一种病原物种特异性序列的筛选方法及系统
文献发布时间:2023-06-19 18:37:28
技术领域
本申请属于生物信息学技术领域,具体涉及一种病原物种特异性序列的筛选方法及系统。
技术背景
病原感染仍然是威胁全世界人类健康的巨大问题,给临床诊断和治疗带来了沉重负担。临床对于病原微生物的主要检测方式为临床培养或生化检测,随着下一代测序技术的不断进步,宏基因组测序(mNGS)极大的提高了病原检测的效率,并有助于识别难以培养的病原微生物。然而,mNGS在临床应用方面仍面临诸多障碍,尤其是当检验样品总量少、样本微生物含量低时,如咽拭子样品、支气管肺泡灌洗液样品、血液样品和脑脊液样品等。同时,由于宿主细胞和核苷酸在这些样本中往往占比很大(通常>90%的宿主成分),极大地降低了微生物鉴定的测序效率。
基于PCR的检测方法以及由常规PCR衍生出来的多重PCR,由于其高灵敏度和低成本而被广泛用于诊断和监测应用。病原靶向测序(tNGS)通过超多重PCR扩增与高通量测序两种技术的结合,能够应对检测样本中存在的几十至上百种病原体。对低浓度的病原微生物的检测,由于通过了PCR的正向富集病原体,同时可以排除宿主核酸对于检测的影响,使其具有更高的灵敏度,可以检测更低浓度的病原微生物。与病原宏基因组测序(mNGS)相比,tNGS具有病原谱范围明确、测序成本低、检测周期短等优势。而基于tNGS进行物种鉴定需要针对物种的特异性区域设计引物,物种鉴定的PCR引物的关键因素之一在于既要寻找物种的特异性区域,又要求寻找出的特异性片段能够能够尽量的覆盖物种内的菌株。两者之间存在一定的矛盾与冲突,因此对于物种鉴定所需要特异性片段筛选具有较高的技术要求。
目前,针对于物种鉴定的特异性区域或者PCR引物设计模板主要依赖于经验、文献检索等方法,该方法存在效率低、试错成本高、特异性序列的种内保守性未知等不足,本申请中的方法建立了一套病原微生物物种特异性片段筛选的有效方法,能够帮助快速的筛选出一套使用最少的引物序列覆盖到最多的物种基因组的特异性序列组合,解决了对于特异性区域查找的难点。
有鉴于此,提出本申请。
发明内容
为解决上述技术问题,本申请通过生物信息学分析,建立了一套快速获取物种特异性序列的方法,能够用于多物种的特异性区域的筛选,相较于文献检索,均有准确性高、时间成本低等优点。另外传统的多重PCR针对16S,18S等保守性较高的片段进行特异性扩增,很难对病原体做到精确至种甚至亚种的分型,而本申请中方法可以用于亚种和菌株的特异性区域的筛选,其适用范围更加广泛。并且本申请提供了一种使用最少的引物对确保物种种内保守且种间特异的序列筛选方法,能够帮助快速的建立起一组适合进行病原微生物鉴定的引物集合。
具体的,本申请提出如下技术方案:
本申请首先提供一种病原物种特异性序列的筛选方法,其特征在于,包括如下步骤:
1)病原比对数据库构建;
基于公共数据库进行病原物种序列的筛选和过滤,构建病原比对数据库;
2)目标物种基因组筛选;
从病原比对数据库中筛选高质量目标物种的基因组序列;
3)物种共有/保守区域筛选;
对筛选出的基因组序列打断成序列片段,合并所有序列片段,并进行聚类;
4)物种间特有序列筛选;
基于聚类结果,从每个聚类cluster从中随机挑选1条序列作为代表序列,将代表序列与病原比对数据库进行比对,得到初步比对结果;将序列相似度M高于阈值,并且该序列除比对到目标物种外未比对到其他物种的代表序列,作为该物种的特异性序列;
进一步的,所述1)中,所述数据库为公共数据库Refseq和/或GenBank数据库。
优选的,所述2)中,所述基因组序列选自完整基因组序列或染色体级别基因组序列;
进一步的,所述3)具体为:基于筛选出的基因组序列,将基因组序列打断为长度为L划窗为N的序列片段,将所有的序列片段合并,得到片段化后的基因组的fasta序列;聚类要求为同一个cluster内的序列的identity>95%;
进一步的,所述L和N的取值为50-100000bp。
进一步的,所述3)进一步包括,选择性去除聚类cluster中包含的基因组数量远小于总基因组数量的cluster。
进一步的,所述步骤4)中,
所述相似度M计算如下:M=A/L
其中,A为一条序列正确比对上参考序列的碱基数,L为该序列的总长度;
所述阈值为0.95。
进一步的,所述方法进一步包括如下步骤:
5)覆盖最多物种基因组的最小特异性序列集合筛选:
基于步骤4)获得的特异性序列所在的cluster,按照每个cluster中包含的菌株数量、每个cluster中拷贝数>2的菌株数量、和每个cluster中包含的序列数量依次进行排序,选取排名第一的cluster,在剩余的cluster结果中去除第一组cluster中已经筛选出的基因组,重新基于以上规则进行排序,进行迭代挑选排名第一的cluster,直到下一次筛选没有新的基因组可以被筛选出。
进一步的,所述步骤5)进一步包括:
将筛选出的每一组cluster对齐基因组片段,确认同一组cluster的相似度以及序列的SNP/INDEL情况,确认是否能在该位点设计引物及是否需要设计兼并引物。
进一步的,所述病原物种为细菌类病原物种。
本申请还提供上述任一所述方法在病原引物设计中的应用。
本申请还提供一种病原物种特异性序列的筛选系统,包括如下模块:
模块1)病原比对数据库构建模块:用于基于公共数据库进行病原物种序列的筛选和过滤,构建病原比对数据库;
模块2)目标物种基因组筛选模块:用于从病原比对数据库中筛选高质量目标物种的基因组序列;
模块3)物种共有/保守区域筛选模块:用于对筛选出的基因组序列打断成序列片段,合并所有序列片段,并进行聚类;
模块4)物种间特有序列筛选模块:用于基于聚类结果,从每个聚类cluster从中随机挑选1条序列作为代表序列,将代表序列与病原比对数据库进行比对,得到初步比对结果;将序列相似度M高于阈值,并且该序列除比对到目标物种外未比对到其他物种的代表序列,作为该物种的特异性序列;
进一步的,模块1)中,所述数据库为公共数据库Refseq和/或GenBank数据库。
进一步的,模块2)中,所述基因组序列选自完整基因组序列或染色体级别基因组序列;
进一步的,模块3)具体为:基于筛选出的基因组序列,将基因组序列打断为长度为L划窗为N的序列片段,将所有的序列片段合并,得到片段化后的基因组的fasta序列;聚类要求为同一个cluster内的序列的identity>95%;
进一步的,所述L和N的取值为50-100000bp。
进一步的,所述模块3)中进一步包括,用于选择性去除聚类cluster中包含的基因组数量远小于总基因组数量的cluster。
进一步的,所述模块3)中,
所述相似度M计算如下:M=A/L
其中,A为一条序列正确比对上参考序列的碱基数,L为该序列的总长度;
所述阈值为0.95。
进一步的,所述方法进一步包括如下模块:
模块6):用于覆盖最多物种基因组的最小特异性序列集合筛选的模块:
基于步骤4)获得的特异性序列所在的cluster,按照每个cluster中包含的菌株数量、每个cluster中拷贝数>2的菌株数量、和每个cluster中包含的序列数量依次进行排序,选取排名第一的cluster,在剩余的cluster结果中去除第一组cluster中已经筛选出的基因组,重新基于以上规则进行排序,进行迭代挑选排名第一的cluster,直到下一次筛选没有新的基因组可以被筛选出。
进一步的,所述模块5)进一步执行包括:
将筛选出的每一组cluster对齐基因组片段,确认同一组cluster的相似度以及序列的SNP/INDEL情况,确认是否能在该位点设计引物及是否需要设计兼并引物。
进一步的,所述病原物种为细菌类病原物种。
本申请还提供一种电子设备,包括:处理器和存储器;所述处理器和存储器相连,其中,所述存储器用于存储计算机程序,所述处理器用于调用所述计算机程序,以执行上述任一项所述的方法。
本申请还提供一种计算机存储介质,所述计算机存储介质存储有计算机程序,所述计算机程序包括程序指令,所述程序指令当被处理器执行时,执行上述任一项所述的方法。
本申请有益技术效果:
1)本申请通过筛选物种间特异且物种内保守的区域,建立了一套快速获取物种特异性序列的方法,同时该方法还能实现使用最少引物对即可保证物种种内保守且种间特异。
2)本申请可以用于多物种的筛选,相较于传统方法,准确性高并且时间成本低。
3)本申请可以用于亚种/菌株型的特异性区域筛选,适用范围更加广泛。
附图说明
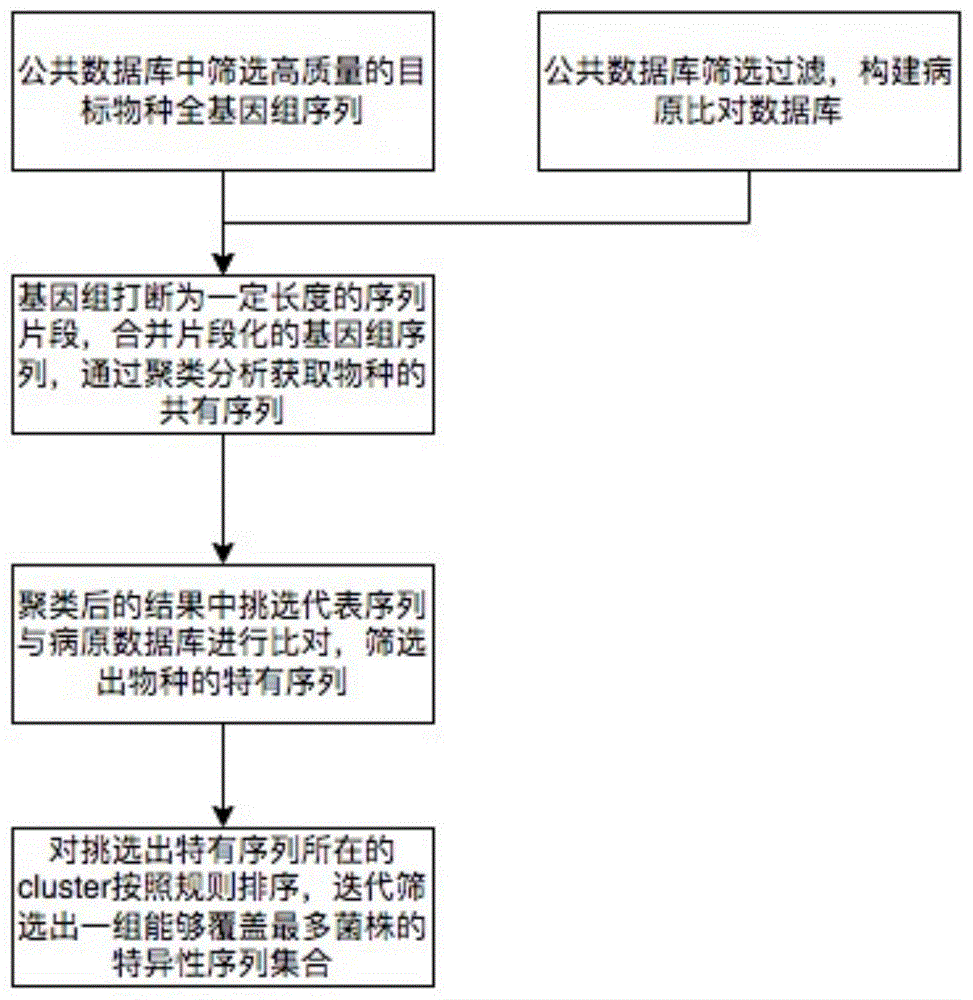
图1、物种特异性序列筛选流程图;
图2、特异性序列覆盖情况。
具体实施方式
下面将结合实施例对本申请的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本申请,而不应视为限制本申请的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市场购买获得的常规产品。
部分术语定义,除非在下文中另有定义,本申请具体实施方式中所用的所有技术术语和科学术语的含义意图与本领域技术人员通常所理解的相同。虽然相信以下术语对于本领域技术人员很好理解,但仍然阐述以下定义以更好地解释本申请。
本申请中的术语“大约”表示本领域技术人员能够理解的仍可保证论及特征的技术效果的准确度区间。该术语通常表示偏离指示数值的±10%,优选±5%。
如本申请中所使用,术语“包括”、“包含”、“具有”、“含有”或“涉及”为包含性的(inclusive)或开放式的,且不排除其它未列举的元素或方法步骤。术语“由…组成”被认为是术语“包含”的优选实施方案。如果在下文中某一组被定义为包含至少一定数目的实施方案,这也应被理解为揭示了一个优选地仅由这些实施方案组成的组。
此外,说明书和权利要求书中的术语第一、第二、第三、(a)、(b)、(c)以及诸如此类,是用于区分相似的元素,不是描述顺序或时间次序必须的。应理解,如此应用的术语在适当的环境下可互换,并且本申请描述的实施方案能以不同于本申请描述或举例说明的其它顺序实施。
本申请所述的一种病原物种特异性序列的筛选方法,包括如下步骤:
1)病原比对数据库构建:基于公共数据库进行病原物种序列的筛选和过滤,构建病原比对数据库;
2)目标物种基因组筛选:从病原比对数据库中筛选高质量目标物种的基因组序列;
3)物种共有/保守区域筛选:对筛选出的基因组序列打断成序列片段,合并所有序列片段,并进行聚类;
4)物种间特有序列筛选:基于聚类结果,从每个聚类cluster从中随机挑选1条序列作为代表序列,将代表序列与病原比对数据库进行比对,得到初步比对结果;将序列相似度M高于阈值,并且该序列除比对到目标物种外未比对到其他物种的代表序列,作为该物种的特异性序列;
在一些实施方式中,步骤1中,所述数据库为公共数据库Refseq和/或GenBank数据库。
在一些实施方式中,步骤2)中,所述基因组序列选自完整基因组序列或染色体级别基因组序列;
在一些实施方式中,所述3)具体为:基于筛选出的基因组序列,将基因组序列打断为长度为L划窗为N的序列片段,将所有的序列片段合并,得到片段化后的基因组的fasta序列;聚类要求为同一个cluster内的序列的identity>95%;
在一些实施方式中,所述L和N的取值为50-100000bp。
在一些实施方式中,所述3)进一步包括,选择性去除聚类cluster中包含的基因组数量远小于总基因组数量的cluster。
在一些实施方式中,所述步骤4)中,
所述相似度M计算如下:M=A/L
其中,A为一条序列正确比对上参考序列的碱基数,L为该序列的总长度;
所述阈值为0.95。
在一些实施方式中,所述方法进一步包括如下步骤:
5)覆盖最多物种基因组的最小特异性序列集合筛选:
基于步骤4)获得的特异性序列所在的cluster,按照每个cluster中包含的菌株数量、每个cluster中拷贝数>2的菌株数量、和每个cluster中包含的序列数量依次进行排序,选取排名第一的cluster,在剩余的cluster结果中去除第一组cluster中已经筛选出的基因组,重新基于以上规则进行排序,进行迭代挑选排名第一的cluster,直到下一次筛选没有新的基因组可以被筛选出。
在一些实施方式中,所述步骤5)进一步包括:
将筛选出的每一组cluster对齐基因组片段,确认同一组cluster的相似度以及序列的SNP/INDEL情况,确认是否能在该位点设计引物及是否需要设计兼并引物。
在一些实施方式中,所述病原物种为细菌类病原物种。
本申请所述的病原物种特异性序列的筛选系统,包括如下模块:
模块1)病原比对数据库构建模块:用于基于公共数据库筛选和过滤,构建病原比对数据库;
模块2)目标物种基因组筛选模块:用于从病原比对数据库中筛选高质量目标物种的基因组序列;
模块3)物种共有/保守区域筛选模块:用于对筛选出的基因组序列打断成序列片段,合并所有序列片段,并进行聚类;
模块4)物种间特有序列筛选模块:用于基于聚类结果,从每个聚类cluster从中挑选1条序列作为代表序列,将代表序列与病原比对数据库进行比对,得到初步比对结果;将序列相似度M高于阈值,并且该序列除比对到目标物种外未比对到其他物种的代表序列,作为该物种的特异性序列;
在一些实施方式中,模块1)中,所述数据库为公共数据库Refseq和/或GenBank数据库。
在一些实施方式中,模块2)中,所述基因组序列选自完整基因组序列或染色体级别基因组序列;
在一些实施方式中,模块3)具体为:基于筛选出的基因组序列,将基因组序列打断为长度为L划窗为N的序列片段,将所有的序列片段合并,得到片段化后的基因组的fasta序列;聚类要求为同一个cluster内的序列的identity>95%;
在一些实施方式中,所述L和N的取值为50-100000bp。
在一些实施方式中,所述模块3)中进一步包括,用于选择性去除聚类cluster中包含的基因组数量远小于总基因组数量的cluster。
在一些实施方式中,所述模块3)中,
所述相似度M计算如下:M=A/L
其中,A为一条序列正确比对上参考序列的碱基数,L为该序列的总长度;
所述阈值为0.95。
在一些实施方式中,所述方法进一步包括如下模块:
模块5):用于覆盖最多物种基因组的最小特异性序列集合筛选的模块:
基于步骤4)获得的特异性序列所在的cluster,按照每个cluster中包含的菌株数量、每个cluster中拷贝数>2的菌株数量、和每个cluster中包含的序列数量依次进行排序,选取排名第一的cluster,在剩余的cluster结果中去除第一组cluster中已经筛选出的基因组,重新基于以上规则进行排序,进行迭代挑选排名第一的cluster,直到下一次筛选没有新的基因组可以被筛选出。
在一些实施方式中,所述模块5)进一步执行包括:
将筛选出的每一组cluster对齐基因组片段,确认同一组cluster的相似度以及序列的SNP/INDEL情况,确认是否能在该位点设计引物及是否需要设计兼并引物。
在一些实施方式中,所述病原物种为细菌类病原物种。
下面结合具体实施例来阐述本申请。
实验例本申请方法体系的建立
本申请通过前期设计和优化,建立的方法体系大体如附图1所示流程,具体的:
1.比对数据库构建和目标物种基因组的获取
基于公共数据库(Refseq和GenBank)数据库,进行筛选和过滤构建病原比对数据库,从中筛选出高质量的目标物种的全基因组序列;对于基因组的选择,优先选择完整基因组(Complete Genome)及染色体级别(Chromosome)水平的基因组序列;
2.物种共有/保守区域筛选
基于筛选出的基因组序列,将基因组序列打断为长度为L划窗为N的序列片段,L和N的取值为50-100000bp,将所有的序列片段合并,得到片段化后的基因组的fasta序列,使用cd-hit-est对上述的fasta序列进行聚类,聚类要求为同一个cluster内的序列的identity>95%,可以选择性的去除聚类的cluster中包含的基因组数量<总基因组数量一定占比的cluster。所述identity为软件参数自动计算得出。
3.物种间特有序列筛选
基于上述聚类完成的结果,对于每一个cluster从中随机挑选出1条序列作为代表序列(因为序列相似度比较高,采用的随机挑选),使用bwa mem将代表序列与病原微生物基因组数据库进行比对,得到初步的比对结果,再将相似度M高于阈值,并且该序列除比对到目标物种外未比对到其他物种的代表序列保留作为该物种的特异性序列,相似度通过如下方式计算得出:
M=A/L
其中A为一条序列正确比对上参考序列的碱基数,L为该序列的总长度;
4.筛选覆盖最多物种基因组的最小特异性序列集合
基于上述特异性序列筛选的cluster,按照每个cluster包含的菌株的数量、拷贝数copy>2的菌株的数量、包含序列数进行排序,挑选排名第一的cluster,然后在剩余的cluster结果中去除第一组cluster中已经筛选出的基因组,重新基于以上规则进行排序,进行迭代挑选排名第一的cluster,直到下一次筛选没有新的基因组可以被筛选出。所述Copy>2的定义为cluster中存在某一个菌株的两个或者多个片段。将筛选出的每一组cluster使用clustalw对齐基因组片段,用于确认同一组cluster的相似度以及序列的SNP/INDEL情况,用于引物设计时考量是否能在该位点设计引物及是否需要设计兼并引物。
实施例1方法体系的验证
(1)从公共数据库下载7种常见病原体的高质量基因组序列,使用本申请上述方法分别进行筛选,具体方法为:将基因组打断成片段,根据将相似性>0.95的片段聚类为1个cluster,最终得到多个cluster,然后从每个cluster中随机挑选1条序列与病原比对数据库进行比对,对于满足相似性>0.95并且该序列除比对到目标物种外未比对到其他物种,作为物种的特异性序列,再从中筛选出了覆盖最多物种基因组的最小特异性序列集合,将挑选的特异性序列与NT数据库进行了二次比对,用于确认筛选的序列为特异性序列,所述的nt库为Nucleotide Sequence Database(https://www.ncbi.nlm.nih.gov/nucleotide/),筛选及二次比对的结果如下表:
基于本申请方法,7个物种中共挑选出92条特异性序列,这些特异性序列与NT库进行比确认挑选出的序列为物种特异性序列。表中所述的nt库比对特异性比例为挑选的特异性cluster的代表序列在比对NT库后仍然为特异性序列的比例。
(2)对于筛选出的特异性序列集,按照迭代出的特异性序列的次序,计算每迭代筛选出1条特异性序列,对于菌株覆盖占比的影响。以肠道沙门氏菌为例:肠道沙门氏菌一共有1319个基因组,按照方法描述中筛选覆盖最多物种基因组的最小特异性序列集合的方法,筛选出1条特异性序列时,能够覆盖肠道沙门氏菌的712(54%)个基因组,筛选出第2条序列时,第一条和第二条序列组成的集合可以覆盖肠道沙门氏菌的1015(77%)个基因组,以此方法进行迭代,直至筛选出的新的特异性序列不会增加菌株覆盖占比。计算挑选出的特异性序列集能够覆盖到物种基因组的占比。
菌株覆盖占比计算方法如下:
菌株覆盖占比=特异序列覆盖的菌株数量/物种总菌株数量。
从图2可知道,对于肠道沙门氏菌、粪肠球菌、李斯特菌、肠道沙门氏菌和EB病毒,<=10个特异性序列能够覆盖97%的物种基因组,其中肠道沙门氏菌的筛选效果最为明显,14个特异性序列能够覆盖该物种的1319个基因组序列。铜绿假单胞菌和金黄色葡萄球菌分别需要16和25个特异性序列能够覆盖97%的基因组序列,相较于其953和505个基因组的数量,同样具有高效性。
(3)基于上述筛选的特异性序列的结果,使用本申请中介绍的方法与随机从总特异性序列中抽取出的序列对于抽取出的特异性序列能够覆盖的菌株的比例进行比较。随机筛选的方法为从总特异性序列中随机抽取与本申请方法挑选的特异性序列一致数量的序列,重复100次并对菌株覆盖占比取平均值,具体数据展示如下:
从结果中看出,在挑选相同数量的特异性序列时,目标物种通过筛选的特异性序列能够覆盖该物种下>=97%的菌株,本申请中的方法的菌株覆盖占比优于随机筛选。从另一个方面也表明了如果通过随机筛选达到和本申请一致的菌株覆盖占比需要更多的特异性cluster。
最后应说明的是:以上各实施例仅用以说明本申请的技术方案,而非对其限制;尽管参照前述各实施例对本申请进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本申请各实施例技术方案的范围。