氮掺杂碳包覆铂镍合金纳米材料及其制备方法和应用
文献发布时间:2024-04-18 19:53:33
技术领域
本发明涉及电催化领域,具体涉及一种氮掺杂碳包覆铂镍合金纳米材料及其制备方法和应用。
背景技术
目前,由于能源危机和环境污染逐渐成为制约人类社会发展的主要障碍之一,人们对发展可再生清洁能源技术有着迫切的需求。燃料电池是通过电化学反应直接将燃料的化学能转变为电能的装置,具有能量转化效率高、燃料来源广泛、污染小等优点,在可再生清洁能源领域扮演着重要的角色,已被逐步应用到交通、通讯、航空等领域。燃料电池的核心部件之一是电极材料,主要涉及的反应为:阳极的氧化反应(例如:氢气氧化HOR,H
采用铂合金催化剂以提高本征活性、降低铂的用量是目前的主要研究方向之一。通常,铂基催化剂主要为铂或者铂合金纳米颗粒负载在碳载体上,这种方法可以将活性位点充分暴露出来,但是无法抑制在长循环过程中铂的团聚和溶解。同时,商业燃料电池采用离子聚合物Nafion,其中的磺酸基团与Pt直接接触会造成活性位点失活。已有研究采用修饰碳载体的方法增强载体与铂/铂合金纳米颗粒的相互作用,抑制铂的团聚和溶解,但是这种方法不能阻止电解质(如Nafion)与铂/铂合金纳米颗粒的直接接触。采用碳包覆铂/铂合金纳米颗粒的方法是一种更有前景的制备高稳定性铂基电催化剂的方法。同时,在碳笼中引入氮元素,利用氮对金属的锚定作用,可以增强碳笼对金属团聚和溶解的抑制作用;碳笼中的氮元素还可以提供M-N-C活性位点,增强材料的电催化活性。
发明内容
本发明的目的是为了克服现有技术存在的上述问题,提供一种氮掺杂碳包覆铂镍合金纳米材料及其制备方法和应用,该氮掺杂碳包覆铂镍合金纳米材料具有良好的电催化性能。
为了实现上述目的,本发明一方面提供一种氮掺杂碳包覆铂镍合金纳米材料,其特征在于,该纳米材料具有内核为铂镍合金纳米颗粒、壳层为氮掺杂碳笼的核壳结构,所述铂镍合金纳米颗粒为实心结构和/或多孔结构。
优选地,在所述纳米材料中,铂/镍的摩尔比为0.1-3:1,优选为0.2-1.5:1。
优选地,在所述纳米材料中,氮含量为1-15重量%,碳含量为5-70重量%,铂含量为5-70重量%,镍含量为5-70重量%,氢含量为0.1-3重量%,氧含量为0.5-20重量%。
优选地,在所述纳米材料的XRD谱图中2θ为39.7°~44.7°的范围内存在至少一个铂镍合金峰。
优选地,所述铂镍合金颗粒的粒径为2-200nm,优选5-100nm。
优选地,所述纳米材料的比表面积为100-500m
优选地,所述纳米材料的稀硫酸酸洗保留率为80%以上。
优选地,所述纳米材料的电阻率为0.01-10000Ω·m,优选为0.01-100Ω·m。
本发明第二方面提供一种氮掺杂碳包覆铂镍合金纳米材料的制备方法,该方法包括如下步骤:
(1)前驱体制备:将含有金属前驱体、碳源、氮源和溶剂的均相溶液中的溶剂除去,得到前驱体材料,其中,所述金属前驱体包括铂源和镍源,所述碳源为酸性有机还原剂;
(2)焙烧:在惰性气氛中,将步骤(1)得到的前驱体材料进行高温热解,得到热解产物,其中,所述高温热解的温度为400-1100℃;
(3)酸洗:将步骤(2)得到的热解产物与酸溶液接触反应,然后依次进行固液分离、洗涤和干燥。
优选地,步骤(1)中,所述碳源为柠檬酸、抗坏血酸、乙二胺四乙酸、2,5-吡啶二羧酸、苯甲酸和对苯二甲酸中的一种或多种;优选地,以金属元素计的所述金属前驱体与所述碳源的摩尔比为1:0.5-5。
优选地,步骤(1)中,所述氮源为乙二胺四乙酸、氨水和六亚甲基四胺中的一种或多种,优选地,以金属元素计的所述金属前驱体与所述氮源的摩尔比为1:0.01-10。
优选地,步骤(1)中,所述铂源为氯铂酸、醋酸四氨合铂、乙酰丙酮铂、氯铂酸盐和氯化铂中的一种或多种。
优选地,步骤(1)中,所述镍源为乙酸镍、六水合二氯化镍、乙酰丙酮镍、碱式碳酸镍、碳酸镍和硫酸镍中的一种或多种。
优选地,步骤(1)中,以铂计的所述铂源与以镍计的所述镍源的摩尔比为1:0.5-20,优选为1:1-10。
优选地,步骤(1)中,所述溶剂为水、醇类溶剂和N,N-二甲基甲酰胺中的一种或多种。
优选地,步骤(1)中,所述醇类溶剂为乙醇。
优选地,步骤(2)中,所述高温热解的升温速率为2-10℃/min。
优选地,步骤(2)中,所述高温热解的恒温时间为1-6h。
优选地,步骤(3)中,所述酸溶液为硫酸溶液、硝酸溶液和盐酸溶液中的一种或多种。
优选地,步骤(3)中,相对于步骤(2)得到的热解产物中的镍元素1mol,所述酸溶液的用量以H+计为2mol以上。
优选地,步骤(3)中,所述酸溶液为硫酸溶液时,酸浓度为0.5-2mol/L,接触反应的温度为25-90℃;所述酸溶液为硝酸溶液时,酸浓度为0.5-15mol/L,接触反应的温度为25-60℃;所述酸溶液为盐酸溶液时,酸浓度为0.5-2mol/L,接触反应的温度为25-90℃。
优选地,步骤(3)中,接触反应的时间为3-50h,优选为3-24h。
本发明第三方面提供利用上述本发明的制备方法得到的氮掺杂碳包覆铂镍合金纳米材料。
本发明第四方面提供一种催化剂,其含有上述本发明的氮掺杂碳包覆铂镍合金纳米材料和导电炭黑。
优选地,所述氮掺杂碳包覆铂镍合金纳米材料与所述导电炭黑的重量比为1:0.1-5。
本发明第五方面提供本发明催化剂的一种制备方法,该制备方法包括:在溶剂的存在下,将上述本发明的氮掺杂碳包覆铂镍合金纳米材料与导电炭黑混合后,将得到的混合物中的溶剂除去并干燥。
优选地,所述氮掺杂碳包覆铂镍合金纳米材料与所述导电炭黑的重量比为1:0.1-5。
优选地,所述混合包括超声、机械搅拌和研磨中的一种或多种,优选超声的时间为0.5-2h,优选机械搅拌的时间为8-24h,优选研磨的条件包括:在惰性气氛中球磨,转速100-500rpm,球磨时间2-24h。
本发明第六方面提供本发明催化剂的另一种制备方法,该制备方法包括:将上述本发明的氮掺杂碳包覆铂镍合金纳米材料与导电炭黑进行固相混合。
优选地,所述氮掺杂碳包覆铂镍合金纳米材料与所述导电炭黑的重量比为1:0.1-5。
优选地,所述固相混合的条件包括:在惰性气氛中球磨,转速100-500rpm,球磨时间2-24h。
本发明第七方面提供上述本发明的氮掺杂碳包覆铂镍合金纳米材料、催化剂、或者上述本发明的制备方法制得的催化剂作为燃料电池催化剂的应用。
通过上述技术方案,本发明的氮掺杂碳包覆铂镍合金纳米材料制备方法简单,还原金属的同时原位生成氮掺杂包覆碳笼;化学组分含量易于调控,具有优异的电催化性能。铂镍合金可以调节铂对含氧物种的吸附能,提高催化剂的本征活性,碳笼中的氮元素与金属的键合作用较强,可以增加对铂和镍的锚定作用,从而增强包覆碳笼对铂镍合金纳米颗粒团聚和溶解的抑制作用,提高稳定性;此外,碳笼中的氮元素还可以提供Ni-N-C活性位点,增强材料的电催化活性。本发明的纳米材料可以直接用作电催化剂,或者与导电碳黑混合后用作电催化剂,催化阴极氧还原反应(ORR)时,电化学循环5000圈后的电化学活性面积(ECSA)稳定性良好,变化率不高于20%。
与现有技术相比较,本发明的氮掺杂碳包覆铂镍合金纳米材料具有以下优点:一是氮掺杂碳包覆铂镍合金纳米材料的组分含量易于调控;二是催化氧还原反应时,其电化学活性面积(ECSA)稳定性良好,循环扫描5000圈后的ECSA变化率不高于20%。
附图说明
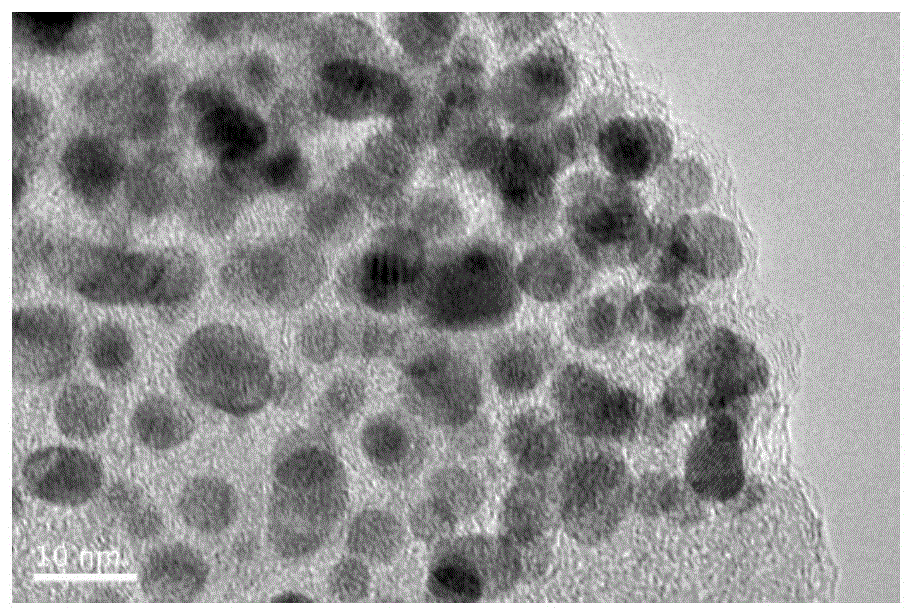
图1是实施例1制备的氮掺杂碳包覆铂镍合金纳米材料的TEM图谱;
图2是实施例1制备的氮掺杂碳包覆铂镍合金纳米材料的XRD图谱;
图3是实施例1制备的氮掺杂碳包覆铂镍合金纳米材料的XPS全谱图;
图4是实施例1A制备的氮掺杂碳包覆铂镍合金催化剂用作ORR催化剂循环扫描5000圈前后的LSV图;
图5是实施例1A制备的氮掺杂碳包覆铂镍合金催化剂用作ORR催化剂循环扫描5000圈前后的ECSA图;
图6是对比例1制备的碳载铂镍催化剂(PtNi/C)用作ORR催化剂循环扫描5000圈前后的LSV图;
图7是对比例1制备的碳载铂镍催化剂(PtNi/C)用作ORR催化剂循环扫描5000圈前后的ECSA图。
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
本发明第一方面提供一种氮掺杂碳包覆铂镍合金纳米材料,该纳米材料具有内核为铂镍合金纳米颗粒、壳层为氮掺杂碳笼的核壳结构,所述铂镍合金纳米颗粒为实心结构和/或多孔结构。
在本发明中,所述铂镍合金纳米颗粒主要为实心结构,具体地,实心结构的铂镍合金纳米颗粒占总颗粒数目的50%以上,优选为60%以上,更优选为70%以上,例如可以为50-95%。
根据本发明,优选地,在所述纳米材料中,铂/镍的摩尔比为0.1-5:1,优选为0.2-3:1,更优选为0.2-2:1,进一步优选为0.3-0.5:1。
在所述纳米材料中,优选地,铂含量为5-70重量%,镍含量为5-70重量%,碳含量为5-70重量%,氢含量为0.1-3重量%,氧含量为0.5-20重量%,氮含量为1-15重量%。更优选地,铂的含量为10-60重量%,优选为20-55重量%,更优选为30-55重量%,镍含量为15-50重量%,优选为15-35重量%,更优选为17-32重量%,碳含量为10-40重量%,更优选为10-30重量%,进一步优选为15-30重量%,氢的含量为0.5-2重量%,更优选为0.5-1.6重量%,氧含量为1-15重量%,更优选为3-12重量%,氮含量为2-10重量%,更优选为2-5重量%。
在所述纳米材料中,优选地,在所述纳米材料的XRD谱图中2θ为39.7°~44.7°的范围内存在至少一个铂镍合金峰,更优选存在1-2个铂镍合金峰。
根据本发明,优选的情况下,所述铂镍合金颗粒的粒径为2-200nm,优选5-100nm。
根据本发明,本发明的纳米材料中壳层碳笼具有以介孔为主的孔结构。优选的情况下,所述纳米材料的比表面积为100-500m
根据本发明,优选的情况下,所述纳米材料的稀硫酸酸洗保留率为80%以上。
优选的情况下,所述纳米材料的电阻率为0.01-10000Ω·m,优选为0.01-100Ω·m。
本发明的纳米材料用于催化燃料电池阴极氧还原反应时,半波电位大于0.80V,0.9V下的质量比活性大于0.10A/mg
本发明第二方面提供一种氮掺杂碳包覆铂镍合金纳米材料的制备方法,该方法包括如下步骤:
(1)前驱体制备:将含有金属前驱体、碳源、氮源和溶剂的均相溶液中的溶剂除去,得到前驱体材料,其中,所述金属前驱体包括铂源和镍源,所述碳源为酸性有机还原剂;
(2)焙烧:在惰性气氛中,将步骤(1)得到的前驱体材料进行高温热解,得到热解产物,其中,所述高温热解的温度为400-1100℃;
(3)酸洗:将步骤(2)得到的热解产物与酸溶液接触反应,然后依次进行固液分离、洗涤和干燥。
在本发明中,所述氮掺杂碳包覆铂镍合金纳米材料的制备方法采用酸性有机还原剂作为碳源,在惰性气氛中热解催化剂前驱体,镍盐在被还原的过程中作为碳笼的模板和催化剂,原位生成包覆碳笼,前驱体中的氮元素在热解过程中掺杂进入碳笼,从而一步法制备出氮掺杂碳包覆铂镍合金纳米材料。
根据本发明,优选地,步骤(1)中,所述碳源同时作为还原剂,只要是具有还原性的酸性有机化合物即可,优选为还原性的有机酸、还原性的多羟基化合物等,具体可以为柠檬酸、抗坏血酸、乙二胺四乙酸、2,5-吡啶二羧酸、苯甲酸和对苯二甲酸中的一种或多种。优选地,以金属元素计的所述金属前驱体与所述碳源的摩尔比可以为1:0.5-5,优选为1:0.5-3。通过采用上述金属前驱体和碳源的比例,更有利于得到碳笼包覆的铂合金颗粒结构。
根据本发明,优选地,步骤(1)中,所述氮源为用于形成氮掺杂碳笼,只要能够提供形成氮掺杂碳笼的氮元素即可,没有特别的限定。具体可以为乙二胺四乙酸、氨和六亚甲基四胺中的一种或多种。优选地,以金属元素计的所述金属前驱体与所述氮源的摩尔比可以为1:0.01-10,优选为1:0.1-5,更优选为1:1-5。
另外,本发明的碳源和氮源可以为同一化合物,例如使用乙二胺四乙酸时,其即可以作为碳源,也可以作为氮源;这样的碳源和氮源还可以进一步与其他碳源和氮源配合使用。
根据本发明,优选地,步骤(1)中,所述铂源只要是在所述溶剂中可溶的含铂化合物即可,例如可以为含铂的无机酸、无机酸盐或有机酸盐等,例如可以为氯铂酸、醋酸四氨合铂、乙酰丙酮铂、氯铂酸盐和氯化铂中的一种或多种,优选为氯铂酸、醋酸四氨合铂等。
根据本发明,优选地,步骤(1)中,所述镍源只要是在所述溶剂中可溶的含镍化合物即可,例如可以为含镍的有机酸盐或无机酸盐等,例如可以为所述镍源为乙酸镍、六水合二氯化镍、乙酰丙酮镍、碱式碳酸镍、碳酸镍和硫酸镍中的一种或多种,优选为乙酸镍、六水合二氯化镍、碱式碳酸镍等。
根据本发明,所述铂源与所述镍源可以根据所需要的铂合金组成进行适当调整,优选地,以铂计的所述铂源与以镍计的所述镍源的摩尔比为1:0.5-20,优选为1:1-10,更优选为1:1-5,例如可以为1:1、1:1.2、1:2、1:3、1:4.5、1:5、1:6.5、1:7.5、1:8、1:9或者1:10等。
本发明的制备方法中,除上述的铂源和镍源之外,优选不使用其他金属源。上述其他金属源,可以为其他金属盐,例如碱金属盐或碱土金属盐。
根据本发明,步骤(1)中,所述前驱体材料是将金属前驱体和碳源在溶剂中溶解形成均相溶液,然后除去均相溶液中的溶剂而得到的前驱体材料。对所述溶剂的种类没有特别的限定,以能够形成均相溶液为准,优选地,所述溶剂为水、醇类溶剂和N,N-二甲基甲酰胺(DMF)中的一种或多种,更优选为水。本发明对所述溶剂的用量也没有特别的限定,同样以能够形成均相溶液为准。
在本发明中,步骤(1)中,对所述均相溶液的形成方式没有特别的限定,例如可以通过搅拌方式以形成所述均相溶液。本发明对搅拌的速率和时间也没有特别的限定,能够形成所述均相溶液即可。另外,为了形成所述均相溶液,也可以进一步通过加热的方式加速溶解。
作为除去所述均相溶液中的溶剂的方法,可以采用蒸发(例如旋蒸或者油浴加热蒸发)的方式除去所述均相溶液中的溶剂,蒸发的温度和工艺可以采用本领域技术人员所公知的现有技术,例如,可以采用加热蒸干或者旋蒸的方式除去所述均相溶液中的溶剂。根据本发明的具体的实施方式,可以在60-120℃真空烘箱中干燥12-24h以除去所述均相溶液中的溶剂。
根据本发明,在进行步骤(2)的高温热解前,优选对催化剂前驱体材料进行适当研磨,得到催化剂前驱体粉末,以利于热解的进行。研磨的方式和程度可以适当选择。
根据本发明,优选地,步骤(2)中,所述惰性气氛可以为氮气、氩气、氖气和氦气中的至少一种。高温热解可以在提供上述高温热解条件的任意装置中进行,例如可以在管式炉中进行。
作为高温热解的条件,优选地,所述高温热解的温度可以为400-1100℃,优选为500-800℃,恒温时间可以为1-6h,优选为2-5h。高温热解的升温速率优选为2-10℃/min,优选为2-5℃/min。
上述高温热解后,优选将热解产物在惰性气氛中自然冷却后,根据需要适当研磨后,再进行后续的酸洗。
根据本发明,步骤(3)中,所述酸溶液可以为本领域常规使用的酸,只要能够适当除去热解产物中的镍元素即可。优选地,所述酸为无机酸溶液和/或有机酸溶液,优选为硫酸溶液、硝酸溶液和盐酸溶液中的一种或多种,进一步优选为硫酸溶液;优选地,所述酸溶液的浓度为0.5-2mol/L。
根据本发明,酸洗的目的在于去除热解产物中的镍元素,通过上述用量的酸溶液,可以使得未被碳层严密包覆的镍被除去。优选地,相对于步骤(2)得到的热解产物中的镍元素1mol,所述酸溶液的用量以H
根据本发明优选的实施方式,所述酸溶液为硫酸溶液时,酸浓度为0.5-2mol/L,接触反应的温度为25-90℃;所述酸溶液为硝酸溶液时,酸浓度为0.5-15mol/L,接触反应的温度为25-60℃;所述酸溶液为盐酸溶液时,酸浓度为0.5-2mol/L,接触反应的温度为25-90℃。
根据本发明,优选地,将热解产物与酸溶液接触反应时,接触反应的时间为3-50h,优选为3-24h。
根据本发明,所述洗涤用于除去由酸洗过程导致的残留在热解产物上的酸,因此,各种能够使热解产物洗至中性的水洗方式均适用于本发明。优选洗涤至洗涤液pH为中性。
根据本发明,干燥用于除去酸洗产物上的水。干燥可以采用常压干燥或减压干燥,优选采用真空干燥。干燥的条件可以包括:温度为60-80℃,时间为2-10h。
根据本发明一种特别优选的实施方式,将铂前驱体、镍前驱体与还原剂混合,以总金属计的金属前驱体与还原剂的摩尔比为1:0.5-5,加入去离子水后搅拌,形成透明的前驱体溶液,除去溶剂后干燥,将所得粉末在惰性气氛(N
本发明第三方面提供利用上述本发明第二方面的制备方法得到的氮掺杂碳包覆铂镍合金纳米材料。本发明第二方面的制备方法可以用于制备第一方面的氮掺杂碳包覆铂镍合金纳米材料,因此制得的氮掺杂碳包覆铂镍合金纳米材料具有第一方面相关的性质,在此不再赘述。
本发明第四方面提供一种催化剂,其含有上述本发明第二方面的氮掺杂碳包覆铂镍合金纳米材料和导电炭黑。
优选地,所述氮掺杂碳包覆铂镍合金纳米材料与所述导电炭黑的重量比为1:0.1-5。
本发明第五方面提供本发明催化剂的一种制备方法,该制备方法包括:在溶剂的存在下,将本发明第一方面或第三方面的氮掺杂碳包覆铂镍合金纳米材料与导电炭黑混合后,将得到的混合物中的溶剂除去并干燥。
本发明第六方面提供本发明催化剂的另一种制备方法,该制备方法包括:将本发明第一方面或第三方面的氮掺杂碳包覆铂镍合金纳米材料与导电炭黑进行固相混合。
根据本发明如上的催化剂和催化剂的制备方法,可以通过将本发明的氮掺杂碳包覆铂镍合金纳米材料与导电炭黑混合后,制得用于燃料电池阴极氧还原反应的催化剂。混合的方法没有特别的限定,可以为液相混合或固相混合,只要能够混合均匀即可。
根据本发明,作为导电炭黑,没有特别的限定,只要是可以用于燃料电池阴极氧还原反应的导电炭黑即可,例如可以为科琴黑(例如ECT-600JD)、卡博特碳黑(例如Vulcan XC72)等。
根据本发明优选的实施方式,优选地,所述氮掺杂碳包覆铂镍合金纳米材料与所述导电炭黑的重量比可以为1:0.1-5,优选为1:0.2-2。另外,优选所述催化剂中导电炭黑含量为10-80重量%,优选为15-65重量%。
作为液相混合的方法,如上第五方面所述,可以在溶剂的存在下,将氮掺杂碳包覆铂镍合金纳米材料与导电炭黑混合后,将得到的混合物中的溶剂除去并干燥。优选地,所述混合包括超声、机械搅拌和研磨中的一种或多种,优选先超声再搅拌混合。超声、机械搅拌和研磨的条件可以适当选择,优选超声的时间为0.5-2h,优选机械搅拌的时间为8-24h,优选研磨的条件包括:在惰性气氛中球磨,转速100-500rpm,球磨时间2-24h。
作为固相混合的方法,如上第六方面所述,可以直接将氮掺杂碳包覆铂镍合金纳米材料与导电炭黑进行固相混合,从而得到催化剂。固相混合的方法和条件没有特别的限定,优选地,所述固相混合的条件包括:在惰性气氛中球磨,转速100-500rpm,球磨时间2-24h,优选2-14h。
本发明第七方面提供本发明第一方面或第三方面的氮掺杂碳包覆铂镍合金纳米材料、本发明第四方面的催化剂、或者本发明第五方面或第六方面的制备方法得到的催化剂作为燃料电池催化剂的应用。
制造碳包覆铂纳米粒子核壳结构是困难的,本发明通过上述技术方案,提供了一种包含氮掺杂碳包覆铂镍合金纳米粒子核壳结构的新材料。
如上述,本发明的氮掺杂碳包覆铂镍合金纳米材料可以直接作为燃料电池阴极氧还原反应的催化剂,也可以与导电炭黑混合制成燃料电池阴极氧还原反应的催化剂。
以下将通过实施例对本发明进行详细描述。如无特殊说明,本发明所采用试剂均为分析纯,所用试剂均为市售可得。
通过高分辨透射电镜(HRTEM,日本电子株式会社JEM-2100)表征材料的表面形貌,加速电压为200kV。
通过X射线衍射(XRD,荷兰马尔文帕纳科公司,Empyrean锐影)表征材料的晶体结构。
通过X射线光电子能谱(XPS,Thermo Scientific,ESCALab250型)测定材料表面各元素的含量和价态。
通过Brunauer-Emmett-Taller法(BET,Quantachrome AS-6B型分析仪)测定材料的比表面积。
通过粉末电阻率测试仪(苏州晶格,ST-2722型半导体粉末电阻率测试仪)测得材料的电阻率。
碳、氢、氧、氮元素的含量测试在ElementarVario EL Cube元素分析仪上进行,具体操作方法如下:样品在锡杯中称量5mg左右,放入自动进样盘,通过球阀进入燃烧管燃烧,燃烧温度1000℃(为排除进样时大气干扰,采用氦气吹扫),经过铜还原后,样品中的C、H、N分别转化为二氧化碳、水、氮气。混合气体通过色谱柱进行分离,最后通过热导池检测。氧元素测定时,样品在装有碳粉的高温裂解管中裂解,样品中的氧转化为一氧化碳,载气携带裂解产物进入串联的洗涤器中,以除去酸气和水蒸气,最后进入红外检测器进行检测。
铂、镍元素的含量测定采用电感耦合等离子体发射光谱法(ICP-OES),具体方法如下:(1)硝解:量取催化剂样品10mg放入烧瓶里,加入新鲜配置的16mL王水,加入磁力搅拌子,将烧瓶放入油浴锅中,120℃冷凝回流12h,冷却至室温后,玻璃注射器吸取溶液,用孔径0.22μm的一次性滤头过滤,滤液加入500mL容量瓶中,超纯水定容。(2)含量测试:取定容后的溶液10mL,采用仪器Agilent 5110进行金属含量测试。
电化学测试方法:(1)电极的制备:称取一定重量的催化剂样品,分散到水、乙醇/异丙醇、全氟磺酸(nafion)的混合溶液中,在冰水中超声1小时以形成均匀的ink,移液枪吸取一定量的ink滴涂在玻碳电极上,自然干燥后即可用于电化学测试,Pt的负载量控制在10-30μg/cm
实施例1
将2.6g六水合氯铂酸、2.2g碱式碳酸镍(其中Ni含量为40.33重量%)、5.9g乙二胺四乙酸混合,加入200mL去离子水,磁力搅拌1h使其溶解完全,然后在80℃油浴中加热搅拌直至溶剂蒸干,放入120℃真空烘箱中干燥12h。将催化剂前驱体粉末研磨后,在氮气气氛中以5℃/min的升温速率升至500℃,保温4h后自然冷却至室温。研磨后,对于1g热解产物用80mL的0.5mol/L稀硫酸在90℃下酸洗12h,过滤,去离子水洗涤至溶液pH为中性,放入60℃真空烘箱中8h,烘干后得到氮掺杂碳包覆铂镍合金纳米材料。
BET法测得材料的比表面积为141.1m
实施例1A
将实施例1的氮掺杂碳包覆铂镍合金纳米材料与科琴黑(ECY-600JD)按照5:2的重量比混合后,放入球磨罐中,在氮气气氛保护下以200rpm的速率球磨4h(每个循环为球磨5min后,停止2min),得到氮掺杂碳包覆铂镍合金催化剂。
实施例2
将51.2g的8重量%氯铂酸水溶液、7.5g四水合乙酸镍、8.7g乙二胺四乙酸混合,加入150mL去离子水,磁力搅拌1h使其溶解完全,旋蒸后放入60℃真空烘箱中干燥12h。将催化剂前驱体粉末研磨后,在氩气气氛中以5℃/min的升温速率升至700℃,保温3h后自然冷却至室温。研磨后,对于1g热解产物用80mL的0.5mol/L稀硫酸在80℃下酸洗36h,过滤,去离子水洗涤至溶液pH为中性,放入60℃真空烘箱中8h,烘干后得到氮掺杂碳包覆铂镍合金纳米材料。
实施例3
将2.4g醋酸四氨合铂、3.2g四水合乙酸镍、2.4g柠檬酸混合,加入200mL去离子水,磁力搅拌1h使其溶解完全,在80℃油浴中加热搅拌的条件下,逐滴加入氨水至溶液pH为8左右,继续在80℃油浴中加热搅拌使溶剂逐渐蒸干,放入60℃真空烘箱中干燥12h。将催化剂前驱体粉末研磨后,在氮气气氛中以2℃/min的升温速率升至600℃,保温5h后自然冷却至室温。研磨后,对于1g热解产物用80mL的0.5mol/L稀硫酸在80℃下酸洗24h,过滤,去离子水洗涤至溶液pH为中性,放入60℃真空烘箱中8h,烘干后得到氮掺杂碳包覆铂镍合金纳米材料。
实施例3A
称取实施例3的氮掺杂碳包覆铂镍合金纳米材料20mg,加入10mL乙醇/水(V/V=3/1)的混合溶剂,超声1h;同时,量取8mg科琴黑(ECT-600JD),加入4mL乙醇/水(V/V=3/1)的混合溶剂,超声1h;将上述两种溶液混合,超声1h,然后进行机械搅拌24h,转速为800rpm,抽滤并在60℃真空烘箱中6h烘干,得到氮掺杂碳包覆铂镍合金催化剂。
对比例1
向氯铂酸和氯化镍摩尔比为1:1的混合物中加入去离子水,搅拌使其溶解完全,然后将该水溶液逐滴加入Vulcan XC72中,超声分散使其混合均匀后,放入120℃真空烘箱中12h以彻底烘干,研磨后将前驱体粉末放入管式炉中,在H
对比例2
按照实施例1的方法制备未掺氮的碳包覆铂镍合金纳米材料,区别仅在于,制备前驱体溶液时用3.8g无水柠檬酸代替乙二胺四乙酸。
对比例2A
利用对比例2的未掺氮的碳包覆铂镍合金纳米材料,按照实施例1A的方法制备未掺氮的碳包覆铂镍合金催化剂。
测试例1
实施例1得到的氮掺杂碳包覆铂镍合金纳米材料的TEM和XRD图谱分别如图1和图2所示。由图1可以看出,该氮掺杂碳包覆铂镍合金纳米材料具有类球形形貌,碳层包覆金属颗粒,合金颗粒以实心结构为主,直径为5-10nm左右。由图2可以看出,该材料包含两组PtNi合金峰和一组Ni单质峰,其中,两组合金峰中有一组偏向Pt单峰,另一组偏向Ni单峰。
同样测定实施例2-3中得到的氮掺杂碳包覆铂镍合金纳米材料的TEM和XRD图谱,其分别类似于图1和图2。
测试例2
利用ICP-OES法测定实施例1-3和对比例2的纳米材料的金属含量,元素分析仪测定材料中碳、氢、氧、氮元素的含量,结果如表1所示。此外,图3为实施例1得到的氮掺杂碳包覆铂镍合金纳米材料的XPS全谱图。由图3可以看出,实施例1制得的纳米材料表面含有Pt、Ni、C、O、N元素。
表1
测试例3
测定上述实施例和对比例制备的纳米材料和催化剂的电化学性能,结果如表2所示。实施例1A得到的氮掺杂碳包覆铂镍合金催化剂用于催化ORR的LSV曲线和ECSA曲线分别如图4和图5所示。对比例1得到的碳载铂镍合金催化剂于催化ORR的LSV曲线和ECSA曲线分别如图6和图7所示。
表2
/>
通过上述结果可以看出,本发明的纳米材料和催化剂具有良好的ORR活性。
通过表2可以看出,对比例1所制备的PtNi合金直接负载在导电炭黑上,在酸性电解质中经过5000圈循环扫描后ORR活性下降严重,半波电位由初始的0.85V下降到0.79V,质量比活性由初始的0.086A/mg
并且,对比例2A催化剂的半波电位为0.85V,质量比活性为0.15A/mg
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。