一种无肌间刺鲫品系培育方法
文献发布时间:2023-06-19 11:59:12
技术领域
本发明涉及一种水产养殖品种培育方法。
背景技术
鲤科鱼类是我国的主要养殖鱼类,年产量约2000多万吨,为我国老百姓提供了近1/3的优质动物蛋白。但因为鲤科鱼类含有的肌间刺多,造成了人们在食用过程中的不便,甚至可能导致卡噪子等身体上的伤害,同时鱼肉产品的加工(如鱼丸等)也受到阻碍,因此鲤科鱼类肌间刺的遗传改良成为我国水产遗传育种的重要目标之一。目前有一些关于肌间刺选育的报道,如徐晓锋等(2015)在草鱼雌核发育的群体中发现一尾生长正常且肌间刺缺失的突变体,但并未获得无或少肌间刺草鱼突变群体的报道;巴西科学家(2017)采用X射线在大盖巨脂鲤中筛选到一个肌间刺缺失的群体,但在后期育种中也未获得少或无肌间刺的群体(Stokstad,2020)。Guo等(2018)通过三角鲂(Megalobrama terminalis)与翘嘴鲌(Culter alburnus)的杂交实验表明,三角鲂♀×翘嘴鲌♂杂交子代的肌间刺数量(127)小于父本(137),大于母本(124),但显著性小于团头鲂♀×翘嘴鲌♂的杂交子代肌间刺数量(133),说明可以通过杂交育种的方法获得少肌间刺数量的品种,并申请了发明专利(公布号CN107347747A)。黎玲等(2013)报道通过人工培育的杂交鲫比野生鲫肌间刺数量少,可通过人工培育和杂交育种方法减少鲫肌间刺数量。鲍宝龙等通过敲除MSTN基因,发明了使肌间刺变粗的方法(发明专利公布号CN111560401A,CN111549030A,CN111549031A,CN111500581A)。高泽霞等通过敲除斑马鱼scxa基因,获得了肌间刺数量减少70%以上的突变体,但也造成了肋骨等其它骨骼发育不良(发明专利申请号:201911104496.6;Nie etal.,2020;Kague et al.,2019),同时也未有在其它鲤科鱼类中进行应用的报道。综上,虽然有一些方法可以减少鲤科鱼类肌间刺的数量,但目前仍未有无肌间刺鲤科鱼类新品种/系出现。
鲫是我国的主要养殖品种之一,年产量约300万吨,其肉质鲜美,深受老百姓喜爱。同时也由于肌间刺(不同品种鲫肌间刺数量平均约71~84)的原因使其品质受到了影响,因此肌间刺也是其遗传育种的重要目标。虽然有报道称可以通过杂交育种和人工选育减少鲫肌间刺数量,但并未有肌间刺数量减少50%以上的品系或品种出现。
发明内容
为对鲫肌间刺进行遗传改良,本发明提出了一种通过基因敲除bmp6基因培育少或无肌间刺鲫的方法。
本发明无肌间刺鲫品系培育方法按以下步骤进行无肌间刺鲫品系培育:
针对bmp6基因在鲫基因组中的2个拷贝bmp6a和bmp6b分别设计敲除靶位点,然后通过2轮基因敲除和筛选,获得F2代bmp6a和bmp6b双基因突变纯合系,然后利用F2代bmp6a和bmp6b双基因突变纯合系进行扩繁形成无肌间刺鲫新品系;
其中,用bmp6a或bmp6b或bmp6a和bmp6b上对应的sgRNA与Cas9蛋白混合后显微注射鲫单细胞期胚胎,进行第一轮基因敲除,构建F0代群体,然后F0代群体养殖3~5个月进行PIT标记和DNA提取,再进行测序确定鲫个体体细胞突变的等位基因及突变率,选取体细胞突变系且体细胞突变率为95%以上的F0代个体作为亲本制备0代受精卵;
用bmp6a或bmp6b或bmp6a和bmp6b上对应的sgRNA与Cas9蛋白混合后显微注射0代受精卵,进行第二轮基因敲除,构建F1代群体,然后F1代群体养殖3~5个月进行PIT标记和DNA提取,再进行测序确定鲫个体体细胞突变的等位基因及突变率,选取体细胞bmp6a和bmp6b双基因突变系且体细胞突变率为95%以上的F1代作为亲本进行繁殖,构建F2代,再从F2代中选取bmp6a和bmp6b双基因突变纯合系。
本发明方法获得肌间刺数量在20枚以下和无肌间刺的鲫品种。
因鲫基因组经历了第4轮基因组复制事件(Chen et al.2019),因此bmp6基因在鲫基因组中存在2个拷贝。
附图说明
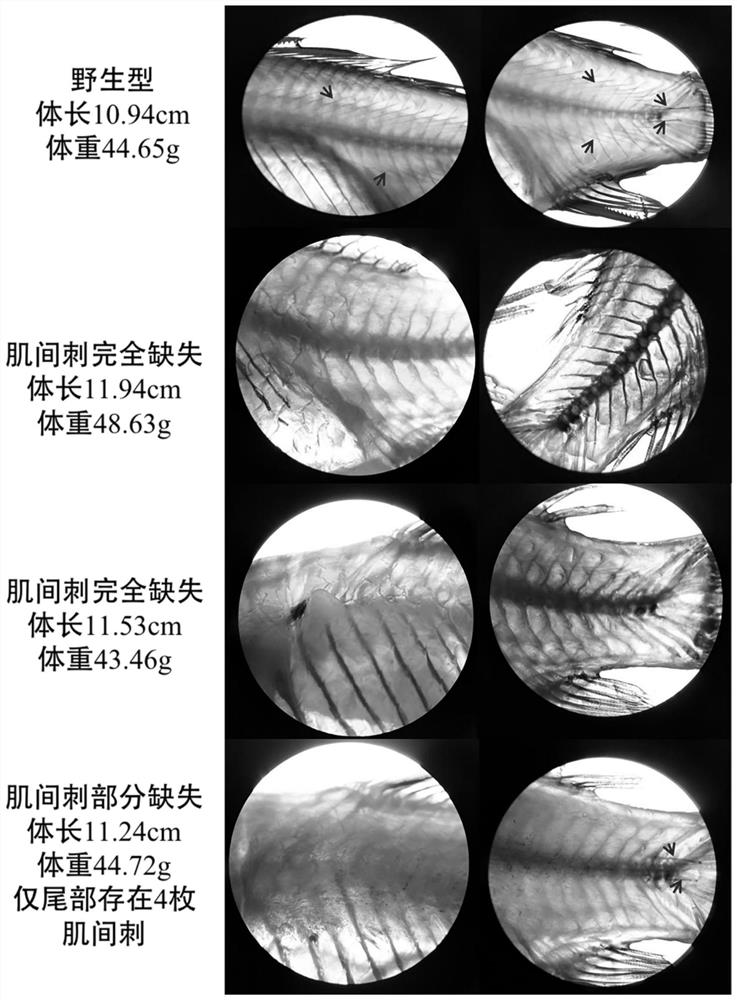
图1是实施例1中典型个体骨骼染色观察对比图,图中红色箭头所指为肌间刺;
图2是实施例1中野生型鲫骨骼X光射线图,图中红色箭头所指为肌间刺;
图3是实施例1中突变体尾部骨骼X光射线图,由图中可见尾部肌肉组织中无肌间刺存在;
图4是实施例1中突变体躯干部骨骼X光射线图,由图中可见躯干部背部肌肉组织中无肌间刺存在;
图5是实施例1中新品系鲫bmp6a的1号外显子测序结果图;其中CAA-1、CAA-2、CAA-3为bmp6a 1号外显子靶位点;WT为野生型,序列编号中数字为突变体PIT标记号,字母为突变等位基因编号;
图6是实施例1中新品系鲫bmp6b的1号外显子测序结果图;其中CAA-7、CAA-8、CAA-9为bmp6b 1号外显子靶位点;WT为野生型,序列编号中数字为突变体PIT标记号,字母为突变等位基因编号;
图7是实施例1中新品系鲫bmp6a的1号外显子蛋白序列对比图;其中WT为野生型,序列编号中数字为突变体PIT标记号,字母为突变等位基因编号;
图8是实施例1中新品系鲫bmp6b的1号外显子蛋白序列对比图;其中WT为野生型,序列编号中数字为突变体PIT标记号,字母为突变等位基因编号。
具体实施方式
下面结合实例对本发明作进一步的详细说明。以下实例旨在说明本发明,并不限制本发明的范围。
下述实施例中所使用的实验方法如无特殊说明均为常规方法。所用材料、试剂、方法和仪器,未经特殊说明,均为本领域常规材料、试剂、方法和仪器,本领域技术人员均可通过商业渠道获得。
具体实施方式一:本实施方式无肌间刺鲫品系培育方法:
针对bmp6基因在鲫基因组中的2个拷贝bmp6a和bmp6b分别设计敲除靶位点,然后通过2轮基因敲除和筛选,获得F2代bmp6a和bmp6b双基因突变纯合系,然后利用F2代bmp6a和bmp6b双基因突变纯合系进行扩繁形成无肌间刺鲫新品系;
其中,用(bmp6a)或(bmp6b)或(bmp6a和bmp6b)上对应的sgRNA与Cas9蛋白混合后显微注射鲫单细胞期胚胎,进行第一轮基因敲除,构建F0代群体,然后F0代群体养殖3~5个月进行PIT标记和DNA提取,再进行测序确定鲫个体体细胞突变的等位基因及突变率,选取体细胞突变系且体细胞突变率为95%以上的F0代个体作为亲本制备0代受精卵;
用bmp6a或bmp6b或bmp6a和bmp6b上对应的sgRNA与Cas9蛋白混合后显微注射0代受精卵,进行第二轮基因敲除,构建F1代群体,然后F1代群体养殖3~5个月进行PIT标记和DNA提取,再进行测序确定鲫个体体细胞突变的等位基因及突变率,选取体细胞bmp6a和bmp6b双基因突变系且体细胞突变率为95%以上的F1代作为亲本进行繁殖,构建F2代,再从F2代中选取bmp6a和bmp6b双基因突变纯合系。
其中,所述的DNA提取和测序方法为:在0.3~0.5cm
其中,突变体检测采用Sanger测序:采用表2所示的引物对样本进行PCR扩增,体系设置为25μL:基因组DNA上清液2μL、10μM上下游目标引物各1μL、2×DreamTaq Master Mix(Thermo Fisher,CA,USA)12.5μL、无酶水补充至25μL;PCR反应程序为95℃3min,95℃30s,60℃30s,72℃30s,35个循环,72℃5min。PCR产物用1.5%琼脂糖凝胶电泳检测。检测后纯化回收,进行TA克隆,菌落PCR扩增后取2μL PCR产物,用8%的聚丙烯检测,挑选与对照条带大小有差异的菌落进行Sanger测序。
具体实施方式二:本实施方式与具体实施方式一的不同点在于:bmp6基因的敲除靶位点如表1所示,
表1
其它与具体实施方式一相同。
表2
具体实施方式三:本实施方式与具体实施方式一或二的不同点在于:采用如表1所示的sgRNA上游引物和序列为5’-GATCCGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3’的sgRNA下游引物,进行sgRNA体外合成;sgRNA体外扩增体系为:10μM sgRNA上游和下游引物各2.5μL、2×Dream Taq Master mix(ThermoFisher,CA,USA)25μL、无酶水补充至50μL;共扩增2管共100μL;sgRNA体外扩增程序为95℃变性3min,之后30个循环设置为95℃30s、58℃30s、72℃30s,72℃延伸5min;然后对扩增产物进行纯化回收,之后进行体外转录,每个靶位点建立30μl反应体系:sgRNA PCR回收产物1μg、NTP Buffer Mix 10μL、T7 RNA Polymerase Mix 2μL,用无酶水补足30μL;37℃转录4h,反应结束加入20μl无酶的水,混匀后加入2μl DNase I,在37℃消化15min,去除未反应的DNA。其它与具体实施方式一或二相同。
sgRNA体外PCR产物用1.5%琼脂糖凝胶电泳检测,条带为120bp。检测后用PCR产物纯化回收试剂盒(Exygen)将PCR产物纯化回收,采用Qubit 3试剂盒(Thermo Fisher,CA,USA)测定浓度待用。sgRNA PCR产物回收浓度在100~160ng/μl之间。
将体外转录的sgRNA用RNA纯化试剂盒(Qiagen)纯化回收。采用Qubit 3试剂盒(Thermo Fisher,CA,USA)测定回收产物浓度,于-80℃冰箱中保存待用。体外转录sgRNA回收浓度在800~3000ng/μL之间。
具体实施方式四:本实施方式与具体实施方式一或二或三的不同点在于:显微注射的方法是:体外合成的sgRNA和Cas9蛋白(NEB M0646,MA,USA)按3:1的摩尔浓度比混合后室温孵育10min,再加入25%的酚红,注射到鲫单细胞期的胚胎中;其中,每个外显子上的靶位点sgRNA等量混合后进行注射,每个sgRNA终浓度均不低于50ng/μL,对照组注射25%的酚红,每粒受精卵注射量1nL±0.02nL。其它与具体实施方式一或二或三相同。
实施例1
无肌间刺鲫品系培育方法:
针对bmp6基因在鲫基因组中的2个拷贝bmp6a和bmp6b分别设计敲除靶位点,然后通过2轮基因敲除和筛选,获得F2代bmp6a和bmp6b双基因突变纯合系,然后利用F2代bmp6a和bmp6b双基因突变纯合系进行扩繁形成无肌间刺鲫新品系;
其中,用(bmp6a)或(bmp6b)或(bmp6a和bmp6b)上对应的sgRNA与Cas9蛋白混合后显微注射鲫单细胞期胚胎,进行第一轮基因敲除,构建F0代群体,然后F0代群体养殖3~5个月进行PIT标记和DNA提取,再进行测序确定鲫个体体细胞突变的等位基因及突变率,选取体细胞突变系且体细胞突变率为95%以上的F0代个体作为亲本制备0代受精卵;
用bmp6a或bmp6b或bmp6a和bmp6b上对应的sgRNA与Cas9蛋白混合后显微注射0代受精卵,进行第二轮基因敲除,构建F1代群体,然后F1代群体养殖3~5个月进行PIT标记和DNA提取,再进行测序确定鲫个体体细胞突变的等位基因及突变率,选取体细胞bmp6a和bmp6b双基因突变系且体细胞突变率为95%以上的F1代作为亲本进行繁殖,构建F2代,再从F2代中选取bmp6a和bmp6b双基因突变纯合系。
其中,所述的DNA提取和测序方法为:在0.3~0.5cm
其中,突变体检测采用Sanger测序:采用表2所示的引物对样本进行PCR扩增,体系设置为25μL:基因组DNA上清液2μL、10μM上下游目标引物各1μL、2×DreamTaq Master Mix(Thermo Fisher,CA,USA)12.5μL、无酶水补充至25μL;PCR反应程序为95℃3min,95℃30s,60℃30s,72℃30s,35个循环,72℃5min。PCR产物用1.5%琼脂糖凝胶电泳检测。检测后纯化回收,进行TA克隆,菌落PCR扩增后取2μL PCR产物,用8%的聚丙烯检测,挑选与对照条带大小有差异的菌落进行Sanger测序。
其中,bmp6基因的敲除靶位点如表1所示。
采用如表1所示的sgRNA上游引物和序列为5’-GATCCGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3’的sgRNA下游引物,进行sgRNA体外合成;sgRNA体外扩增体系为:10μM sgRNA上游和下游引物各2.5μL、2×DreamTaq Master mix(Thermo Fisher,CA,USA)25μL、无酶水补充至50μL;共扩增2管共100μL;sgRNA体外扩增程序为95℃变性3min,之后30个循环设置为95℃30s、58℃30s、72℃30s,72℃延伸5min;然后对扩增产物进行纯化回收,之后进行体外转录,每个靶位点建立30μl反应体系:sgRNA PCR回收产物1μg、NTP Buffer Mix 10μL、T7 RNA Polymerase Mix 2μL,用无酶水补足30μL;37℃转录4h,反应结束加入20μl无酶的水,混匀后加入2μl DNase I,在37℃消化15min,去除未反应的DNA。
sgRNA体外PCR产物用1.5%琼脂糖凝胶电泳检测,条带为120bp。检测后用PCR产物纯化回收试剂盒(Exygen)将PCR产物纯化回收,采用Qubit 3试剂盒(Thermo Fisher,CA,USA)测定浓度待用。sgRNA PCR产物回收浓度在100~160ng/μl之间。
将体外转录的sgRNA用RNA纯化试剂盒(Qiagen)纯化回收。采用Qubit 3试剂盒(Thermo Fisher,CA,USA)测定回收产物浓度,于-80℃冰箱中保存待用。体外转录sgRNA回收浓度在800~3000ng/μL之间。
其中显微注射的方法是:体外合成的sgRNA和Cas9蛋白(NEB M0646,MA,USA)按3:1的摩尔浓度比混合后室温孵育10min,再加入25%的酚红,注射到鲫单细胞期的胚胎中;其中,每个外显子上的靶位点sgRNA等量混合后进行注射,每个sgRNA终浓度均不低于50ng/μL,对照组注射25%的酚红,每粒受精卵注射量1nL±0.02nL。
本实施例中所采用的二倍体鲫来自于中国水产科学研究院黑龙江水产研究所呼兰试验场。亲鱼于700m
对本实施例形成的无肌间刺鲫新品系进行突变体骨骼染色观察,发现5尾样本肌间刺完全缺失,4尾样本中肌间刺数量仅4~20枚(如图1~4所示)。对上述这些样本在bmp6a的1号外显子和bmp6b的1号外显子进行检测发现1号外显子的3个靶位点均有突变(如图5和图6所示),基因敲除造成了蛋白翻译提前终止(如图7和图8所示)。
- 一种无肌间刺鲫品系培育方法
- 发育正常无肌间刺鱼类新品种培育方法