一种稠环化合物的晶型、其组合物、制备方法及其应用
文献发布时间:2023-06-19 09:35:27
技术领域
本发明属于药物化学的技术领域,具体涉及一种稠环化合物的晶型、其组合物、制备方法及其应用。
背景技术
恶性肿瘤是一类严重威胁人类健康的重大疾病,临床上应用的大部分小分子抗肿瘤药物或是影响DNA的结构和功能、或是干扰核酸的合成与修复、或是抑制某种管家蛋白(如微管蛋白)的合成与功能,因而具有普遍的细胞毒性,临床应用中存在毒副作用大、易产生耐药性等缺点。
PI3K小分子抑制剂作为新一类的分子靶向药物,潜力巨大,前景广阔。因此,需要更多结构新颖、生物活性高、成药性良好的PI3K抑制剂,用于肿瘤的靶向治疗,以及用于抗炎或治疗自身免疫性疾病。
新一代PI3K小分子抑制剂,对多种肿瘤细胞有明显的增殖活性抑制作用。但是,药物多晶型现象是影响药品质量与临床疗效的重要因素之一,不同晶型会导致稳定性、吸收和生物利用度的差异,从而影响药物临床疗效。因此该稠环化合物的多晶型及其制备技术对该药物应用具有非常重要的意义。
发明内容
本发明目的在于提供一种稠环化合物的晶型、其组合物、制备方法及其应用。该稠环化合物晶型的可用于肿瘤的靶向治疗,以及用于抗炎或治疗自身免疫性疾病。
本发明提供了一种稠环化合物的多晶型:
稠环化合物的结构式为:
所述多晶型包括:晶型I,使用X射线衍射法,2θ角在约11.3°、17.2°、21.1°处有特征衍射峰;晶型II,使用X射线衍射法,2θ角在约25.1°、21.2°、14.1°处有特征衍射峰;晶型III,使用X射线衍射法,2θ角在约6.6°、13.4°、8.0°处有特征衍射峰;晶型IV,使用X射线衍射法,2θ角在约11.8°、13.3°、16.7°处有特征衍射峰;晶型V,使用X射线衍射法,2θ角在约6.5°、13.3°、20.0°处有特征衍射峰;其中,2θ角的误差范围为±0.5°。
进一步的,晶型I,使用X射线衍射法,2θ角还约11.3°、17.2°、21.1°、22.7°、18.5°、13.8°处有特征衍射峰;晶型II,使用X射线衍射法,2θ角在约25.1°、21.2°、14.1°、16.0°、7.0°、18.4°处有特征衍射峰;晶型III,使用X射线衍射法,2θ角在约6.6°、13.4°、8.0°、20.0°、21.1°、10.5°处有特征衍射峰;晶型IV,使用X射线衍射法,2θ角在约11.8°、13.3°、16.7°、17.8°、21.8°、24.4°处有特征衍射峰;晶型V,使用X射线衍射法,2θ角在约6.5°、13.3°、20.0°、10.4°、24.0°、24.8°处有特征衍射峰;其中,2θ角的误差范围为±0.5°。
具体的,晶型I,使用X射线衍射法,2θ角在11.3°、13.8°、17.2°、18.5°、20.2°、21.1°、22.7°、25.2°、28.0°、29.6°、31.2°、33.2°处有特征衍射峰;晶型II,使用X射线衍射法,2θ角在5.7°、7.0°、9.9°、11.5°、12.2°、14.1°、16.0°、17.3°、18.4°、21.2°、22.9°、25.1°、31.7°处有特征衍射峰;晶型III,使用X射线衍射法,2θ角在6.6°、8.0°、8.8°、10.5°、11.9°、13.4°、14.3°、17.9°、20.0°、21.1°、22.8°处有特征衍射峰;晶型IV,使用X射线衍射法,2θ角在6.6°、8.2°、9.7°、11.8°、13.3°、15.0°、16.7°、17.8°、20.2°、21.8°、24.4°、27.9°、29.1°、30.4°处有特征衍射峰;晶型V,使用X射线衍射法,2θ角在6.5°、8.0°、10.4°、13.3°、16.2°、17.7°、20.0°、21.0°、24.0°、24.8°、26.7°、29.1°处有特征衍射峰。
本发明的另一目的在于提供一种稠环化合物多晶型的组合物,用于肿瘤的靶向治疗,以及用于抗炎或治疗自身免疫性疾病。
一种稠环化合物多晶型的组合物,所述晶型I和晶型IV占晶体组合物重量在50%以上。
一种稠环化合物晶型的组合物,包括所述晶型I、晶型II、晶型III、晶型IV和晶型V中的一种或两种以上的组合;且所述晶型I和晶型IV占晶体组合物重量在50%以上。
本发明的另一目的在于提供一种稠环化合物多晶型的组合物,用于肿瘤的靶向治疗,以及用于抗炎或治疗自身免疫性疾病。
一种稠环化合物多晶型的组合物,所述晶型I和晶型IV占晶体组合物重量在80%以上。
一种稠环化合物晶型的组合物,包括所述晶型I、晶型II、晶型III、晶型IV和晶型V中的一种或两种以上的组合;且所述晶型I和晶型IV占晶体组合物重量在80%以上。
本发明的另一目的在于提供一种稠环化合物晶型的组合物,用于肿瘤的靶向治疗,以及用于抗炎或治疗自身免疫性疾病。
一种稠环化合物多晶型的组合物,所述晶型I和晶型IV占晶体组合物重量在90%以上。一种稠环化合物晶型的组合物,包括所述晶型I、晶型II、晶型III、晶型IV和晶型V中的一种或两种以上的组合;且所述晶型I和晶型IV占晶体组合物重量在90%以上。
本发明的另一目的在于提供一种含有上述晶型的药物组合物,用于肿瘤的靶向治疗,以及用于抗炎或治疗自身免疫性疾病。
一种含有所述的晶型I、晶型II、晶型III、晶型IV或者晶型V及药学上可接受的赋形剂作为成分。
本发明的另一目的在于提供一种晶型I的制备方法,向稠环化合物中加入第一有机溶剂,加热溶解,再加入第二有机溶剂;再搅拌析晶;再过滤,干燥。
进一步的,所述第一有机溶剂为二氯甲烷、三氯甲烷、四氢呋喃中的一种或者几种,所述第一有机溶剂的用量为稠环化合物重量的3-5倍,所述第二有机溶剂为丙酮、丁酮、乙醇、乙酸乙酯中的一种或者几种,所述第二有机溶剂的用量为稠环化合物重量的6-12倍。
进一步的,所述搅拌析晶的温度为0-40℃,所述析晶的时间为4-15小时,所述干燥的温度为60-150℃,所述干燥的时间为4-15小时。
本发明的另一目的在于提供一种晶型IV的制备方法,将所述稠环化合物溶于有机溶剂和/或水中,加热溶解;再搅拌析晶;再过滤,干燥。
进一步的,所述有机溶剂和/或水为乙腈-水,所述乙腈的用量为稠环化合物重量的30-90倍,所述水的用量是稠环化合物重量的0-20倍。
进一步的,所述搅拌析晶的温度为0-40℃,所述析晶的时间为0.5-30小时,所述干燥的温度为60-150℃,所述干燥的时间为1-20小时。
本发明的另一目的在于提供一种上述晶型在制备抑制磷脂酰肌醇3-激酶的药物中的应用。该药物可用于肿瘤的靶向治疗,以及用于抗炎或治疗自身免疫性疾病。
一种上述晶型在制备抑制磷脂酰肌醇3-激酶的药物中的应用,所述药物为抗肿瘤药物。
进一步的,所述肿瘤可以是:脑癌、头颈癌、食管癌、肺癌、肝癌、胃癌、肾癌、胰腺癌、前列腺癌、结直肠癌、卵巢癌、乳腺癌、甲状腺癌、皮肤癌、白血病、骨髓异常增生综合症、肉瘤、骨肉瘤或横纹肌瘤。
进一步的,所述药物为抗炎药物或治疗自身免疫性疾病的药物。
进一步的,所述抗炎药物是用于治疗慢性阻塞性肺病或哮喘的药物,所述自身免疫性疾病可以是风湿性关节炎、银屑病或系统性红斑狼疮。
本发明的有益效果为:
本申请的晶型I、II、III、IV、V均具有明显的晶型衍射特征峰,能够克服无定型没有规则的晶体结构的技术缺陷。本发明的晶型I、晶型II、晶型IV在高湿试验、高温试验5天条件下稳定性显著优于参考文献(CN 201610235304.5)的无定型。本发明晶型I、晶型IV在不同溶液中的溶解度和稳定性良好,易于人体吸收,在治疗疾病上具备优异的效果。
附图说明
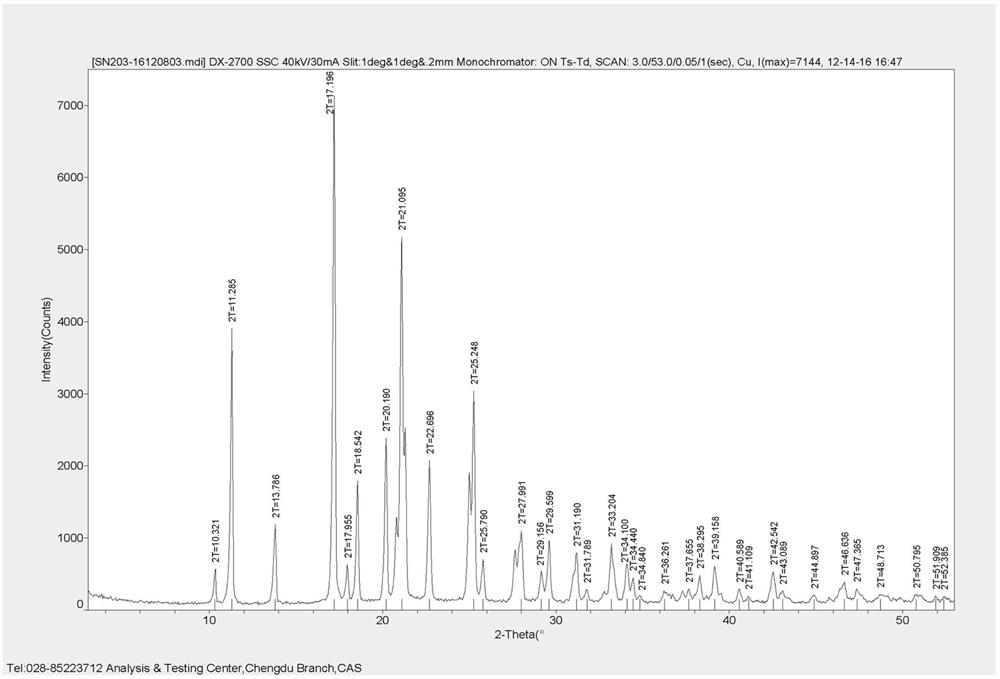
图1A为本发明中的晶型I的X射线衍射图。
图1B为本发明中的晶型I的X射线衍射峰数据图。
图1C为本发明中的晶型I的DSC图。
图2A为实施例7所得晶型II的X射线衍射图。
图2B为实施例7所得晶型II的X射线衍射峰数据图。
图3A为实施例9所得晶型III的X射线衍射图。
图3B为实施例9所得晶型III的X射线衍射峰数据图。
图4A为实施例10所得晶型V的X射线衍射图。
图4B为实施例10所得晶型V的X射线衍射峰数据图。
图5A为本发明中的晶型IV的X射线衍射图。
图5B为本发明中的晶型IV的X射线衍射峰数据图。
图5C为本发明中的晶型IV的DSC图。
图6为实施例6所得晶型I的X-射线衍射图。
图7为实施例8所得晶型II的X-射线衍射图。
图8为实施例11所得无定型的X-射线衍射图。
图9为实施例12中无定型高湿5天试验后的X-射线衍射图。
图10为实施例12中晶型IV高湿5天试验后的X-射线衍射图。
图11为实施例12中晶型I高湿5天试验后的X-射线衍射图。
图12为实施例12中晶型II高湿5天试验后的X-射线衍射图。
图13为实施例13中无定型高温5天试验后的X-射线衍射图。
图14为实施例13中晶型IV高温5天试验后的X-射线衍射图。
图15为实施例13中晶型I高温5天试验后的X-射线衍射图。
图16为实施例13中晶型II高温5天试验后的X-射线衍射图。
图17为实施例14中晶型IV加速试验30天后的X-射线衍射图。
图18为实施例14中晶型I加速试验30天试验后的X-射线衍射图。
图19为实施例14中晶型II加速试验30天试验后的X-射线衍射图。
具体实施方式
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
本发明所有数值指定(例如温度、时间、浓度及重量等,包括其中每一者的范围)通常可是适当以0.1或1.0的增量改变(+)或(-)的近似值。所有数值指定均可理解为前面有术语“约”。
一种稠环化合物的多晶型及其制备方法和应用,稠环化合物的结构式为:
晶型I,使用X射线衍射法,2θ角在约11.3°、17.2°、21.1°处有特征衍射峰;晶型II,使用X射线衍射法,2θ角在约25.1°、21.2°、14.1°处有特征衍射峰;晶型III,使用X射线衍射法,2θ角在约6.6°、13.4°、8.0°处有特征衍射峰;晶型IV,使用X射线衍射法,2θ角在约11.8°、13.3°、16.7°处有特征衍射峰;晶型V,使用X射线衍射法,2θ角在约6.5°、13.3°、20.0°处有特征衍射峰;其中,2θ角的误差范围为±0.5°。
进一步的,晶型I,使用X射线衍射法,2θ角在约11.3°、17.2°、21.1°、22.7°、18.5°、13.8°处有特征衍射峰;晶型II,使用X射线衍射法,2θ角在约25.1°、21.2°、14.1°、16.0°、7.0°、18.4°处有特征衍射峰;晶型III,使用X射线衍射法,2θ角在约6.6°、13.4°、8.0°、20.0°、21.1°、10.5°处有特征衍射峰;晶型IV,使用X射线衍射法,2θ角在约11.8°、13.3°、16.7°、17.8°、21.8°、24.4°处有特征衍射峰;晶型V,使用X射线衍射法,2θ角在约6.5°、13.3°、20.0°、10.4°、24.0°、24.8°处有特征衍射峰;其中,2θ角的误差范围为±0.5°。
实施例1
晶型I的制备方法:
称取实施例11制备的稠环化合物10g、二氯甲烷30g加至反应瓶中,加热溶解,加入90g乙醇,浓缩至剩余重量约为100g,20℃搅拌析晶4小时,过滤,100℃干燥得到晶型I。
由上述方法制得的晶型I熔点207-209℃;X-射线衍射图中,2θ角在11.3°、13.8°、17.2°、18.5°、20.2°、21.1°、22.7°、25.2°、28.0°、29.6°、31.2°、33.2°处有特征衍射峰(见图1A和图1B)。图1C为晶型I的热分析图,TGA显示晶型I在145℃左右开始失重,DSC图显示在209℃左右有吸收峰。
实施例2
晶型I在不同环境下的溶解性(μg/mL)如表1所示:
表1:
实施例3
晶型IV的制备方法:
称取实施例11制备的稠环化合物10g、乙腈300g加至反应瓶中,加热溶解,25℃搅拌析晶4小时,过滤,80℃干燥10小时得到晶型IV。
由上述方法制得的晶型IV熔点185-188℃;X-射线衍射图中,2θ角在6.6°、8.2°、9.7°、11.8°、13.3°、15.0°、16.7°、17.8°、20.2°、21.8°、24.4°、27.9°、29.1°、30.4°处有特征衍射峰(见图5A和图5B)。图5C为晶型IV的热分析图,TGA显示晶型IV在250℃左右开始失重,DSC图显示在188℃左右有吸收峰。
实施例4
晶型IV的制备方法:
称取实施例11制备的稠环化合物10g、乙腈200g加至反应瓶中,加热溶解,然后加入50g水,5℃搅拌析晶20小时,过滤,120℃干燥2小时得到晶型IV。
由上述方法制得的晶型IV熔点185-187℃;X-射线衍射图中,2θ角在6.6°、8.2°、9.7°、11.8°、13.2°、15.0°、16.7°、17.8°、20.2°、21.8°、24.4°、27.9°、29.1°、30.4°处有特征衍射峰。
实施例5
晶型IV在不同环境下的溶解性(μg/mL)如表2所示:
表2:
实施例6
晶型I的制备方法:
将1.0g实施例11制备的稠环化合物加入烧瓶,加入30mL溶剂(乙醇),加热,油浴温度85℃,搅拌溶清后,静置于室温,冷却析晶,过滤并收集固体,常温真空干燥。
由上述方法制得的晶型I的X-射线衍射图中,2θ角在10.4°、11.3°、13.9°、17.3°、18.0°、18.6°、20.3°、20.8°、21.3°、22.8°、25.3°、28.0°、29.7°、31.3°处有特征衍射峰(见图6)。
实施例7
晶型II的制备方法:
称取4.0g实施例11制备的稠环化合物,加入12.0g二氯甲烷,加热溶解,加入36.0g无水甲醇,15℃搅拌16小时,过滤,少许甲醇洗涤滤饼,滤饼于60℃减压干燥,烘干得3.90g淡黄色固体。
由上述方法制得的晶型II的表征数据见图2A和图2B。X-射线衍射图中,2θ角在5.7°、7.0°、9.9°、11.5°、12.2°、14.1°、16.0°、17.3°、18.4°、21.2°、22.9°、25.1°、31.7°处有特征衍射峰(见图2A)。
实施例8
晶型II的制备方法:
将1.0g实施例11制备的稠环化合物加入烧瓶,加入45mL溶剂(EA:MeOH=1:1),加热,油浴温度70℃,搅拌溶清后,静置于室温,冷却析晶,过滤并收集固体,常温真空干燥。
由上述方法制得的晶型II的X-射线衍射图中,2θ角在5.7°、7.0°、9.9°、11.5°、12.2°、14.2°、15.9°、17.2°、18.3°、21.2°、23.0°、25.1°、31.5°处有特征衍射峰(见图7)。
实施例9
晶型III的制备方法:
称取4.0g实施例11制备的稠环化合物,加入200mL乙酸乙酯,加热回流至固体溶解,降温析晶,15℃搅拌16小时,过滤,滤饼于60℃减压干燥,烘干得3.55g淡黄色固体。
由上述方法制得的晶型III的表征数据见图3A、图3B。X-射线衍射图中,2θ角在6.6°、8.0°、8.8°、10.5°、11.9°、13.4°、14.3°、17.9°、20.0°、21.1°、22.8°处有特征衍射峰(见图3A)。
实施例10
晶型V的制备方法
称取1.0g实施例11制备的稠环化合物,加入6.0g四氢呋喃,加热回流至固体溶解,降温析晶,15℃搅拌16小时,过滤,滤饼于60℃减压干燥,烘干得0.29g淡黄色固体。
由上述方法制得的晶型V的表征数据见图4A、图4B。X-射线衍射图中,2θ角在6.5°、8.0°、10.4°、13.3°、16.2°、17.7°、20.0°、21.0°、24.0°、24.8°、26.7°、29.1°处有特征衍射峰(见图4A)。
实施例11
参考文献(CN 201610235304.5)制备稠环化合物:(R)-N-(5-(3-氰基-4-(3-甲基吗啉基)喹啉-6-基)-2-甲氧基吡啶-3-基)甲磺酰胺。
步骤1:将6-溴-4-氯-喹啉-3-甲腈(1.605g,6.0mmol)和(R)-3-甲基吗啉(1.821g,18.0mmol,3eq)于二氧六环(30.0mL)中、在100℃下搅拌。反应结束后真空浓缩,浓缩液用水(60mL)稀释,乙酸乙酯(60mL×3)萃取。合并有机层,依次用水(20mL)和食盐水(20mL)洗涤,无水硫酸钠干燥,过滤,浓缩,经快速柱色谱(硅胶,石油醚/乙酸乙酯=3:1,v/v)纯化得到淡黄色固体(R)-6-溴-4-(3-甲基吗啉)喹啉-3-腈(1.566g,产率为78.5%)。
步骤2:将(R)-6-溴-4-(3-甲基吗啉)喹啉-3-腈(1.0g,3mmol),N-(2-甲氧基-5-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶-3-基)甲磺酰胺(1.18g,3.6mmol,1.2eq)和2N碳酸钾水溶液(4.5mL,3.0eq)于二氧六环(20mL)中的混合物脱气,然后加入[1,1'-双(二苯基膦基)二茂铁]二氯化钯(110mg,0.15mmol,0.05eq)。将产生的反应混合物脱气并回充氮气(三个循环),然后在100℃下氮气气氛中搅拌5小时。将反应混合物冷却至室温,用(30mL)水稀释,用乙酸乙酯(30mL×3)萃取。合并有机层并用食盐水(30mL)洗涤,无水硫酸钠干燥,过滤,浓缩,经快速柱色谱(硅胶,二氯甲烷/甲醇=200:1,v/v)纯化得到白色固体(1.056g,产率为77.3%)。
所得白色固体的XRD如图8所示,从图8可看出采用参考文献(CN 201610235304.5)制备方法得到的(R)-N-(5-(3-氰基-4-(3-甲基吗啉基)喹啉-6-基)-2-甲氧基吡啶-3-基)甲磺酰胺为无定型。
实施例12
考察晶型对稳定性的影响—高湿5天试验
将实施例11所得无定型、实施例3所得晶型IV、实施例6所得晶型I、实施例8所得晶型II各取30mg×3,分别装入西林瓶中,常温下敞口放入底部盛有饱和硝酸钾的干燥器中,于5日后取样,送XRD检测。
无定型高湿5天试验后XRD如图9所示,相较于高湿实验前(图8)在2θ角为7.2°、10.3°、11.3°、16.9°、17.3°、21.2°处有明显新增峰,峰形和峰强度变化幅度大,无定型有明显转晶现象。
晶型IV高湿5天试验后XRD如图10所示,相较于高湿实验前晶型IV(图5A)基本无变化。
晶型I高湿5天试验后XRD如图11所示,相较于高湿实验前晶型I(图6)基本无变化。
晶型II高湿5天试验后XRD如图12所示,相较于高湿实验前晶型II(图7)基本无变化。
实施例13
考察晶型对稳定性的影响—高温5天试验
将实施例11所得无定型、实施例3所得晶型IV、实施例6所得晶型I、实施例8所得晶型II各取30mg×3,分别装入西林瓶,敞口放入QG 2003ba培养干燥箱,调节温度至60℃,于5日后取样,送XRD检测。
无定型高温5天试验后XRD如图13所示,相较于高温实验前(图8)在2θ角为11.3°、17.2°、18.5°、20.2°、21.1°、22.7°、25.3°处有明显新增峰,峰形和峰强度变化幅度大,无定型有明显转晶现象。
晶型IV高温5天试验后XRD如图14所示,相较于高温实验前晶型IV(图5A)基本无变化。
晶型I高温5天试验后XRD如图15所示,相较于高温实验前晶型I(图6)基本无变化;
晶型II高温5天试验后XRD如图16所示,相较于高温实验前晶型II(图7)基本无变化。
实施例14
晶型I、II、IV加速稳定性考察
将实施例3所得晶型IV、实施例6所得晶型I、实施例8所得晶型II各取30mg×3,分别装入封口袋中,放入BPN-80CH二氧化碳培养箱,设定温度40℃,于30日后取样,送XRD检测。
晶型IV加速稳定性试验30天后XRD如图17所示,相较于加速试验前晶型IV(图5A)基本无变化。
晶型I加速稳定性试验30天后XRD如图18所示,相较于加速试验前晶型I(图6)基本无变化。
晶型II加速稳定性试验30天后XRD如图19所示,相较于加速试验前晶型II(图7)基本无变化。
综上所述,参考文献(CN201610235304.5)制备得到的(R)-N-(5-(3-氰基-4-(3-甲基吗啉基)喹啉-6-基)-2-甲氧基吡啶-3-基)甲磺酰胺为无定型,该无定型在高湿试验、高温试验5天条件下有明显的转晶现象,不适合作为药物晶型进行开发。而本申请的晶型I、II、III、IV、V均具有明显的晶型衍射特征峰,能够克服无定型没有规则的晶体结构的技术缺陷。本发明中的晶型I、晶型II、晶型IV在高湿试验、高温试验5天条件下稳定,晶型I、晶型II、晶型IV加速试验30天晶型稳定,本发明的晶型I、晶型II、晶型IV稳定性显著优于参考文献(CN 201610235304.5)的无定型。本发明中的晶型I、晶型IV在可以用于制备多种药物,且根据晶型I和晶型IV的在不同环境下的溶解度及24小时内的稳定性可知,本发明中的多晶型制备的药物易于人体吸收,在治疗疾病上具备优异的效果。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
- 一种稠环嘧啶类化合物的盐、晶型及其制备方法和应用
- 一种稠三环类化合物及其制备方法、以及含该类化合物的药物组合物及其应用