全基因组致病SNP精细定位的因果关联分析方法
文献发布时间:2023-06-19 13:45:04
技术领域
本发明涉及生物基因工程技术领域,特别涉及一种全基因组致病SNP精细定位的因果关联分析方法。
背景技术
本部分的陈述仅仅是提供了与本发明相关的背景技术,并不必然构成现有技术。
自全基因组关联分析(Genome Wide Association Study,GWAS)方法被提出至今,已发现遍布全基因组14多万个SNP与4000多种常见疾病存在统计学关联。然而,很少的SNP得到实验室功能验证,难以阐明其遗传机制。如此高的假阳性率为后续验证带来困难的同时,也会导致非遗传学家对GWAS结果的怀疑。如何进一步精细定位真正的致病SNP,降低假阳性率,是当今研究者广泛探讨的问题。
发明人发现,现已有很多研究者从多角度提出各种精细定位的算法,可大致分为以下四类:
(1)启发式精细定位法,常见分析思路为通过广义线性回归模型和广义线性混合模型筛选边际相关的SNP后,根据top SNP与周围LD的结构,筛选R2大于某一阈值的SNP。但当区域内SNP间高度相关时,这类方法无法有效降低假阳性率。
(2)以条件回归分析方法为代表的条件回归法,给定top SNP后,判断其余SNP的条件P值是否仍有意义,很多时候区域内的top SNP并不一定就是真的致病位点,所以当两个SNP高度相关时,将影响模型的判断结果。
(3)以LASSO为代表的惩罚回归模型,通过判断回归系数是否为0进行变量筛选。这种策略下构建的稀疏模型,在SNP高度相关的区域上,仅保留少数几个SNP,容易误删真正的致病SNP。
(4)以贝叶斯变量选择模型为代表的贝叶斯模型,通过计算SNP为致病位点的后验概率进行精细定位,这类方法常需要预先设定致病位点个数,错误设定将影响分析结果。
发明内容
为了解决现有技术的不足,本发明提供了一种全基因组致病SNP精细定位的因果关联分析方法,在因果推断框架的指导下,构建了面向全基因组致病位点精细定位的因果GWAS分析策略与方法(Causal Diagram-based Stepwise Fine-Mapping,CDSFM算法),降低GWAS结果的假阳性率,提高捕获致病SNP的命中率。
为了实现上述目的,本发明采用如下技术方案:
本发明第一方面提供了一种全基因组致病SNP精细定位的因果关联分析方法。
全基因组致病SNP精细定位的因果关联分析方法,包括以下过程:
获取待分析的基因组数据;
使用单因素回归模型对基因组数据进行全基因组关联分析,筛选P值低于预设阈值的显著SNP,定义显著的SNP为第一备选基因集,将第一备选基因集中的SNP按照P值从小到大进行排序;
固定第一备选基因集中P值最小的SNP
如若被删除的SNP为SNP
若第二备选基因子集中SNP的个数小于或等于2,计算结束,所得第二备选基因子集中的所有SNP为筛选的致病位点;否则,继续计算,直至第m备选基因子集中SNP的个数小于或等于m+1,则运算停止。
该过程中SNP的剔除条件为,若两个SNP之间的LD等于1,则这两个SNP都保留在第一备选基因子集中,或分析两个条件P值,若其中一个P值为缺失值,则将两个SNP都保留在第一备选基因子集中;
若两个P值都不缺失,将P值与定义的预设阈值作比较,如果两个P值都大于或都小于预设阈值,两个SNP都保留在第一备选基因子集中;若两个P值中一个大于预设阈值,一个小于预设阈值,则将P值大于预设阈值的SNP从第一备选基因子集中剔除。
进一步的,采用m元回归分析进行SNP的条件独立性计算获取第m备选基因子集的过程。
使用m元回归模型对备选基因集S
此过程中对应的剔除条件为:如果所得的m个条件P值均大于或小于0.05,则m个SNP均保留在S
单因素或多因素回归模型为线性回归或logistic回归模型。
条件独立检验可能的因果关系时,所依据的统计学原理包括:
考虑LD结构和条件P值,真正的致病位点不会因虚假关联的位点而与结局条件独立;
给定真正的致病位点后,虚假关联的位点与结局条件独立;
当两个致病位点存在强LD时,可能会同时条件独立于结局;
当没有真正的致病SNP时,与致病SNP间LD较大的SNP更容易被保留。
本发明第二方面提供了一种全基因组致病SNP精细定位的因果关联分析系统。
全基因组致病SNP精细定位的因果关联分析系统,包括:
数据获取模块,被配置为:获取待分析的基因组数据;
因果GWAS模块,被配置为:
使用单因素回归模型对基因组数据进行全基因组关联分析,筛选P值低于预设阈值的显著SNP,定义被选择的SNP为第一备选基因集,将第一备选基因集中的SNP按照P值从小到大进行排序;
固定第一备选基因集中P值最小的SNP
如若被删除的SNP为SNP
若第二备选基因子集中SNP的个数小于或等于2,计算结束,所得第二备选基因子集中的所有SNP为筛选的致病位点;否则,继续计算,直至第m备选基因子集中SNP的个数小于或等于m+1,则运算停止。
本发明第三方面提供了一种计算机可读存储介质,其上存储有程序,该程序被处理器执行时实现如本发明第一方面所述的全基因组致病SNP精细定位的因果关联分析方法中的步骤。
本发明第四方面提供了一种电子设备,包括存储器、处理器及存储在存储器上并可在处理器上运行的程序,所述处理器执行所述程序时实现如本发明第一方面所述的全基因组致病SNP精细定位的因果关联分析方法中的步骤。
与现有技术相比,本发明的有益效果是:
1、本发明所述的方法、系统、介质或电子设备,在因果推断框架的指导下,构建面向全基因组的致病位点精细定位的因果GWAS分析策略(即CDSFM算法),在脱离具体的因果图模型的束缚下,通过逐步条件独立调整策略,有效地降低了假阳性率(FalseDiscoveryRate,FDR),提升了真阳性率(True Discovery Rate,TDR),将捕获致病SNP的命中率提高到90%以上,且具有较高的检验效能。
2、本发明所述的方法、系统、介质或电子设备,突破了统计关联的局限性,整体表现明显优于广义线性回归模型、LASSO回归模型、GCTA模型及贝叶斯变量选择回归模型等现有的精细定位方法,为全基因组遗传易感位点精细定位提供了新策略和新方法。
附图说明
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
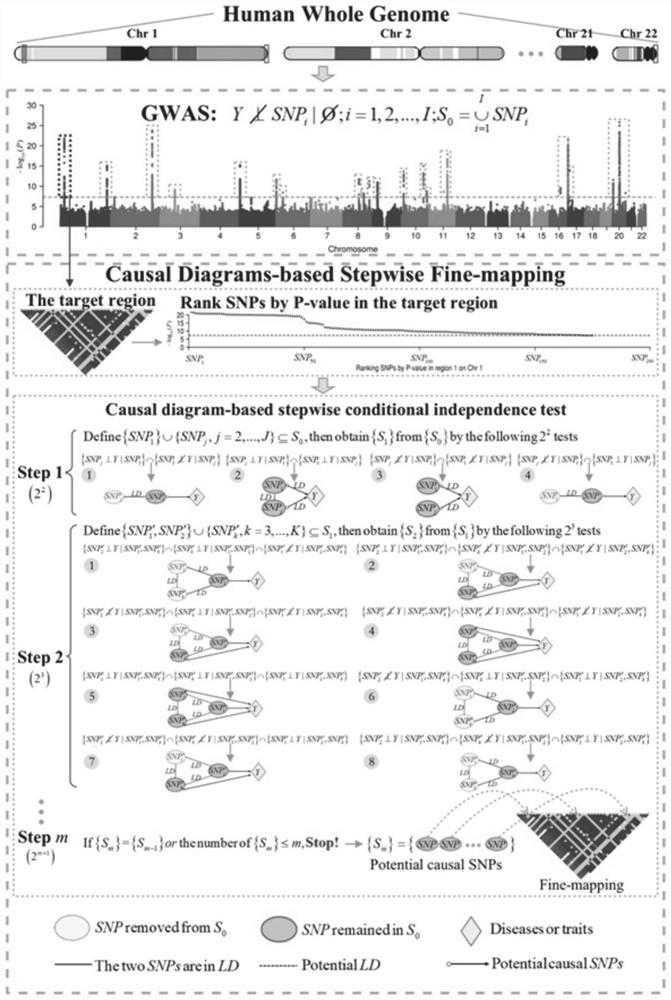
图1为本发明实施例提供的CDSFM算法的统计学原理图。
图2为本发明实施例1提供的CDSFM算法示例图。
图3为本发明实施例2提供的面向全基因组易感位点精细定位的因果GWAS方法框架示意图。
图4为本发明实施例2提供的结局为数量性状时CDSFM算法与其他精细定位算法比较模拟结果示意图。
图5为本发明实施例2提供的结局为质量性状时CDSFM算法与其他精细定位算法比较模拟结果示意图。
具体实施方式
下面结合附图与实施例对本发明作进一步说明。
应该指出,以下详细说明都是例示性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
本发明构建了精细定位算法CDSFM,该算法同时考虑LD结构和条件P值,基于的统计原理为真正的致病位点不会因虚假关联的位点而与结局条件独立;给定真正的致病位点后,虚假关联的位点与结局条件独立;当两个致病位点存在强LD时,可能会同时条件独立于结局;当模型中没有真致病SNP时,与致病SNP间LD较大的SNP更容易被保留。
上述四条原理与图1中的因果图情形一一对应,也概括了条件独立检验时可能的所有因果关系。算法的判断标准为借助回归模型分析,某一SNP的条件P值>0.05,将其从备选集中剔除;若模型中的SNP的条件P值均大于或小于0.05,同时保留模型中的所有SNP。
实施例1:
以一个小的数据集为例,假设数据中SNP与Y的真实因果关系如图2中所示。假设原始数据集中有7个SNP,为筛选结局Y的致病位点,首先使用一元回归模型判断各SNP是否与Y边际独立,剔除SNP
实施例2:
如图3所示,本发明实施例2提供了一种面向全基因组的致病SNP精细定位的因果GWAS方法,包括以下步骤:
(1)首先判断基因组的每个SNP与结局Y是否独立。本模型中使用单因素回归模型(如线性回归或logistic回归模型)对样本进行全基因组关联分析,基于分析结果筛选P值低于某阈值(如P<5×10
将基因集S
(2)固定S
SNP
考虑到共线性问题,如果模型中的两个SNP之间的LD等于1,则这两个SNP都保留在基因集S
(3)若备选基因集S
(4)需要注意的是,当使用m元回归模型对备选基因集S
本实施例所述的方法突破了统计关联的局限性,整体表现明显优于广义线性回归模型,LASSO回归模型,GCTA模型及贝叶斯变量选择回归模型等现有的精细定位方法(图4和图5),为全基因组遗传致病位点精细定位提供了新策略和新方法。
实施例3:
本发明实施例3提供了一种全基因组致病SNP精细定位的因果关联分析系统,包括:
数据获取模块,被配置为:获取待分析的基因组数据;
因果GWAS模块,被配置为:
使用单因素回归模型对基因组数据进行全基因组关联分析,筛选P值低于预设阈值的显著SNP,定义被选择的SNP为第一备选基因集(即S
固定第一备选基因集中P值最小的SNP
如若被删除的SNP为SNP
若第二备选基因子集中SNP的个数小于或等于2或者第二备选基因子集等于第一备选基因子集,计算结束,所得第二备选基因子集中的所有SNP为筛选的致病位点;否则,继续计算,直至第m备选基因子集中SNP的个数小于或等于m+1,则运算停止。
所述系统的工作方法与实施例1提供的全基因组致病SNP精细定位的因果关联分析方法相同,这里不再赘述。
实施例4:
本发明实施例4提供了一种计算机可读存储介质,其上存储有程序,该程序被处理器执行时实现如本发明实施例1所述的全基因组致病SNP精细定位的因果关联分析方法中的步骤。
实施例5:
本发明实施例5提供了一种电子设备,包括存储器、处理器及存储在存储器上并可在处理器上运行的程序,所述处理器执行所述程序时实现如本发明实施例1所述的全基因组致病SNP精细定位的因果关联分析方法中的步骤。
本领域内的技术人员应明白,本发明的实施例可提供为方法、系统、或计算机程序产品。因此,本发明可采用硬件实施例、软件实施例、或结合软件和硬件方面的实施例的形式。而且,本发明可采用在一个或多个其中包含有计算机可用程序代码的计算机可用存储介质(包括但不限于磁盘存储器和光学存储器等)上实施的计算机程序产品的形式。
本发明是参照根据本发明实施例的方法、设备(系统)、和计算机程序产品的流程图和/或方框图来描述的。应理解可由计算机程序指令实现流程图和/或方框图中的每一流程和/或方框、以及流程图和/或方框图中的流程和/或方框的结合。可提供这些计算机程序指令到通用计算机、专用计算机、嵌入式处理机或其他可编程数据处理设备的处理器以产生一个机器,使得通过计算机或其他可编程数据处理设备的处理器执行的指令产生用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的装置。
这些计算机程序指令也可存储在能引导计算机或其他可编程数据处理设备以特定方式工作的计算机可读存储器中,使得存储在该计算机可读存储器中的指令产生包括指令装置的制造品,该指令装置实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能。
这些计算机程序指令也可装载到计算机或其他可编程数据处理设备上,使得在计算机或其他可编程设备上执行一系列操作步骤以产生计算机实现的处理,从而在计算机或其他可编程设备上执行的指令提供用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的步骤。
本领域普通技术人员可以理解实现上述实施例方法中的全部或部分流程,是可以通过计算机程序来指令相关的硬件来完成,所述的程序可存储于一计算机可读取存储介质中,该程序在执行时,可包括如上述各方法的实施例的流程。其中,所述的存储介质可为磁碟、光盘、只读存储记忆体(Read-Only Memory,ROM)或随机存储记忆体(RandomAccessMemory,RAM)等。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 全基因组致病SNP精细定位的因果关联分析方法
- 一种结合随机森林和Relief-F的全基因组SNP位点分析方法