2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及农药合成技术领域,具体涉及2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法。
背景技术
五氟磺草胺是一种广泛使用的的苗后用除草剂,化学名称为:3-(2,2-二氟乙氧基)-N-(5,8-二甲氧基-[1,2,4]三唑并[1,5-C]嘧啶-2-基)-α,α,α-三氟甲苯基-2-磺酰胺,属于三唑并嘧啶磺酰胺类除草剂,其作用机制同目前的磺胺脲类及咪唑啉酮类一样,均通过抑制乙酰乳酸合成酶(ALS)而起作用,对多种稗草包括大龄稗草都有良好的防效,而且杀草谱很广,可用于水稻田防除稗草及一年生莎草和多种阔叶杂草。残留的五氟磺草胺易被光解及微生物降解,所以,五氟磺草胺由于其低毒、高效、环境友好受到广泛关注。五氟磺草胺的通过2-(2,2-二氟乙氧基)-6-(三氟甲基)苯磺酰氯与5,8-二甲氧基-[1,2,4]三唑[1,5-c]嘧啶-2-胺在有机溶剂中经碱作用反应制备得到。
作为合成五氟磺草胺的重要原料,2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的合成路线往往较为繁琐,涉及的反应步骤较多,总产率较低。
因此,需要提供一种更加合适的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的合成方法。
发明内容
(一)要解决的技术问题
鉴于上述技术问题,为了解决现有技术中存在的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的合成路线繁琐,总产率较低的问题,本发明提供2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法。
(二)技术方案
为了达到上述目的,本发明采用的主要技术方案包括:
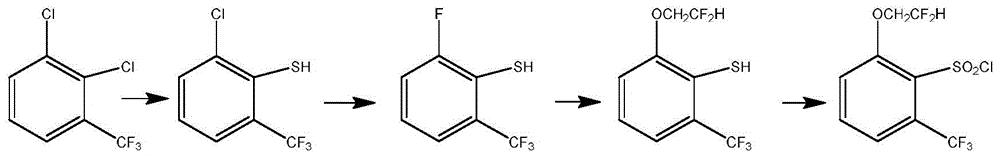
2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,包括如下步骤:
S1:将式I化合物与3-巯基丙酸2-乙基己酯、叔丁醇钠在非质子试剂中反应得式II化合物;
S2:将式II化合物与氟化试剂溶解在第一溶剂中,加入第一催化剂反应得式III化合物;
S3:将式III化合物与2,2-二氟乙醇在碱作用下在第二溶剂中进行反应得式Ⅳ化合物;
S4:将式Ⅳ化合物溶于第三溶剂,然后在一定温度条件下与氯气反应,得到目标产物2-(2,2-二氟乙氧基)-6-(三氟甲基)苯磺酰氯。
如上所述的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,优选地,步骤S1中,式I化合物、3-巯基丙酸2-乙基己酯以及叔丁醇钠的摩尔比为1:2.5-4.2:0.3-0.8。
如上所述的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,优选地,步骤S1中,式I化合物、3-巯基丙酸2-乙基己酯以及叔丁醇钠的反应温度为120-180℃,反应时间为12-24h。
如上所述的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,优选地,步骤S2中,加入第一催化剂后,升温至100-220℃反应2-3h后得式III化合物;
式II化合物、氟化试剂以及第一催化剂的摩尔比为1:1.5-1.8:0.3-0.7。
如上所述的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,优选地,步骤S2中,第一溶剂为1,3-二甲基咪唑啉酮,氟化试剂为氟化钾或者氟化钠,第一催化剂为三苯基溴化磷或者二异丙基乙胺。
如上所述的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,优选地,步骤S3中,式III化合物、2,2-二氟乙醇以及碱的摩尔比为1:0.8-1.2:1.5-2.7;
式II化合物与氟化试剂在50-90℃的温度下反应3-6h。
如上所述的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,优选地,步骤S3中,所述碱为氢氧化钠、氨基钠、氢化钠中的一种或者几种;所述第二溶剂为二甲基甲酰胺、二甲基亚砜、乙腈中的一种或者几种。
如上所述的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,优选地,步骤S4中,式Ⅳ化合物与氯气的摩尔比为1:5-15。
如上所述的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,优选地,步骤S4中,将式Ⅳ化合物溶于第三溶剂,然后在30-40℃下与氯气反应2-3h,得到目标产物2-(2,2-二氟乙氧基)-6-(三氟甲基)苯磺酰氯。
如上所述的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,优选地,步骤S4中,第三溶剂为氯代烷烃、水、乙酸中的一种或者几种。
(三)有益效果
本发明的2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的合成方法反应步骤较少,反应路线简洁,操作方便,目标产物2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的总产率能够稳定维持在85%以上,收率较高。
附图说明
图1为本发明中2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的合成路线图。
具体实施方式
为了更好的解释本发明,以便于理解,下面结合附图以及具体实施方式,对本发明作详细描述。
实施例1
如图1所示,本实施例提供2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,包括如下步骤:
S1:式Ⅱ化合物的合成:
将0.1mol式I化合物、0.3mol的3-巯基丙酸2-乙基己酯、0.05mol叔丁醇钠加入200ml四氯化碳中,将体系加热至160℃,反应16h。经计算,式Ⅱ化合物的收率为88%。
S2:式Ⅲ化合物的合成:
将0.1mol式Ⅱ化合物、0.16mol氟化钠、0.05mol三苯基溴化磷和3mol 1,3-二甲基咪唑啉酮加入装有精馏柱的三口瓶,搅拌升温回流反应,保持反应温度为160℃,反应2.5h。反应结束后蒸出无色透明液体,即式Ⅲ化合物,经计算,收率为90%。
S3:式Ⅳ化合物的合成:
将0.3mol NaNH
S4:式Ⅴ化合物的合成:
将0.1mol式Ⅳ化合物溶于100mL乙酸与10mL水的混合溶剂中,升温至30℃,通入1mol氯气后,升温至35℃,在该温度下保温2.5小时。反应结束后将反应体系倒入到300mL冰水中,用乙酸乙酯萃取后,合并脱溶得白色固体2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯,经计算,本实施例中2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的收率为91%。
实施例2
本实施例提供2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,包括如下步骤:
S1:式Ⅱ化合物的合成:
将0.1mol式I化合物、0.25mol的3-巯基丙酸2-乙基己酯、0.03mol叔丁醇钠加入200ml乙醚中,将反应体系加热至120℃,反应12h。经计算,式Ⅱ化合物的收率为86%。
S2:式Ⅲ化合物的合成:
将0.1mol式Ⅱ化合物、0.15mol氟化钠、0.03mol二异丙基乙胺和4mol 1,3-二甲基咪唑啉酮加入装有精馏柱的三口瓶,搅拌升温回流反应,保持反应温度为110℃,反应2h。反应结束后蒸出无色透明液体,即式Ⅲ化合物,经计算,收率为87%。
S3:式Ⅳ化合物的合成:
将0.225mol氢氧化钠悬浮于250mL二甲基亚砜中,降温至5℃,加入0.12mol二氟乙醇,升温至50℃,搅拌1小时。然后缓慢滴加0.15mol式Ⅲ化合物,滴毕反应2小时。反应结束后将反应液倒入300mL冰水中,用乙酸乙酯萃取,合并有机相水洗至中性,脱溶得式Ⅳ化合物,经计算,式Ⅳ化合物的收率为90%。
S4:式Ⅴ化合物的合成:
将0.1mol式Ⅳ化合物溶于100mL乙酸与10mL水的混合溶剂中,升温至25℃,通入0.5mol氯气后,升温至30℃,在该温度下保温2小时。反应结束后将反应体系倒入到300mL冰水中,用乙酸乙酯萃取后,合并脱溶得白色固体2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯,经计算,本实施例中2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的收率为89%。
实施例3
本实施例提供2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,包括如下步骤:
S1:式Ⅱ化合物的合成:
将0.1mol式I化合物、0.42mol的3-巯基丙酸2-乙基己酯、0.08mol叔丁醇钠加入200ml四氯化碳中,将体系加热至180℃,反应24h。经计算,式Ⅱ化合物的收率为85%。
S2:式Ⅲ化合物的合成:
将0.1mol式Ⅱ化合物、0.18mol氟化钾、0.07mol二异丙基乙胺和2.5mol 1,3-二甲基咪唑啉酮加入装有精馏柱的三口瓶,搅拌升温回流反应,保持反应温度为220℃,反应3h。反应结束后蒸出无色透明液体,即式Ⅲ化合物,经计算,收率为89%。
S3:式Ⅳ化合物的合成:
将0.405mol氢化钠悬浮于250mL乙腈中,降温至5℃,加入0.18mol二氟乙醇,升温至90℃,搅拌2小时。然后缓慢滴加0.15mol式Ⅲ化合物,滴毕反应4小时。反应结束后将反应液倒入300mL冰水中,用乙酸乙酯萃取,合并有机相水洗至中性,脱溶得式Ⅳ化合物,经计算,式Ⅳ化合物的收率为88%。
S4:式Ⅴ化合物的合成:
将0.1mol式Ⅳ化合物溶于100mL乙酸与10mL水的混合溶剂中,升温至30℃,通入1.5mol氯气后,升温至40℃,在该温度下保温3小时。反应结束后将反应体系倒入到300mL冰水中,用乙酸乙酯萃取后,合并脱溶得白色固体2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯,经计算,本实施例中2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的收率为90%。
实施例4
本实施例提供2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,包括如下步骤:
S1:式Ⅱ化合物的合成:
将0.1mol式I化合物、0.3mol的3-巯基丙酸2-乙基己酯、0.05mol叔丁醇钠加入200ml四氯化碳中,将体系加热至170℃,反应14h。经计算,式Ⅱ化合物的收率为86%。
S2:式Ⅲ化合物的合成:
将0.1mol式Ⅱ化合物、0.16mol氟化钾、0.06mol二异丙基乙胺和3mol 1,3-二甲基咪唑啉酮加入装有精馏柱的三口瓶,搅拌升温回流反应,保持反应温度为140℃,反应2h。反应结束后蒸出无色透明液体,即式Ⅲ化合物,经计算,收率为87%。
S3:式Ⅳ化合物的合成:
将0.15mol氢氧化钠和0.1mol氢化钠悬浮于250mL二甲基甲酰胺中,降温至5℃,加入0.15mol二氟乙醇,升温至60℃,搅拌2小时。然后缓慢滴加0.15mol式Ⅲ化合物,滴毕反应3小时。反应结束后将反应液倒入300mL冰水中,用乙酸乙酯萃取,合并有机相水洗至中性,脱溶得式Ⅳ化合物,经计算,式Ⅳ化合物的收率为89%。
S4:式Ⅴ化合物的合成:
将0.1mol式Ⅳ化合物溶于120mL氯代烷烃中,升温至30℃,通入1.5mol氯气后,升温至35℃,在该温度下保温2.5小时。反应结束后将反应体系倒入到300mL冰水中,用乙酸乙酯萃取后,合并脱溶得白色固体2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯,经计算,本实施例中2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的收率为88%。
实施例5
本实施例提供2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的制备方法,包括如下步骤:
S1:式Ⅱ化合物的合成:
将0.1mol式I化合物、0.3mol的3-巯基丙酸2-乙基己酯、0.05mol叔丁醇钠加入200ml四氯化碳中,将体系加热至170℃,反应18h。经计算,式Ⅱ化合物的收率为87%。
S2:式Ⅲ化合物的合成:
将0.1mol式Ⅱ化合物、0.16mol氟化钠、0.03mol三苯基溴化磷和3mol 1,3-二甲基咪唑啉酮加入装有精馏柱的三口瓶,搅拌升温回流反应,保持反应温度为210℃,反应2h。反应结束后蒸出无色透明液体,即式Ⅲ化合物,经计算,收率为86%。
S3:式Ⅳ化合物的合成:
将0.3mol NaNH
S4:式Ⅴ化合物的合成:
将0.1mol式Ⅳ化合物溶于150mL水中,升温至30℃,通入1mol氯气后,升温至35℃,在该温度下保温2.5小时。反应结束后将反应体系倒入到300mL冰水中,用乙酸乙酯萃取后,合并脱溶得白色固体2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯,经计算,本实施例中2-(2,2-二氟乙氧基)-6-三氟甲基苯磺酰氯的收率为87%。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。