AKT抑制剂化合物和阿比特龙的组合及使用方法
文献发布时间:2023-06-19 11:21:00
发明的优先权
本申请要求2011年4月1日提交的美国临时申请61/470,803和2011年4月1日提交的美国临时申请61/470,624的优先权。将这些临时申请的全部内容引入本申请作为参考。
技术领域
本发明大体涉及化合物的药物组合,所述化合物具有针对过度增殖性病症(例如癌症)的活性并且包括抑制AKT激酶活性的化合物。本发明还涉及将所述组合用于体外、原位和体内诊断或者治疗哺乳动物细胞或者有关病理学病症的方法。
背景技术
蛋白激酶(PK)是通过转移ATP上的末端(γ)磷酸酯,催化蛋白的酪氨酸、丝氨酸和苏氨酸残基上羟基的磷酸化的酶。通过信号转导途径,这些酶调节细胞生长、分化和增殖,即细胞周期的几乎所有方面以某种或者其它方式取决于PK活性(Hardie,G.and Hanks,S.(1995)The Protein Kinase Facts Book.I and II,Academic Press,San Diego,CA)。此外,PK活性异常与许多病症有关,范围从相对无生命危险的疾病例如牛皮癣至极其致命的疾病例如恶性胶质瘤(脑癌)。蛋白激酶是治疗调节的重要靶向类别(Cohen,P.(2002)Nature Rev.Drug Discovery 1:309)。
国际专利申请公开WO 2008/006040详述了一系列式I的AKT抑制剂:
目前,仍然需要可用于治疗过度增殖性疾病(例如癌症)的改善的方法和组合物。
发明内容
已经确定的是,体外和体内抑制癌细胞生长的累加或者协同效应可通过给予式I的化合物或者其药用盐与其它某些具体的化学治疗剂的组合来实现。所述组合和方法可用于过度增殖性病症(例如癌症)的治疗。
本发明的一个方面提供了治疗哺乳动物中过度增殖性病症的方法,其包括对哺乳动物给予:a)式I的化合物或者其药用盐;以及b)一种或者多种药剂,所述式I为:
所述药剂选自:5-FU、铂剂(卡铂、顺铂、奥沙利铂等等)、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。
式I的化合物或者其药用盐和化学治疗剂可共同配制用于作为药物组合物的组合给药或者它们可作为治疗性组合交替(先后)分开给药。
本发明的一个方面提供了治疗哺乳动物中由AKT激酶所调节的疾病或者病症的方法,其包括对所述哺乳动物给予:a)式I的化合物或者其药用盐;以及b)一种或者多种药剂,所述药剂选自:5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。
本发明的一个方面提供了用于治疗过度增殖性病症的组合,所述组合包含:a)式I的化合物或者其药用盐;以及b)一种或者多种药剂,所述药剂选自:5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。
本发明的一个方面提供了用于治疗由AKT激酶所调节的疾病或者病症的组合,所述组合包含:a)式I的化合物或者其药用盐;以及b)一种或者多种药剂,所述药剂选自:5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。
本发明的一个方面提供了式I的化合物或者其药用盐在制备用于治疗哺乳动物中过度增殖性病症的药物中的用途,其中对所述哺乳动物给予一种或者多种药剂,所述药剂选自:5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。
本发明的一个方面提供了式I的化合物或者其药用盐在制备用于治疗哺乳动物中由AKT激酶所调节的疾病或者病症的药物中的用途,其中对所述哺乳动物给予一种或者多种药剂,所述药剂选自:5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。
本发明的一个方面提供了试剂盒,其包含式I的化合物或者其药用盐、容器以及包装说明书或者标签,所述包装说明书或者标签指明用于治疗过度增殖性病症的式I的化合物或者其药用盐与一种或者多种药剂的给药,所述药剂选自:5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。
本发明的一个方面提供了产品,其为在过度增殖性病症的治疗中分开、同时或者先后使用的组合制剂,包含具有式I的化合物或者其药用盐和化学治疗剂,所述化学治疗剂选自5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。
本发明的一个方面提供了产品,其作为在过度增殖性病症(例如前列腺癌)的治疗中分开、同时或者先后使用的组合制剂,包含具有式I的化合物或者其药用盐和阿比特龙或者其药用盐。
除了提供为给定的过度增殖性病症提供改善的治疗之外,与接受不同治疗的相同患者所经历的生活质量相比,本发明某些组合的给药可改善患者的生活质量。例如,如果相同患者仅接受化学治疗剂作为治疗,则与他们将会经历的生活质量相比,对患者给予本申请所述式I的化合物或者其药用盐和化学治疗剂的组合可提供改善的生活质量。例如,用本申请所述组合的组合疗法可降低所需化学药物的剂量,由此减轻与高剂量化学治疗剂有关的副作用(例如,恶心、呕吐、脱发、皮疹、食欲减退、重量减轻等等)。所述组合还可引起降低的肿瘤负荷和有关的不良事件,例如,疼痛、器官功能障碍、重量减轻等等。相应地,本发明的一个方面提供了式I的化合物或者其药用盐,其治疗性用于改善用药剂治疗过度增殖性病症的患者的生活质量,所述药剂选自5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。相应地,本发明的另一个方面提供了式I的化合物或者其药用盐,用于改善患者生活质量的治疗性用途,所述患者用阿比特龙或者其药用盐治疗过度增殖性病症。
附图说明
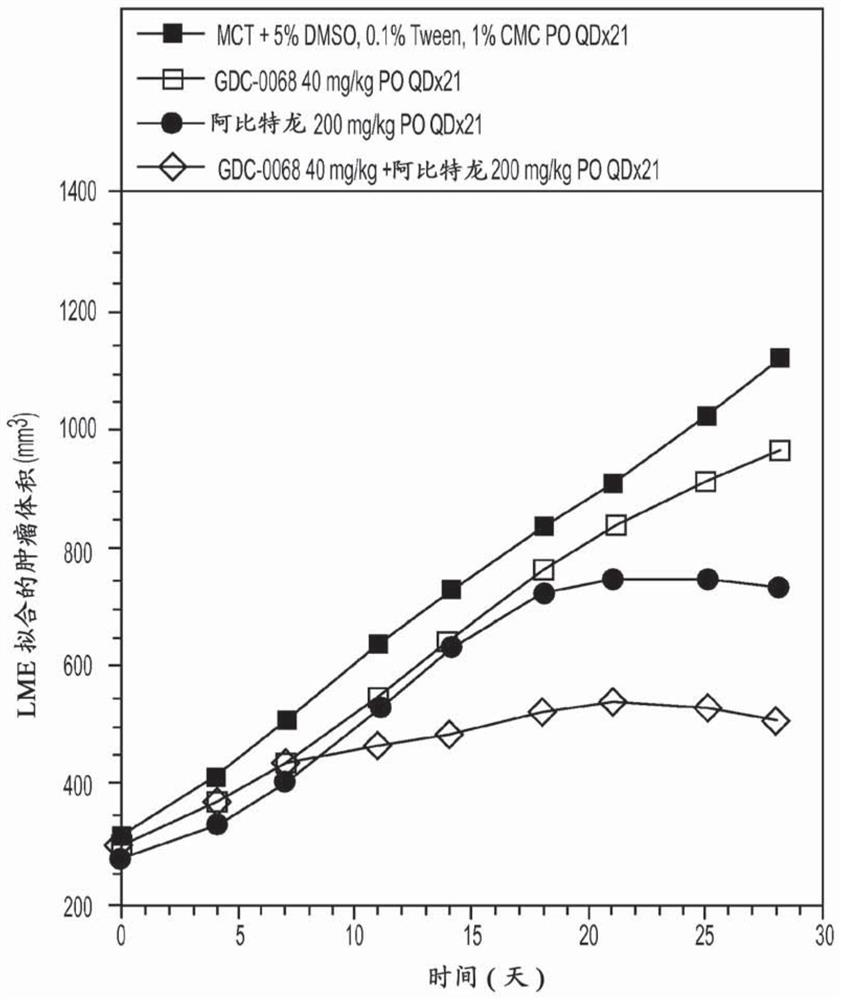
图1示例说明在LuCaP35V原发性前列腺肿瘤中PO给药的式Ia的化合物(GDC-0068)+阿比特龙的数据。
图2示例说明在DU-145.x1原发性前列腺肿瘤中PO给药的式Ia的化合物(GDC-0068)+阿比特龙的数据。
具体实施方式
单词“包含(comprise)”、“包含(comprising)”、“包括(include)”、“包括(including)”和“包括(includes)”,当在说明书和下面的权利要求中使用时,意欲具体说明所叙述的性能、整体、组分或步骤的存在,但不排除存在或添加一或多种其它性能、整体、组分、步骤或基团。
包含比卡鲁胺、GnRH拮抗剂和阿比特龙的抗雄激素疗法的活性,已经导致改善了具有前列腺癌的患者的生存率。然而,几乎所有目前具有激素敏感的晚期前列腺癌的患者都进行至CRPC并需要其它形式的疗法。常由PTEN丧失所显示的PI3K/Akt信号传导的活化是CRPC的常见标志。该途径的脱调节导致涉及生存、增殖、细胞周期进展、生长、迁移和血管发生的下游靶标(例如,PRAS40、MTOR、GSK3b、FOXO等等)的活化。值得注意的是,小鼠模型中PTEN的前列腺特异性缺失可靠地重现人前列腺的特征,而在条件性Pten敲除模型中Akt1缺失明显降低前列腺癌(Chen et al.2006;Guertin et al.2009;Nardella et al.2009)。另外,Pten缺失促进前列腺癌的细胞系和小鼠模型中的雄激素独立性(Gao et al.2006;Jiaoet al.2007)。在前列腺癌患者中,PTEN丧失与较高的Gleason评分、前列腺切除术后复发(recurrence post-prostectomy)、骨转移和向去雄抗性的进行有关。另外,PTEN丧失与总体生存率的下降有关。总起来说,这些结果暗示PI3K/Akt途径的活化是前列腺癌的重要驱动器。
最近的非临床数据暗示AR和PI3K/Akt途径之间的相互串扰(reciprocalcrosstalk)出现在PTEN-缺乏的CRPC中。具体而言,PI3K/Akt途径的活化与被抑制的雄激素信号传导有关,并且PI3K/Akt途径的抑制恢复了PTEN-缺乏的前列腺细胞中的AR信号。提出的解释这些观察结果的机制包括PI3K/Akt抑制导致经HER激酶增量调节的AR的反馈活化以及由磷酸酶PHLPP解除Akt反馈抑制的AR的抑制(Carver et al.2011)。PI3K/Akt和AR途径之间的所述相互协同性(cooperativity)暗示只有一条途径能导致次优的临床效力。因此,AR和PIK3/Akt途径的组合抑制可导致更加完全的肿瘤细胞存活的消退和更加持久的临床益处。
本申请所用的术语“烷基”是指1至12个碳原子的饱和直链或支链一价烃基,其中烷基可以任选被一个或多个如下所述的取代基独立地取代。烷基的实例包括,但不限于,甲基(Me、-CH
术语“烯基”是指2至12个碳原子、且具有至少一个不饱和位点(即碳-碳sp
术语“炔基”是指2至12个碳原子、且具有至少一个不饱和位点(即碳-碳sp叁键)的直链或支链一价烃基,其中炔基可以任选被一个或多个本申请所述的取代基独立地取代。实例包括,但不限于,乙炔基(-C≡CH)和丙炔基(炔丙基,-CH
术语“碳环(carbocycle)”、“碳环基(carbocycly)”、“碳环(carbocyclic ring)”和“环烷基”指作为单环的环具有3至12个碳原子或者作为二环的环具有7至12个碳原子的单价非芳族的、饱和的或者部分不饱和的环。具有7至12个原子的二环碳环可排列(例如)成二环[4,5]、二环[5,5]、二环[5,6]或者二环[6,6]系统,而具有9或10个环原子的二环碳环可排列成二环[5,6]或者二环[6,6]系统,或者排列成桥连系统如二环[2.2.1]庚烷、二环[2.2.2]辛烷和二环[3.2.2]壬烷。单环碳环的实例包括,但不限于,环丙基、环丁基、环戊基、1-环戊-1-烯基、1-环戊-2-烯基、1-环戊-3-烯基、环己基、1-环己-1-烯基、1-环己-2-烯基、1-环己-3-烯基、环己二烯基、环庚基、环辛基、环壬基、环癸基、环十一基、环十二基等。
“芳基”是指6-20个碳原子的一价芳族烃基,其通过从母体芳环体系的单一碳原子上除去一个氢原子而得到。一些芳基在示例性结构中表示为“Ar”。芳基包括包含与饱和、部分不饱和的环,或芳族碳环或杂环稠合的芳环的二环基团。典型的芳基包括,但不限于,衍生自下列的基团:苯(苯基)、取代的苯、萘、蒽、联苯、茚、茚满、1,2-二氢萘、1,2,3,4-四氢萘等。芳基可以任选被一个或多个本申请描述的取代基独立地取代。
术语“杂环(heterocycle)”、“杂环基”和“杂环(heterocyclic ring)”在本申请中互换使用并指饱和的或者部分不饱和的(即,环内具有一个或者多个双键和/或三键)具有3至20个环原子的碳环基,其中至少一个环原子为选自氮、氧和硫的杂原子,剩余的环原子为C,其中一个或者多个环原子任选地独立取代有一个或者多个以下所述的取代基。杂环可以为具有3至7个环成员的单环(2至6个碳原子和1至4个选自N、O、P和S的杂原子)或者具有7至10个环成员的二环(4至9个碳原子和1至6个选自N、O、P和S的杂原子),例如:二环[4,5]、二环[5,5]、二环[5,6]或者二环[6,6]系统。杂环在Paquette,Leo A.;″Principles ofModern Heterocyclic Chemistry″(W.A.Benjamin,New York,1968),particularlyChapters 1,3,4,6,7,and 9;″The Chemistry of Heterocyclic Compounds,A series ofMonographs〞(John Wiley&Sons,New York,1950to present),in particular Volumes13,14,16,19,and 28;以及J.Am.Chem.Soc.(1960)82:5566中有述。术语“杂环”包括杂环烷氧基。“杂环基”还包括其中杂环基与饱和的、部分不饱和的环或者芳族碳环或者杂环稠合的基团。杂环的实例包括,但不限于,吡咯烷基、四氢呋喃基、二氢呋喃基、四氢噻吩基、四氢吡喃基、二氢吡喃基、四氢噻喃基、哌啶子基、吗啉代、硫吗啉代、噻噁烷基(thioxanyl)、哌嗪基、高哌嗪基、氮杂环丁烷基、氧杂环丁烷基、硫杂环丁烷基、高哌啶基、氧杂环庚烷基、硫杂环庚烷基、氧氮杂
术语“杂芳基”指5-、6-或者7-元环的单价芳族基团,并包括5-20个原子的稠合的环系(其中至少一个是芳族的),所述环系含有一个或者多个独立选自氮、氧和硫的杂原子。杂芳基的实例为吡啶基(包括,例如,2-羟基吡啶基)、咪唑基、咪唑并吡啶基、嘧啶基(包括,例如,4-羟基嘧啶基)、吡唑基、三唑基、吡嗪基、四唑基、呋喃基、噻吩基、异噁唑基、噻唑基、噁唑基、异噻唑基、吡咯基、喹啉基、异喹啉基、吲哚基、苯并咪唑基、苯并呋喃基、噌啉基、吲唑基、吲嗪基、酞嗪基、哒嗪基、三嗪基、异吲哚基、蝶啶基、嘌呤基、噁二唑基、三唑基、噻二唑基、噻二唑基、呋咱基、苯并呋咱基、苯并噻吩基、苯并噻唑基、苯并噁唑基、喹唑啉基、喹喔啉基、萘啶基和呋喃并吡啶基。杂芳基任选地独立取代有一个或者多个本申请所述的取代基。
所述杂环或者杂芳基在适当连接时可以是碳(碳联的)、氮(氮联的)或者氧(氧联的)连接的。作为实例且非限制,碳键合的杂环或者杂芳基在以下位置键合:吡啶的2、3、4、5或者6位,哒嗪的3、4、5或者6位,嘧啶的2、4、5或者6位,吡嗪的2、3、5或者6位,呋喃、四氢呋喃、噻吩(thiofuran)、噻吩(thiophene)、吡咯或者四氢吡咯的2、3、4或者5位,噁唑、咪唑或者噻唑的2、4或者5位,异噁唑、吡唑或者异噻唑的3、4或者5位,氮杂环丙烷的2或者3位,氮杂环丁烷的2、3或者4位,喹啉的2、3、4、5、6、7或者8位或者异喹啉的1、3、4、5、6、7或者8位。
作为实例而非限制,氮键合的杂环或者杂芳基在以下位置键合:氮杂环丙烷、氮杂环丁烷、吡咯、吡咯烷、2-吡咯啉、3-吡咯啉、咪唑、咪唑烷、2-咪唑啉、3-咪唑啉、吡唑、吡唑啉、2-吡唑啉、3-吡唑啉、哌啶、哌嗪、吲哚、二氢吲哚、1H-吲唑的1位,异吲哚或者二氢异吲哚的2位,吗啉的4位,以及咔唑或者β-咔啉的9位。
术语“治疗(treat)”和“治疗(treatment)”指的是治疗性处置以及预防或阻止性措施,其中目的是预防或延迟(衰减)不希望的生理变化或异常,诸如癌症的生长、发展或者扩散。对本发明来说,有利的或目标临床效果包括,但不限于,症状减轻,疾病程度降低,疾病状态稳定化(即未恶化),延迟或减缓疾病发展,改善或减缓疾病状态,和症状缓解(不论是部分的或全部的),不论是否是可检测或不可检测的。“治疗”还可以是指:与不得到医治而期望的存活相比,可以延长存活。需要治疗的那些包括:患有病症或障碍的那些,以及易于患所述病症或障碍的那些或者其中所述病症或障碍应该被预防的那些。
短语“治疗有效量”表示本发明化合物的量,所述化合物(i)治疗具体疾病、病症或者障碍,(ii)减弱、改善或者消除所述具体疾病、病症或者障碍的一种或者多种症状,或者(iii)预防或者延迟本申请所述具体疾病、病症或者障碍的一种或者多种症状的发作。在癌症的情况下,治疗有效量的药物可减少癌细胞的数量;减小肿瘤大小;抑制(即,在某种程度上减慢并优选停止)癌细胞向外周器官的浸润;抑制(即,在某种程度上减慢并优选停止)肿瘤转移;在某种程度上抑制肿瘤生长;和/或在某种程度上减轻一种或者多种与癌症有关的症状。为了达到药物可以预防所存在癌细胞的生长和/或将其杀死的程度,它可以是细胞生长抑制性的和/或细胞毒性的。对于癌症治疗,可以例如通过评价疾病进展的时间(TTP)和/或测定反应率(RR)来测定其效果。
术语“癌症(cancer)”和“癌的(cancerous)”是指哺乳动物中特征通常为未调节的细胞生长的生理条件或者描述所述生理条件。“肿瘤”包含一种或者多种癌细胞。癌症的实例包括但不限于癌(carcinoma)、淋巴瘤、母细胞瘤、肉瘤以及白血病(leukemia)或者淋巴样恶性肿瘤(lymphoid malignancy)。所述癌症的更具体的实例包括鳞状细胞癌(例如上皮鳞状细胞癌);肺癌,包括小细胞肺癌、非小细胞肺癌(“NSCLC″)、肺腺癌(adenocarcinomaof the lung)和肺鳞状细胞癌(squamous carcinoma of the lung);腹膜癌;肝细胞癌;胃癌(gastric or stomach cancer),包括胃肠癌;胰腺癌;成胶质细胞瘤;子宫颈癌;卵巢癌;肝癌(liver cancer);膀胱癌;肝细胞瘤(hepatoma);乳腺癌(breast cancer);结肠癌;直肠癌;结肠直肠癌;子宫内膜癌或者子宫癌;唾液腺癌;肾癌或者肾癌;前列腺癌;外阴癌(vulval cancer);甲状腺癌;肝脏癌(hepatic carcinoma);肛门癌;阴茎癌;以及头颈癌。本申请所用的胃癌(Gastric cancer)包括胃癌(stomach cancer),其可在胃的任何部分发展并可在胃各处蔓延并蔓延至其它器官;具体而言为食道、肺、淋巴结和肝。
“化学治疗剂”是可用于治疗癌症的生物学的(大分子)或者化学的(小分子)化合物,而与其作用机理无关。化学治疗剂包括,但是不限于:烷化剂、抗代谢药、纺丝体毒植物生物碱(spindle poison plant alkaloids)、细胞毒性/抗肿瘤抗生素(cytotoxic/antitumor antibiotics)、拓扑异构酶抑制剂(topoisomerase inhibitors)、蛋白、抗体、光敏性药物和激酶抑制剂。化学治疗剂包括在“靶向疗法”和非靶向常规化学疗法中使用的化合物。
术语“哺乳动物”包括,但不限于,人、小鼠、大鼠、豚鼠、猴、狗、猫、马、牛、猪、羊以及家禽。
术语“包装说明书”是指通常包括在治疗产品的市售包装中的说明书,其含有关于适应症、用法、剂量、给药、禁忌症和/或者注意事项的信息,这些信息涉及上述治疗产品的使用。
本申请使用的短语“药用盐”是指本发明化合物的药用有机或者无机盐。示例性盐包括但不限于硫酸盐、枸橼酸盐、乙酸盐、草酸盐、盐酸盐、氢溴酸盐、氢碘酸盐、硝酸盐、硫酸氢盐、磷酸盐、酸式磷酸盐、异烟酸盐、乳酸盐、水杨酸盐、酸式枸橼酸盐、酒石酸盐、油酸盐、鞣酸盐(tannate)、泛酸盐(pantothenate)、酒石酸氢盐、抗坏血酸盐、琥珀酸盐、马来酸盐、龙胆酸盐(gentisinate)、富马酸盐、葡糖酸盐、葡糖醛酸盐、糖二酸盐(saccharate)、甲酸盐、苯甲酸盐、谷氨酸盐、甲磺酸盐(mesylate)、乙磺酸盐、苯磺酸盐、对甲苯磺酸盐和扑酸盐(即1,1′-亚甲基-二-(2-羟基-3-萘甲酸盐))。药用盐可涉及另一种分子如乙酸根离子、琥珀酸根离子或者其它抗衡离子的包合物(inclusion)。抗衡离子可以是稳定母体化合物电荷的任何有机或者无机部分。此外,药用盐可在其结构中具有多于一个带电原子。多个带电原子为药用盐的部分的情况可具有多个抗衡离子。因此,药用盐可具有一个或者多个带电原子和/或者一个或者多个抗衡离子。
若本发明化合物为碱,则期望的药用盐可通过本领域可得的任何合适方法来制备,例如用无机酸(如盐酸、氢溴酸、硫酸、硝酸、甲磺酸、磷酸等)或者用有机酸(如乙酸、马来酸、琥珀酸、扁桃酸、富马酸、丙二酸、丙酮酸、草酸、羟乙酸、水杨酸、吡喃糖基酸(pyranosidyl acid)如葡萄糖醛酸或者半乳糖醛酸、α-羟基酸如枸橼酸或者酒石酸、氨基酸如天冬氨酸或者谷氨酸、芳族酸如苯甲酸或者肉桂酸、磺酸如对甲苯磺酸或者乙磺酸等)处理游离碱。例如,由P.Stahl et al,Camille G.(eds.)Handbook of PharmaceuticalSalts.Properties,Selection and Use.(2002)Zurich:Wiley-VCH;S.Berge et al,Journal of Pharmaceutical Sciences(1977)66(1)1 19;P.Gould,International J.ofPharmaceutics(1986)33 201 217;Anderson et al,The Practice of MedicinalChemistry(1996),Academic Press,New York;Remington’s Pharmaceutical Sciences,18
若本发明化合物为酸,则期望的药用盐可通过任何合适方法来制备,例如用无机或者有机碱(如胺(伯胺、仲胺或者叔胺)、碱金属氢氧化物或者碱土金属氢氧化物等)处理游离酸。合适盐的示例性实例包括但不限于从以下物质得到的有机盐:氨基酸如甘氨酸和精氨酸、氨、伯胺、仲胺和叔胺以及环状胺如哌啶、吗啉和哌嗪;以及从以下物质得到的无机盐:钠、钙、钾、镁、锰、铁、铜、锌、铝和锂。
短语“药用的”表示所述物质或者组合物必须与制剂包含的其它成分和/或者用其治疗的哺乳动物在化学上和/或者毒理学上是相容的。
“溶剂化物”是指一种或者多种溶剂分子与本发明化合物的缔合物(association)或者络合物(complex)。化合物可以非溶剂化物以及溶剂化物形式存在。形成溶剂化物的溶剂的实例包括但不限于水、异丙醇、乙醇、甲醇、DMSO、乙酸乙酯、乙酸和乙醇胺。术语“水合物”是指当溶剂分子是水时的络合物。该物理缔合物涉及多种程度的离子键合和共价键合,其包括氢键键合。在某些实例中所述溶剂化物将能够分离,例如当一个或多个溶剂分子被掺到结晶固体的晶格中时。溶剂化物的制备通常是已知的,例如,M.Caira et al,J.Pharmaceutical Sci.,93(3),601 611(2004)。E.C.van Tonder et al,AAPSPharmSciTech.,5(1),article 12(2004);以及A.L.Bingham et al,Chem.Commun.,603604(2001)描述了溶剂化物、半溶剂化物、水合物等的类似制备。典型的、非限制性的方法涉及在高于环境温度时将发明的化合物溶于期望量的期望溶剂(有机溶剂或者水或者其混合物)中,并以足够形成晶体的速率冷却所述溶液,然后通过标准方法分离所述晶体。分析技术,例如I.R.光谱,显示作为溶剂化物(或者水合物)的晶体中溶剂(或者水)的存在。
本申请所用的术语“协同的”指比两种或更多种单个药剂累加效应更有效的治疗组合。式I的化合物或者其药用盐与一种或者多种化学治疗剂之间协同性相互作用的确定可基于从本申请所述的测定中获得的结果。为了获得组合指数,可使用Chou和Talalay组合方法以及用CalcuSyn软件的剂量-效果分析来分析这些测定的结果(Chou and Talalay,1984,Adv.Enzyme Regul.22:27-55)。已经在若干测定系统中评估了该发明所提供的组合,并且可采用在抗癌剂中的定量协同作用、相加作用和拮抗作用的标准程序分析所述数据。所采用的程序是Chou和Talalay在″New Avenues in Developmental CancerChemotherapy,″Academic Press,1987,Chapter 2中描述的程序。小于0.8的组合指数值指示协同作用,大于1.2的值指示拮抗作用而0.8至1.2的值指示累加效应。所述组合疗法可提供“协同作用”并证明是“协同的”,即,当所述活性成分一起使用时所实现的效应大于由分开使用所述化合物产生的效应的总和。当所述活性成分为以下成分时,可达到协同效应:(1)在组合的单位剂量制剂中共同配制并同时给药或者递送;(2)作为分开制剂交替或平行递送;或者(3)通过一些其它用药方案。当在交替疗法中递送时,例如通过分开注射器中的不同注射液,当先后给予或者递送所述化合物时可达到协同效应。一般而言,在交替疗法期间,有效剂量的各活性成分是先后给药的,即,连续地,而在组合疗法中,有效剂量的两种或者更多种活性成分是一起给药的。在一些实例中,使用BLISS独立模型和最高单个药剂(HSA)模型(Lehár et al.2007,Molecular Systems Biology 3:80)评估组合效应。BLISS评分定量来自单个药剂的增强程度并且BLISS评分>0表明大于单纯的相加作用。HAS评分>0表明组合效应大于相应浓度时单个药剂响应的最大值。
在一个方面中,本发明提供了治疗过度增殖性病症的方法,其中式I的化合物或者其盐和一种或者多种药剂的给药提供了治疗过度增殖性病症的协同效应,所述药剂选自:5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。在另一个方面中,所述协同效应具有小于约0.8的组合指数。
在一个方面中,本发明提供了治疗过度增殖性病症的方法,其中式I的化合物或者其盐与阿比特龙或者其盐的组合给药提供了治疗过度增殖性病症的协同效应。在另一个方面中,所述协同效应具有小于约0.8的组合指数。
在一个方面中,给药GDC-0068或者其盐与阿比特龙或者其盐的组合(并且还任选地与泼尼松或者泼尼松龙组合)以治疗癌症。在一个实例中,所述癌症为前列腺癌。在另一个实例中,所述癌症为转移性前列腺癌。在一个实例中,所述癌症为前列腺腺癌(prostateadenocarcinoma)。
式I化合物
式I化合物包含式I的化合物及其药用盐,所述式I为:
其中:
R
R
R
A为
G为任选地被一至四个R
R
或者R
R
或者R
R
或者R
R
或者R
各个R
R
m、n和p独立地为0或者1。
式I的具体化合物为化合物,其中A为
式I的具体化合物为式Ia化合物或者其药用盐,所述式Ia为:
在本发明的一个方面中,式I的化合物不包含式Ia化合物(S)-2-(4-氯苯基)-1-(4-((5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-基)-3-(异丙基氨基)丙-1-酮或者其药用盐(该化合物也可称作GDC-0068),所述式Ia为:
式I化合物的制备
本发明化合物可通过包括与化学领域中众所周知的方法类似的方法的合成路线来合成,特别是根据本申请所含有的描述。起始原料通常可从商业来源例如AldrichChemicals(Milwaukee,WI)得到或者使用本领域技术人员所熟知的方法容易制备(例如,通过Louis F.Fieser and Mary Fieser,Reagents for Organic Synthesis,v.1-19,Wiley,N.Y.(1967-1999ed.),或者Beilsteins Handbuch der organischen Chemie,4,Aufl.ed.Springer-Verlag,Berlin,包括附录中通常所描述的方法制备)。
式I的化合物可逐一制备,或者作为包含至少2个,例如5至1,000个化合物或者10至100化合物的化合物库制备。通过本领域技术人员已知的操作,使用液相或者固相化学,式I化合物的库可通过组合的‘裂分和混合’途径或者通过多个平行合成来制备。因此根据本发明的另一个方面,提供有包含至少2个式I的化合物或者其盐的化合物库。
为了用作示例说明,方案1-4和方案A-J显示制备本发明化合物以及关键中间体的通用方法。对于个别反应步骤更加详细的描述,见下面实施例部分。本领域的技术人员应当理解的是,其它合成路线可用于合成发明的化合物。虽然在方案中描述并在下面讨论了具体的起始原料和试剂,但是其它起始原料和试剂可容易地被替换,从而提供多种衍生物和/或反应条件。除此之外,多种通过下面所述方法制备的化合物可根据本公开使用本领域技术人员所熟知的常规化学来进一步修饰。
方案1
方案1显示制备式I的化合物10的方法,其中R
方案2
方案2显示制备式I的化合物22、25和27的方法,其中R
可选择地,化合物24的7-羟基可在碱(如NaH或者KOH)存在下用烷基化试剂(如烷基卤化物)进行烷基化得到其中R
方案3
方案3显示制备化合物73和74的可选择的方法。根据方案3,使用氨合成子的14的胺化得到63。在50℃-250℃和/或在高压在甲酰胺存在下使用例如甲酸铵进行嘧啶的形成得到二环单元64。使用例如POCl
方案4
向化合物1引入手性助剂(例如,Evans噁唑烷酮等等)可通过标准酰化操作来完成,得到结合物2。例如,在-20℃至100℃在胺类碱存在下,用活化剂(例如,COCl
相应地,本发明的另一个方面提供了制备式I的化合物的方法,其包括:
使得化合物与氨基酸反应,
所述化合物具有以下式:
其中R
所述氨基酸具有以下式:
其中R
在方案1-4和实施例所述的式I化合物的合成中所使用的氨基酸或者是可商购的或者可根据本申请所公开的方法制备。例如,在某些实施方案中,用于制备式I化合物的氨基酸包括具有式1A的β-苯基甘氨酸氨基酸、具有式2A的γ-苯基甘氨酸氨基酸、具有式3A的β-苯基丙氨酸氨基酸和具有式4A的γ-苯基丙氨酸氨基酸。
制备式1A-4A的氨基酸的方法显示在方案A-J中。
方案A
方案A说明制备任选取代的式1A的β-苯基甘氨酸氨基酸25和26的方法,其中R
可选择地,所述活化的烯烃24可在适当温度(例如,-20℃至回流温度)在适合溶剂(如THF)中用仲胺(例如,二乙胺)处理,得到氨基酯中间体(未显示)。在其中化合物24具有富电子芳族环或者缺电子/大体积仲胺的情况下,可需要加热(例如,在密封管中30-240℃)或者微波化学。使用适于酯的条件(例如,对于甲基的含水LiOH、对于苄基的氢化、对于叔丁酯的酸等等)可完成所述酯的皂化,形成氨基酸26。
在方案A的替代方案中,Pg可用化合物23和25中的R
方案A1
方案A1显示方案1的替代方案,其中活化的烯烃24反应形成氨基酸26A。
方案B
方案B显示制备任选取代的式1A的β-苯基甘氨酸氨基酸30和31的方法,其中R
方案C
方案C显示制备任选取代的式1A的β-苯基甘氨酸氨基酸36的方法,其中R
在方案C的一种替代方案中,R
在方案C的另一种替代方案中,Pg可用化合物35和36中的R
方案D
方案D显示制备任选取代的式2A的γ-苯基甘氨酸氨基酸40的方法,其中R
在方案D的一种替代方案中,Boc可用化合物39和40中的R
方案D1
方案D1显示形成γ氨基酸40d和40e的单个对映异构体的方法,其中R
方案E
方案E显示制备任选取代的式2A的γ-苯基甘氨酸氨基酸44的方法,其中R
在方案E的一种替代方案中,Pg可用化合物43和44中的R
方案F
方案F显示制备任选取代的式3A的β-苯基丙氨酸氨基酸48、49和50的方法,其中R
在方案F的一个替代方案中,Pg可用化合物49或者50中的R
方案G
方案G显示制备任选取代的式4A的α-苯基丙氨酸氨基酸54的方法,其中R
在方案G的一种替代方案中,在化合物53脱保护之后,Pg可用化合物54中的R
方案H
方案H显示制备任选取代的式2A的γ-苯基甘氨酸氨基酸56的方法,其中R
在方案H的一种替代方案中,Pg可用化合物56中的R
方案I
方案I显示制备任选取代的式3A的β-苯基丙氨酸氨基酸61和62的方法,其中R
方案J
使用方案J中所示的操作可制备任一种b-氨基酸的对映异构体。可将与具有适当立体化学以产生在氨基酸b-位期望的化学的适当手性助剂(R*)(例如,Evans的助剂或者磺内酰胺)偶联的乙酸2-苯酯用亚胺或者亚胺离子合成子(例如,在-100℃至50℃在路易斯酸(例如,TiCl
在制备式I的化合物中,中间体远距离官能团(例如,伯胺或者仲胺等等)的保护可以是必需的。这种保护的需要将取决于远距离官能团的性质和制备方法的条件而变化。适合的氨基-保护基(NH-Pg)包括乙酰基、三氟乙酰基、叔丁氧基羰基(BOC)、苄基氧基羰基(CBz)和9-芴基亚甲基氧基羰基(Fmoc)。这种保护的需要由本领域的技术人员容易地确定。对于保护基以及其使用的一般描述,见T.W.Greene,Protective Groups in OrganicSynthesis,John Wiley&Sons,New York,1991.
在制备式I的化合物的任意合成方法中,将反应产物彼此分离和/或者与起始原料分离可能是有益的。通过本领域常见技术将每步或者多步中期望的产物分离和/或者纯化(以下称为分离)为期望的同质性程度。通常所述分离涉及多相萃取、从溶剂或者溶剂混合物中结晶、蒸馏、升华或者色谱法。色谱法可涉及任何数目的方法,包括例如:反相和正相;尺寸排阻(size exclusion);离子交换;高、中和低压液相色谱方法和装置;小规模分析;模拟移动床(SMB)和制备性薄层或者厚层色谱,以及小规模薄层和快速色谱技术。
另一类分离方法涉及用所选择的试剂处理混合物,从而与期望的产物、未反应的起始原料、反应副产物等结合或者使得期望的产物、未反应的起始原料、反应副产物等分离。所述试剂包括吸附剂(adsorbent)或者吸收剂(absorbent),如活性炭、分子筛、离子交换介质等。可供选择地,所述试剂可以是酸(在碱性物质的情况下),碱(在酸性物质的情况下),结合剂如抗体、结合蛋白,选择性螯合剂如冠醚,液/液离子交换试剂(LIX)等。
对适当的分离方法的选择依赖于所涉及的物质的性质。例如,沸点和分子量(在蒸馏和升华中)、存在或者不存在极性官能团(在色谱法中)、在酸性和碱性介质中物质的稳定性(在多相萃取中)等等。本领域技术人员将采用最有可能实现期望的分离的技术。
可通过本领域技术人员公知的方法(如色谱法和/或者分级结晶),基于非对映异构体的物理化学差别,将非对映异构混合物分离为其单独的非对映异构体。对映异构体可通过以下方式分离:通过使对映异构体混合物与适当的光学活性的化合物(例如,手性助剂如手性醇或者Mosher′s酰氯)反应将其转化为非对映异构混合物,分离非对映异构体,然后将单独的非对映异构体转化(例如,水解)为相应的纯的对映异构体。此外,一些本发明化合物可以是阻转异构体(例如,取代的联芳(biaryl))并视为本发明的部分。对映异构体也可通过使用手性HPLC柱分离。
单一的立体异构体,例如,基本上不含其立体异构体的对映异构体,可通过以下方式获得:使用诸如形成非对映异构体的方法,用光学活性的拆分剂来拆分外消旋混合物(Eliel,E.and Wilen,S.″Stereochemistry of Organic Compounds,″John Wiley&Sons,Inc.,New York,1994;Lochmuller,C.H.,J.Chromatogr.,(1975)113(3):283-302)。本发明的手性化合物的外消旋混合物可通过任何合适的方法分开和离析,所述方法包括:(1)与手性化合物形成离子性非对映异构的盐,然后通过分级结晶或者其它方法分离,(2)与手性衍生试剂形成非对映异构的化合物,分离所述非对映异构体,然后转化为纯的立体异构体,(3)在手性条件下直接分离基本上纯的或者富含的立体异构体。参见:″DrugStereochemistry,Analytical Methods and Pharmacology,″IrvingW.Wainer,Ed.,Marcel Dekker,Inc.,New York(1993)。
在方法(1)的情况下,非对映异构的盐可通过以下方式形成:使对映异构纯的手性碱如马钱子碱(brucine)、奎宁、麻黄碱、番木鳖碱(strychnine)、α-甲基-β-苯基乙胺(安非他明)等与带有酸性官能团的不对称化合物如羧酸和磺酸反应。可通过分级结晶或者离子色谱法诱导非对映异构体的盐分离。对于氨基化合物的光学异构体的分离而言,加入手性羧酸或者磺酸如樟脑磺酸、酒石酸、扁桃酸或者乳酸可引起非对映异构体的盐的形成。
可供选择地,通过方法(2),使待拆分的底物与手性化合物的一种对映异构体反应,形成非对映异构对(E.and Wilen,S.″Stereochemistry of Organic Compounds″,JohnWiley&Sons,Inc.,1994,p.322)。非对映异构化合物可通过以下方式形成:使不对称化合物与对映异构纯的手性衍生试剂如薄荷基衍生物反应,接着分离非对映异构体,然后水解得到纯的或者富集的对映异构体。确定光学纯度的方法涉及制备外消旋混合物的手性酯,如在碱的存在下制备薄荷基酯例如(-)氯甲酸薄荷基酯,或者Mosher酯,乙酸α-甲氧基-α-(三氟甲基)苯基酯(Jacob III.J.Org.Chem.,(1982)47:4165),然后就两种阻转异构的对映异构体或者非对映异构体的存在而分析
化学治疗剂
某些化学治疗剂与式I的化合物或者其药用盐组合已经证明具有令人惊讶的及意想不到的体外和体内抑制细胞增殖性质。所述化学治疗剂包括:5-FU、铂剂、伊立替康、多西他赛、多柔比星、吉西他滨、SN-38、卡培他滨、替莫唑胺、厄洛替尼、PD-0325901、紫杉醇、贝伐珠单抗、帕妥珠单抗、他莫昔芬、雷帕霉素、拉帕替尼、PLX-4032、MDV3100、阿比特龙和GDC-0973。
5-FU(氟尿嘧啶,5-氟尿嘧啶,CAS登记号51-21-8)为胸苷酸合酶抑制剂并已经用于癌症治疗数十年,所述癌症包括结肠直肠癌和胰腺癌(US 2802005;US 2885396;Duschinsky et al(1957)J.Am.chem.Soc.79:4559;Hansen,R.M.(1991)Cancer Invest.9:637-642)。5-FU的命名为5-氟-1H-嘧啶-2,4-二酮。
卡铂(CAS登记号41575-94-4)是用于针对卵巢癌、肺癌、头颈部癌的化学治疗药物(US 4140707;Calvert et al(1982)Cancer Chemother.Pharmacol.9:140;Harland et al(1984)Cancer Res.44:1693)。卡铂的命名为环丁烷-1,1-二羧酸二氨铂(azanide;cyclobutane-1,1-dicarboxylic acid;platinum)。
顺铂(Cisplatin)、顺铂(cisplatinum)或者顺式-二氨二氯化亚铂(II)(CAS登记号15663-27-1)为用于治疗多种类型癌症的化学治疗药物,所述癌症包括肉瘤、一些癌瘤(例如,小细胞肺癌和卵巢癌)、淋巴瘤以及生殖细胞肿瘤(germ cell tumors)。它是含有铂的抗癌药物种类中的第一个成员,该种类目前还包括卡铂和奥沙利铂。顺铂具有如下结构:顺式-PtCl
奥沙利铂(CAS登记号63121-00-6)为用于癌症化学治疗的配位络合物(美国专利4,169,846)。已经在晚期癌症(胃癌、卵巢癌)中将奥沙利铂与其它铂化合物(顺铂、卡铂)作比较。奥沙利铂通常在称为FOLFOX的组合中与氟尿嘧啶和甲酰四氢叶酸给药用于结肠直肠癌的治疗。
伊立替康(CAS登记号97682-44-5)为拓扑异构酶1抑制剂,其阻止DNA解旋。伊立替康经水解活化成拓扑异构酶I的抑制剂SN-38。所述活性代谢物SN-38对拓扑异构酶I的抑制最终导致对DNA复制和转录的抑制。它主要用于结肠癌,具体而言,与其它化学治疗剂组合用于结肠癌。所述组合包括用药方案FOLFIRI,其由输注的5-氟尿嘧啶、甲酰四氢叶酸和伊立替康组成。
多柔比星(CAS登记号23214-92-8)为蒽环类抗生素。同所有蒽环类一样,它通过嵌入DNA起作用。多柔比星常用于多种癌症(包括血液恶性肿瘤)、多种癌瘤以及软组织肉瘤的治疗。多柔比星的命名为(8S,10S)-10-(4-氨基-5-羟基-6-甲基-四氢-2H-吡喃-2-基氧基)-6,8,11-三羟基-8-(2-羟基乙酰基)-1-甲氧基-7,8,9,10-四氢并四苯-5,12-二酮。
多西他赛(CAS登记号114977-28-5)用于治疗乳腺癌、卵巢癌以及NSCLC(US4814470;US 5438072;US 5698582;US 5714512;US 5750561;Mangatal et al(1989)Tetrahedron 45:4177;Ringel et al(1991)J.Natl.Cancer Inst.83:288;Bissery et al(1991)Cancer Res.51:4845;Herbst et al(2003)Cancer Treat.Rev.29:407-415;Davieset al(2003)Expert.Opin.Pharmacother.4:553-565)。多西他赛的命名为(2R,3S)-N-羧基-3-苯基异丝氨酸,N-叔丁酯,13-酯与5,20-环氧-1,2,4,7,10,13-六羟基紫杉-11-烯-9-酮4-乙酸酯2-苯甲酸酯,三水合物((2R,3S)-N-carboxy-3-phenylisoserine,N-tert-butyl ester,13-ester with 5,20-epoxy-1,2,4,7,10,13-hexahydroxytax-11-en-9-one4-acetate2-benzoate,trihydrate)(US 4814470;EP 253738;CAS登记号114977-28-5)。
吉西他滨(CAS登记号95058-81-4)为阻断DNA复制的核苷类似物,用于治疗多种癌瘤,所述癌瘤包括胰腺癌、乳腺癌、NSCLC和淋巴瘤(US 4808614;US 5464826;Hertel et al(1988)J.Org.Chem.53:2406;Hertel et al(1990)Cancer Res.50:4417;Lund et al(1993)Cancer Treat.Rev.19:45-55)。吉西他滨的命名为4-氨基-1-[3,3-二氟-4-羟基-5-(羟基甲基)四氢呋喃-2-基]-1H-嘧啶-2-酮。
SN-38(CAS登记号86639-52-3)为伊立替康的活性代谢物(见上文)。它比伊立替康本身活性强200倍。它的命名为7-乙基-10-羟基-喜树碱。
卡培他滨(CAS登记号154361-50-9)为口服给药的用于转移性乳腺癌和结肠直肠癌治疗的化学治疗剂。卡培他滨为在肿瘤中被酶解转化成5-氟尿嘧啶的前药,其中它抑制DNA合成并减慢肿瘤组织的生长。卡培他滨的活化采取具有三种酶解步骤和两种中间代谢物的途径形成5-氟尿嘧啶,所述代谢物为5′-脱氧-5-氟胞嘧啶(5′-DFCR)和5′-脱氧-5-氟尿嘧啶(5′-DFUR)。卡培他滨的命名为[1-(3,4-二羟基-5-甲基-四氢呋喃-2-基)-5-氟-2-氧代-1H-嘧啶-4-基]氨基甲酸戊酯。
替莫唑胺(CAS登记号85622-93-1)为用于治疗IV级星形细胞瘤(也称作多形性胶质母细胞瘤)以及黑色素瘤(皮肤癌的形式)的烷化剂。替莫唑胺的命名为4-甲基-5-氧代-2,3,4,6,8-五氮杂二环[4.3.0]壬-2,7,9-三烯-9-甲酰胺。
厄洛替尼(CAS登记号183321-74-6,
PD-0325901(CAS登记号391210-10-9,Pfizer)为治疗癌症的可能的口服片剂的第二代、非-ATP竞争性、变构MEK抑制剂(US 6960614;US 6972298;US 2004/147478;US 2005/085550)。已经进行II期临床试验,可能治疗乳腺癌、结肠癌和黑色素瘤。PD-0325901的命名为(R)-N-(2,3-二羟基丙氧基)-3,4-二氟-2-(2-氟-4-碘苯基氨基)苯甲酰胺,并具有以下结构:
紫杉醇(CAS登记号33069-62-4,
贝伐珠单抗(CAS登记号216974-75-3,
贝伐珠单抗和其它人源化的抗-VEGF抗体在US 6884879中有更多描述。另外的抗-VEGF抗体包括G6或者B20系列抗体,例如,G6-31,B20-4.1,(WO 2005/012359;WO 2005/044853;US 7060269;US 6582959;US 6703020;US 6054297;WO 98/45332;WO 96/30046;WO94/10202;EP 0666868B1;US 2006/009360;US 2005/0186208;US 2003/0206899;US 2003/0190317;US 2003/0203409;20050112126;Popkov et al(2004)Journal ofImmunological Methods 288:149-164。“B20系列抗体”为根据WO 2005/012359的图27-29的任一项从B20抗体或者B20-衍生的抗体的序列衍生的抗-VEGF抗体,将其整个公开特地引入本申请作为参考。在一个实施方案中,所述B20系列抗体结合至人VEGF上包含残基F17、M18、D19、Y21、Y25、Q89、I91、K101、E103和C104的功能表位。其它抗-VEGF抗体包括结合至人VEGF上包含残基F17、M18、D19、Y21、Y25、Q89、I91、K101、E103和C104或者可选择地包含残基F17、Y21、Q22、Y25、D63、I83和Q89的功能表位的抗体。
曲妥珠单抗(
帕妥珠单抗(OMNITARG
替莫唑胺(CAS登记号85622-93-1,
他莫昔芬(CAS登记号10540-29-1,
雷帕霉素(CAS登记号53123-88-9,西罗莫司,
拉帕替尼(CAS登记号388082-78-8,
PLX-4032(CAS登记号1029872-55-5)已经显示在黑色素瘤细胞系中引起程序性细胞死亡。PLX-4032中断B-Raf/MEK/ERK途径上的B-Raf/MEK步骤-如果所述B-Raf具有常见的V600E突变。PLX-4032在黑色素瘤患者中起作用,他们的肿瘤具有V600E BRAF突变(即,在B-RAF蛋白上的氨基酸位置号600,正常的缬氨酸被谷氨酸所替代)。大约60%的黑色素瘤具有所述V600E BRAF突变。PLX-4032具有下列结构:
MDV3100(CAS登记号915087-33-1)为开发用于激素难治性前列腺癌治疗的雄激素受体拮抗剂药物。已经报道在服用所述药物一个月后前列腺特异性抗原血清水平降低高达89%。与比卡鲁胺相反的是,MDV3100不能促进AR向细胞核的易位并且另外还阻止AR对DNA以及AR对共活化物蛋白的结合。发现MDV 3100对正在进行的I和II期试验中的转移性去雄抗性(castration-resistant)前列腺癌患者是临床有效的。MDV3100的命名为4-(3-(4-氰基-3-(三氟甲基)苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-N-甲基苯甲酰胺。
阿比特龙(CAS登记号154229-19-3;见美国专利5,604,213和5,618,807)及其盐醋酸阿比特龙是正在研究用于去雄抗性前列腺癌的药物。它通过抑制CYP17A1(CYP450c17)(也称作17α-羟化酶/17,20裂解酶的酶)阻断睾酮的形成。该酶涉及DHEA和雄烯二酮的形成,其最终可代谢成睾酮。阿比特龙的命名为(3S,8R,9S,10R,13S,14S)-10,13-二甲基-17-(吡啶-3-基)-2,3,4,7,8,9,10,11,12,13,14,15-十二氢-1H-环戊二烯并[a]菲-3-醇。它还作为以下乙酸酯前药注册:乙酸(3S,8R,9S,10R,13S,14S)-10,13-二甲基-17-(吡啶-3-基)-2,3,4,7,8,9,10,11,12,13,14,15-十二氢-1H-环戊二烯并[a]菲-3-基酯。
GDC-0973为MEK的选择性抑制剂,MEK也称作丝裂原活化蛋白激酶激酶(MAPKK),所述酶是常在人肿瘤中活化的RAS/RAF/MEK/ERK途径的关键组分。在缺少外源性生长因子的情况下,MEK/ERK途径的不适当活化促进细胞生长。评估GDC-0973用于实体瘤的I期临床试验正在进行中。GDC-0973可如国际专利申请公开WO2007044515(A1)中所述来制备。GDC-0973的命名为:(S)-(3,4-二氟-2-(2-氟-4-碘苯基氨基)苯基)(3-羟基-3-(哌啶-2-基)氮杂环丁烷-1-基)甲酮,并具有以下结构:
药物组合物
本发明的药物组合物或者制剂包含式I化合物、化学治疗剂以及一种或者多种可药用的载体、助流剂、稀释剂或者赋形剂的组合。
本发明的式I化合物和化学治疗剂可以非溶剂化形式以及与可药用的溶剂(如水、乙醇等)的溶剂化形式存在,并且意图是本发明包括所有溶剂化的和非溶剂化的形式。
本发明的式I化合物和化学治疗剂还可以呈不同的互变异构形式存在,并且所有这些形式都包括在本发明的范围中。术语“互变异构体”或者“互变异构形式”是指可通过低能垒互相转化的不同能量的结构异构体。例如,质子互变异构体(也称为质子移变互变异构体)包括通过质子迁移进行的互相转化,如酮-烯醇异构化和亚胺-烯胺异构化。价键互变异构体包括通过一些成键电子的重组进行的互相转化。
药物组合物包括由多于一种(例如,两种)药学活性剂与任何无药学活性的赋形剂、稀释剂、载体或者助流剂一起组成的大量的(bulk)组合物和单独的剂量单位,所述药学活性剂包括式I化合物和选自本申请所述另外药剂名单的化学治疗剂。所述大量的组合物和各单独剂量单位可含有固定量的上述药学活性剂。所述大量的组合物是尚未形成单独剂量单位的物质。示例性剂量单位为口服剂量单位如片剂、丸剂、胶囊剂等。类似地,本申请所述的通过给予本发明的药物组合物治疗患者的方法也意在包括大量的组合物和单独的剂量单位的给药。
药物组合物还包括同位素标记的本发明化合物,其与本申请所述的化合物相同,但是事实上一个或者多个原子被原子质量或者质量数不同于自然界中常见原子质量或者质量数的原子代替。预期任何具体原子或者元素的所有同位素都包括在本发明化合物和它们的用途的范围中。可引入本发明化合物中的示例性同位素包括氢、碳、氮、氧、磷、硫、氟、氯和碘的同位素,如
式I化合物和化学治疗剂可按照标准制药规范来配制以用于治疗哺乳动物(其包括人)中过度增殖性病症的治疗性处置(其包括预防性治疗)的治疗性组合。本发明提供了包含式I化合物与一种或者多种可药用的载体、助流剂、稀释剂或者赋形剂的药物组合物。
合适的载体、稀释剂和赋形剂是本领域技术人员公知的,并且包括以下物质,如碳水化合物、蜡、水溶性聚合物和/或者水可溶胀聚合物(swellable polymer)、亲水性物质或者疏水性物质、明胶、油、溶剂、水等。所用的具体载体、稀释剂或者赋形剂将取决于应用本发明化合物的方式和目的。通常基于本领域技术人员认为给予哺乳动物安全的溶剂(GRAS)来选择溶剂。一般而言,安全溶剂为无毒性含水溶剂如水和可在水中溶解或者混溶的其它无毒性溶剂。合适的含水溶剂包括水、乙醇、丙二醇、聚乙二醇(例如,PEG 400、PEG 300)等及其混合物。制剂还可包括以下物质中的一种或者多种:缓冲剂、稳定剂、表面活性剂、润湿剂、润滑剂、乳化剂、助悬剂、防腐剂、抗氧化剂、遮光剂(opaquing agent)、助流剂、加工助剂(processing aid)、着色剂、增甜剂、芳香剂、矫味剂和提供药物(即本发明化合物或者其药物组合物)的优质外观或者辅助制造药物产品(即药物)的其它已知添加剂。
制剂可使用常规溶出和混合操作制备。例如,将大块的药品(即,本发明化合物或者化合物的经稳定形式(例如,与环糊精衍生物或者其它已知复合剂(complexationagent)的复合物))在一种或者多种上述的赋形剂存在下溶于合适的溶剂中。通常将本发明化合物配制成提供容易可控制药物的剂量且使患者能够依从所给出的方案的药物剂型。
取决于用于给药药物的方法,用于施用的药物组合物(或者制剂)可按多种方式包装。一般地,用于分配的物品包括容器,容器内存放有适当形式的药物制剂。合适的容器是本领域技术人员公知的,并且包括以下物质,如瓶(塑料的和玻璃的)、小袋(sachet)、安瓿、塑料袋、金属圆筒等。容器还可包括防止不慎重取得包装中的内含物的的防干扰装置(tamper-proof assemblage、)。此外,在容器上具有描述容器中的内含物的标签。所述标签还可包括适当的注意事项。
可制备本发明化合物的药物制剂用于多种给药途径和类型。例如,具有期望的纯度的式I的化合物可任选与药用稀释剂、载体、赋形剂或者稳定剂(Remington′sPharmaceutical Sciences(1995)18th edition,Mack Publ.Co.,Easton,PA)以冻干制剂、磨细的粉末剂或者水溶液剂形式混合。配制可如下进行:在环境温度在适当的pH以及在适当的纯度与生理学可接受的载体(即在采用的剂量和浓度下对受体是无毒性的载体)混合。制剂的pH主要取决于具体用途和化合物的浓度,但范围可为约3至约8。
所述药物制剂优选为无菌的。特别地,用于体内给药的制剂必须为无菌的。所述灭菌通过滤过经无菌滤膜容易地完成。
药物制剂通常可储存为固体组合物、冻干制剂或者水溶液剂。
所述药物制剂将按照与良好医学实践一致的方式(即量、浓度、时间表、过程、媒介物和给药途径)来确定剂量和给药。在此背景下考虑的因素包括所治疗的具体病症、所治疗的具体哺乳动物、个体患者的临床情况、病症的起因、药物的递送位点、给药方法、给药的时间表和医学实践者已知的其它因素。所给药的化合物的“治疗有效量”将由这些考虑因素控制,并且是预防、改善或者治疗凝结因子介导的病症所需的最小量。所述量优选低于对宿主有毒的或者使宿主显著地更易于出血的量。
作为通常的建议,每剂量口服或者肠胃外给药的式I化合物的初始药学有效量应当在约0.01-1000mg/kg的范围,即约0.1至20mg/kg的患者体重/天的范围,所用化合物的通常起始范围为0.3至15mg/kg/天。所要给药的式I化合物的剂量和化学治疗剂的剂量各自可以为约1mg至约1000mg/单位剂量形式,或者为约10mg至约100mg/单位剂量形式。式I化合物和化学治疗剂的剂量可以约1∶50至约50∶1以重量计的比率或者以约1∶10至约10∶1以重量计的比率给药。
可接受的稀释剂、载体、赋形剂和稳定性在所用的剂量和浓度下对受体是无毒性的,并且包括缓冲剂如磷酸盐、枸橼酸盐和其它有机酸;抗氧化剂,包括抗坏血酸和蛋氨酸(methionine);防腐剂(如十八烷基二甲基苄基氯化铵;氯化六甲双铵(hexamethoniumchloride);苯扎氯铵、苄索氯胺;苯酚、丁醇或者苄醇;对羟基苯甲酸烷基酯,如对羟基苯甲酸甲酯或者对羟基苯甲酸丙酯;儿茶酚;间苯二酚(resorcinol);环己醇;3-戊醇和间甲酚);低分子量(少于约10个残基)多肽;蛋白质,如血清白蛋白、明胶或者免疫球蛋白;亲水性聚合物,如聚乙烯吡咯烷酮;氨基酸,如甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、精氨酸或者赖氨酸;单糖、二糖和其它碳水化合物,包括葡萄糖、甘露糖或者糊精;螯合剂,如EDTA;糖如蔗糖、甘露醇、海藻糖或者山梨醇;成盐的抗衡离子,如钠;金属络合物(例如,Zn-蛋白质络合物);和/或者非离子型表面活性剂,如TWEEN
可制备式I的化合物的缓释制剂。缓释制剂的合适实例包括含有式I的化合物的固态疏水性聚合物的半渗透性基质,其中基质以成形的物品形式(例如薄膜或者微胶囊)存在。缓释基质的实例包括聚酯、水凝胶(例如,聚(甲基丙烯酸2-羟基乙酯)或者聚(乙烯醇))、聚交酯(US专利3,773,919)、L-谷氨酸和γ-乙基-L-谷氨酸的共聚物、非降解性乙烯-乙酸乙烯酯、降解性乳酸-羟乙酸共聚物如LUPRON DEPOT
所述药物制剂包括适于本申请详述的给药途径的制剂。制剂可适宜地以单位剂量形式存在并可通过药学领域公知的任何方法制备。技术和制剂通常参见Remington′sPharmaceutical Sciences 18
可将适于口服给药的式I的化合物和/或化学治疗剂的制剂制备成离散的单位如丸剂、硬的或者软的(例如)明胶胶囊、扁胶囊、糖锭(troches)、锭剂(lozenges)、含水或者油混悬剂、可分散粉剂或者颗粒剂、乳剂、糖浆剂或者酏剂,其各自含有预设量的式I化合物和/或化学治疗剂。式I化合物的量和化学治疗剂的量可作为组合制剂在丸剂、胶囊剂、溶液剂或者混悬剂中配制。可选择地,式I化合物和化学治疗剂可交替在丸剂、胶囊剂、溶液剂或者混悬剂中单独配制。
制剂可根据制备药物组合物的领域已知的任何方法制备,所述组合物可含有一种或者多种试剂,包括增甜剂、矫味剂、着色剂和防腐剂,以提供适口的制剂。压制片可如下制备:在合适的机器中对自由流动形式(如粉末或者颗粒)的活性成分以及任选混合的粘合剂、润滑剂、惰性稀释剂、防腐剂、表面活性剂或者分散剂进行压制。模制片可如下制备:在合适的机器中对用惰性液态稀释剂润湿的粉末状活性成分的混合物进行模制。可任选对片剂进行包衣或者刻痕,并任选进行配制以提供活性成分从其中缓慢或者控制释放。
药物制剂的片剂赋形剂可包括:填充剂(或者稀释剂),增加组成片剂的粉末状药物的大体积;崩解剂,当它被消化时促进片剂分裂成小碎片,理想地是个体药物颗粒,并且促进药物的快速溶出和吸收;粘合剂,确保颗粒剂和片剂可以需要的机械强度形成,并在已经被压缩后保持片剂在一起,防止在包装、运输和日常搬运期间分裂成它的组分粉末;助流剂,改善生产期间组成片剂的粉末的流动性;润滑剂,确保生产期间压片粉末不粘附于用于压片的设备,它们通过压缩改善粉末状混合物的流动并且当从完成的片剂设备中推出时最小化摩擦力和破裂;抗粘附剂,其具有与助流剂类似的功能,减少生产期间组成片剂的粉末与用于冲出片剂形状的设备之间的粘着力;调味剂,其掺入片剂给予其更加令人愉快的味道或者掩蔽不愉快的味道;以及着色剂,有助于识别和患者顺应性。
含有活性成分以及混合有适于制造片剂的无毒性生理学可接受的赋形剂的片剂是可接受的。这些赋形剂可以是,例如,惰性稀释剂,如碳酸钙或者碳酸钠、乳糖、磷酸钙或者磷酸钠;成粒剂和崩解剂(granulating and disintegrating agent),如玉米淀粉或者海藻酸;粘合剂,如淀粉、明胶或者阿拉伯胶;以及润滑剂,如硬脂酸镁、硬脂酸或者滑石。片剂可以是未包衣的或者可通过已知技术(包括微胶囊化)包衣,以延迟在胃肠道的崩解和吸收,由此在较长的时间提供持续的作用。例如,可采用定时延迟物质,如单独的或者与蜡结合的单硬脂酸甘油酯或者二硬脂酸甘油酯。
对于治疗眼部或者其它外部组织如嘴和皮肤而言,所述制剂优选应用为局部软膏剂(ointment)或者乳膏剂(cream),其含有的活性成分的量为例如,0.075至20%w/w。当配制成软膏剂时,活性成分可与石蜡(paraffinic)或者可与水混溶的软膏基质一起使用。可供选择地,活性成分可与水包油性乳膏基质一起配制成乳膏。
如果期望的话,乳膏基质的水相可包括多元醇,即,具有两个或者更多个羟基的醇,如丙二醇、丁-1,3-二醇、甘露醇、山梨醇、甘油和聚乙二醇(包括PEG 400)及这些醇的混合物。局部制剂可包括增强活性成分通过皮肤或者其它作用区域吸收或者渗透的化合物。所述皮肤渗透增强剂的实例包括二甲基亚砜和相关类似物。
本发明的乳剂的油相可由已知成分以已知方式构成,包含至少一种乳化剂与脂肪或者油或者与脂肪和油两者的混合物。优选地,还包括亲水性乳化剂以及作为稳定剂的亲脂性乳化剂。同时,含有或者不含有稳定剂的乳化剂构成了所谓的乳化蜡(emulsifyingwax),所述蜡和油和脂肪一起构成了形成软膏制剂的油性分散相的所谓乳化软膏基质。适用于本发明制剂的乳化剂和乳化稳定剂包括
所述药物制剂的含水混悬剂含有活性物质以及混合有适于制备水性混悬剂的赋形剂。所述赋形剂包括助悬剂,如羧甲基纤维素钠、交联羧甲基纤维素、聚维酮、甲基纤维素、羟丙基甲基纤维素、海藻酸钠、聚乙烯吡咯烷酮、西黄蓍胶和阿拉伯胶,以及分散或者润湿剂(dispersing or wetting agent),如天然存在的磷脂(例如,卵磷脂)、烯化氧与脂肪酸的缩合产物(例如,聚氧乙烯硬脂酸酯)、环氧乙烷与长链脂肪醇的缩合产物(例如,十七亚乙氧基十六醇(heptadeca ethyleneoxycetanol))、环氧乙烷与衍生自脂肪酸和己糖醇脱水物(hexitol anhydride)的偏酯的缩合产物(例如,聚氧乙烯脱水山梨糖醇单油酸酯(polyoxyethylene sorbitan monooleate))。水性混悬剂还可含有一种或者多种防腐剂如对羟基苯甲酸乙酯或者对羟基苯甲酸正丙酯、一种或者多种着色剂、一种或者多种矫味剂和一种或者多种增甜剂如蔗糖或者糖精。
药物组合物可呈无菌注射制剂,如无菌注射水性混悬剂或者油性混悬液制剂形式存在。该混悬液可使用上文已提及的合适的分散剂或者润湿剂和助悬剂根据本领域已知方法配制。无菌注射制剂还可以是于无毒性的肠胃外可接受的稀释剂或者溶剂中的无菌注射溶液或者混悬液,如于1,3-丁二醇中的溶液,或者制备为冻干粉末。可使用的可接受媒介物和溶剂包括水、林格氏溶液(Ringer′s solution)和等张氯化钠溶液。此外,无菌不挥发性油(sterile fixed oil)通常可用作溶剂或者助悬介质。出于该目的,可采用任何温和的不挥发性油,包括合成性甘油一酯或者甘油二酯。此外,脂肪酸如油酸同样可用于制备注射剂。
可与载体物质结合以产生单一剂量形式的活性成分的量将随着所治疗的宿主和具体的给药模式而变化。例如,意在对人类口服给药的定时释放制剂可含有约1至1000毫克活性物质,以及混合有适当和适宜量的载体物质,所述载体其可占总组合物(重量:重量)的约5至约95%。可制备药物组合物以提供给药时容易测量的量。例如,意在用于静脉输注的水溶液每毫升溶液可含有约3至500μg活性成分,从而合适体积的输注以约30毫升/hr的速率出现。
适于肠胃外给药的制剂包括水性和非水性无菌注射溶液剂,其可含有抗氧化剂、缓冲剂、抑菌剂和使得制剂与预期受体的血液等张的溶质;以及水性和非水性无菌混悬剂,其可包括助悬剂和增稠剂。
适于局部给药至眼部的制剂还包括滴眼剂,其中将活性成分溶于或者悬浮于合适的载体(尤其是活性成分的含水溶剂)中。在所述制剂中存在的活性成分的浓度优选为约0.5至20%w/w,例如约0.5至10%w/w,例如约1.5%w/w。
适于在口内局部给药的制剂包括糖锭(lozenge),其含有于矫味基质(通常是蔗糖和阿拉伯胶或者西黄蓍胶)中的活性成分;锭剂(pastille),其含有于惰性基质(如明胶和甘油,或者蔗糖和阿拉伯胶)中的活性成分;以及漱口剂,其包含于液态载体中的活性成分。
适于直肠给药的制剂可呈现为栓剂形式,其具有合适基质(其包含例如可可脂或者水杨酸酯)。
适于肺内或者经鼻给药的制剂具有例如为0.1至500微米的粒度(包括在0.1和500微米之间,增量为例如0.5、1、30微米、35微米等的粒度),其通过鼻道经快速吸入给药或者通过口经吸入给药,以便到达肺泡囊(alveolar sacs)。合适的制剂包括活性成分的水性或者油性溶液剂。适于气雾剂或者干粉给药的制剂可根据常规方法制备,并可与其它治疗药物(如迄今用于治疗或者预防下文所述的病症的化合物)一起递送。
适于阴道给药的制剂可呈现为阴道栓剂、棉塞(tampon)、乳膏剂、凝胶剂、糊剂、泡沫或者喷雾制剂,这些制剂除了活性成分外还含有本领域已知为适当的载体。
制剂可包装在单位剂量或者多剂量容器例如密封安瓿或者小瓶中,并且可在冷冻干燥(冻干)条件下储存,在立即使用前仅需要加入无菌液态载体例如水,用于注射。即时注射溶液剂(Extemporaneous injection solutions and suspension)和混悬剂从前述种类的无菌粉末、颗粒和片剂制备。优选的单位剂量制剂是含有本申请上文所述的日剂量或者单位日亚剂量(sub-dose)或者其适当分数的活性成分的制剂。
本发明还提供了兽用组合物(veterinary composition),由此其含有上文定义的至少一种活性成分以及兽用载体。兽用载体是用于给药所述组合物目的的物质,并可为固态、液态或者气态物质,这些物质在兽医领域要么是惰性的要么是可接受的,并且与活性成分相容。这些兽用组合可经肠胃外、口服或者经任何其它期望的途径给药。
组合疗法
式I的化合物或者其药用盐可与其它化学治疗剂组合用于过度增殖性疾病或者病症的治疗,所述过度增殖性疾病或者病症包括肿瘤、癌症和新生物组织,以及恶化前和非新生物的或者非恶性的过度增殖性病症。在某些实施方案中,式I的化合物或者其药用盐在作为组合疗法的剂量用药方案中与具有抗过度增殖性质或者用于治疗过度增殖性病症的第二种化合物组合。所述剂量用药方案的第二种化合物优选具有与式I的化合物或者其药用盐互补的活性,从而使得它们不能不利地影响彼此。这种化合物可以对预期目的有效的量给药。在一个实施方案中,所述治疗性组合通过剂量用药方案给药,其中所述治疗有效量的式I化合物或者其药用盐在每日两次至每三周一次(q3wk)的范围内给药,并且治疗有效量的化学治疗剂在每日两次至每三周一次的范围内给药。
所述组合疗法可作为同时或者先后用药方案给药。当先后给药时,所述组合可以在两次或者更多次给药中给予。所述组合给药包括使用分开制剂的共同给药,以及以任一次序的连续给药,其中优选有两种(或者所有)活性剂同时发挥它们生物学活性的时间段。
在本发明的一个具体方面中,可在一种或者多种药剂给药开始之后给予式I的化合物或者其药用盐,历时约1至约10天。在本发明的另一个具体方面,可在组合给药开始之前给予式I的化合物或者其药用盐,历时约1至约10天。在本发明的另一个具体方面,式I的化合物或者其药用盐的给药与所述化学治疗剂的给药在同一天开始。
任何上述共同给药药剂的适合剂量为目前所使用的剂量并且可由于新近鉴定的药剂和其它化学治疗剂或者治疗组合作用(协同作用)而降低,如增加治疗指数或者减轻毒性或者其它副作用或者后果。
在抗癌疗法的具体实施方案中,式I的化合物或者其药用盐,可与化学治疗剂组合,以及与手术疗法和放射疗法组合。为了实现期望的组合治疗效应,应选择式I的化合物或者其药用盐和其它一种或者多种药学活性化学治疗剂的量和给药的相对时机。
药物组合物的给药
所述化合物可通过任何适于所要治疗病症的途径给药。适合的途径包括口服途径、肠胃外途径(其包括皮下、肌内、静脉内、动脉内、吸入、皮内、鞘膜内、硬膜外和输注技术)、经皮途径、直肠途径、鼻途径、局部途径(其包括口腔含服和舌下途径)、阴道途径、腹膜内途径、肺内途径和鼻内途径。局部给药还可涉及经皮给药如经皮贴剂或者离子电渗疗法装置的使用。
药物的制剂在Remington′s Pharmaceutical Sciences,18
治疗人类患者的剂量可以为约20mg至约1600mg/天的式I的化合物或者其药用盐。通常的剂量可以为约50mg至约800mg的所述化合物。取决于包括所述具体化合物的吸收、分布、代谢和排泄的药代动力学(PK)和药效学(PD)性质,可每日一次(QD)、每日两次(BID)或者更频繁地给予剂量。除此之外,毒性因素可影响剂量和给药用量方案。当口服给药时,所述丸剂、胶囊剂或者片剂可每日两次、每日一次或者更不频繁地(如对于具体的时间周期,每周一次或者每两周或者三周一次)摄入。所述用药方案可重复多个治疗周期。
治疗方法
(1)式I的化合物或者其药用盐和(2)化学治疗剂的治疗性组合用于治疗疾病、病症和/或障碍,其包括,但不限于,哺乳动物中由AKT激酶所调节的疾病、病症和/或障碍。可根据本发明的方法治疗的癌症包括,但不限于,间皮瘤、子宫内膜癌、乳腺癌、肺癌、卵巢癌、前列腺癌(其包括去雄抗性前列腺癌“CRPC”)、胰腺癌、黑色素瘤、胃癌、结肠癌、神经胶质瘤、头颈部癌。
制品
在本发明的另一个实施方案中,提供了用于治疗上述疾病和病症的含有式I的化合物或者其药用盐的制品或者“试剂盒”。在一个实施方案中,所述试剂盒包含容器和式I的化合物或者其药用盐。
所述试剂盒还可包含附在容器上或者容器中的标签或者包装说明书。术语“包装说明书”用来指通常包括在治疗产品的市售包装中的说明书,其含有关于适应症、用法、剂量、给药、禁忌症和/或者注意事项的信息,这些信息涉及所述治疗产品的使用。合适的容器包括,例如,瓶、小瓶、注射器、发泡包装(blister pack)等。容器可从多种材料(如玻璃或者塑料)形成。容器可装有有效治疗所述病症的式I的化合物或其药用盐或者其制剂,并可具有无菌入口(例如,容器可为静脉注射溶液袋或者具有可由皮下注射针头刺穿的塞子的小瓶)。在组合物中至少一种活性药物是式I的化合物或其药用盐。标签或者包装说明书指示所述组合物用于治疗选择的病症如癌症。在一个实施方案中,标签或者包装说明书指示包含式I的化合物或其药用盐的组合物可用于治疗起因于异常细胞生长的病症。标签或者包装说明书还可指示所述组合物可用于治疗其它病症。可供选择地或者另外地,所述制品还可包含第二种容器,所述容器包含药用缓冲液,如抑菌性注射用水(BWFI)、磷酸盐缓冲生理盐水、林格氏溶液和葡萄糖溶液。试剂盒还可包括从商业和使用者角度看是期望的其它物质,包括其它缓冲液、稀释剂、滤器、针头和注射器。
试剂盒还可包含给药式I的化合物或其药用盐以及第二种药物制剂(如果存在)的说明。例如,若试剂盒包含第一种组合物(含有式I的化合物或其药用盐)和第二种药物制剂,则试剂盒还可包含将第一种和第二种药物组合物同时、先后或者分开给予需要所述制剂的患者的说明。
在另一实施方案中,试剂盒适于递送固态口服形式的式I的化合物或其药用盐,如片剂或者胶囊剂。这样的试剂盒优选包括多个单位剂量。所述试剂盒可以包括针对预期用途为目的的剂量卡片。这样的试剂盒的一个实例是“泡罩包装”。泡罩包装在包装工业中是公知的,并且广泛用于包装药物单位剂量形式。如果期望的话,可提供记忆辅助装置(memory aid),其可呈例如数字、字母或者其它标记形式,或者具有日历插入物,所述记忆辅助装置指定在可对所述剂量进行给药的治疗时间表中的天数。
根据一实施方案,试剂盒可包含(a)在其中含有式I的化合物或其药用盐的第一个容器;以及任选地(b)在其中含有第二种药物制剂的第二个容器,其中所述第二种药物制剂包含具有抗过度增殖活性的第二种化合物。可供选择地或者另外地,所述试剂盒还可包含第三个容器,其包含药用缓冲液,如抑菌性注射用水(BWFI)、磷酸盐缓冲生理盐水、林格氏溶液和葡萄糖溶液。其还可包括从商用和使用者角度来看是期望的其它物质,包括其它缓冲液、稀释剂、滤器、针头和注射器。
在试剂盒包含式I的化合物或其药用盐和第二种治疗药物(即化学治疗剂)的组合物的某些其它实施方案中,所述试剂盒可包含用于容纳分开的组合物的容器,如分开的瓶或者分开的箔包装(foil packet),然而,分开的组合物还可容纳在单一的未分开的容器中。典型地,试剂盒包含给药分开的组分的说明。当分开的组分优选以不同剂量形式(例如口服和肠胃外)给药时,当以不同剂量间隔给药时,或者当对联用的单独组分进行滴定对主治医师是期望之时,试剂盒形式是特别有益的。
本发明的具体方面
在本发明的一个具体方面中,所述过度增殖性病症为癌症。
在本发明的一个具体方面中,所述癌症与PTEN突变有关。
在本发明的一个具体方面中,所述癌症与AKT突变、过度表达或者扩增有关。
在本发明的一个具体方面中,所述癌症与PI3K突变有关。
在本发明的一个具体方面中,所述癌症选自乳腺癌、肺癌、卵巢癌、前列腺癌(例如,去雄抗性的前列腺癌)、黑色素瘤、胃癌、结肠癌、肾癌、头颈部癌和神经胶质瘤。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和5-FU。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐、5-FU和奥沙利铂并且所述癌症为胃癌、卵巢癌或者结肠癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐、5-FU和奥沙利铂并且所述癌症为胃癌、前列腺癌、头部或者颈部癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐、5-FU、奥沙利铂和亚叶酸并且所述癌症为胃癌、卵巢癌或者结肠癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐、5-FU、奥沙利铂和亚叶酸并且所述癌症为胃癌、前列腺癌、头部或者颈部癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和卡铂。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和卡铂并且所述癌症为乳腺癌、肺癌或者前列腺癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和卡铂并且所述癌症为乳腺癌、肺癌、前列腺癌、头部或者颈部癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和伊立替康。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和伊立替康并且所述癌症为结肠癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和多西他赛。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和多西他赛并且所述癌症为乳腺癌、神经胶质瘤、肺癌、黑色素瘤、卵巢癌或者前列腺癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和多西他赛并且所述癌症为乳腺癌、卵巢癌或者前列腺癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和多柔比星。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和多柔比星并且所述癌症为乳腺癌、肺癌、卵巢癌、神经胶质瘤或者前列腺癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和SN-38。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和SN-38并且所述癌症为结肠癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和替莫唑胺。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和替莫唑胺并且所述癌症为神经胶质瘤。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和铂剂。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和铂剂并且所述癌症为卵巢癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和GDC-0973或者其药用盐。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和GDC-0973或者其药用盐并且所述癌症为胰腺癌、前列腺癌、黑色素瘤或者乳腺癌。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和PLX-4032或者其药用盐。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和PLX-4032或者其药用盐并且所述癌症为黑色素瘤。
在本发明的一个具体方面中,对所述哺乳动物给予式I的化合物或者其药用盐和阿比特龙或者其药用盐并且所述癌症为前列腺癌。在一个实例中,所述组合还包含给予泼尼松。
在本发明的一个具体方面中,对所述哺乳动物给予GDC-0068或者其药用盐和阿比特龙或者其药用盐并且所述癌症为前列腺癌。在一个实例中,所述组合还包含给予泼尼松龙或者泼尼松。
在本发明的一个具体方面中,可口服给予式I的化合物或者其药用盐。
在本发明的一个具体方面中,将式I的化合物或者其药用盐配制成片剂。
通用的制备操作
实施例
为了示例说明本发明,包括下列实施例。然而,应当理解的是,这些实施例不限制本发明并且仅仅意在提出实施本发明的方法。本领域的技术人员应当认识到所述化学反应可容易地适于制备多种本发明的其它AKT抑制剂,并且认为制备本发明化合物的可选方法在本发明的范围之内。例如,根据本发明合成非示例性化合物可通过对本领域技术人员显而易见的修改来顺利进行,所述修改例如,通过适当保护干扰基团,通过采用不同于所述试剂的本领域已知的其它适合试剂和/或通过进行反应条件的路线修改。可选择地,应当认为本申请所公开的或者本领域已知的其它反应具有制备本发明其它化合物的适用性。
实施例1
步骤3:向(2R)-2-甲基-5-氧代环戊羧酸乙酯(20g,117.5mmol)和硫脲(9.2g,120.9mmol)的混合物在乙醇(100mL)中的溶液加入KOH(8.3g,147.9mmol)在水(60mL)中的溶液。将混合物回流10小时。冷却后,除去溶剂并在0℃将残余物用浓HCl(12mL)中和,然后用DCM(3x150mL)萃取。除去溶剂并通过硅胶色谱(用己烷/乙酸乙酯(2∶1)洗脱)将残余物纯化,得到(R)-2-巯基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-醇(12g,56%).MS(APCI+)[M+H]+183.
步骤4:(R)-2-巯基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-醇(12g,65.8mmol)在蒸馏水(100mL)中的混悬液加入兰尼镍(15g)和NH
步骤5:将(R)-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-醇(5.8g,38.62mmol)在POCl3(20mL)中的混合物回流5分钟。真空除去过量的POCl3并将残余物溶于DCM(50mL)中。然后将混合物加入饱和的NaHCO3(200mL)中。将水相用DCM(3x100mL)萃取,并将合并的有机相干燥并浓缩。通过硅胶色谱(用乙酸乙酯洗脱)将残余物纯化,得到(R)-4-氯-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶(3.18g,49%)。1H NMR(CDCl3,400MHz)δ8.81(s,1H),3.47(m,1H),3.20(m,1H),3.05(m,1H),2.41(m,1H),1.86(m,3H),1.47(m,3H).
步骤6:以三份向(R)-4-氯-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶(2.5g,14.8mmol)在CHCl3(60mL)中的溶液加入MCPBA(8.30g,37.0mmol)。将混合物在室温搅拌2天。将混合物冷却至0℃并向其中先后滴加Na2S2O3(10g)在水(60mL)中的溶液、Na2CO3(6g)在水(20mL)中的溶液。将反应混合物搅拌20分钟。将水相用CHCl3(2x200mL)萃取,并将合并的有机相在低温(<25℃)浓缩。通过硅胶色谱(用乙酸乙酯-DCM/MeOH(20∶1)洗脱)将残余物纯化,得到(R)-4-氯-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-氧化物(1.45g,53%)。1H NMR(CDCl3,400MHz)δ8.66(s,1H),3.50(m,1H),3.20(m,2H),2.44(m,1H),1.90(m,1H),1.37(d,J=7.2Hz,3H).
步骤7:将(R)-4-氯-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-氧化物(1.45g,7.85mmol)在乙酸酐(20mL)中的溶液加热至110℃持续2小时。冷却后,真空除去过量的溶剂。通过硅胶色谱(用己烷/乙酸乙酯(3∶1)洗脱)将残余物纯化,得到乙酸(5R)-4-氯-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-7-基酯(1.25g,70%)。1H NMR(CDCl3,400MHz)δ8.92(m,1H),6.30-6.03(m,1H),3.60-3.30(m,1H),2.84(m,1H),2.40-2.20(m,1H),2.15(d,J=6Hz,2H),1.75(m,2H),1.47(d,J=6.8,2H),1.38(d,J=7.2,1H).MS(APCI+)[M+H]+227.
步骤8:向乙酸(5R)-4-氯-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-7-基酯(0.5g,2.2mmol)在NMP(10mL)中的溶液加入1-Boc-哌嗪(0.9g,4.8mmol)。将反应混合物加热至110℃持续12小时。冷却后,将反应混合物用乙酸乙酯(200mL)稀释并用水(6x100mL)洗涤。将有机相干燥并浓缩。通过硅胶色谱(用乙酸乙酯洗脱)将残余物纯化,得到4-((5R)-7-乙酰氧基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-羧酸叔丁酯(0.6g,72%)。1H NMR(CDCl3,400MHz)δ8.60(d,1H),6.05-5.90(m,1H),3.80-3.30(m,9H),2.84(m,1H),2.20-(m,1H),1.49(s,9H),1.29-1.20(m,3H).MS(APCI+)[M+H]+377.通过手性分离HPLC(Chiralcel ODH柱,250x20mm,己烷/EtOH 60:40,21mL/分钟)将所得非对映异构体的混合物纯化。第一个峰(RT=3.73分钟)为4-((5R,7R)-7-乙酰氧基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-羧酸叔丁酯(0.144g,24%)。第二个峰(RT=5.66分钟)为4-((5R,7S)-7-乙酰氧基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-羧酸叔丁酯(0.172g,29%)。MS(APCI+)[M+H]+377.
步骤9:向4-((5R,7R)-7-乙酰氧基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-羧酸叔丁酯(0.144g,0.383mmol)在THF(4mL)中的溶液加入LiOH(3M,2mL)。将混合物在室温搅拌6小时,然后用2N HCl(3mL)淬灭。除去溶剂并通过硅胶色谱(用乙酸乙酯洗脱)将残余物纯化,得到4-((5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-羧酸叔丁酯(89mg,70%)。1H NMR(CDCl3,400MHz)δ8.52(s,1H),5.48(br,1H),5.14(m,1H),3.82-3.40(m,9H),2.20(m,2H),1.49(s,9H),1.19(d,J=6.8Hz,3H).MS(APCI+)[M+H]+335.
步骤10:在DCM(5mL)中将4-((5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-羧酸叔丁酯用HCl(4M的二噁烷溶液,2mL)处理6小时,得到(5R,7R)-5-甲基-4-(哌嗪-1-基)-6,7-二氢-5H-环戊二烯并[d]嘧啶-7-醇二盐酸盐。MS(APCI+)[M+H]+235.
步骤11:将2,4-二甲氧基苄基氨基甲酸叔丁酯(3.96g,14.8mmol)溶于THF(74mL)并冷却至-78℃。历时五分钟滴加丁基锂(7.44mL,16.3mmol)处理该溶液,得到淡黄色溶液。将溶液搅拌15分钟后滴加氯(甲氧基)甲烷(1.35mL,17.8mmol)(纯态的)。将反应混合物在-78℃搅拌10分钟,然后慢慢温热至环境温度过夜。将反应混合物真空浓缩,得到黄色凝胶状物,将其在半饱和NH4Cl溶液和乙醚之间分配。萃取水相一次,并合并有机相。将有机层用先后用水、盐水洗涤,分离,以Na2SO4干燥,过滤并真空浓缩。1H NMR证实了期望的接近纯的(>90%)2,4-二甲氧基苄基(甲氧基甲基)氨基甲酸叔丁酯(4.81g,104%产率),为淡黄色油状物,其无需纯化即可使用。
步骤12:将(R)-4-苄基-3-(2-(4-氯苯基)乙酰基)噁唑烷-2-酮(3.00g,9.10mmol)溶于DCM(91mL)并冷却至-78℃。先后向溶液中加入TiCl4的1M甲苯溶液(11.4mL,11.4mmol)、DIEA(1.66mL,9.55mmol),得到暗紫色反应混合物。将该反应混合物搅拌15分钟之后滴加2,4-二甲氧基苄基(甲氧基甲基)氨基甲酸叔丁酯(3.40g,10.9mmol)在DCM(10mL)中的溶液。将反应混合物在-78℃搅拌15分钟,然后在盐水-冰浴中将其温热至-18℃持续一小时。历时2.5小时将该反应混合物慢慢温热至0℃。然后加入饱和的NH4Cl溶液(100mL)将反应混合物淬灭。分离各相,并将有机层用DCM萃取一次。将合并的有机层以MgSO4干燥,过滤并真空浓缩,得到黄色油状物。通过色谱(用4∶1己烷∶乙酸乙酯洗脱的硅胶)将残余物纯化,得到纯物质,为无色油状物2,4-二甲氧基苄基((S)-3-((R)-4-苄基-2-氧代噁唑烷-3-基)-2-(4-氯苯基)-3-氧代丙基)氨基甲酸叔丁酯(4.07g,73.5%产率)。在环境温度将所述2,4-二甲氧基苄基((S)-3-((R)-4-苄基-2-氧代噁唑烷-3-基)-2-(4-氯苯基)-3-氧代丙基)氨基甲酸叔丁酯(680mg,1.12mmol)溶于DCM(10.6mL)和水(560uL;19∶1DCM∶水)。用DDQ(380mg,1.67mmol)处理该溶液,并将反应混合物搅拌一天,通过TLC和LCMS分析得到反应完成。将反应混合物用DCM稀释并用半饱和的NaHCO3溶液洗涤。将有机层以MgSO4干燥,过滤并真空浓缩,得到黄橙色油状物。通过色谱(用9∶1己烷∶乙酸乙酯洗脱的硅胶)将残余物纯化,得到醛副产物和(S)-3-((R)-4-苄基-2-氧代噁唑烷-3-基)-2-(4-氯苯基)-3-氧代丙基氨基甲酸叔丁酯的混合物(不可分离的),为淡黄色油状物(729mg合并重量)。LC/MS(APCI+)m/z 359.1[M-BOC+H]+.
步骤13:向LiOH-H2O(0.0978g,2.33mmol)在2∶1 THF∶H2O(33mL)中的溶液加入35%H2O2(0.240mL,2.91mmol)。将反应混合物在室温搅拌35分钟,然后冷却至0℃。通过滴液漏斗滴加含有(S)-3-((R)-4-苄基-2-氧代噁唑烷-3-基)-2-(4-氯苯基)-3-氧代丙基氨基甲酸叔丁酯(0.535g,1.17mmol)和2,4-二甲氧基苯甲醛(0.194g,1.17mmol)的混合物在THF(7mL)中的溶液。将冰浴慢慢温热,并将反应混合物搅拌过夜。然后将反应混合物冷却至0℃,加入1M Na2SO3(7mL)。将混合物搅拌5分钟,然后温热至室温并搅拌另外20分钟。然后将反应混合物转移至分液漏斗并用乙醚(3X)洗涤。用KHSO4(固体)酸化水层,并用DCM(2X)萃取该混合物。将合并的萃取物干燥(Na2SO4),过滤并浓缩,得到(S)-3-(叔丁氧基羰基氨基)-2-(4-氯苯基)丙酸(0.329g,94.2%产率),为白色残余物。LC/MS(APCI+)m/z 200[M-BOC+H]+.
步骤14:向(S)-3-(叔丁氧基羰基氨基)-2-(4-氯苯基)丙酸(0.329g,1.10mmol)在2∶1二噁烷∶DCM(10mL)中的溶液加入4M HCl/二噁烷(5.49ml,22.0mmol)。将反应混合物在室温搅拌过夜(16小时),此后将其浓缩至1/3体积。将所得混浊的混合物用乙醚稀释,并将混合物再浓缩至1/3体积。将混合物再用乙醚(20mL)稀释,并通过具有氮气压的中号玻璃料漏斗(medium frit funnel)过滤将固体分离,用乙醚(5X10mL)冲洗,在氮气压干燥,并真空干燥,得到(S)-3-氨基-2-(4-氯苯基)丙酸盐酸盐(0.199g,76.8%产率),为白色固体。HPLC>99面积%纯。LC/MS(APCI+)m/z 200.
步骤15:向(S)-3-氨基-2-(4-氯苯基)丙酸盐酸盐(0.199g,0.843mmol)和四甲基氢氧化铵五水合物(0.382g,2.11mmol)在10∶1MeCN∶H2O(7.7mL)中的溶液加入Boc2O(0.368g,1.69mmol)。将反应混合物在室温搅拌过夜(12小时),此后在旋转蒸发仪上除去MeCN。将混合物用水稀释并用乙醚(2X)洗涤。用KHSO4(固体)酸化水层,并用DCM萃取该混合物,并将合并的萃取物干燥(Na2SO4),过滤并浓缩,得到(S)-3-(叔丁氧基羰基氨基)-2-(4-氯苯基)丙酸(0.229g,90.6%产率),为泡沫状物。HPLC>99面积%纯。LC/MS(APCI+)m/z200[M-BOC+H]+.
步骤16:向(5R,7R)-5-甲基-4-(哌嗪-1-基)-6,7-二氢-5H-环戊二烯并[d]嘧啶-7-醇二盐酸盐(88mg,0.29mmol)和(S)-3-(叔丁氧基羰基氨基)-2-(4-氯苯基)丙酸(86mg,0.29mmol)在DCM(10mL)和二异丙基乙基胺(0.22mL,1.3mmol)的溶液中加入HBTU(110mg,0.29mmol)。将反应混合物在室温搅拌1小时。除去溶剂并将残余物溶于乙酸乙酯(100mL),用水(6x50ml)洗涤。将有机相干燥并浓缩,得到(S)-2-(4-氯苯基)-3-(4-((5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-基)-3-氧代丙基氨基甲酸叔丁酯(116mg,78%).1H NMR(CDCl3,400MHz)δ8.51(s,1H),7.34-7.20(m,4H),5.15-5.09(m,2H),4.15-4.05(m,1H),3.87-3.85(m,2H),3.78-3.38(m,7H),3.22-3.19(m,1H),2.20-2.10(m,2H),1.48(s,9H),1.41(s,9H),1.14-1.12(d,J=7.2Hz,3H).MS(APCI+)[M+H]+516.
步骤17:在DCM(5mL)中将(S)-2-(4-氯苯基)-3-(4-((5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-基)-3-氧代丙基氨基甲酸叔丁酯用HCl(4M的二噁烷溶液,2mL)处理6小时,得到(S)-3-氨基-2-(4-氯苯基)-1-(4-((5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-基)丙-1-酮二盐酸盐。1H NMR(D2O,400MHz)δ8.38(s,1H),7.37-7.35(d,J=8.4Hz,2H),7.23-7.21(d,J=8.4Hz,2H),5.29-5.25(m,1H),4.64(s,9H),4.31-4.28(m,1H),4.11(m,1H),3.88-3.79(m,2H),3.70-3.20(m,10H),2.23-2.17(m,1H),2.07-1.99(m,1H),1.22-1.20(m,2H),0.98-0.96(d,J=6.8Hz,2H).MS(APCI+)[M+H]+416.
实施例2
步骤16:在室温将3-(叔丁氧基羰基(异丙基)氨基)-2-(4-氯苯基)丙酸钾(77.2g,203mmol)溶于THF(515mL)并用特戊酰氯(26.3mL,213mmol)处理。将混合物搅拌3小时,形成混合物酸酐。在分开的烧瓶中将(S)-4-苄基噁唑烷-2-酮(46.1g,260mmol)溶于THF(600mL)并冷却至-78℃。将溶液用n-BuLi(102mL的2.50M己烷溶液,254mmol)处理并搅拌一小时。经套管向搅拌的Li-噁唑烷酮中加入制备的酸酐溶液,并将混合物温热至室温过夜。加入饱和的氯化铵溶液将混合物淬灭,然后在更多的水和乙酸乙酯之间分配。萃取水相若干次,并将有机物合并。将有机层先后用水、盐水洗涤,分离,以MgSO
表1中所示实施例3-9也可根据上述方法制得。
表1
实施例10
步骤1:将环丙基溴化镁(64.0mL,32.00mmol)的THF溶液用氯化锌(II)(64.00mL,32.00mmol)在THF中的溶液处理。将混合物在室温搅拌20分钟。加入2-(4-溴苯基)乙腈(5.228g,26.67mmol)和二[三叔丁基膦]钯(0.6814g,1.333mmol)在THF(2mL)中的溶液。氮气下将反应混合物在环境温度搅拌12小时。用饱和的NH4Cl淬灭反应混合物,用二氯甲烷稀释并分离。用二氯甲烷(2X)洗涤水层,然后用水(3X)洗涤合并的有机层,以Na2SO4干燥并真空浓缩。将粗产物经SiO2色谱处理(用25∶1己烷/乙酸乙酯洗脱),得到2-(4-环丙基苯基)乙腈(2.76g,66%)。1H NMR(CDCl3,400MHz)7.20(d,J=8.2,2H),7.07(d,J=8.2,2H),3.70(s,2H),1.94-1.85(m,1H),1.01-0.95(m,2H),0.71-0.66(m,2H).
步骤2:将甲醇(65mL)冷却至0℃并用HCl(气体)饱和。用2-(4-环丙基苯基)乙腈(2.76g,17.56mmol)在甲醇(6mL)中的溶液处理该溶液。在含有CaSO4的干燥管下将反应混合物加热至回流过夜。将反应混合物冷却并真空浓缩。将粗混合物再混悬于乙酸乙酯和水,然后分离。将有机层用饱和的NaHCO3、饱和的NaCl洗涤,以Na2SO4干燥并真空浓缩,得到2-(4-环丙基苯基)乙酸甲酯,为油状物(3.10g,93%)。1H NMR(CDCl3,400MHz)δ7.16(d,J=8.3,2H),7.02(d,2H),3.68(s,3H),3.58(s,2H),1.92-1.83(m,1H),0.97-0.91(m,2H),0.70-0.64(m,2H).
步骤3:将2-(4-环丙基苯基)乙酸甲酯(3.10g,16.30mmol)溶于THF/MeOH/水的混合物(2∶2∶1,80mL)中,并用氢氧化锂水合物(0.8548g,20.37mmol)处理该溶液。然后将混合物在环境温度搅拌4小时。用3N HCl将反应混合物中和至pH为4并真空浓缩。将该固体再溶于乙酸乙酯和水。用3N HCl将pH再调节到约3至约4的pH。然后分离各层。用乙酸乙酯(2X)洗涤水层。然后将合并的有机层用饱和的NaCl洗涤,以Na2SO4干燥并浓缩,得到2-(4-环丙基苯基)乙酸(2.82g,98%)。1H NMR(CDCl3,400MHz)7.16(d,J=8.2,2H),7.03(d,2H),3.60(s,2H),1.92-1.83(m,1H),098-0.91(m,2H),0.70-0.64(m,2H).
步骤4:在甲苯(14mL)中将2-(4-环丙基苯基)乙酸(2.82g,16.003mmol)与(R)-4-苄基噁唑烷-2-酮(3.4030g,19.204mmol)合并。将该混悬液用三乙胺(6.6917mL,48.010mmol)处理,然后加热至80℃。滴加特戊酰氯(1.9893mL,16.003mmol)在甲苯(3.5mL)中的溶液处理该溶液。将反应混合物在80℃加热过夜。将反应混合物冷却并用2N HCl洗涤,然后分离。用甲苯洗涤水层,然后用2N HCl、水、饱和的NaHCO3(2X)、饱和的NaCl洗涤合并的有机物,以Na2SO4干燥并真空浓缩。将粗产物经SiO2色谱处理(用9∶1己烷/乙酸乙酯洗脱),得到(R)-4-苄基-3-(2-(4-环丙基苯基)乙酰基)噁唑烷-2-酮(3.43g,64%)。1H NMR(CDCl3,400MHz)7.33-7.20(m,5H),7.16-7.11(m,2H),7.05(d,J=8.2,2H),4.70-4.63(m,1H),4.32-4.14(m,4H),3.26(dd,J1=3.2,J2=13.3,1H),2.75(dd,J1=9.5,J2=13.3,1H),1.93-1.85(m,1H),0.98-0.92(m,2H),0.72-0.66(m,2H).
步骤5:根据实施例1中所述操作,使用(R)-4-苄基-3-(2-(4-环丙基苯基)乙酰基)噁唑烷-2-酮,制备(S)-2-((S)-1-(叔丁氧基羰基)吡咯烷-2-基)-2-(4-环丙基苯基)乙酸(0.287g,26%)。MS(ESI+)[M+H]345.7.
步骤6:根据实施例3中所述操作,使用(S)-2-((S)-1-(叔丁氧基羰基)吡咯烷-2-基)-2-(4-环丙基苯基)乙酸,制备(S)-2-((S)-1-(4-环丙基苯基)-2-(4-((5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-基)-2-氧代乙基)吡咯烷-1-羧酸叔丁酯(0.199g,94%)。MS(ESI+)[M+H]562.1.
步骤7:根据实施例3中所述操作,使用(S)-2-((S)-1-(4-环丙基苯基)-2-(4-((5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-基)-2-氧代乙基)吡咯烷-1-羧酸叔丁酯,制备(S)-2-(4-环丙基苯基)-1-(4-((5R,7R)-7-羟基-5-甲基-6,7-二氢-5H-环戊二烯并[d]嘧啶-4-基)哌嗪-1-基)-2-((S)-吡咯烷-2-基)乙酮(0.145g,77%)。MS(ESI+)[M+H]462.2.1H NMR(CD3OD,400MHz)8.56(s,1H),7.26(d,2H),7.13(d,2H),5.29(dd,1H),5.32-5.26(dd,1H),4.32(d,1H),4.29-4.18(m,1H),4.12-3.95(m,2H),3.88-3.61(m,6H),3.51-3.38(m,1H),3.35-3.30(m,1H),2.32-2.24(m,1H),2.22-2.03(m,2H),1.95-1.85(m,2H),1.82-1.73(m,2H),1.40-1.34(m,1H),1.16(d,3H),1.01-0.95(m,2H),0.69-0.64(m,2H).
表2中所示实施例也可根据上述方法制得。
表2
实施例14体外细胞增殖测定
实施例2的化合物与某些具体化学治疗剂的组合的体外效能可使用
所述均质″加入-混合-测量(add-mix-measure)″模式导致细胞溶解并产生与存在的ATP的量成比例的荧光信号。ATP的量与在培养物中存在的细胞数目直接成比例。所述
使用
实施例15体内肿瘤异种移植物效力
本发明的代表性组合的效能通过将癌细胞的同种异体移植物或者异种移植物植入啮齿动物中并将荷瘤动物用所述组合处理来进行体内测量。可变结果可取决于细胞系、在肿瘤细胞中存在或者不存在某些突变、给予实施例2化合物和化学治疗剂的顺序、给药方案以及其它因素而为预期的。将受试小鼠用药物或者对照(媒介物)处理并历时若干周或者更长时间监测以测量肿瘤体积倍增的时间、细胞死亡对数值和肿瘤抑制。
在该模型中测试的本发明代表性组合的效果呈现在图1-2中。
图中的数据证明,与各药剂单独给药相比,代表性组合提供了改善的效果。
已经确定的是,本发明的某些组合提供针对某些癌症表型的改善的效应。例如,本发明的某些组合提供针对与PTEN突变、AKT突变(例如过度表达或者扩增水平)、PI3K突变或者Her2/ErbB2扩增有关的癌症的改善的效应。相应地,本申请所述的某些组合针对这些类型的癌症特别有用。例如,在胃癌中,PTEN-丧失预示着本发明的某些组合(例如,式I的化合物与5-FU/顺铂)具有更好的效力,并且在前列腺癌PTEN-无效细胞系中可看到式I的化合物和多西他赛的组合具有更强的效应。
可通过本领域已知的任何适合的方法测量PTEN状态。在一个实例中,使用IHC。可选择地,可使用蛋白质印迹分析。针对PTEN的抗体是可商购的(Cell SignalingTechnology,Beverly,MA,Cascade Biosciences,Winchester,MA)。PTEN状态的IHC和蛋白质印记分析的实例操作在Neshat,M.S.et al.Enhanced sensitivity of PTEN-deficienttumors to inhibition of FRAP/mTOR,Proc.Natl cad.Sci.USA 98,10314-10319(2001)和Perren,A.,et.al.Immunohistochemical Evidence of Loss of PTEN Expression inPrimary Ductal Adenocarcinomas of the Breast,American Journal of Pathology,Vol.155,No.4,October 1999中有描述。另外,可使用本领域已知的技术鉴定与AKT突变、PI3K突变以及与Her2/ErbB2扩增或者突变有关的癌症。在一个实例中,使用IHC确定患者或者组织样本的PTEN状态,并且对所述样本或者患者进行组织学评分(histo score)或者组织学评分(HScore)。使用公式计算HScore实例方法为:HScore=(%1+细胞x 1)+(%2+细胞x 2)+(%3+细胞x 3)(见Shoman,N,et.al,Mod Path(2005)18,250-259)。来自相同患者或者一批患者的非癌性组织的平均PTEN HScore可用于确定患者或者样本HScore是否低或者无效。在一个实例中,认为小于约200的HScore是低的并对应于PTEN低,而认为约0的HScore是无效的。
一个方面包括在患有癌症的患者中肿瘤生长抑制(TGI)的方法,所述癌症包含PTEN突变、AKT突变(例如,过度表达或者扩增)、PI3K突变或者Her2/ErbB2扩增或者突变,所述方法包括对所述患者给予GDC-0068或者其药用盐和阿比特龙或者其药用盐。在某些实施方案中,所述组合是协同的。在某些实施方案中,所述组合的TGI高于GDC-0068或者所述化学治疗剂任一单独的TGI。在某些实施方案中,所述组合的TGI比GDC-0068或者化学治疗剂任一单独的TGI高约10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%或者75%。
测量TGI的方法是本领域已知的。在一个实例方法中,测定平均肿瘤体积并在治疗之前和之后比较来自患者的平均肿瘤体积。可使用本领域中的任何方法以两个尺寸(长度和宽度)测量肿瘤体积,所述方法例如UltraCal IV calipers(Fred V.Fowler Company)或者通过PET(正电子断层扫描),或者通过其它一些方法。可使用公式肿瘤体积(mm
使用该公式,100%的TGI值指示肿瘤停滞,大于约1%但小于约100%指示肿瘤生长抑制,而大于约100%指示肿瘤消退。
在某些实施方案中,所述癌症包含一种或者多种AKT、PI3k、PTEN和HER2突变或者AKT、PI3k、PTEN或者HER2异常信号传导。在一个实例中,所述癌症为含有高pAKT活性和PTEN低的或者无效状态的胃癌。
在一个具体方面,本发明提供了治疗患有癌症的患者的方法,所述癌症与PTEN突变或者表达丧失、AKT突变或者扩增、PI3K突变或者扩增或者Her2/ErbB2扩增有关,所述方法包括对所述患者给予本发明的组合。在另一个方面中,本发明提供了鉴定患有癌症的患者的方法,所述癌症可用本发明的组合治疗,所述方法包括确定患者的癌症是否与PTEN突变或者表达丧失、AKT突变或者扩增、PI3K突变或者扩增或者Her2/ErbB2扩增有关,其中患者的癌症与PTEN突变或者表达丧失、AKT突变或者扩增、PI3K突变或者扩增或者Her2/ErbB2扩增的关联是可用本发明的化合物治疗的癌症的征兆。在再一个方面中,本发明提供了还包括用本发明的组合治疗所鉴定的患者的方法。
在另一个实例中,所要治疗的癌症与PTEN阳性、低或无效状态与HER2阳性或者阴性状态的组合有关。实例包括胃癌,其为(i)PTEN阴性的(HScore小于约10或者0)且Her2阴性的,(ii)PTEN低(HScore小于约200)且Her2阴性的,(iii)PTEN阴性的且Her2阳性的,或者(iv)PTEN阳性的且Her2阴性的。在该实例中,可将所述癌症用式I的化合物(例如GDC-0068或者其盐)和阿比特龙或者其药用盐治疗。
此外,因为对于本领域的技术人员而言,多种修改和变化应当是显而易见的,所以不期望将本发明限制于上述确切的组成和方法。相应地,可认为所有适合的修改和等价形式都落入由下述的权利要求所定义的本发明的范围之内。
- AKT抑制剂化合物和阿比特龙的组合及使用方法
- AKT抑制剂化合物和阿比特龙的组合及使用方法