基硅烷键合硅胶为填充剂;以甲醇-水(20:80)为流动相;检测波长为265nm。理论板数按紫丁香苷峰计算应不低于2000。
4.供试品溶液制备方法考察
4.1提取方式考察
取刺五加对照提取物(批号CWJBT180918)4份各0.5g,精密称定,置具塞锥形瓶中,加70%甲醇25ml,称定重量,2份超声处理、2份回流处理,提取时间均为20min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。各取10μl注入色谱仪,分析计算不同提取方式下紫丁香苷含量。见下表28。
表28不同提取方式分析结果
结果表明,回流提取与超声提取无明显差异,故选择更为便捷的超声提取。
4.2提取溶剂考察
取刺五加对照提取物(批号CWJBT180918)6份各0.5g,精密称定,置具塞锥形瓶中,分别加入水、70%甲醇、甲醇25ml,称定重量,超声处理20min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。各取10μl注入色谱仪,分析计算不同提取方式下紫丁香苷含量。见下表29。
表29不同提取溶剂的分析结果
结果表明,以甲醇、70%甲醇提取时的提取效率比水提取时相对较大,且为了与特征图谱提取溶剂保持一致,故以70%甲醇作其提取溶剂。
4.3提取时间考察
取刺五加对照提取物(批号CWJBT180918)6份各0.5g,精密称定,置具塞锥形瓶中,加入70%甲醇25ml,称定重量,分别超声处理20min、30min、40min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。各取10μl注入色谱仪,分析计算不同提取方式下紫丁香苷含量。见下表30。
表30不同提取时间的分析结果
结果表明,在提取时间为20min时即可充分提取,且为与特征图谱提取时间保持一致,选择提取时间为20min。
4.4取样量考察
取刺五加对照提取物(批号CWJBT180918)6份,两份0.2g,两份0.5g,两份1.0g,精密称定,置具塞锥形瓶中,加入70%甲醇25ml,称定重量,超声处理20min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。各取10μl注入色谱仪,分析计算不同提取方式下紫丁香苷含量。见下表31。
表31不同取样量分析结果
结果表明,取样量为0.5g时提取效率较高,选择取样量0.5g。
4.5供试品溶液制备方法确定
取刺五加对照提取物0.5g,精密称定,置具塞锥形瓶中,精密加入70%甲醇25ml,称定重量,超声处理(功率600W,频率40kHz)20min,放冷,再称定重量,用70%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
5.方法学考察
5.1专属性实验
按拟定方法制备供试品溶液及阴性对照溶液,进行检测。结果见图30。
结果表明,阴性溶液色谱图对待测峰的测定无干扰,表明该方法专属性好。
5.2精密度考察
取对照品溶液连续进样6次,记录紫丁香苷的峰面积,计算RSD值。见下表32。
表32精密度考察结果
结果表明,精密度考察中紫丁香苷的峰面积RSD值为0.14%,该仪器精密度良好。
5.3线性关系
精密称取紫丁香苷对照品适量,加甲醇制备成浓度为89.1072μg/ml和161.8091μg/ml的对照品储备液。分别进样1μl、2μl、4μl、6μl、8μl、10μl、12μl和8μl、10μl、12μl注入液相色谱仪,按拟定实验方法进行测定,分析得到峰面积,以对照品进样量(μg)为横坐标X,相应的峰面积为纵坐标Y,绘制响应曲线。见图31和下表33。
表33紫丁香苷标准曲线分析结果
结果表明:紫丁香苷进样量范围在89.1072~1941.6228μg,线性关系为Y=2626.2977X+12037.4196,R
5.4稳定性实验
取同一供试品溶液(批号:CWJBT180918),分别在0,3,6,9,12,24h测定紫丁香苷色谱峰面积。见下表34。
表34紫丁香苷稳定性实验结果
结果表明,在本实验条件下,紫丁香苷的峰面积RSD值为1.69%,供试品溶液在24小时内稳定性良好。
5.5重复性
取同一供试品(批号:CWJBT180918)0.5g,精密称定6份,由同一操作人员按照拟定的方法制成供试品溶液,计算6份供试品的含量。见下表35。
表35重复性实验结果
结果表明,紫丁香苷含量的RSD值为1.74%,表明本方法重复性良好。
5.6中间精密度
取同一供试品(批号:CWJBT180918)由不同人员(A、B)在不同时间(T1、T2)制备供试品溶液分别在安捷伦1260、岛津LC-20AD、Waters e2695型高效液相色谱仪上进行分析,计算供试品中紫丁香苷的含量。见下表36和表37。
表36中间精密度实验结果—不同人员、不同时间
表37中间精密度实验结果—不同仪器
结果表明,紫丁香苷的含量由不同人员、不同时间测定的RSD值为0.71%,不同仪器测定的RSD值为1.72%,表明本方法中间精密度良好。
5.7准确度
取已知含量的供试品(批号CWJBT180918,紫丁香苷的含量0.814%)0.25g,共6份,精密称定,分别精密加入一定量的紫丁香苷对照品(纯度:95.2%),按拟定的方法进行供试品溶液的制备并测定,计算回收率。见下表38。计算公式如下:
表38加样回收实验结果
结果表明,紫丁香苷加样回收率在100.84%~103.55%,回收率结果的RSD值为1.11%,本方法准确度良好。
5.8耐用性考察
采用不同品牌C18色谱柱(Agilent 5TC-C18(2)250*4.6mm、岛津Shim-pack GIST4.6*250mm、Diamosil 5um C18(2)250*4.6mm)对同一供试品(批号:CWJBT180918)进行检测。见下表39。
表39耐用性考察结果
结果表明,色谱柱耐用性考察中,紫丁香苷测量结果的RSD值为1.66%,本方法色谱柱耐用性良好。
6.样品含量测定验证
以18批刺五加对照提取物为供试品,按拟定的方法制备供试品溶液并测定,记录峰面积,计算紫丁香苷的含量。见图32和下表40。
表40 18批刺五加对照提取物紫丁香苷含量测定结果
结果表明,18批刺五加对照提取物紫丁香苷的含量实测范围为0.47~1.20%,平均含量为0.80%,SD为0.22%。平均值的70~130%限量范围为含紫丁香苷(C
结合以上数据,确定含量限度为:刺五加对照提取物按干燥瓶计算,含紫丁香苷(C
以上所述,仅是本发明的较佳实施例,并非对本发明做任何形式上的限制,凡是依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化,均落入本发明的保护范围之内。
- 刺五加对照提取物的制备工艺及其质量控制方法
- 地榆及地榆炭对照提取物的制备工艺及其质量控制方法