一种提高L-赖氨酸产量的大肠杆菌工程菌的构建方法及应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及一种提高L-赖氨酸产量的大肠杆菌工程菌的构建方法及应用,属于生物工程技术领域。
背景技术
L-赖氨酸参与生物体中的蛋白质合成、能量代谢和脂肪代谢,是参与人体代谢平衡的八大必需氨基酸之一。目前商品化的L-赖氨酸主要以L-赖氨酸盐酸盐和L-赖氨酸硫酸盐的形式广泛应用于饲料、食品及医药行业中。微生物发酵法凭借其污染小、产率高、成本低的优势,广泛应用于L-赖氨酸工业化生产。
谷氨酸棒杆菌(Corynebacterium glutamicum)及其亚种的突变菌是早期应用于L-赖氨酸工业生产的菌株。目前,国内外用于工业化生产L-赖氨酸的菌株多为谷氨酸棒杆菌(Coryneb acterium glutamicum)及其亚种和大肠杆菌(Escherichia coli)的改造型菌株。
在谷氨酸棒杆菌(Corynebacterium glutamicum)和大肠杆菌(Escherichiacoli)中,二氨基庚二酸(dap)途径是其主要的L-赖氨酸生物合成途径。二氨基庚二酸(dap)途径包含多个酶促反应,增强L-赖氨酸合成过程中关键酶的表达可以促进L-赖氨酸的合成,如中国发明专利CN108504617A(申请号:201810313577.6)公开了一种高产L-赖氨酸的大肠杆菌重组菌及其构建方法,公布了一种通过增强大肠杆菌重组菌株中meso-DAP合成能力来提高菌株积累L-赖氨酸的方法。
大肠杆菌中的L-赖氨酸主要参与蛋白质的合成以及通过cadA和ldcC基因编码的L-赖氨酸脱羧酶I和Ⅱ来催化分解生成戊二胺和二氧化碳。目前,利用表达质粒介导的氨基酸合成途径中关键性酶基因的过表达是对L-赖氨酸生产菌进行基因改造的主要手段。中国专利文献CN104878034A(申请号:201510185330.7)公开了L-赖氨酸基因工程生产菌,主要是增强L-赖氨酸合成途径的相关基因,弱化分支代谢途径,筛选获得一株L-赖氨酸高产菌种,构建方法为敲除原始菌株中与L-赖氨酸代谢途径相关的基因,所述敲除的基因为cadA、ldcC、gltI、thrC、cynT、thrL和maeB;增强所述基因敲除菌株中与L-赖氨酸代谢途径相关的基因,获得基因增强菌株,所述增强的基因为ybjE和cyo operon。
然而通过减少L-赖氨酸参与蛋白质的合成,从而提高L-赖氨酸产量的方法,可能会因为减少了L-赖氨酸参与蛋白质的合成,对菌体本身产生某些副作用,影响菌体的正常生长代谢,因此该方法极少报道。
发明内容
本发明针对现有技术的不足,提供了一种提高L-赖氨酸产量的大肠杆菌工程菌的构建方法及应用。
发明人研究了多种实验方案,最终得到了本发明的技术方案:敲除大肠杆菌基因组中的5个赖氨酸tRNA基因,构建大肠杆菌工程菌。本发明首次发现利用该方法构建的大肠杆菌工程菌相较于原始菌菌株,不仅L-赖氨酸的产量提高,菌体生物量也有提高。
本发明的技术方案如下:
基因在制备产L-赖氨酸的大肠杆菌工程菌中的应用,所述基因的核苷酸序列如SEQ ID NO.1所示,具体为大肠杆菌(Escherichia coli)中敲除核苷酸序列如SEQ ID NO.1所示的基因得到大肠杆菌工程菌。
根据本发明优选的,上述应用中,大肠杆菌(Escherichia coli)为大肠杆菌CGMCC1.366。
根据本发明优选的,上述大肠杆菌工程菌构建方法,包括步骤如下:
(1)以大肠杆菌(Escherichia coli)基因组DNA为模板,PCR扩增敲除基因的上下游基因片段作为同源臂序列,上游同源臂t-Lys1核苷酸序列如SEQ ID NO.2所示,下游同源臂t-Lys2核苷酸序列如SEQ ID NO.3所示;敲除基因的核苷酸序列如SEQ ID NO.1所示;
(2)通过PCR扩增质粒pKD13的抗性标签基因片段FRT-Kan-FRT,核苷酸序列如SEQID NO.4所示;
(3)利用无缝克隆技术将步骤(1)制得的上游同源臂t-Lys1、下游同源臂t-Lys2片段与步骤(2)制得的FRT-Kan-FRT片段与线性化载体pET-28a(+)连接,制得重组质粒;
(4)制备大肠杆菌感受态细胞,将质粒pKD46转化入感受态细胞,得到重组菌株E.coli-pKD46;
(5)将步骤(4)获得的重组菌株制备为感受态细胞,将步骤(3)制得的重组质粒转化入感受态细胞,得到重组菌株E.coli-kanR;
(6)将步骤(5)获得的重组菌株制备为感受态细胞,将质粒pCP20转化入感受态细胞,即得大肠杆菌工程菌E.coli-ΔtLys。
根据本发明优选的,所述步骤(1)中,PCR扩增以大肠杆菌(Escherichia coli)的基因组DNA为模板,获得上游同源臂t-Lys1,核苷酸序列如SEQ ID NO.2所示;
PCR扩增引物的核苷酸序列如下:
t-Lys1-F:gtggtggtggtggtgctcgagGTGCAGGATAAATCCCGCC SEQ ID NO.5;
t-Lys1-R:ggtccacggagaattcACTTTTTCGTTGCTTTCGGTTT SEQ ID NO.6。
根据本发明优选的,PCR扩增的反应体系如下,总体系为50μl:
PCR扩增程序如下:
95℃预变性5min;94℃变性30sec,55℃退火30sec,72℃延伸30sec,30个循环;72℃延伸10min,4℃保存。
根据本发明优选的,所述步骤(1)中,PCR扩增以大肠杆菌(Escherichia coli)的基因组DNA为模板,获得下游同源臂t-Lys2,核苷酸序列如SEQ ID NO.3所示;
PCR扩增引物的核苷酸序列如下:
t-Lys2-F cgagctcggtaccATGTAAAAAAGCGCCCTAAAGG SEQ ID NO.7;
t-Lys2-R:cagcaaatgggtcgcggatccATGCTTAACGGCGTCGGC SEQ ID NO.8。
根据本发明优选的,PCR扩增的反应体系如下,总体系为50μl:
PCR扩增程序如下:
95℃预变性5min;94℃变性30sec,55℃退火30sec,72℃延伸30sec,30个循环;72℃延伸10min,4℃保存。
根据本发明优选的,所述步骤(2)中,PCR扩增模板为质粒pKD13的DNA,获得带有FRT位点的卡那霉素抗性片段FRT-Kan-FRT,核苷酸序列如SEQ ID NO.4所示;
PCR扩增引物的核苷酸序列如下:
Kan-F:aagtGAATTCTCCGTGGACCTGCA SEQ ID NO.9;
Kan-R:tttacatGGTACCGAGCTCGGATCCG SEQ ID NO.10。
根据本发明优选的,PCR扩增的反应体系如下,总体系为50μl:
所述的PCR扩增程序如下:
95℃预变性5min;94℃变性30sec,55℃退火30sec,72℃延伸2min,30个循环;72℃延伸10min,4℃保存。
根据本发明优选的,所述步骤(3)中,多片段无缝克隆体系如下,总体系为10μl:
所述的无缝克隆程序如下:
多片段重组反应,50℃,15min;降至4℃或立即置于冰上冷却。
根据本发明优选的,所述步骤(4)的具体步骤如下:
(i)挑取大肠杆菌(Escherichia coli)单菌落,在种子培养基中培养至菌体浓度OD
(ii)将pKD46质粒高压电击转化至步骤(i)制得的感受态细胞中,移入液体复苏培养基,在28~32℃培养12~16h后,筛选,即得重组菌株E.coli-pKD46。
进一步优选的,所述步骤(i)中,种子培养基,每升组分如下:
蛋白胨8~12g、酵母粉4~6g、氯化钠8~12g,余量水。
进一步优选的,所述步骤(i)中,电转缓冲液,每升组分如下:
山梨醇85~96g、甘露醇85~96g、甘油95~105mL,余量水。
进一步优选的,所述步骤(i)中,高压电击转化的条件为:1800V电击5ms。
进一步优选的,所述步骤(ii)中液体复苏培养基,每升组分如下:
蛋白胨8~12g、酵母粉4~6g、氯化钠8~12g、山梨醇85~96g、甘露醇65~73g,余量水。
根据本发明优选的,所述步骤(5)的具体步骤如下:
①挑取步骤(4)获得的大肠杆菌(Escherichia coli)单菌落,在种子培养基中培养至菌体浓度OD
②将步骤(3)制得的重组质粒高压电击转化至步骤①制得的感受态细胞中,移入液体复苏培养基,在28~32℃培养12~16h后,筛选,即得重组菌株E.coli-kanR。
进一步优选的,所述步骤①中,种子培养基,每升组分如下:
蛋白胨8~12g、酵母粉4~6g、氯化钠8~12g,余量水。
进一步优选的,所述步骤①中,电转缓冲液,每升组分如下:
山梨醇85~96g、甘露醇85~96g、甘油95~105mL,余量水。
进一步优选的,所述步骤①中,高压电击转化的条件为:1800V电击5ms。
进一步优选的,所述步骤②中液体复苏培养基,每升组分如下:
蛋白胨8~12g、酵母粉4~6g、氯化钠8~12g、山梨醇85~96g、甘露醇65~73g,余量水。
根据本发明优选的,所述步骤(6)的具体步骤如下:
Ⅰ挑取步骤(5)获得的大肠杆菌(Escherichia coli)单菌落,在种子培养基中培养至菌体浓度OD
Ⅱ将质粒pCP20高压电击转化至步骤Ⅰ制得的感受态细胞中,移入液体复苏培养基,在28~32℃培养12~16h后,筛选,即得大肠杆菌工程菌E.coli-ΔtLys。
根据本发明进一步优选的,所述步骤Ⅰ中,种子培养基,每升组分如下:
蛋白胨8~12g、酵母粉4~6g、氯化钠8~12g,余量水。
根据本发明进一步优选的,所述步骤Ⅰ中,电转缓冲液,每升组分如下:
山梨醇85~96g、甘露醇85~96g、甘油95~105mL,余量水。
根据本发明进一步优选的,所述步骤Ⅰ中,高压电击转化的条件为:1800V电击5ms。
根据本发明进一步优选的,所述步骤Ⅱ中液体复苏培养基,每升组分如下:
蛋白胨8~12g、酵母粉4~6g、氯化钠8~12g、山梨醇85~96g、甘露醇65~73g,余量水。
上述构建方法制得的大肠杆菌工程菌E.coli-ΔtLys在生产L-赖氨酸中的应用。
有益效果
本发明提供一种提高L-赖氨酸产量的大肠杆菌工程菌的构建方法,发明人通过研究发现大肠杆菌中赖氨酸tRNA基因敲除后,不仅能够提高L-赖氨酸的产量,而且菌体的生物量也有提高。构建的大肠杆菌工程菌,与原始菌相比,大肠杆菌工程菌株的L-赖氨酸产量提高,降低生产成本,菌体生物量也有一定提高。
附图说明
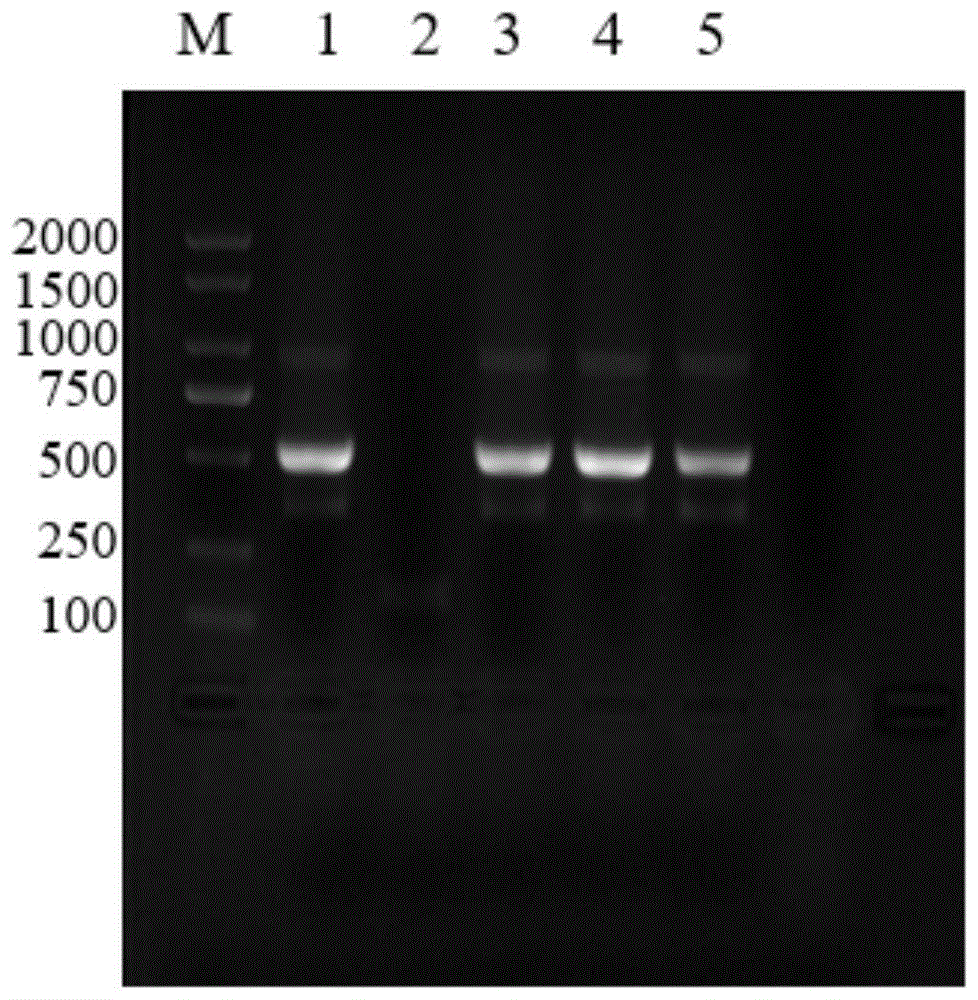
图1为实施例1中基因片段同源臂t-Lys1、同源臂t-Lys2的电泳图;
图中:泳道1为t-Lys1,泳道3、4、5为t-Lys2。
图2为实施例1中基因片段FRT-Kan-FRT电泳图。
图3为实施例3中基因片段bet电泳图。
图4为实施例4中基因片段t-Lys1-FRT-Kan-FRT-t-Lys2电泳图。
图5为原始菌与大肠杆菌工程菌24h生长曲线。
具体实施方式
下面结合实施例对本发明的技术方案做进一步阐述,但本发明所保护范围不限于此。
生物材料的来源
大肠杆菌CGMCC 1.366可由中国微生物菌种保藏管理委员会普通微生物中心获得。
质粒pET-28a(+)来源于武汉淼灵生物科技有限公司。
质粒pKD13购自杭州宝赛生物科技有限公司。
实施例中使用的药品及试剂,若无特殊说明,则为以上市普通产品,实施例中未详加说明的内容,均按本领域现有技术
实施例1
基因敲除片段构建
(i)提取大肠杆菌(Escherichia coli)CGMCC 1.366基因组DNA,以该基因组DNA为模板,进行PCR扩增,得到同源臂t-Lys,核苷酸序列如SEQ ID NO.2所示;
所述的PCR引物序列如下:
t-Lys1-F:gtggtggtggtggtgctcgagGTGCAGGATAAATCCCGCC SEQ ID NO.5;
t-Lys1-R:ggtccacggagaattcACTTTTTCGTTGCTTTCGGTTT SEQ ID NO.6;
所述的PCR扩增体系,见表1:
表1
所述的PCR扩增程序如下:
95℃预变性5min;94℃变性30sec,55℃退火30sec,72℃延伸30sec,30个循环;72℃延伸10min,4℃保存;
提取大肠杆菌(Escherichia coli)基因组DNA,以该基因组DNA为模板,进行PCR扩增,得到同源臂t-Lys2,核苷酸序列如SEQ ID NO.3所示;
所述的PCR引物序列如下:
t-Lys2-F cgagctcggtaccATGTAAAAAAGCGCCCTAAAGG SEQ ID NO.7;
t-Lys2-R:cagcaaatgggtcgcggatccATGCTTAACGGCGTCGGC SEQ ID NO.8;
所述的PCR扩增体系,见表2:
表2
所述的PCR扩增程序如下:
95℃预变性5min;94℃变性30sec,55℃退火30sec,72℃延伸30sec,30个循环;72℃延伸10min,4℃保存;
琼脂糖凝胶电泳检验PCR产物,长度为约500bp,见图1,与理论值500bp接近,使用SanPrep柱式DNA胶回收试剂盒(上海生工)进行胶回收,回收产物-20℃保存,备用;
(ii)提取质粒pKD13的DNA,以DNA为模板,进行PCR扩增,得到FRT-Kan-FRT片段,核苷酸序列如SEQ ID NO.4所示;
所述的PCR引物序列如下:
Kan-F:aagtGAATTCTCCGTGGACCTGCA SEQ ID NO.9;
Kan-R:tttacatGGTACCGAGCTCGGATCCG SEQ ID NO.10;
所述的PCR扩增体系,见表3:
表3
所述的PCR扩增程序如下:
95℃预变性5min;94℃变性30sec,55℃退火30sec,72℃延伸2min,30个循环;72℃延伸10min,4℃保存;
琼脂糖凝胶电泳检验PCR产物,长度为约1300bp,见图2,与理论值1324bp接近,使用SanPrep柱式DNA胶回收试剂盒(上海生工)进行胶回收,回收产物-20℃保存,备用;(iii)使用无缝克隆试剂盒将步骤(i)制得的t-Lys1、t-Lys2片段与步骤(ii)制得的FRT-Kan-FRT片段与线性化载体pET-28a(+)连接,获得重组质粒。
所述的重叠无缝克隆连接体系见,表4:
表4
所述的无缝克隆扩增程序如下:
多片段重组反应,50℃,15min;降至4℃或立即置于冰上冷却,备用;
实施例2
制备大肠杆菌感受态
(i)挑取大肠杆菌(Escherichia coli)CGMCC 1.366单菌落,接种于10mL种子培养基中,37℃、220r/min,过夜培养;
种子培养基,每升组分如下:
蛋白胨10g、酵母粉5g、氯化钠10g,余量水。
(ii)取1mL上述菌液转接到100mL种子培养基中,37℃、220r/min培养至OD
(iii)将菌液转移至100mL离心管,冰浴15-20min,使菌体停止生长;
(iv)冰浴后4℃、5000g、5min离心,收集菌体;
(v)离心后的菌体用预冷的电转缓冲液(ETM)洗涤3次;
电转缓冲液,每升组分如下:
山梨醇91g、甘露醇91g、甘油100mL,余量水。
(vi)洗涤结束后,使用1000μL电转缓冲液重悬菌体;
(vii)将制备好的感受态细胞分装100μL每管,-80℃保存,备用。
实施例3
质粒pKD46电转化实施例2制备的大肠杆菌(Escherichia coli)感受态细胞
(i)首先利用核酸超微量分光光度计测定质粒pKD46浓度,达到300μg/mL浓度后1800V电击5ms,进行电转化,得到的细胞使用复苏培养基30℃复苏培养1h后,取100μL涂布在含100μg/mL氨苄霉素的LB固体培养基上,在30℃培养1天,筛选具有氨苄抗性的转化子,得到大肠杆菌重组菌E.coli-pKD46。
挑取在氨苄霉素LB固体培养基生长的单菌落为模板,以pKD46-F和pKD46-R为引物进行PCR扩增,扩增产物利用琼脂糖凝胶电泳进行验证;
所述的PCR引物序列如下:
pKD46-F:ATGAGTATTCAACATTTCCGTGTCG SEQ ID NO.11;
pKD46-R:TTACCAATGCTTAATCAGTGAGGC SEQ ID NO.12;
所述的PCR扩增体系为20μl,表5:
表5
所述的PCR扩增程序如下:
95℃预变性5min;94℃变性30sec,55℃退火45sec,72℃延伸4min,30个循环;72℃延伸10min,4℃保存;
琼脂糖凝胶电泳检验PCR产物,结果显示,使用引物pKD46-F和pKD46-R能够扩增出一条特异性基因条带,大小约为750bp,见图3,与理论值786bp接近,表明敲除框转化成功,得到大肠杆菌工程菌E.coli-pKD46。
液体复苏培养基,每升组分如下:
蛋白胨10g、酵母粉5g、氯化钠10g、山梨醇91g、甘露醇69.4g,余量水。
实施例4
实施例1制备的重组质粒电转化实施例3制备的大肠杆菌重组菌E.coli-pKD46
(i)首先利用核酸超微量分光光度计测定重组质粒浓度,达到300μg/mL浓度后1800V电击5ms,进行电转化,得到的细胞使用复苏培养基37℃复苏培养1h后,取100μL涂布在含100μg/mL氨苄霉素的LB固体培养基上,在37℃培养1天,筛选具有卡那霉素抗性的转化子,得到大肠杆菌重组菌E.coli-KanR。
挑取在卡那霉素LB固体培养基生长的单菌落为模板,t-Lys1-F和t-Lys2-R为引物进行PCR扩增,扩增产物利用琼脂糖凝胶电泳进行验证;
所述的PCR引物序列如下:
t-Lys1-F:gtggtggtggtggtgctcgagGTGCAGGATAAATCCCGCC SEQ ID NO.5;
t-Lys2-R:cagcaaatgggtcgcggatccATGCTTAACGGCGTCGGC SEQ ID NO.8;
所述的PCR扩增体系为20μl,表6:
表6
所述的PCR扩增程序如下:
95℃预变性5min;94℃变性30sec,55℃退火45sec,72℃延伸4min,30个循环;72℃延伸10min,4℃保存;
琼脂糖凝胶电泳检验PCR产物,结果显示,使用引物t-Lys1-F和t-Lys2-R能够扩增出一条特异性基因条带,大小约为2300bp,见图4,与理论值2324bp接近,表明敲除框转化成功,得到大肠杆菌工程菌E.coli-KanR。
液体复苏培养基,每升组分如下:
蛋白胨10g、酵母粉5g、氯化钠10g、山梨醇91g、甘露醇69.4g,余量水。
实施例5
质粒pCP20电转化实施例4制备的得到大肠杆菌重组菌E.coli-KanR。
(i)首先利用核酸超微量分光光度计测定质粒pCP20浓度,达到300μg/mL浓度后1800V电击5ms,进行电转化,得到的细胞使用复苏培养基37℃复苏培养1h后,取100μL涂布在LB固体培养基上,在37℃培养1天,筛选转化子。
液体复苏培养基,每升组分如下:
蛋白胨10g、酵母粉5g、氯化钠10g、山梨醇91g、甘露醇69.4g,余量水。
实施例6
阳性菌落筛选
挑取实施例5阳性重组菌落(筛选转化子),依次接种到含100μg/mL氨苄霉素抗性和不含抗性的固体LB培养基中37℃培养过夜,培养完成后,挑取只在不含抗性的LB固体培养基生长的单菌落为模板,t-Lys1-F和t-Lys2-R为引物进行PCR扩增,扩增产物利用琼脂糖凝胶电泳进行验证;
所述的PCR引物序列如下:
t-Lys1-F:gtggtggtggtggtgctcgagGTGCAGGATAAATCCCGCC SEQ ID NO.5;
t-Lys2-R:cagcaaatgggtcgcggatccATGCTTAACGGCGTCGGC SEQ ID NO.8;
所述的PCR扩增体系为20μl,表7:
表7
所述的PCR扩增程序如下:
95℃预变性5min;94℃变性30sec,55℃退火45sec,72℃延伸4min,30个循环;72℃延伸10min,4℃保存;
琼脂糖凝胶电泳检验PCR产物,结果显示,使用引物t-Lys1-F和t-Lys2-R能够扩增出一条特异性基因条带,大小约为1100bp,与理论值1085bp接近,表明赖氨酸tRNA基因成功敲除,得到大肠杆菌工程菌E.coli-ΔtLys,该大肠杆菌工程菌敲除了基因核苷酸序列如SEQ ID NO.1所示。
实施例7
L-赖氨酸发酵测试
将制备的实施例6制备的大肠杆菌工程菌E.coli-ΔtLys、原始菌CGMCC 1.366分别接种至100mL LB培养基(蛋白胨10g/L,酵母膏5g/L,NaCl 10g/L,余量水)中220rpm 37℃下进行种子培养12h,其后按体积百分比2%接种量分别接种至100mL发酵培养基(葡萄糖100g/L,蛋白胨20g/L,玉米浆30mL/L,尿素5g/L,(NH4)
表8大肠杆菌工程菌与原始菌L-赖氨酸的产量
结果显示与原始菌相比,在发酵50h,大肠杆菌工程菌发酵液中L-赖氨酸的含量达到3.2±0.15g/L,而原始菌在50h时的产量基本为2.48±0.08g/L,大肠杆菌工程菌L-赖氨酸产量较原始菌提高了29%;与原始菌相比,大肠杆菌工程菌株生物量也有一定提高;这表明敲除大肠杆菌5个赖氨酸tRNA基因后可以提高大肠杆菌L-赖氨酸发酵产酸水平,并且与原始菌相比,大肠杆菌工程菌体生物量也有一定提高。
发明人也进行了敲除谷氨酸棒状杆菌CICC 23604中赖氨酸tRNA基因序列的实验,构建谷氨酸棒状杆菌工程菌未成功,发明人发现敲除谷氨酸棒状杆菌赖氨酸tRNA基因序列后,菌体不生长。进一步说明并不是所有的微生物菌体中敲除赖氨酸tRNA基因序列,都可以提高L-赖氨酸的产量,有的微生物菌体中敲除赖氨酸tRNA基因序列,会影响菌体的正常生长。
本发明提供一种提高L-赖氨酸产量的大肠杆菌工程菌的构建方法,发明人通过研究发现赖氨酸tRNA基因敲除后,能够增加L-赖氨酸产量,菌体的生物量也有提高。构建的大肠杆菌工程菌,与原始菌相比,大肠杆菌工程菌株的L-赖氨酸产量提高,降低生产成本,菌体生物量也有一定提高。