一种拉坦前列素高级中间体的制备方法及其用途
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于有机合成技术领域,特别涉及一种拉坦前列素高级中间体的制备方法及其用途。
背景技术
前列腺素(Prostaglandins, PGs)是一类有生理活性的不饱和脂肪酸,广泛分布于身体各组织和体液中,1934年,由von Euler教授发现并证明在精子、人的生殖腺和某些动物的提取物中有平滑肌刺激的存在。因为它被认为来自前列腺,所以被von Euler教授命名为前列腺素。在1957年,Bergström教授和他的同事首次在羊精囊中分离得到两种前列腺素的结晶纯品:PGE
因此,对这些天然化合物进行化学修饰,以提高其化学和代谢稳定性是非常可取的。例如,拉坦前列素就是对PGF
前列腺素同样在疼痛和发热的起源中起作用,还能调节炎症、血压、凝血、生殖功能与睡眠等。由于其广泛的生物活性,被广泛认为是迄今发现的最重要的天然分离株之一和独特的结构。到目前为止,已有20多种前列腺素类似物在世界范围内销售。近50年来,开发高效的PGs合成方法一直是合成化学家的目标。
近年来,虽然药物拉坦前列素已有部分制备报道。但是,已报道的拉坦前列素制备方法复杂,而且合成路线冗长,效率很低,不具备进一步放大和实用价值。
发明内容
为了解决拉坦前列素制备方法复杂、效率低的问题,本发明提供了一种拉坦前列素高级中间体的制备方法,该方法步骤简单、效率高,利于拉坦前列素的大量生产,也为拉坦前列素的制备提供一种新的途径。
本发明还提供了一种拉坦前列素高级中间体的制备方法在合成拉坦前列素中的用途。
本发明通过以下技术方案实现:
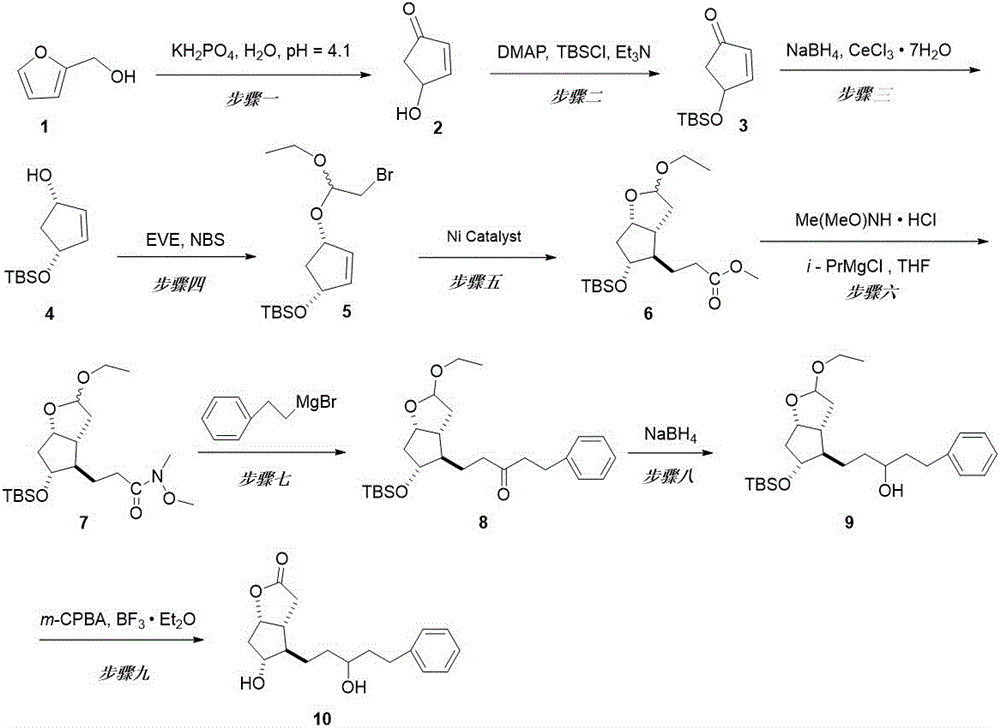
本发明提供了一种拉坦前列素高级中间体的制备方法,所述拉坦前列素高级中间体为化合物10,所述化合物10的结构式如下:
;
所述制备方法包括:
将糠醇和磷酸二氢钾溶解于水进行氧化重排,获得烯酮;
采用叔丁基二甲基硅基将所述烯酮的羟基保护起来,获得化合物3;
通过卢奇还原反应将化合物3的酮羰基还原成醇羟基,获得化合物4;
将乙烯基乙醚与N-溴代琥珀酰亚胺溶解于有机溶剂形成溴鎓离子,再加入化合物4进行亲核取代,获得化合物5;
以吡啶作为溶剂,丙烯酸甲酯在锌粉和NiCl
将化合物6溶解到无水四氢呋喃中,与Me(MeO)NH•HCl发生酰胺化反应,获得化合物7;
将化合物7与苯乙基溴化镁发生格氏反应,获得化合物8;
将化合物8的ω-链上酮羰基用NaBH
化合物9在三氟化硼乙醚和
其中,化合物3~9的结构式分别如式3~9所示:
。/>
进一步的,所述将糠醇和磷酸二氢钾溶解于水进行氧化重排,获得烯酮,具体包括:
将糠醇和结晶状态的磷酸二氢钾溶解于蒸馏水,后用磷酸调节pH至4.1,90℃下回流反应39 h,获得烯酮。
进一步的,所述采用叔丁基二甲基硅基将所述烯酮的羟基保护起来,获得化合物3,具体包括:
将4-二甲氨基吡啶溶解到三乙胺中,后加入溶解于无水二氯甲烷中的烯酮,在0℃下缓慢加入溶解在无水二氯甲烷中的叔丁基二甲基氯硅烷,加热至42 ℃回流反应4h,获得化合物3。
进一步的,所述通过卢奇还原反应将化合物3的酮羰基还原成醇羟基,获得化合物4,具体包括:
将七水合三氯化铈和化合物3溶解于甲醇中,后在-15 ℃下缓慢加入硼氢化钠粉末,移至室温反应30min,获得化合物4。
进一步的,所述将乙烯基乙醚与N-溴代琥珀酰亚胺溶解于有机溶剂形成溴鎓离子,再加入化合物4进行亲核取代,获得化合物5,具体包括:
将N-溴代琥珀酰亚胺溶解于无水二氯甲烷中,在-20 ℃后缓慢滴加乙烯基乙醚,后缓慢加入溶解于无水二氯甲烷的化合物4,再逐步升至常温,反应2h,获得化合物5。
进一步的,所述以吡啶作为溶剂,丙烯酸甲酯在锌粉和NiCl
将锌粉和NiCl
进一步的,所述将化合物6溶解到无水四氢呋喃中,与Me(MeO)NH•HCl发生酰胺化反应,获得化合物7,具体包括:
将Me(MeO)NH•HCl溶解于无水四氢呋喃后加入溶解于无水四氢呋喃的化合物6,搅拌5-10min,在-78℃缓慢滴加作为催化剂的异丙基氯化镁,滴加完成后移至常温反应20-30min,获得化合物7。
进一步的,所述将化合物7与苯乙基溴化镁发生格氏反应,获得化合物8,具体包括:
格氏试剂苯乙基溴化镁的制备:将打磨光亮的镁屑加入到无水四氢呋喃中搅拌2min,加入碘单质做引发剂,后在80℃油浴下加入2-苯基溴乙烷反应30min,获得苯乙基溴化镁;
将化合物7溶解在无水四氢呋喃中,0℃下缓慢滴加新制备好的格氏试剂苯乙基溴化镁,转至常温反应2h,获得化合物8。
进一步的,所述将化合物8的ω-链上酮羰基用NaBH
将化合物8溶解在无水甲醇中,在-15℃下加入NaBH
进一步的,所述化合物9在三氟化硼乙醚和
将化合物9和
进一步的,所述制备方法中,获得烯酮、化合物3、化合物4、化合物6、化合物7、化合物8、化合物9和化合物10的反应均在惰性气体环境下进行。
基于同一发明构思,本发明还提供一种拉坦前列素高级中间体的制备方法在合成拉坦前列素中的用途。
本发明实施例中的一个或多个技术方案,至少具有如下技术效果或优点:
1.本发明一种拉坦前列素高级中间体的制备方法,该方法以镍催化还原串联反应为关键,一步构建Corey内脂骨架及连续四个手性立体中心,丰富了合成该骨架的方法,发展了构建该结构单元的新途径,相比现有拉坦前列素高级中间体合成方法而言,本发明制备方法高效、简洁、反应条件温和且简单,能够大幅降低拉坦前列素高级中间体的合成成本,利于拉坦前列素的大量生产,解决了现有合成技术中合成路线冗长、效率低,不适合进一步放大的问题。
2.本发明一种拉坦前列素高级中间体的制备方法在合成拉坦前列素中的用途,将本发明拉坦前列素高级中间体的制备方法应用于拉坦前列素药物的合成制备,该反应原料价廉易得、反应条件温和、步骤简单、效率高,利于拉坦前列素的大量生产,为拉坦前列素的制备提供一种新的途径,也为后续前列腺素类药物的合成做准备。
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
图1为本发明实施例拉坦前列素高级中间体的合成路径图。
具体实施方式
下文将结合具体实施方式和实施例,具体阐述本发明,本发明的优点和各种效果将由此更加清楚地呈现。本领域技术人员应理解,这些具体实施方式和实施例是用于说明本发明,而非限制本发明。
在整个说明书中,除非另有特别说明,本文使用的术语应理解为如本领域中通常所使用的含义。因此,除非另有定义,本文使用的所有技术和科学术语具有与本发明所属领域技术人员的一般理解相同的含义。若存在矛盾,本说明书优先。
除非另有特别说明,本发明中用到的各种原材料、试剂、仪器和设备等,均可通过市场购买得到或者可通过现有方法制备得到。
本发明整体思路如下:
由于现有的拉坦前列素制备方法复杂、合成路线冗长,效率低,不具备进一步放大和实用价值。
而本发明的目的在于克服现有技术的缺点,发展一种高效、简洁、反应条件温和且简单、可大幅降低成本的新方法,将其应用于药物拉坦前列腺素的高级中间体的制备;并为后续的生理和药理活性测试做准备。本发明通过有机合成化学的手段,以目标拉坦前列素的不对称合成为导向,设计以镍催化还原串联反应为关键步骤,一步构建Corey内脂骨架及连续四个手性立体中心,丰富了合成该骨架的方法,发展了构建该结构单元的新方法。同时,将该方法应用于拉坦前列素药物的合成制备,反应条件温和且简单,为后续前列腺素类药物的合成做准备,体现该方法的应用价值,具备一定的意义。
下面将结合实施例及实验数据对本申请一种拉坦前列素高级中间体的制备方法及其用途进行详细说明。
如图1所示,按照步骤一至步骤九进行反应,下面对各个步骤进行详细描述。
步骤一:
1000 mL双口瓶备用,加入磁子后搭建回流装置,除湿置换氩气三次,准确称取糠醇(即化合物1)(98%, 20.00 g, 199.90 mmol)加入反应瓶中,结晶状的磷酸二氢钾(99%,1.02 g, 7.44 mmol, 0.0365 e.q)溶于500 ml 蒸馏水中并用磷酸调pH至4.1后加入到反应瓶中,再在回流装置上加上氩气气球进行保护换气一次。最后将装置转移至油浴锅中,设置回流温度90℃反应39 h。反应结束后,用乙酸乙酯(150 ml×3)进行萃取,水相减压浓缩后用乙酸乙酯溶解,加无水硫酸钠干燥后,再减压浓缩后得烯酮(即化合物2)红棕色油状物7.20 g(Yield = 52%)。无需纯化直接投下一步使用。R
步骤二:
取100 mL双口瓶备用,加入磁力搅拌子并除湿置换氩气三次,称取4-二甲氨基吡啶(DMAP) (99%, 0.91 g, 7.34 mmol, 0.1 e.q),搭建回流装置,回流装置上加上氩气气球进行保护,再次置换氩气三次,吸取三乙胺(17.08 g, 168.74 mmol, 2.3 e.q)和溶解在二氯甲烷的4-羟基环戊烯酮(7.20 g, 73.37 mmol, 1.0 e.q)加入反应瓶中。先将装置转移至0℃中,并于搅拌下缓慢加入溶解在二氯甲烷中的叔丁基二甲基氯硅烷(TBSCl) (97%,12.54 g, 80.70 mmol, 1.1 e.q),滴加完后移至油浴锅,设置回流温度42℃反应4 h。反应结束后,用NH
步骤三:
取50 mL双口瓶备用,加入磁力搅拌子并除湿置换氩气三次,加入七水合三氯化铈(99%, 2.63 g, 6.99 mmol, 2.0 e.q),再次置换氩气三次,换气后插氩气气球保护。用甲醇溶解化合物3(0.74 g, 3.49 mmol, 1.0 e.q),于常温(25±5℃)下将溶解后的底物转移至双口瓶中。在-15℃下缓慢加入硼氢化钠(0.13 g, 3.49 mmol, 1.0 e.q)至反应瓶中,5min以上。然后逐渐升至室温,反应30 min后,滴加饱和氯化铵进行淬灭当反应液从乳白色变为澄清时停止滴加。减压浓缩去除甲醇溶剂,用蒸馏水(10 mL)洗涤,乙酸乙酯(20 mL×3)萃取,饱和食盐水(10 mL)洗涤,无水硫酸钠干燥,减压浓缩后得到化合物4橘黄色油状物(0.66g, Yield =88%)。R
步骤四:
取50 mL双口瓶备用,加入磁力搅拌子并除湿置换氩气三次,加入
步骤五:
取10 mL双口瓶备用,加入磁力搅拌子并除湿置换氩气,加入锌粉(0.21 g, 3.20mmol, 4.0 e.q)和六水合氯化镍(NiCl
步骤六:
取100 mL双口瓶备用,加入磁力搅拌子并除湿置换氩气,加入Me(MeO)NH•HCl(98%, 0.65 g, 6.68 mmol, 2.0 e.q)后再次置换氩气3次,换气后插氩气气球保护。加入溶解于THF(40 mL)的底物化合物6(1.25 g, 3.34 mmol),搅拌约5 min。移至-78℃缓慢滴加(约10 min)异丙基氯化镁(2M/L)10 ml (9.56 mL, 19.11 mmol, 5.72 eq)滴加完成后移至常温,然后一直维持在室温(25℃)搅拌点板检测反应,待原料反应完全,约20 min。饱和氯化铵溶液(10 mL)进行淬灭,乙酸乙酯(30 mL×3)进行萃取,用饱和食盐水(10 mL)洗涤后,合并有机相,无水硫酸钠进行干燥,减压浓缩后经柱层析分离纯化,洗脱剂的比例为Petroleum ether : EtOAc = 5:1得到化合物7无色油状物1.34 g(Yield = 99%)。R
步骤七:
格氏试剂的制备:取10 mL双口瓶备用,加入磁力搅拌子并除湿置换氩气,加入打磨光亮的镁屑 (0.13 g, 5.40 mmol, 2.1 e.q) ,搭建回流装置,回流装置上加上氩气气球进行保护,再次置换氩气3次,加入4 mL重蒸后的THF,搅拌2 min后,称取I
化合物8的制备:取25 mL双口瓶备用,加入磁力搅拌子并除湿置换氩气,加入底物化合物7(0.40 g, 1.00 mmol, 1.0 e.q) 再次置换氩气3次,换气后插氩气气球保护。加入8 mL无水THF搅拌溶解底物,在0℃中将刚制备好的格氏试剂缓慢滴加至反应瓶中后转至常温。然后一直维持在室温(25℃)搅拌点板检测反应,待原料反应完全,约2 h。饱和氯化铵溶液(10 mL)进行淬灭,乙酸乙酯(15 mL×3)进行萃取,用饱和食盐水(10 mL)洗涤后,合并有机相,无水硫酸钠进行干燥,减压浓缩后经柱层析分离纯化,洗脱剂的比例为Petroleumether : EtOAc =30:1得化合物8无色油状物0.21 g(Yield = 46%). R
步骤八:
取25 mL双口瓶备用,加入磁力搅拌子并除湿置换氩气3次,换气后插氩气气球保护。加入溶解于甲醇(10 mL)的底物化合物8 (异构体) (0.21 g, 0.46 mmol, 1.0 e.q),在-15℃下加入硼氢化钠(0.02 g, 0.51 mmol, 1.1 e.q)搅拌约5 min后移至常温(25℃)搅拌,点板检测反应,待原料反应完全,约30 min。饱和氯化铵溶液(10 mL)进行淬灭,旋干溶剂甲醇,用乙酸乙酯(30 mL×3)进行萃取,用饱和食盐水(10 mL)洗涤后,合并有机相,无水硫酸钠进行干燥,减压浓缩后经柱层析分离纯化,洗脱剂的比例为Petroleum ether : EtOAc= 5:1得到化合物9无色油状物0.21 g(Yield = 96%)。R
步骤九:
取25 mL双口瓶备用,加入磁力搅拌子并除湿置换氩气3次,换气后插氩气气球保护。加入溶解于CH
综上所述, 本发明公开了一种抗青光眼和降低眼内压药物拉坦前列素的高级中间体的制备方法,通过有机合成化学的手段,以目标拉坦前列素的合成为导向,设计以镍催化还原串联反应为关键,一步构建Corey内脂骨架及连续四个手性立体中心(即化合物6),发展了构建该结构单元的新方法。同时,将该方法应用于拉坦前列素药物的合成制备。该反应所需原料价廉易得、条件温和、反应简单,为后续其他类型PGs的合成做奠定基础;体现该方法的优越性,具有较高的的实用价值。
对于得到的化合物10,作为拉坦前列素的高级中间体,可以参考(Eur. J. Org.Chem. 2020, 6221–6227)文献上的方法制备得到拉坦前列素(Latanoprost)。
最后,还需要说明的是,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。