一种普拉洛芬中间体的合成方法及其中间体合成普拉洛芬的方法
文献发布时间:2024-04-18 19:58:30
技术领域
本发明涉及化合物反应技术领域,具体为一种普拉洛芬中间体的合成方法及其中间体合成普拉洛芬的方法。
背景技术
普拉洛芬是一种丙酸类非甾体消炎镇痛药,化学名称:(2RS)-2-(10-氢-9-氧杂-1-氮杂蒽-6-基)丙酸,CAS登记号52549-17-4。其作用比阿司匹林、吲哚美辛、布洛芬强,作用机制为抑制环氧合酶活性,阻断二十碳四烯酸衍生物的合成,减少前列腺素的合成,从而阻断炎症介质的作用而发挥药效。
普拉洛芬的结构式如式0所示:
普拉洛芬一般以2-卤代烟酸和4-羟基苯酮类化合物为原料,在碘化亚铜存在下进行乌尔曼缩合得式I,之后在酸和碘苯二乙酸作用下于原甲酸三甲酯中重排得式II,再经卤代成酰氯后在路易斯酸作用下分子内关环制得式III,再经硼氢化还原得式IV,最后经氯化异丙醇还原和碱水解、后处理酸化得式0。专利WO2022267945A1中公开了其合成方法,反应方程式如下:
现有的合成工艺收率低,合成路线较长,第二步碘苯二乙酸价格昂贵,不具备经济性,第三步卤代成酰氯后分子内关环操作难度较大,第四步硼氢化还原操作危险系数高,综上现有工艺不适合工业化生产。
发明内容
针对现有技术中存在的这些问题,本发明的目的提供一种普拉洛芬中间体的合成方法及其中间体合成普拉洛芬的方法。
一种普拉洛芬中间体的合成方法,所述普拉洛芬中间体为α-甲基-5-氧代-5H-苯并吡喃并[2,3-b]吡啶-7-乙酸,其合成方法路线如下:
S1:将路易斯酸加入有机溶剂I中,滴加丙酰氯,再滴加苯甲醚,控温-5~15℃反应,得到中间体IV;
S2:将中间体ΙV、原甲酸三甲酯、锌粉加入到甲醇中,缓慢加入重排催化剂,搅拌,减压除去甲醇,80~120℃反应,得到中间体III;
S3:将中间体III、催化剂加入到有机溶剂II中,控温25~55℃缓慢滴加2-氯烟酰氯和二氯甲烷的混合溶液,保温反应,得到中间体II;
S4:将中间体II加入到氢溴酸中,80~140℃反应,得到中间体I。
进一步地,步骤S1中,所述有机溶剂I为二氯甲烷、四氢呋喃、甲醇中的一种;所述路易斯酸为无水三氯化铝、三氯化铁中的一种;所述苯甲醚与路易斯酸的投料摩尔比为1:(0.8~1.5)。
进一步地,步骤S2中,所述重排催化剂为碘单质、液溴中的一种;所述中间体IV与重排催化剂的投料摩尔比为1:(1.0~1.5),反应时间为2~4h。
进一步地,步骤S3中,所述有机溶剂II为二氯甲烷、二氯乙烷、无水乙醇、四氯化碳中一种;所述催化剂为三氯化铁、三氯化铝、氯化锌中的一种;所述中间体III与催化剂的投料摩尔比为1:(2.5~5.0);反应时间为1~3h。
进一步地,步骤S4中,所述中间体II与氢溴酸的投料体积比为1:(5.0~10.0)。
更进一步地,所述普拉洛芬中间体的合成方法具体为:
S1:在5℃下,将无水三氯化铝加入到二氯甲烷中,滴加丙酰氯,再滴加苯甲醚,有明显放热,控温-5~5℃,滴毕反应1小时;将反应液滴入预先降温的水中,低温下搅拌5~20min,静置分层,收集二氯甲烷相,用无水硫酸钠干燥、浓缩,得到淡黄色液体,即为中间体IV。
S2:室温下,将中间体ΙV、原甲酸三甲酯、锌粉加入到甲醇中,30~50℃下,滴加液溴,搅拌,减压蒸馏除去甲醇,回流反应2~4h,降至室温,加入水和二氯甲烷,搅拌,静置分层,收集二氯甲烷相,用无水硫酸钠干燥、浓缩得到深棕色液体,即为中间体III。
S3:室温下,将无水三氯化铁、中间体ΙII加入到二氯甲烷中,控温30~45℃缓慢滴加2-氯烟酰氯和二氯甲烷的混合溶液,滴毕,保温反应1~3h,降至室温,加入硅藻土,搅拌,过滤,滤饼用二氯甲烷洗涤,二氯甲烷相用2M盐酸洗涤两次,收集二氯甲烷相,用无水硫酸钠干燥,浓缩得到深棕色液体,即为中间体II。
S4:室温下,将中间体II加入到48%氢溴酸中,在回流反应6~14h,降至室温,加水淬灭反应,用二氯甲烷萃取,收集二氯甲烷相,用无水硫酸钠干燥,浓缩得到黄色固体,固体用乙酸乙酯打浆,过滤,滤饼用重量比为1:1的乙酸乙酯-石油醚混合液洗涤,干燥,得到中间体I。
在公开专利WO2022267945A1的制备方法中,本申请制备的普拉洛芬中间体I是其在制备式III化合物时的一种杂质,本申请通过合成现有制备普拉洛芬工艺中的杂质,然后再通过该杂质(即中间体I)高收率的制备出了普拉洛芬。
一种普拉洛芬中间体合成普拉洛芬的方法,所述合成方法路线如下:
S5:将中间体Ι加入有机溶剂III中,加入还原剂,加热至50~70℃,反应2~5h,降至室温,将反应液加入冰水中,用碱调pH=6~8,过滤、萃取、洗涤,无水硫酸钠干燥,得到中间体0;
S6:将中间体0溶于氢溴酸中,回流反应,即得普拉洛芬。
进一步地,步骤S5中,所述有机溶剂III为四氢呋喃、甲醇、异丙醇中的一种,所述还原剂为硼氢化钠、硼氢化钾、异丙醇铝中一种,所述碱为碳酸氢钠,所述萃取使用的萃取溶剂为乙酸乙酯。
进一步地,步骤S6中,所述氢溴酸的浓度为35~45wt%。
进一步地,步骤S6中,回流反应结束后滴加20~30wt%氨水调pH>10,抽滤,滤饼用水淋洗,干燥得到普拉洛芬。
与现有技术相比,本发明的有益效果是:
第一步以苯甲醚为原料进行傅-克酰基化反应,精简了合成路线,提高原子利用率。第二步避免了碘苯二乙酸的使用,选择了经济实用的锌粉。第三步和第四步避免了复杂且危险的硼氢化还原,而且最后进行分子内关环,得到目标中间体。该中间体再经异丙醇铝还原和氢溴酸在异丙醇溶液中反应即可得到普拉洛芬。本申请的合成方法具有操作简单安全、反应条件温和、收率较高、原材料成本低,适宜工业化和商业化大生产。
附图说明
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
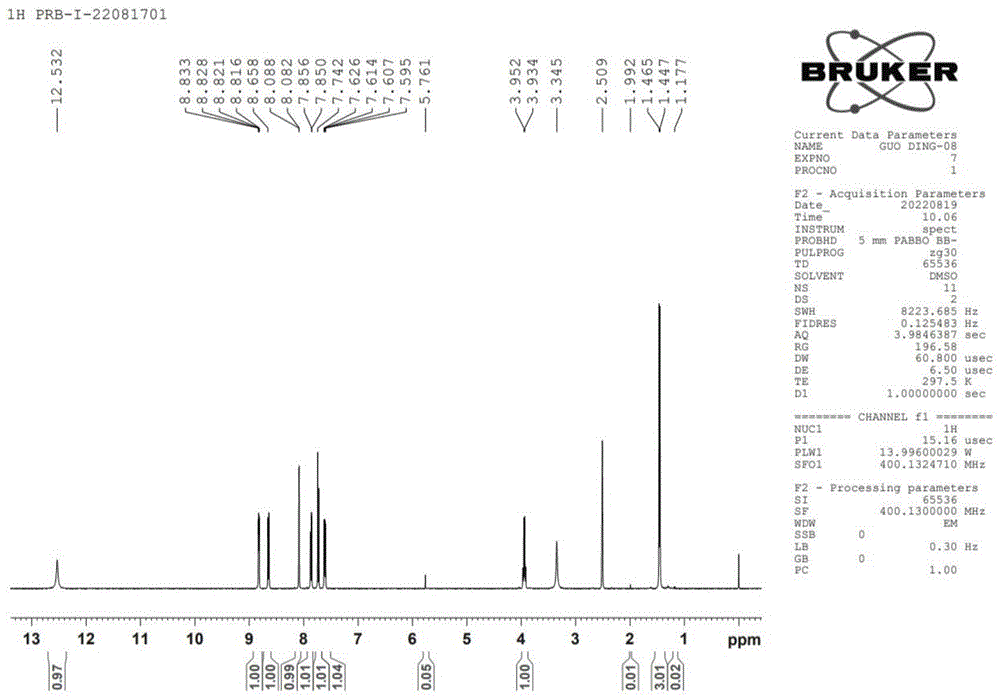
图1为本发明实施例16合成的中间体I的的核磁谱图;
图2为本发明实施例16合成的中间体I的液相色谱图。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
实施例1
一种普拉洛芬中间体IV的合成方法,其具体合成步骤如下:
将无水三氯化铝(135.6g,1.02mol)加入二氯甲烷(700ml)中,滴加丙酰氯(94.1g,1.02mol),控温-5~5℃;滴毕,滴加苯甲醚(100.0g,0.92mol),控温-5~5℃反应;TLC监控原料无剩余,将反应液滴入预先降温至5℃的水(1L)中淬灭,再加入二氯甲烷(100ml)萃取,收集二氯甲烷相,加入无水硫酸钠(250g)干燥,减压浓缩得中间体IV 151.0g,收率99.4%,纯度:95.50%。
实施例2
普拉洛芬中间体IV的合成方法与实施例1基本相同,其不同之处在于:将有机溶剂二氯甲烷替换为四氢呋喃。中间体IV的收率为83.2%,纯度:81.64%。
实施例3
普拉洛芬中间体IV的合成方法与实施例1基本相同,其不同之处在于:将有机溶剂二氯甲烷替换为甲醇。中间体IV的收率为75.4%,纯度82.96%。
实施例4
普拉洛芬中间体IV的合成方法与实施例1基本相同,其不同之处在于:将反应温度区间调整为5~10℃。中间体IV的收率为64.0%,纯度:76.29%。
实施例5
普拉洛芬中间体IV的合成方法与实施例1基本相同,其不同之处在于:将三氯化铝当量替换为1.0。中间体IV的收率为74.9%,纯度:81.70%。
实施例6
普拉洛芬中间体IV的合成方法与实施例1基本相同,其不同之处在于:将三氯化铝当量替换为1.5。中间体IV的收率为76.2%,纯度:89.37%。
实施例7
一种普拉洛芬中间体III的合成方法,其具体合成步骤如下:
室温下,将中间体IV(151g,0.92mol)、原甲酸三甲酯(583.5g,2.30mol)、锌粉(6.0g,0.09mol)加入甲醇(753mL)中,外温45℃下滴加液溴(154.3g,0.97mol),滴毕搅拌30min,减压浓缩除去甲醇,120℃下回流反应3h,TLC监控,无原料剩余,体系降至室温,加入水(800ml)和二氯甲烷(1L),搅拌5min,静置分层,收集下层二氯甲烷相,用无水硫酸钠干燥,减压浓缩得到中间体III 168.3g,收率94.2%,纯度:68.73%。
实施例8
普拉洛芬中间体III的合成方法与实施例7基本相同,其不同之处在于:将缓慢滴加液溴替换为分批加入等摩尔量的碘单质。中间体III的收率为72.6%,纯度:61.34%。
实施例9
普拉洛芬中间体III的合成方法与实施例7基本相同,其不同之处在于:将液溴当量替换为1.5。中间体III的收率为75.4%,纯度60.48%。
实施例10
普拉洛芬中间体III的合成方法与实施例7基本相同,其不同之处在于:将反应温度区间调整为85-90℃。中间体III的收率为70.4%,纯度:61.37%。
实施例11
一种普拉洛芬中间体II的合成方法,其具体合成步骤如下:
室温下,将无水三氯化铁(425.9g,2.63mol)、中间体ΙII(170.0g,0.88mol)加入到二氯甲烷(1.5L)中,控温35-40℃缓慢滴加2-氯烟酰氯(200.2g,1.14mol)和二氯甲烷的混合溶液,滴毕,保温反应2h,TLC监控,无原料剩余,体系降至室温,加入硅藻土充分搅拌20min,过滤,滤饼用二氯甲烷(1L)洗涤,二氯甲烷相用2M盐酸(1.5L)洗涤两次,收集二氯甲烷相,用无水硫酸钠干燥,减压浓缩得到中间体II 234.1g,收率80.2%,纯度:75.18%。
实施例12
普拉洛芬中间体II的合成方法与实施例11基本相同,其不同之处在于:将溶剂二氯甲烷替换为二氯乙烷。中间体II的收率为71.1%,纯度70.59%:。
实施例13
普拉洛芬中间体II的合成方法与实施例11基本相同,其不同之处在于:将催化剂三氯化铁替换为三氯化铝。中间体II的收率为65.0%,纯度:69.61%。
实施例14
普拉洛芬中间体II的合成方法与实施例11基本相同,其不同之处在于:将反应温度区间调整为50-55℃。中间体II的收率为63.8%,纯度67.88%。
实施例15
普拉洛芬中间体II的合成方法与实施例11基本相同,其不同之处在于:将三氯化铁当量替换为5.0。中间体II的收率为75.20%,纯度:69.53%。
实施例16
一种普拉洛芬中间体I的合成方法,其具体合成步骤如下:
室温下,将中间体II(234.1g,223ml,0.70mol)加入到48%氢溴酸(1.1L)中,回流反应11h;降至室温,加入水(1.3L)淬灭反应,用二氯甲烷(2L)萃取,收集二氯甲烷相,用无水硫酸钠(500g)干燥,浓缩得到黄色固体,固体用乙酸乙酯(100ml)打浆,过滤,滤饼用乙酸乙酯:石油醚=1:1(体积比,300ml)洗涤,鼓风干燥,得到中间体I 172.8g,收率91.5%,液相纯度99.49%。
所得中间体I的核磁氢谱如图1所示,其氢谱数据如下:
1
液相检测结果如下表和图2所示,采用本发明制备的普拉洛芬中间体I产物杂质较少,纯度为99.49%。
实施例17
普拉洛芬中间体I的合成方法与实施例16基本相同,其不同之处在于:将反应温度区间调整为80-85℃。中间体I的收率为83.0%,纯度:92.44%。
实施例18
普拉洛芬中间体I的合成方法与实施例16基本相同,其不同之处在于:将打浆试剂乙酸乙酯替换为等体积的甲基叔丁基醚。中间体I的收率为84.5%,纯度:98.38%。
实施例19
普拉洛芬中间体I的合成方法与实施例16基本相同,其不同之处在于:将48%氢溴酸体积比替换为10V(2.2L)。中间体I的收率为82.7%,纯度:98.69%。
实施例20
一种普拉洛芬中间体0的合成方法,其具体合成步骤如下:
室温下,将中间体Ι(160.0g,0.60mol)用异丙醇(1.5L)溶解,加入异丙醇铝(118.0g,0.06mol),加热至60℃,反应4h,降至室温,将反应液加入冰水中,用碳酸氢钠固体调pH=6~8,过滤,滤液用乙酸乙酯萃取两次(500ml*2),合并有机相,用饱和碳酸氢钠溶液(300ml)、水(300ml)、饱和食盐水(300ml)洗涤,无水硫酸钠干燥,减压浓缩得到黄色固体136.2g,收率84.5%,纯度:99.63%。
实施例21
普拉洛芬中间体0的合成方法与实施例20基本相同,其不同之处在于:将有机溶剂异丙醇替换为等体积的四氢呋喃,将还原剂异丙醇铝替换为等重量的硼氢化钠。中间体0的收率为70.6%,纯度:99.12%。
实施例22
普拉洛芬中间体0的合成方法与实施例20基本相同,其不同之处在于:将有机溶剂异丙醇替换为等体积的甲醇,将还原剂异丙醇铝替换为等重量的硼氢化钾。中间体0的收率为73.0%,纯度:99.30%。
实施例23
一种普拉洛芬的合成方法,其具体合成步骤如下:
将中间体0黄色固体(125.5g,0.46mol)溶于40%氢溴酸(100ml)中,加热至回流5h,滴加25%氨水调pH=11,抽滤,滤饼用水(200ml)淋洗,50℃鼓风干燥得褐色固体即普拉洛芬112.4g,收率95.2%,纯度:99.96%。
实施例24
普拉洛芬的合成方法与实施例23基本相同,其不同之处在于:将40%氢溴酸溶液替换为氢溴酸的异丙醇溶液(氢溴酸/异丙醇=1/5(V/V))。收率为83.3%,纯度:99.95%。
前述的实例仅是说明性的,用于解释本发明所述方法的一些特征。本发明所述的室温为25℃。所附的权利要求旨在要求可以设想的尽可能广的范围,且本文所呈现的实施例仅是根据所有可能的实施例的组合的选择的实施方式的说明。因此,申请人的用意是所附的权利要求不被说明本发明的特征的示例的选择限制。在权利要求中所用的一些数值范围也包括了在其之内的子范围,这些范围中的变化也应在可能的情况下解释为被所附的权利要求覆盖。
- 一种用于合成抗肿瘤药物niraparib的中间体的制备方法及中间体
- 一种用于合成乳酸脱氢酶A抑制剂的关键中间体的合成方法
- 一种艾曲波帕中间体的合成方法
- 一种普拉洛芬中间体化合物、其制备方法及用途
- 一种新的普拉洛芬重要中间体的制备方法