3CLpro蛋白酶抑制剂的药物组合物
文献发布时间:2024-04-18 19:58:26
本申请要求申请日为2022年5月31日的专利申请CN202210606143.1的优先权,本申请引用上述中国专利申请的全文。
技术领域
本发明属于药物制剂领域,具体涉及3CLpro蛋白酶抑制剂或其药学上可接受的盐的药物组合物。
背景技术
3CLPro(3C-like protease,又称3C样蛋白酶)是新型冠状病毒(COVID-19,SARS-CoV-2)产生的主要蛋白酶,冠状病毒大多数功能蛋白(非结构蛋白)由ORF1ab基因编码,先翻译成一个多蛋白体(7096aa),再由3CLPro切割成多个有活性的蛋白如病毒复制蛋白RdRp。此外,该蛋白可能切割胞内蛋白NEMO从而抑制干扰素信号途径的活化。因此抑制3CLPro能够有效抑制病毒的感染与复制。
WO2023036140A记载了一种3CLPro蛋白酶抑制剂,其中化合物(5-溴-2-((R)-1-(5-(甲氨基)烟碱酰)哌啶-3-基)氨基)-3-硝基苯基((2R,6S)-2,6-二甲基吗啉基)甲酮对SARS-CoV-2 3CLpro/Mpro蛋白酶具有良好的抑制作用,且在小鼠体内具有较高的暴露量和生物利用度,有望开发为临床药物,其结构如下所示:
特别地,式(I)的盐酸盐形式将用作治疗新型冠状病毒的药物,优选给药途径是通过口腔,这一途径提供给药的最大舒适性和便利性。临床前动物实验研究,式(I)化合物达峰时间快,Cmax高易产生毒性,为降低药物毒性,提高药物安全性,稳定血药浓度,,因此需开发一种口服缓释制剂以满足临床用药需求。
发明内容
本发明目的在于提供一种药物组合物,其含有:活性剂和缓释材料,其中所述的活性剂为式(I)化合物(5-溴-2-((R)-1-(5-(甲氨基)烟碱酰)哌啶-3-基)氨基)-3-硝基苯基((2R,6S)-2,6-二甲基吗啉基)甲酮或其药学上可接受的盐,
所述的缓释材料为纤维素醚。
本发明一些实施方案,所述的纤维素醚选自羟丙甲纤维素或羟乙基纤维素的一种或多种组合。
本发明一些实施方案,其药学上可接受的盐为一盐酸盐。
本发明一些实施方案,其还含有填充剂,所述的填充剂选自无水乳糖或微晶纤维素的一种或多种组合。
本发明一些实施方案,其还含有助流剂,所述的助流剂为胶态二氧化硅。
本发明一些实施方案,其还含有润滑剂,所述的润滑剂为硬脂酸镁。
本发明一些实施方案,其任选含有pH调节剂,所述的pH调节剂为枸橼酸钠。
本发明一些实施方案,所述缓释材料占组合物总重量的15~40%,优选15%~30%,更优选20%。
本发明一些实施方案,其中填充剂为无水乳糖和微晶纤维素的混合物,且无水乳糖:微晶纤维素的质量比为1:3~3:1。
本发明一些实施方案,其中填充剂为无水乳糖和微晶纤维素的混合物,且无水乳糖:微晶纤维素的质量比为1:1。
本发明还提供一种药物组合物,其含有:
(a)含量10%~30%的活性剂,所述活性剂为(5-溴-2-((R)-1-(5-(甲氨基)烟碱酰)哌啶-3-基)氨基)-3-硝基苯基((2R,6S)-2,6-二甲基吗啉基)甲酮一盐酸盐;
(b)含量15%~40%的缓释材料,所述缓释材料选自羟丙甲纤维素或羟乙基纤维素。
(c)含量20%~80%的填充剂,所述填充剂选自无水乳糖和微晶纤维素的一种或多种组合;
(d)含量0%~2%的助流剂,所述助流剂为胶态二氧化硅;
(e)含量0.5%~3%的润滑剂,所述润滑剂为硬脂酸镁;
(f)含量0~5%的pH调节剂,所述pH调节剂为枸橼酸钠;
其中,所有含量以重量计,且含量的总和为100%。
本发明一些实施方案,所述活性剂含量为15%~20%。
本发明一些实施方案,所述缓释材料含量为15%~30%,优选地,缓释材料含量为20%~30%;进一步地,缓释材料含量为20%。
本发明一些实施方案,所述填充剂含量为50%~70%,优选地,缓释材料含量为60%~70%。
本发明一些实施方案,所述助流剂含量为0.5%~1%,优选地,助流剂含量为0.5%。
本发明一些实施方案,所述润滑剂含量为0.5%~2%,优选地,润滑剂含量为1%~2%,进一步地,润滑剂含量为1%。
本发明一些实施方案,所述pH调节剂含量为0~2%;优选地,pH调节剂含量为0。
本发明一些实施方案,所述的药物组合物含有式(Ia)所示的(5-溴-2-((R)-1-(5-(甲氨基)烟碱酰)哌啶-3-基)氨基)-3-硝基苯基((2R,6S)-2,6-二甲基吗啉基)甲酮一盐酸盐、羟丙甲纤维素、无水乳糖、微晶纤维素、胶态二氧化硅和硬脂酸镁
本发明一些实施方案,所述羟丙甲纤维素的含量为15%~30%,优选20%~30%,更进一步优选20%。
本发明一些实施方案,所述的药物组合物含有:
(a)含量15%~20%的式(Ia)所示的(5-溴-2-((R)-1-(5-(甲氨基)烟碱酰)哌啶-3-基)氨基)-3-硝基苯基((2R,6S)-2,6-二甲基吗啉基)甲酮一盐酸盐;
(b)含量20%~30%的羟丙甲纤维素;
(c)含量60%~70%的无水乳糖和微晶纤维素;
(d)含量0.5%的胶态二氧化硅;
(e)含量1%的硬脂酸镁,所有含量以重量计,且含量的总和为100%。
本发明一些实施方案,其是一种经口服给药的缓释制剂。
本发明一些实施方案,缓释制剂为片剂、胶囊或颗粒剂。
本发明还提供药物组合物用于制备治疗由冠状病毒引起的疾病的药物中的应用。
本发明一些实施方案,所述疾病为呼吸道传染病。
本发明一些实施方案,所述疾病为严重急性呼吸综合征。
本发明一些实施方案,所述冠状病毒为SARS-CoV-2。
本发明还提供药物组合物的方法,其特征在于:将活性剂、填充剂、缓释材料、助流剂和润滑剂混合,采用直混或干压造粒后压片。
本发明中,rpm是转速单位,是指转/分钟;
本发明中,原料药是指式(Ia)化合物。
本发明某些实施方案,干压造粒的参数根据本领域技术人员水平进行常规测定,参数范围:水平进料速度0-12rpm,垂直进料速度150-300rpm,压轮速度3-12rpm,粉碎速度188-300rpm,压轮压力3-12KN/cm,压轮间隙1-2mm。
本发明某些实施方案,混合转速10-20rpm,混合时间5-20min。
本发明某些实施方案,活性剂、填充剂、缓释材料、助流剂与润滑剂混合后,可通过孔径为1.0-2.5mm的筛网,进行过筛混合。
本发明某些实施方案,制剂制备方法中助流剂和润滑剂可分批加入,先加入一部分与活性剂、填充剂、缓释材料混合,再加入剩余处方量助流剂和润滑剂,助流剂和润滑剂的总量为总处方量。
附图说明
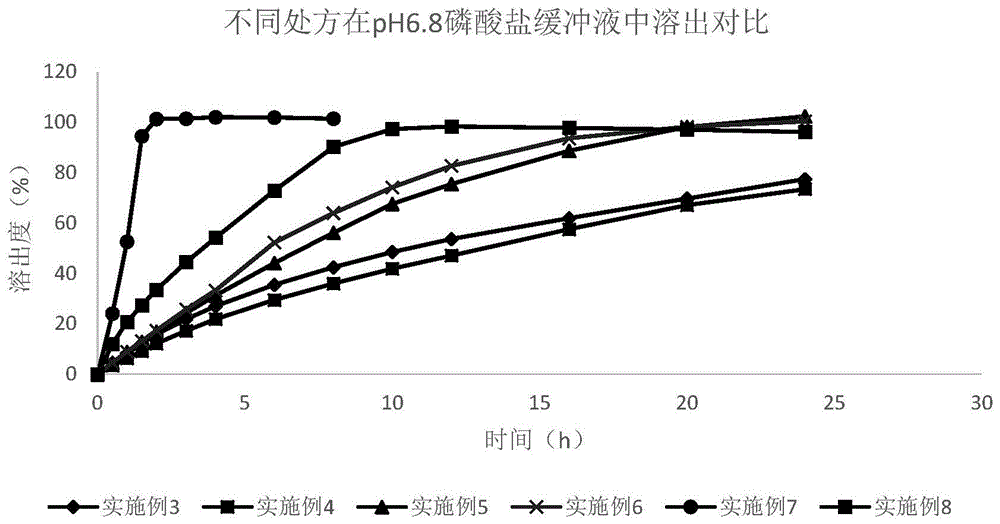
图1为不同处方在pH6.8磷酸盐缓冲液的溶出行为;
图2为原料药和实施例8片剂在食蟹猴体内的PK结果。
具体实施方式
下面通过实施例对本发明进行详细描述,但并不意味着对本发明任何不利限制。本发明的化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。对本领域的技术人员而言,在不脱离本发明精神和范围的情况下针对本发明具体实施方式进行各种变化和改进将是显而易见的。
实施例1式(I)化合物的制备方法
(5-溴-2-((R)-1-(5-(甲氨基)烟碱酰)哌啶-3-基)氨基)-3-硝基苯基((2R,6S)-2,6-二甲基吗啉基)甲酮
反应路线:
操作步骤:
步骤A:在室温条件下,氮气保护,将化合物I-1(6g,22.73mmol)溶于N,N-二甲基甲酰胺(113mL)中。随后,将上述溶液降温至0℃,向上述反应液中依次加入N,N-二异丙基乙胺(8.8g,68.19mmol)和2-(7-偶氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(12g,33.82mmol)和(2R,6S)-2,6-二甲基吗啉(3.1g,27.27mmol)。然后该反应体系在0℃下继续搅拌15分钟。
LCMS监测显示原料消失后,向反应液中加入水(200mL)淬灭,混合液用乙酸乙酯(100mL×3次)萃取,合并有机相,有机相用饱和食盐水(100mL×2次)洗涤。然后用无水硫酸钠干燥,过滤,减压浓缩。所得残余物用硅胶柱层析纯化得到化合物I-2(6.6g)。
MS(ESI)M/Z:361.0[M+H]
步骤B:在室温条件下,氮气保护,将化合物I-2(6.6g,18.33mmol)溶于N,N-二甲基甲酰胺(92mL)中。随后,向上述反应液中依次加入(R)-3-氨基哌啶-1-羧酸叔丁酯(4.4g,22.00mmol)和N,N-二异丙基乙胺(7.1g,54.99mmol)。然后该反应体系在80℃下搅拌2小时。
LCMS监测显示原料消失后,向反应液中加入水(200mL)淬灭,混合液用乙酸乙酯(100mL×3次)萃取,合并有机相,有机相用饱和食盐水(100mL×2次)洗涤。然后用无水硫酸钠干燥,过滤,减压浓缩。所得残余物用硅胶柱层析纯化得到化合物I-3(9g)。
MS(ESI)M/Z:441.0[M-99]
步骤C:在室温条件下,氮气保护,将化合物I-3(9g,16.64mmol)溶于4M的盐酸-1,4-二氧六环溶液(83.2mL)。然后该反应体系在室温下搅拌1小时。
LCMS监测显示原料消失后,将反应液减压蒸馏浓缩。得到粗品化合物I-4(8g)。
MS(ESI)M/Z:441.0[M+H]
步骤D:在-15~-10℃条件下,氮气保护,将化合物I-4(1.8g,3.77mmol)溶于N,N-二甲基甲酰胺(20mL)中。维持-15℃~-10℃,依次加入2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(2.6g,6.80mmol,HATU)、N,N-二异丙基乙胺(2.4g,18.84mmol)及5-(甲氨基)烟酸(0.917mg,6.03mmol)。该反应体系在-15℃~-10℃下搅拌20分钟。向反应液中加入水(60mL)淬灭,混合液用乙酸乙酯(120mL×2次)萃取,合并有机相,有机相用饱和食盐水(100mL)洗涤。然后用无水硫酸钠干燥,过滤,减压浓缩纯化得到式(I)化合物(2.0g)。
MS(ESI)M/Z:575.2[M+H]
1H NMR(400MHz,DMSO-d6)δ8.26(s,1H),8.10-7.47(m,4H),6.75(s,1H),6.17(s,1H),4.37(s,1H),4.15-3.35(m,7H),3.30-3.02(m,2h),2.97-2.77(m,1H),2.70(s,3H),2.00-1.30(m,4H),1.15(brs,3H),1.03(brs,3H).
实施例2式(Ia)化合物的制备方法
(5-溴-2-((R)-1-(5-(甲氨基)烟碱酰)哌啶-3-基)氨基)-3-硝基苯基((2R,6S)-2,6-二甲基吗啉基)甲酮一盐酸盐
称取式(I)化合物(607.2mg,实施例1制备得到)于样品瓶中,加入乙酸乙酯(6mL)和6M的盐酸溶液(0.39mL),50℃下搅拌24h,减压浓缩得到固体。加入甲醇和甲基叔丁基醚的混合溶液(1:9,体积比),50℃下搅拌48h后过滤收集固体得到式(Ia)化合物。
实施例3
步骤1、将活性剂、填充剂、缓释材料、助流剂与润滑剂放置料斗混合机中混合。
步骤2、采用干压制粒机,将混合后物料加入干压制粒机中进行干压造粒。
步骤3、向整粒后的颗粒加入助流剂及润滑剂,混合后压片。
步骤4、压片:规格50mg(以游离碱计算),理论片重300mg,冲模9mm圆形浅凹冲,硬度5-25kg。
实施例4
步骤1、将活性剂、填充剂、缓释材料、助流剂与润滑剂放置料斗混合机中混合。
步骤2、采用干压制粒机,将混合后物料加入干压制粒机中进行干压造粒。
步骤3、向整粒后的颗粒加入助流剂及润滑剂,混合后压片。
步骤4、压片:规格50mg(以游离碱计算),理论片重300mg,冲模9mm圆形浅凹冲,硬度5-25kg。
实施例5
步骤1、将活性剂、填充剂、缓释材料、助流剂与润滑剂放置料斗混合机中混合。
步骤2、采用干压制粒机,将混合后物料加入干压制粒机中进行干压造粒。
步骤3、向整粒后的颗粒加入外加助流剂及润滑剂,混合后压片
步骤4、压片:规格50mg(以游离碱计算),理论片重300mg,冲模9mm圆形浅凹冲,硬度5-25kg。
实施例6
步骤1、将活性剂、填充剂、缓释材料、助流剂与润滑剂放置料斗混合机中混合。
步骤2、采用CCS220干压制粒机,将混合后物料加入干压制粒机中进行干压造粒。
步骤3、向整粒后的颗粒加入外加助流剂及润滑剂,混合后压片。
步骤4、压片:规格50mg(以游离碱计算),理论片重300mg,冲模9mm圆形浅凹冲,硬度5-25kg。
实施例7
步骤1、将活性剂、填充剂、缓释材料、助流剂与润滑剂放置料斗混合机中混合。
步骤2、总混:称取处方量的胶态二氧化硅、硬脂酸镁,加入料斗混合机中继续混合;
步骤3、压片:规格50mg(以游离碱计算),理论片重300mg,冲模9mm圆形浅凹冲,硬度5-25kg;
实施例8
步骤1、将活性剂、填充剂、缓释材料、助流剂与润滑剂放置料斗混合机中,混合。
步骤2、总混:称取处方量的胶态二氧化硅、硬脂酸镁,加入料斗混合机中继续混合;
步骤3、压片:规格50mg(以游离碱计算),理论片重300mg,冲模9mm圆形浅凹冲,硬度5-25kg;
实施例9溶出实验
根据《中国药典》2020年版溶出方法(第一法篮法),测试实施例3-8制得的片剂在pH6.8磷酸盐缓冲液的溶出度,转速100转/分钟,具体如表2所示。
表2溶出度结果
结论:对比以上处方,在pH6.8磷酸盐缓冲液中,实施例7缓释片的溶出速度最快,2h释放已达100%。实施例3和实施例4缓释片的溶出速度最慢,24h仍未达到完全释放。实施例5和实施例6缓释片溶出速度接近,24h已完全释放,实施例8缓释片12h可达完全释放。除实施例7处方中缓释材料含量为10%外,其余实施例处方中缓释材料含量为20%-30%均可达到延迟释放的目的。
实施例10缓释材料含量筛选
实施例10a、10b、10c和10d的制备方法参考实施例8的方法,根据实施例9方法评价其在pH6.8磷酸盐缓冲液中溶出行为,具体结果如表3所示:
表3溶出度结果
由上表可以看出,对比以上处方,在pH6.8磷酸盐缓冲液中,实施例10a缓释片的溶出速度最快,6h释放已达99.9%。实施例10b、实施例10c缓释片和实施例10d溶出速度接近,16h可达到完全释放,由此可见,缓释材料含量为15%-30%均可达到延迟释放的目的。
生物学测试评价
测试例1:评价式(I)化合物对SARS-CoV-2 3CL
本实验采用荧光共振能量转移的方法来检测SARS-CoV-2 3CL
新型冠状病毒M
用反应缓冲液配置酶溶液,在各孔中加49.5μL的酶溶液;在Min孔中加49.5μL的反应缓冲溶液。化合物检测IC
实验测得,式(I)化合物的IC
测试例2:CD1小鼠的体内药代动力学实验
以雄性CD1小鼠为受试动物,研究本发明化合物对雄性CD1小鼠在剂量为10mg/kg时经口服给药后的体内药代动力学行为。
供试品:对照化合物和式(I)化合物。
试验动物
种属:雄性CD1小鼠(3只/组)
等级:SPF级
体重:约20~30g
年龄:6~8周
来源:维通利华
雄性CD1小鼠灌胃给予供试品溶液,给药剂量为10mg/kg,给药体积为10mL/kg。
供试品溶液的配制:供试品采用溶媒(10%DMSO+50%PEG400+40%纯净水)溶解,配制时依次加入DMSO、PEG400和纯净水。
对照组:CN113072497A专利化合物2,其结构式如下:
实验组:式(I)化合物。
分别于给药前(0h)和给药后0.25h,0.5h,1h,2h,4h,8h,24h,按照设置的采血时间点,从小鼠足背静脉采集约0.025~0.03mL全血(用EDTA-K2抗凝),置于湿冰条件下,并在采集后30min内以4000g离心力、4℃、离心5min,分离血浆并转移到检测管中。
药代动力学主要参数用WinNonlin(PhoenixTM,version 6.1)计算得到,药代动力学参数参见如表4-1和表4-2。
表4-1CD1小鼠静脉给药的药代动力学参数
表4-2CD1小鼠口服给药的药代动力学参数
备注:NA代表无相应数据。
结果显示:与对照组相比,式(I)化合物在小鼠体内具有较高的暴露量和生物利用度。
测试例3:评价实施例8片剂在食蟹猴体内的PK性质
3.1溶媒和供试品溶液的配制
溶媒:2%吐温80+98%(0.5%(w/v))水溶液
配制方法:
0.5%(w/v)的甲基纤维素(MC)水溶液的配制:称取约10g甲基纤维素(MC),准确量取2000mL加热至60℃左右的纯水至合适容器中,分次少量缓慢加入甲基纤维素(MC),并剧烈搅拌,最后用磁力搅拌器搅拌至澄明,配置成澄清透明的0.5%(w/v)MC水溶液,常温放置备用。
2%吐温80+98%(0.5%(w/v))水溶液的配制:取40mL吐温80,加入到1960mL的0.5%(w/v,重量体积比)MC水溶液,搅拌均匀,常温放置备用。
3.2.灌胃供试品的配制
称取原料药至玻璃容器中,加入适量2%吐温80+98%(0.5%(w/v))混合溶液,边加边搅拌混匀,必要时超声,后用NaOH溶液调节pH至4.0±1.0。
原料药:式(Ia)化合物。
3.3.试验设计
3.3.1.动物分组及给药
食蟹猴8只,随机分成2组,每组4只,雌雄各半。给药信息设计如表5:
表5给药组信息
3.3.2.PK采样时间点
表6各组采样信息
2.3.3.PK样本采集及处理
食蟹猴于前或后肢静脉在相应采样时间点采全血约0.5mL至EDTA-K
表7食蟹猴体内PK结果表
结论:实施例8片剂在食蟹猴体内C
- 取代脲化合物、其药学上可接受的盐或其溶剂合物、其应用、药物及药物组合物
- 用于诱导饱足感和延长饱腹感的基于蜂房哈夫尼菌的药物组合物和食品组合物
- 一种抗结核化合物及其在制备抗结核药物中的应用以及一种抗结核药物组合物
- 用于给药HIV蛋白酶抑制剂的可溶性稳定药物组合物及浓缩的该药物组合物的制备方法
- 包含金属蛋白酶抑制剂的化妆品组合物或药物组合物