电化学表面相图计算方法及系统
文献发布时间:2024-04-18 19:52:40
技术领域
本发明涉及电化学技术领域,具体涉及一种电化学表面相图计算方法及系统。
背景技术
我国的碳中与碳达峰政策指出了回收利用空气或处理工业排放的CO2之重要性,利用电化学方法处理CO2变成其他可利用的资源或捕捉封存变得越来越重要。在电化学反应过程中,各物种与表面的作用非常复杂,不容易通过表征手段获取。理论计算得到的表面相图就成为在电化学条件下洞悉和理解表面吸附物分布情况的合理手段。表面相图(Surface Phase Diagram)是一种用于描述材料表面上各种不同化学物种(如分子、原子、离子等)之间相互作用和相对稳定性的图形表示方式,可以帮助我们了解材料表面在不同条件下的相变和稳定性。表面相图通常是一个二维图表,其纵轴表示吸附物种的吉布斯自由能,而横轴表示不同的电压。在这个二维图表中,各种不同化学物种的相对稳定性可以用颜色或曲线等方式表示,例如,某种化学物种在某一覆盖度下稳定时,一般处于在相图中能量较低的位置。通过表面相图找到能量较低的构形或者物相就可以找出最稳定的构形,从而判断出,在电化学电压下,材料表面的吸附物种的覆盖情况。通过研究表面相图,可以预测在不同条件下材料表面的相变和化学反应,从而指导材料的设计和制备。
一般可以使用量子化学计算方法来计算表面相图,但由于量子化学计算的复杂性,通过手动寻找表面不同的吸附位点和组合确定能量最低的分子构形或者排列组合,这种手动排列计算表面相图的方法耗时、繁杂且容易出错。尤其是在合金或复杂氧化物表面,其独立的吸附位点众多,手动搭建模型难以一一考虑并快速搭建;且可能性众多,需要依次提交量子化学计算,并等待计算结果,这些步骤繁杂且操作量大。
相关技术中,公布号为CN112800609A的专利申请文献提出的获取材料相图的方法中,以第一性原理计算方法为基础结合模拟或近似算法计算材料在不同温度和压强下的吉布斯自由能;根据材料在不同温度和压强下的吉布斯自由能的参数生成材料的初始相图;选取至少一个实验点数据对初始相图进行修正。该方案使用准静态随机SQS方法模拟考虑了磁性对材料能量影响,设置了设置惩罚函数以加速相图计算,获取的是块体材料的相图,没有考虑电化学环境下材料与溶液或分子的相互作用。
发明内容
本发明所要解决的技术问题在于如何提高电化学表面相图计算的效率。
本发明通过以下技术手段解决上述技术问题的:
第一方面,本发明提出了一种电化学表面相图计算方法,所述方法包括:
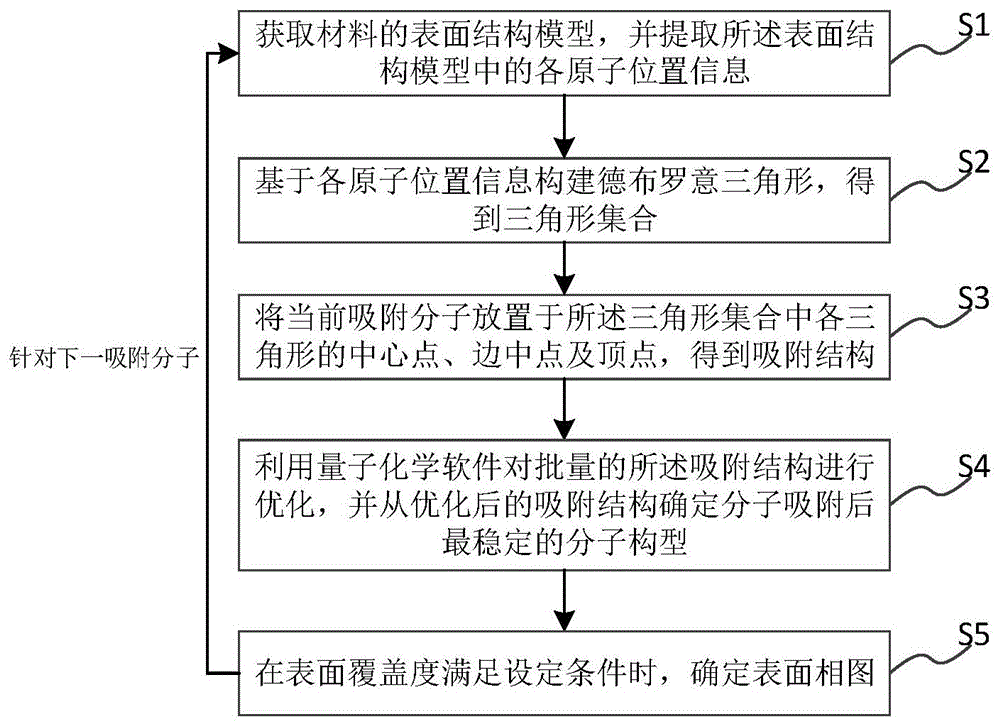
S1、获取材料的表面结构模型,并提取所述表面结构模型中的各原子位置信息;
S2、基于各原子位置信息构建德布罗意三角形,得到三角形集合;
S3、将当前吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构,每个所述吸附结构中的吸附分子间距离大于设定阈值且任一位置仅有一个吸附分子;
S4、利用量子化学软件对批量的所述吸附结构进行优化,并从优化后的吸附结构确定分子吸附后最稳定的分子构型;
S5、针对下一吸附分子重复执行步骤S1~S4,直至表面覆盖度满足设定条件时,确定表面相图。
进一步地,所述提取所述表面结构模型中的各原子位置信息,包括:
采用Python ase包的Atoms方法,提取所述表面结构模型中的各原子位置信息。
进一步地,所述基于各原子位置信息构建德布罗意三角形,得到三角形集合,包括:
基于各所述原子位置信息构建点集;
采用德布罗意三角化方法对所述点集进行处理,以最近邻的三点形成三角形,且各线段皆不相交,构建所述三角形集合;
其中,所述三角形集合中的三角形无重叠,且任何一个三角形都满足一个三角形的外接圆中不包含任何其他点。
进一步地,所述将吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构,包括:
以三角形三边的中点作为桥位吸附、三角形的中心点作为空洞吸附、三角形的顶点作为顶位吸附,将吸附分子置于所述三角形集合中各三角形的中心点、边中点及顶点,得到所述吸附结构。
进一步地,在至少添加两个吸附分子时,在所述将吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构之后,所述方法还包括:
将至少两个吸附分子的几何位置信息置于第一列表中;
利用python的np.unique方法对所述列表中重复的几何位置信息进行排除。
进一步地,在至少添加两个吸附分子时,在所述将吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构之后,所述方法还包括:
计算某吸附结构中两个吸附分子之间的距离;
在两个吸附分子之间的距离小于设定阈值时,则删除该吸附结构。
进一步地,所述利用量子化学软件对批量的所述吸附结构进行优化,并从优化后的吸附结构确定分子吸附后最稳定的分子构型,包括:
将所述吸附结构存储于文件夹中,并将文件夹中的吸附结构批量导入所述量子化学软件中进行优化;
在识别到优化结束标识字符时,输出所述吸附结构的位置及能量至第二列表中;
比较各所述吸附结构的能量,将能量最低的吸附结构的位置作为最稳定的分子构型。
进一步地,所述量子化学软件采用VASP量子化学软件,所述优化结束标识字符为OUTCAR文件中捕捉到的reached required accuracy字段。
第二方面,本发明提出了一种电化学表面相图计算系统,所述系统包括:
获取模块,用于获取材料的表面结构模型,并提取所述表面结构模型中的各原子位置信息;
三角形集合构建模块,用于基于各原子位置信息构建德布罗意三角形,得到三角形集合;
吸附结构构建模块,用于将当前吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构;
优化模块,用于利用量子化学软件对批量的所述吸附结构进行优化,并从优化后的吸附结构确定分子吸附后最稳定的分子构型;
确定模块,用于针对下一吸附分子重复执行获取模块动作,直至表面覆盖度满足设定条件时,确定表面相图。
第三方面,本发明提出了一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序被处理器执行时,实现如上所述的电化学表面相图计算方法。
本发明的优点在于:
(1)本发明基于表面结构模型中各原子位置信息构建德布罗意三角形,得到三角形集合,将当前吸附分子放置于三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构,利用量子化学软件对批量的吸附结构进行优化,并从优化后的吸附结构确定分子吸附后最稳定的分子构型,本发明通过自动排列可以得到表面不同位点吸附分子的不重复排列组合,从而可以批量输入至量子化学计算软件进行计算,从计算的文件中获取最低能量的分子构型并不断重复上述流程进行表面相图的绘制,减少量子化学手段的计算量,解决了使用量子化学计算方法来计算表面相图存在的步骤繁杂且操作量大的问题,可大幅提高表面相图计算的效率,且方法简单易掌握。同时对于实际产业应用具有重要的意义,可为实验或生产人员提供一个不同电压下表面物种的热力学相图从而指导生产过程,通过所计算的表面相图可以帮助了解不同电压下的稳定电极表面是什么吸附物占主导,从而设计催化剂或在生产过程中设计合适的电压提高产品的选择性或产率。
(2)考虑在吸附一个或几个分子的情况下,进行其他可能吸附位置的分子排布时,有些吸附位置会被重复考虑,本发明利用python的np.unique方法对重复吸附几何位置信息进行排除,使用去重后可以降低后续量子化学软件的计算量,从而节约计算成本。
(3)本发明在某一吸附结构的两个吸附分子之间的距离小于设定阈值时,则删除该吸附结构,以只保留分子间距较大的构形,确保得到一个保证了分子间具有一定距离且不重复的表面吸附结构。
本发明附加的方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
图1是本发明实施例提出的电化学表面相图计算方法的流程示意图;
图2是本发明实施例提出的电化学表面相图计算方法的原理框图;
图3是本发明中Cu原子构成的复杂表面示意图;
图4是本发明中获取图3所示复杂表面的吸附位点示意图;
图5是本发明中计算得到的某Cu的表面相图;
图6是本发明实施例提出电化学表面相图计算系统的结构示意图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
如图1所示,本发明第一实施例提出了一种电化学表面相图计算方法,所述方法包括以下步骤:
S1、获取材料的表面结构模型,并提取所述表面结构模型中的各原子位置信息;
需要说明的是,基于所研究的分子或材料的结构,可通过化学建模软件搭建一个合理的表面结构模型。
S2、基于各原子位置信息构建德布罗意三角形,得到三角形集合;
S3、将当前吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构,每个所述吸附结构中的吸附分子间距离大于设定阈值且任一位置仅有一个吸附分子;
需要说明的是,以Cu原子构成的复杂表面为例,吸附分子可为用户设置的任何分子。
S4、利用量子化学软件对批量的所述吸附结构进行优化,并从优化后的吸附结构确定分子吸附后最稳定的分子构型;
S5、针对下一吸附分子重复执行步骤S1~S4,直至表面覆盖度满足设定条件时,确定表面相图。
本实施例通过自动排列可以得到表面不同位点吸附分子的不重复排列组合,从而可以批量输入至量子化学计算软件进行计算,从计算的文件中获取最低能量的分子构型并不断重复上述流程进行表面相图的绘制,解决了使用量子化学计算方法来计算表面相图存在的步骤繁杂且操作量大的问题,且简单易掌握。
在一实施例中,所述步骤S1中,提取所述表面结构模型中的各原子位置信息,具体包括:
采用Python ase包的Atoms方法,提取所述表面结构模型中的各原子位置信息。
需要说明的是,本实施例可定义了一个函数用于选择表结构模型的原子,并利用现有的ase软件包内的Atoms方法获取表面原子的几何位置。
在一实施例中,所述步骤S2:基于各原子位置信息构建德布罗意三角形,得到三角形集合,具体包括以下步骤:
S21、基于各所述原子位置信息构建点集;
S22、采用德布罗意三角化方法对所述点集进行处理,以最近邻的三点形成三角形,且各线段皆不相交,构建所述三角形集合;
其中,所述三角形集合中的三角形无重叠,且任何一个三角形都满足一个三角形的外接圆中不包含任何其他点。
需要说明的是,本实施例利用scipy包的Delaunay方法将点集中的表面原子做Delaunay化,得到一系列互不重叠且互不交叠的三角形。
在一实施例中,所述步骤S3:将吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构,具体包括:
以三角形三边的中点作为桥位吸附、三角形的中心点作为空洞吸附、三角形的顶点作为顶位吸附,将吸附分子置于所述三角形集合中各三角形的中心点、边中点及顶点,得到所述吸附结构。
在一实施例中,如图2所示,在至少添加两个吸附分子时,在步骤S3:将吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构之后,所述方法还包括:
将至少两个吸附分子的几何位置信息置于第一列表中;
利用python的np.unique方法对所述列表中重复的几何位置信息进行排除。
需要说明的是,考虑在吸附一个或几个分子的情况下,进行其他可能吸附位置的分子排布时,有些吸附位置会被重复考虑,本实施例利用python的np.unique方法对已放置吸附分子的几何位置信息进行排除,使用去重后可以降低后续量子化学软件的计算量,从而节约计算成本。
在一实施例中,在至少添加两个吸附分子时,在步骤S3:将吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构之后,所述方法还包括:
计算某吸附结构中两个吸附分子之间的距离;
在两个吸附分子之间的距离小于设定阈值时,则删除该吸附结构。
需要说明的是,本实施例所述设定阈值为本领域技术人员经过大量实验得出的一个用于吸附分子间距离进行比较的常数,该设定阈值也可以通过化学直觉预先设置或测试得到,若吸附分子间的距离小于这个设定阈值则删除这个构形即只保留分子键距较大的构形。
本在某一吸附结构的两个吸附分子之间的距离小于设定阈值时,则删除该吸附结构,确保得到一个保证了分子间具有一定距离且不重复的表面吸附结构。
在一实施例中,所述步骤S4:利用量子化学软件对批量的所述吸附结构进行优化,并从优化后的吸附结构确定分子吸附后最稳定的分子构型,具体包括以下步骤:
S41、将所述吸附结构存储于文件夹中,并将文件夹中的吸附结构批量导入所述量子化学软件中进行优化;
S42、在识别到优化结束标识字符时,输出所述吸附结构的位置及能量至第二列表中;
S43、比较各所述吸附结构的能量,将能量最低的吸附结构的位置作为最稳定的分子构型。
在一实施例中,所述量子化学软件采用VASP量子化学软件,所述优化结束标识字符为OUTCAR文件中捕捉到的reached required accuracy字段。
需要说明的是,本实施例在得到吸附结构会输出到对应文件夹,再批量提交这些吸附结构到量子化学软件优化后,在识别到量子化学软件中对应的优化结束标识字符后,根据优化几何构型计算体系在不同pH和电位条件下的能量,获取吸附结构的位置及能量。通过建立一个包含各吸附结构位置和能量的列表,比较这些构型后得到能量最低的构型,将能量最低的构型下载,通过将吸附结构批量上传和下载操作可进一步节省时间。然后再次返回步骤S1中所建立三角形化的表面,针对下一个吸附分子重复执行流程,确定下一吸附分子最稳定的构型和位置,直至达到用户所期待的表面覆盖度,这样可以防止吸附分子导致的表面重构造成的影响。通过执行本实施例所提方法对如图3所示的铜表面的结构进行表面相图绘制,图4为针对铜表面结构确定的三角形,最终得到的铜表面的表面相图如图5所示。
此外,如图6所示,本发明第二实施例提出了一种电化学表面相图计算系统,所述系统包括:
获取模块10,用于获取材料的表面结构模型,并提取所述表面结构模型中的各原子位置信息;
三角形集合构建模块20,用于基于各原子位置信息构建德布罗意三角形,得到三角形集合;
吸附结构构建模块30,用于将当前吸附分子放置于所述三角形集合中各三角形的中心点、边中点及顶点,得到吸附结构,每个所述吸附结构中的吸附分子间距离大于设定阈值且任一位置仅有一个吸附分子;
优化模块40,用于利用量子化学软件对批量的所述吸附结构进行优化,并从优化后的吸附结构确定分子吸附后最稳定的分子构型;
确定模块50,用于针对下一吸附分子重复执行获取模块动作,直至表面覆盖度满足设定条件时,确定表面相图。
本实施例通过自动排列可以得到表面不同位点吸附分子的不重复排列组合,从而可以批量输入至量子化学计算软件进行计算,从计算的文件中获取最低能量的分子构型并不断重复上述流程进行表面相图的绘制,解决了使用量子化学计算方法来计算表面相图存在的步骤繁杂且操作量大的问题,且表面相图绘制准确度较高。
在一实施例中,所述获取模块10,具体用于采用Python ase包的Atoms方法,提取所述表面结构模型中的各原子位置信息。
在一实施例中,所述三角形集合构建模块20,具体用于:
基于各所述原子位置信息构建点集;
采用德布罗意三角化方法对所述点集进行处理,以最近邻的三点形成三角形,且各线段皆不相交,构建所述三角形集合;
其中,所述三角形集合中的三角形无重叠,且任何一个三角形都满足一个三角形的外接圆中不包含任何其他点。
在一实施例中,所述吸附结构构建模块30,具体用于:以三角形三边的中点作为桥位吸附、三角形的中心点作为空洞吸附、三角形的顶点作为顶位吸附,将吸附分子置于所述三角形集合中各三角形的中心点、边中点及顶点,得到所述吸附结构。
在一实施例中,所述系统还包括第一去重模块,具体用于:
将至少两个吸附分子的几何位置信息置于第一列表中;
利用python的np.unique方法对所述列表中重复的几何位置信息进行排除。
在一实施例中,所述系统还包括第二去重模块,具体用于:
计算某吸附结构中两个吸附分子之间的距离;
在两个吸附分子之间的距离小于设定阈值时,则删除该吸附结构。
在一实施例中,所述优化模块40,具体用于:
将所述吸附结构存储于文件夹中,并将文件夹中的吸附结构批量导入所述量子化学软件中进行优化;
在识别到优化结束标识字符时,输出所述吸附结构的位置及能量至第二列表中;
比较各所述吸附结构的能量,将能量最低的吸附结构的位置作为最稳定的分子构型。
在一实施例中,所述量子化学软件采用VASP量子化学软件,所述优化结束标识字符为OUTCAR文件中捕捉到的reached required accuracy字段。
需要说明的是,本发明所述电化学表面相图计算系统的其他实施例或具有实现方法可参照上述各方法实施例,此处不再赘余。
此外,本发明第三实施例提出了一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序被处理器执行时,实现如上实施例一所述的电化学表面相图计算方法。
需要说明的是,在流程图中表示或在此以其他方式描述的逻辑和/或步骤,例如,可以被认为是用于实现逻辑功能的可执行指令的定序列表,可以具体实现在任何计算机可读介质中,以供指令执行系统、装置或设备(如基于计算机的系统、包括处理器的系统或其他可以从指令执行系统、装置或设备取指令并执行指令的系统)使用,或结合这些指令执行系统、装置或设备而使用。就本说明书而言,“计算机可读介质”可以是任何可以包含、存储、通信、传播或传输程序以供指令执行系统、装置或设备或结合这些指令执行系统、装置或设备而使用的装置。计算机可读介质的更具体的示例(非穷尽性列表)包括以下:具有一个或多个布线的电连接部(电子装置),便携式计算机盘盒(磁装置),随机存取存储器(RAM),只读存储器(ROM),可擦除可编辑只读存储器(EPROM或闪速存储器),光纤装置,以及便携式光盘只读存储器(CDROM)。另外,计算机可读介质甚至可以是可在其上打印所述程序的纸或其他合适的介质,因为可以例如通过对纸或其他介质进行光学扫描,接着进行编辑、解译或必要时以其他合适方式进行处理来以电子方式获得所述程序,然后将其存储在计算机存储器中。
应当理解,本发明的各部分可以用硬件、软件、固件或它们的组合来实现。在上述实施方式中,多个步骤或方法可以用存储在存储器中且由合适的指令执行系统执行的软件或固件来实现。例如,如果用硬件来实现,和在另一实施方式中一样,可用本领域公知的下列技术中的任一项或他们的组合来实现:具有用于对数据信号实现逻辑功能的逻辑门电路的离散逻辑电路,具有合适的组合逻辑门电路的专用集成电路,可编程门阵列(PGA),现场可编程门阵列(FPGA)等。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。在本发明的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。