用于高通量分析的微流控细胞捕获和分析设备
文献发布时间:2024-04-18 19:52:40
发明背景
1.发明领域
本发明涉及微流控装置。具体地,本发明涉及微流控装置及其用途,以及用于分析细胞的方法。
2.相关技术描述
单细胞代表基本生物单位。然而,大多数生物知识呈现为研究细胞群而非单细胞的结果。难免存在基础和应用问题,如涉及干细胞分化的转录控制、基因表达中的固有噪声和疾病起源的问题,这只能从单细胞水平上来解决。例如,单细胞分析允许直接测量基因表达动力学或清楚地鉴别共调控的基因,甚至存在会使普通群体测量不清楚的非同步性和异质性时也是如此。类似地,单细胞方法在干细胞研究和癌症生物学中至关重要,其中由于受纯化方案限制而分离的原代细胞群为异质的,并且通常只有少数细胞群是最相关的。因此,高通量单细胞测量技术受关注并在临床和研究环境中具有广泛的应用。
测量单细胞中转录水平的现有方法包括RT-qPCR(1)、使用数字PCR(2)的单分子计数,或杂交探针(3,4)以及新一代测序(5)。在这些方法中,单细胞RT-qPCR提供了灵敏、特异和动态范围的综合优点,但其受低通量、试剂费用高和难以准确测量低丰度转录本的限制(6)。
微流控装置使用活动阀门来定位和分离细胞,允许对单个微生物细胞进行分离和基因组扩增(32)。遗憾的是,由于涉及操作阀门机构的手动操作,该装置不能进行高通量分析。此外,所述装置不能使分离的细胞在处理和分析之前从上清液中洗涤出来。反过来这会产生污染事件,并进一步限制该装置的下游应用。
因此,微流体研究的目标是开发出可对单细胞中的转录进行可扩展分析的综合技术。微流控系统提供了用于单细胞分析的众多优点:测量的经济性、平行化和自动化,以及提高来自小量反应的灵敏度和精确度。过去十年中,人们投入了大量精力用于研发在芯片上集成和可扩展的单细胞遗传分析(7,8)。因此,微流控单细胞基因表达分析的许多基本功能已经在分离包括细胞操作和捕获(9,10)、RNA纯化和cDNA合成(11-13),以及芯片外细胞分离cDNA合成和预扩增后的微流控qPCR(14)中得到展示。具体地,最近已将微流控qPCR装置(Biomark动态阵列,Fluidigm公司)应用到单细胞研究中(15,16)。尽管这些系统提供了高通量qPCR数据读出,但是它们没有进行前段样品制备,且需要通过FACS或微量移液管进行单细胞分离,然后在分析前进行芯片外处理和起始模板的预扩增。将单细胞分析的所有步骤集成于一个能测量大量细胞的强大系统的关键步骤还有待报道。Toriello等人描述了直接测量单细胞基因表达的集成装置的单独示范,组合了RNA捕获、PCR扩增和使用集成毛细管电泳的扩增子末端检测的所有步骤(17)。尽管该系统的工程复杂,通量被限制于每轮4个细胞,细胞捕获需要细胞代谢标记,且所述分析不是定量的。
在许多类型的分析之前,需要分离单细胞或有限数目的细胞,这通常需要使用细胞捕获机构。对于单细胞或少量细胞进行可靠和可扩展分析的目标而言,低捕获通量和低捕获效率成为巨大的挑战。低捕获效率需要成千上万个细胞以使在使用细胞系时少量单细胞测量不成问题,然而,在使用稀少细胞类型如干细胞的原代样品时,这就成为一个大问题。同样,对所捕获细胞和通过所述捕获器的细胞的观察表明,细胞捕获效率取决于细胞大小,其可能潜在地将偏差引入到单细胞测量中。
发明概述
本发明部分基于这样的发现,即相比μL体积的分析,集成微流控装置可用于对每个实验中pL至nL体积(50pL-100nL)的上百(上千)单细胞进行可扩展、高性能、低成本和敏感性分析的高通量分析。所述描述和实例提供了首次实施强大和高通量的单细胞处理和芯片上的核酸扩增,由此取得了微流控生物学分析的主要里程碑。
本文提供了能够进行可扩展的定量单细胞遗传分析的微流控技术。具体地,本文的示例为集成微流控装置,其用于对每个实验中上百至上千通量的单细胞mRNA和miRNA表达进行高通量RT-qPCR分析。本描述显示,相比μL体积的分析,该技术提供了具有改善性能、降低成本和更高灵敏度的可扩展的单细胞基因表达测量的有力工具。
本文提供的实施例公开了完全集成的微流控装置,其能够进行每轮上百单细胞的基因表达的高精度RT-qPCR测量。而且,所述装置的实施方案能够执行所有的单细胞处理步骤,包括细胞捕获、细胞裂解、逆转录和定量PCR。除了更高的通量和降低成本,本文显示纳升(nanoliter)体积处理减少了测量噪声、增加了灵敏度,并提供了单核苷酸特异性。本描述显示将该技术应用于对3300个单细胞进行下述测量:i)K562细胞中miRNA表达,ii)胚胎干细胞中分化期间miRNA和其靶转录本之一的共调控,和iii)原代小叶乳腺癌细胞中单核苷酸变体检测。本文中建立的核心功能提供了这样的基础,各种芯片上的单细胞转录分析将以此为基础发展。在阅读了下述本发明的具体实施方案描述及其附图后,对本领域技术人员来说,本发明的其他方面和特征将变得很明显。
可选择地,处理和分析的细胞类型可选自以下一种或多种:滚环扩增、多重置换扩增、同温DNA和RNA扩增、cDNA末端快速扩增(RACE)、简并性寡聚引物PCR、线粒体DNA PCR、基因组PCR、数字PCR、RT-PCR、测序、免疫化学、邻位连接PCR、免疫PCR、代谢物分析、酶法分析、报告基因表达分析、杂交研究。
根据第一实施方案,提供的微流控装置包括:(a)细胞捕获室,其具有至少一个入口和至少一个出口,其中每个入口和出口具有开启和关闭位置,以此当所述入口处于开启位置时流体能够流入细胞捕获室,而当所述出口处于开启位置时流体能够流出细胞捕获室,并且以此当所述入口处于关闭位置时将阻止流体流入所述细胞捕获室,而当所述出口处于关闭位置时将阻止流体流出所述细胞捕获室,其中流经所述细胞捕获室的方向指示了所述细胞捕获室的上游和下游方向;(b)细胞漏斗,其位于细胞捕获室中且可操作地引导细胞通过所述细胞捕获室流向细胞捕获室中的一个或多个期望的位置;和(c)细胞捕获器,其通常位于所述细胞漏斗的下游,以此将所述细胞捕获器定位以接收从所述细胞漏斗向下游流动的细胞。
所述微流控装置可进一步包括:(a)与流体注入通道流体连通的第二入口,所述上游入口具有开启和关闭位置,其中所述开启位置允许流体从所述流体注入通道进入所述细胞捕获室,而在所述关闭位置阻止流体从所述流体注入通道进入所述细胞捕获室;和(b)与一个或多个辅助室流体连通的第二出口,所述下游出口具有开启和关闭位置,其中所述开启位置允许流体流出所述细胞捕获室并进入一个或多个辅助室,而在所述关闭位置阻止流体从所述细胞捕获室进入一个或多个辅助室。
所述微流控装置可进一步包括与流体注入通道流体连通的第二入口,所述第二入口具有开启和关闭位置,其中所述开启位置允许流体从所述流体注入通道进入所述细胞捕获室,而在所述关闭位置阻止流体从所述流体注入通道进入所述细胞捕获室,并且其中所述细胞捕获室的体积是可膨胀的。
所述细胞捕获室可具有一个入口和一个出口,其中所述入口与流体注入通道流体连通,并且其中所述细胞捕获室的体积是可膨胀的。
所述细胞漏斗可施加力以引导细胞流向所述细胞捕获室中一个或多个期望的位置。通过所述细胞漏斗施加于细胞的力可选自以下一种或多种:机械力、重力、电磁力、静电力、磁力、声学力、水力和光学力。而且,一种或多种上述的力可有助于将细胞引导至期望的位置。例如,物理结构可对流体施加机械力,反过来流体产生水力。通过所述细胞漏斗施加于细胞的力可为水力。
所述细胞捕获器可选自以下一种或多种:机械力捕获器、水力捕获器、介电泳力捕获器、磁力捕获器、声学力捕获器、亲和力捕获器、光学力捕获器和膜片钳捕获器。所述细胞捕获器可为水力捕获器。
所述第二出口可与第一辅助室流体连通。所述第一辅助室可与第二辅助室流体连通,其中在所述第一和第二辅助室之间有阀门,其中所述阀门具有开启位置以允许流体从第一辅助室流向第二辅助室,且具有关闭位置以阻止流体从第一辅助室流向第二辅助室。所述第一辅助室可与第二辅助室流体连通,且所述第二辅助室与第三辅助室流体连通;其中在所述第一和第二辅助室之间有阀门,其中所述阀门具有开启位置以允许流体从第一辅助室流向第二辅助室,且具有关闭位置以阻止流体从第一辅助室流向第二辅助室;其中在所述第二和第三辅助室之间有阀门,其中所述阀门具有开启位置以允许流体从第二辅助室流向第三辅助室,且具有关闭位置以阻止流体从第二辅助室流向第三辅助室。
所述辅助室的体积可为可膨胀的。所述细胞捕获室的体积可为0.1nL-100.0nL。所述可膨胀的细胞捕获室的未膨胀体积可为0.1nL-100.0nL。所述细胞捕获室的体积可为0.6nL。所述未膨胀的细胞捕获室可为0.6nL。通过初始室的膨胀或通过打开阀门以提供流体流入一个或多个辅助室可增加给定室的有效体积。所述第二辅助室和第一辅助室之间的比率可为5:1。所述第二辅助室和第一辅助室之间的比率可为至少5:1。所述膨胀的细胞捕获室和未膨胀的细胞捕获室之间的比率可为5:1,或所述膨胀的第一辅助室和未膨胀的第一辅助室之间的比率可为5:1。所述膨胀的细胞捕获室和未膨胀的细胞捕获室之间的比率为至少5:1,或所述膨胀的第一辅助室和未膨胀的第一辅助室之间的比率可为至少5:1。所述第二辅助室和第一辅助室之间,或所述膨胀的细胞捕获室和未膨胀的细胞捕获室之间,或所述膨胀的第一辅助室和未膨胀的第一辅助室之间的比率可根据所选的反应混合物、该混合物组分的浓度和待分析物质的浓度而变化。可选择地,所述细胞捕获室可为0.05nL-100.0nL。可选择地,所述细胞捕获室可为0.05nL-90.0nL。可选择地,所述细胞捕获室可为0.1nL-95.0nL。可选择地,所述细胞捕获室可为0.1nL-90.0nL.可选择地,所述细胞捕获室可为0.1nL-85.0nL。可选择地,所述细胞捕获室可为0.1nL-80.0nL。可选择地,所述细胞捕获室可为0.1nL-75.0nL。可选择地,所述细胞捕获室可为0.1nL-70.0nL。可选择地,所述细胞捕获室可为0.1nL-65.0nL。可选择地,所述细胞捕获室可为0.1nL-60.0nL。可选择地,所述细胞捕获室可为0.1nL-55.0nL。可选择地,所述细胞捕获室可为0.1nL-50.0nL。可选择地,所述细胞捕获室可为0.1nL-45.0nL。可选择地,所述细胞捕获室可为0.1nL-40.0nL。可选择地,所述细胞捕获室可为0.1nL-35.0nL。可选择地,所述细胞捕获室可为0.1nL-30.0nL。可选择地,所述细胞捕获室可为0.1nL-25.0nL。可选择地,所述细胞捕获室可为0.1nL-20.0nL。可选择地,所述细胞捕获室可为0.1nL-15.0nL。可选择地,所述细胞捕获室可为0.1nL-10.0nL。可选择地,所述细胞捕获室可为0.1nL-9.0nL。可选择地,所述细胞捕获室可为0.1nL-8.0nL.可选择地,所述细胞捕获室可为0.1nL-7.0nL。可选择地,所述细胞捕获室可为0.1nL-6.0nL。可选择地,所述细胞捕获室可为0.1nL-5.0nL。可选择地,所述细胞捕获室可为0.1nL-4.0nL。可选择地,所述细胞捕获室可为0.1nL-3.0nL。可选择地,所述细胞捕获室可为0.1nL-2.0nL。可选择地,所述细胞捕获室可为0.1nL-1.0nL.
所述装置可包括1-10个细胞捕获器和对应的细胞漏斗。可选择地,漏斗可与多于一个的专用捕获器结合使用。所述细胞捕获器也可设计为每一个容纳多于一个细胞,且每一个可由一个或多个细胞漏斗来提供细胞。此外,如果待检测的是混合细胞群,细胞捕获器的大小可使其选择特定的细胞类型。而且,所述细胞捕获器和漏斗可设计为排除某些细胞类型或选择某些细胞类型。
所述细胞漏斗可包括各自具有近端和远端的一对细胞导流板,其中所述近端位于所述捕获室的对侧,且其中所述细胞导流板的每个远端相对于近端在下游方向对角线上成角度,以此所述细胞导流板的远端提供开口,其大小允许在所述细胞导流板的远端之间通过细胞。所述细胞捕获器可大体为杯形或“U”形,或者具有大体为杯形或“U”形的区域,从而细胞可从一侧进入所述“U”形内部而不能通过。所述细胞捕获器可提供流经所述细胞捕获器的流体。可在细胞被捕获于所述捕获器之前、期间和之后流经所述捕获器。该流经可辅助捕获细胞和辅助洗涤所捕获的细胞。而且,所述细胞漏斗或细胞捕获器可具有如任一图1a-x、2a-e、12b-i和24所示的结构。
根据又一个实施方案,提供了细胞捕获和制备方法,所述方法包括:(a)使细胞在流体中流经室;(b)使细胞在流体中通过漏斗流向细胞捕获器;(c)在所述室中捕获预定数目的细胞;(d)中断流体中的细胞流;(e)通过使洗涤液流经所述室来洗涤所捕获的细胞,其中所述洗涤液流将污染物从所述室中去除;和(f)将所述预定数目的细胞封闭于所述室内。
根据又一个实施方案,提供了细胞捕获和制备方法,所述方法包括:(a)使细胞在流体中流经室;(b)使细胞在流体中通过漏斗流向细胞捕获器;(c)在所述室中捕获预定数目的细胞;(d)中断流体中的细胞流;(e)通过使洗涤液流经所述室来洗涤所述捕获的细胞,其中所述洗涤液流将污染物从所述室中去除;和(f)将所述预定数目的细胞封闭于所述室内。
所述方法还可包括处理和分析洗涤后的捕获细胞,其中分析选自以下一种或多种:滚环扩增、多重置换扩增、同温DNA和RNA扩增、cDNA末端快速扩增(RACE)、简并性寡聚引物PCR、线粒体DNA PCR、基因组PCR、数字PCR、RT-PCR、测序、免疫化学、邻位连接PCR、免疫PCR、代谢物分析、酶法分析、报告基因表达分析和杂交研究等。
所述方法还可包括:(a)细胞裂解;(b)逆转录;和(c)扩增。
所述方法还可包括:(a)细胞裂解;(b)逆转录;和(c)定量扩增。
根据又一个实施方案,提供了细胞分析方法,所述方法包括:(a)将细胞引导至细胞捕获室,其中所述室的体积为0.1nL-100nL;(b)在所述室中捕获预定数目的细胞;(c)通过使洗涤液流经所述室来洗涤所捕获的细胞,其中所述洗涤液流将污染物从所述室中去除;和(d)分离所述室中预定数目的细胞。
所述方法还可包括分析所述洗涤后的捕获细胞,其中分析选自以下一种或多种:滚环扩增、多重置换扩增、同温DNA和RNA扩增、cDNA末端快速扩增(RACE)、简并性寡聚引物PCR、线粒体DNA PCR、基因组PCR、数字PCR、RT-PCR、测序、免疫化学、邻位连接PCR、免疫PCR、代谢物分析、酶法分析、报告基因表达分析和杂交研究等。
所述方法还可包括:(a)细胞裂解;(b)逆转录;和(c)扩增。
所述方法还可包括:(a)细胞裂解;(b)逆转录;和(c)定量扩增。
根据又一个实施方案,提供了细胞分析方法,所述方法包括:(a)将细胞引导至细胞捕获室,其中所述室的体积为0.1nL-100nL;(b)在所述室中捕获预定数目的细胞;(c)通过使洗涤液流经所述室来洗涤所捕获的细胞,其中所述洗涤液流将污染物从所述室中去除;(d)分离所述室中预定数目的细胞;(e)裂解所述室中的细胞;(f)逆转录通过所述细胞裂解释放的RNA;和(g)通过聚合酶链式反应(PCR)扩增步骤(f)中所转录的cDNA。
根据又一个实施方案,提供了细胞分析方法,所述方法包括:(a)将细胞引导至细胞捕获室,其中所述室的体积为0.1nL-100nL;(b)在所述室中捕获预定数目的细胞;(c)通过使洗涤液流经所述室来洗涤所捕获的细胞,其中所述洗涤液流将污染物从所述室中去除;(d)分离所述室中预定数目的细胞;(e)裂解所述室中的细胞;(f)逆转录通过所述细胞裂解释放的RNA;和(g)通过定量聚合酶链式反应(PCR)扩增步骤(f)中所转录的cDNA。
根据又一个实施方案,提供了细胞捕获和制备方法,所述方法包括:(a)使细胞在流体中流经室;(b)使细胞在流体中通过漏斗流向细胞捕获器;(c)在所述室中捕获预定数目的细胞;(d)中断流体中的细胞流;和(e)将所述预定数目的细胞封闭于所述室内。
根据又一个实施方案,提供了细胞捕获和制备方法,所述方法包括:(a)使细胞在流体中流经室;(b)使细胞在流体中通过漏斗流向细胞捕获器;(c)在所述室中捕获预定数目的细胞;(d)中断流体中的细胞流;和(e)将所述预定数目的细胞封闭于所述室内。
根据又一个实施方案,提供了细胞分析方法,所述方法包括:(a)将细胞引导至细胞捕获室,其中所述室的体积为0.1nL-100nL;(b)在所述室中捕获预定数目的细胞;和(c)分离所述室中预定数目的细胞。
根据又一个实施方案,提供了细胞分析方法,所述方法包括:(a)将细胞引导至细胞捕获室,其中所述室的体积为0.1nL-100nL;(b)在所述室中捕获预定数目的细胞;(c)分离所述室中预定数目的细胞;(d)裂解所述室中的细胞;(e)逆转录通过所述细胞裂解释放的RNA;和(f)通过聚合酶链式反应(PCR)扩增步骤(f)中所转录的cDNA。
根据又一个实施方案,提供了细胞分析方法,所述方法包括:(a)将细胞引导至细胞捕获室,其中所述室的体积为0.1nL-100nL;(b)在所述室中捕获预定数目的细胞;(c)分离所述室中预定数目的细胞;(d)裂解所述室中的细胞;(e)逆转录通过所述细胞裂解释放的RNA;和(f)通过定量聚合酶链式反应(PCR)扩增步骤(f)中所转录的cDNA。
所述方法还可包括将所述细胞引导至细胞捕获室之前的细胞分选步骤。所述方法还可包括在洗涤之前固定所捕获的细胞。固定还可促进对捕获室中细胞的洗涤。
附图说明
在说明本发明实施方案的附图中:
图1A-1L为根据本发明涉及水力细胞捕获器的各种实施方案的细胞漏斗和对应的细胞笼(cell cage)的示意图。
图1M-1X为根据本发明涉及水力细胞捕获器的各种实施方案的细胞笼的示意图。
图2A为细胞捕获检测装置示意图。
图2B为根据本发明一个实施方案的细胞漏斗和细胞捕获器的几何体示意图。
图2C为根据本发明一个实施方案的优选细胞漏斗和细胞捕获器的几何体示意图。
图2D为接近具有图2C所示几何体的捕获器的K562单细胞光学显微图。
图2E为图2C中K562细胞被捕获后的光学显微图。
图3描绘了化学裂解法和热裂解法相比较的结果图。
图4为背景信号与来自单细胞的信号的比较图。
图5描绘了细胞裂解物浓度对RT-qPCR的抑制作用的图。
图6显示了不同RT-PCR稀释物对qPCR性能影响的图。
图7为根据本发明一个实施方案的微流控装置的示意图。
图8为描绘了对9个单细胞检测miRNA16和一个无细胞对照的qPCR结果图。
图9为描绘了来自实验得到的部分被洗脱两次后三个室的洗脱效率测量结果图。
图10为根据本发明一个实施方案用于单细胞捕获分析的微流控装置的示意图。
图11为概述图10的微流控装置操作的示意图。
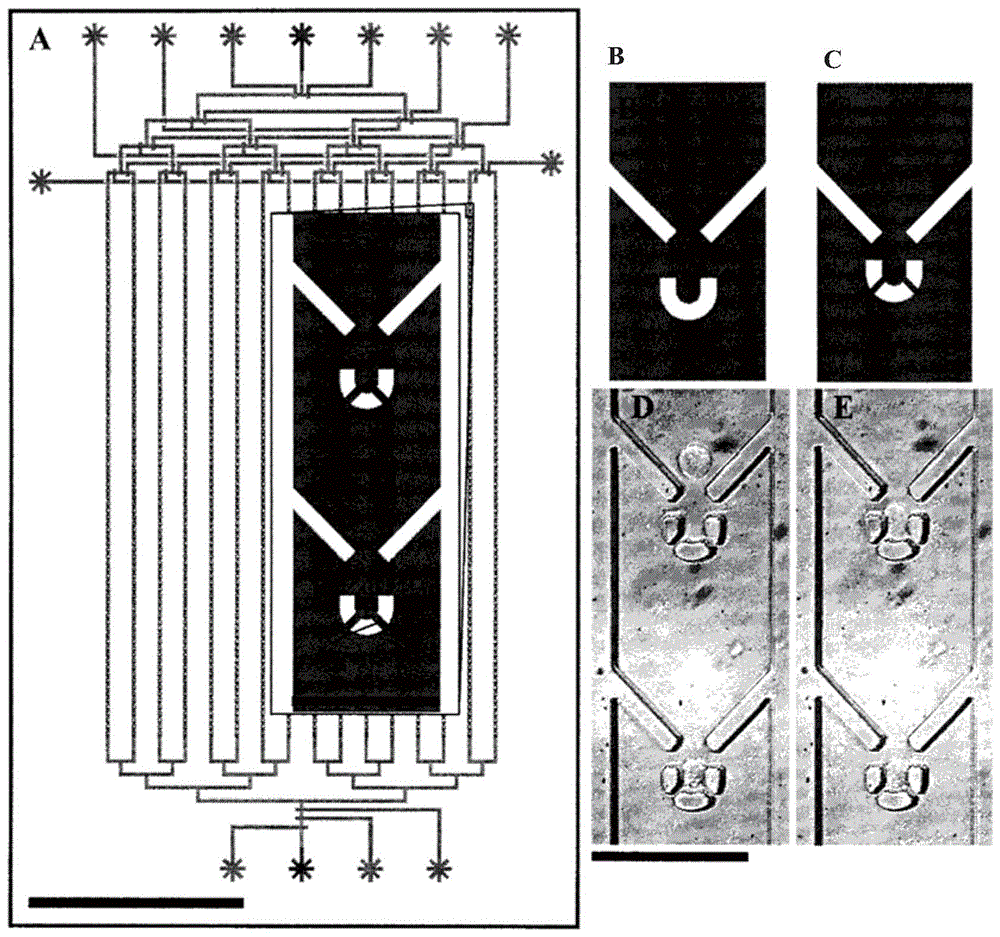
图12A为根据本发明一个实施方案的微流控装置。比例尺:4mm。所述装置特征为6个样品输入通道,每一个分为50个化合物反应室,总共300个RT-qPCR反应,使用约20μL试剂。长方形框表示B中描绘的区域。
图12B为对应于图12A中黑色长方形内区域的阵列单元的光学显微图。
图12C为具有捕获的单细胞的两个细胞捕获室的光学显微图。
图12D为注入至图12A中所描绘装置的单细胞悬浮液的示意图。
图12E为将单细胞从液流中分离以允许洗涤该细胞的细胞捕获器的示意图。
图12F为在热裂解之前启动气动阀门以使得单细胞分离的示意图。
图12G为注入逆转录(RT)反应试剂的示意图。
图12H为试剂注入管线后用PCR试剂冲洗的示意图。
图12I为在50nL qPCR室中将qPCR试剂与RT产物组合的示意图。
图13A为图12A显示6个通道中的300个反应的整个装置的荧光图。
图13B为类似于图12A中图像的处理序列产生的300个实时扩增曲线图。
图13C为8倍系列稀释的纯化总RNA中GAPDH的芯片上和芯片外RT-qPCR的图。
图13D描绘了在K562细胞裂解稀释物中GAPDH的实时扩增曲线图。
图13E描绘了在细胞裂解物稀释系列物中GAPDH的测量CT值的图。
图14比较了来自K562细胞裂解物的GAPDH在所述装置中进行qPCR之前在图12A的微流控装置中进行RT或在管中进行RT的测量结果图。
图15A为沿着图12A装置的所有6个通道的每个室中细胞位置的示意图。
图15B为显示图14A所示实验的CT测量结果的散点图。
图15C为在K562单细胞(N=233)中测量的GAPDH转录本数目的柱状图。
图16A为显示由Cedex测量的原始样品中细胞大小分布的柱状图。
图16B为显示由微流控捕获器分离的细胞大小分布的柱状图。
图17A为在注入微流控装置之前,芯片外PBS洗涤的细胞中GAPDH测量结果的散点图。
图17B为在注入微流控装置之前,芯片上PBS洗涤的细胞中GAPDH测量结果的散点图。
图17C为来自上样的纯化RNA且洗涤或未洗涤所述细胞捕获室的GAPDH测量结果图。
图18A为沿着装置中所有6个通道的每个室中细胞的位置的示意图。
图18B为对应于图18A)的实验在30个PCR循环后整个装置的荧光图像。
图19为在hESC细胞聚集体中miR16测量结果的散点图。
图20A为在K562细胞和hESC中miR-16表达的单细胞测量结果的柱状图。
图20B为K562细胞和hESC之间miR-223差异表达的柱状图。
图20C为相比来自大块细胞裂解物的数字PCR测量结果,根据本发明一个实施方案的微流控装置中由RT-qPCR测量的单细胞miRNA平均拷贝数的图。
图20D为K562细胞和hESC之间miR-223差异表达的柱状图
图21A为K562细胞和hESC中miR-16表达的单细胞测量结果的柱状图。
图21B,C为显示miR-145和OCT4转录本分布的柱状图。
图21D为显示分离自小叶乳腺癌样品的原代细胞中SP1野生型和SNV突变体转录本的共表达测量结果的散点图。
图22A为FBS分化7天后CA1S细胞中OCT4的mRNA-FISH的代表性图像。
图22B为未分化的CA1S细胞的代表性图像。
图23为显示根据本发明一个实施方案构建的miRNA序列标签文库中每个标签长度处比对标签的百分比的柱状图。
图24为根据本发明一个实施方案的用于测序和表征单细胞转录组的微流控装置的示意图。
图25A为说明根据本发明一个实施方案,为了纯化和固定模板(RNA)将功能化的珠子和细胞混合的图像。
图25B为说明根据本发明一个实施方案,将功能化的珠子堆积于室中的图像。
图26为根据本发明一个实施方案,用于磁性固定功能化的珠子以结合模板(DNA/RNA)并使多步反应在单个室中发生的微流控装置。
图27为集成了单细胞处理和微阵列点样以将不同试剂递送给不同反应的微流控装置的示意图。
图28为根据本发明一个实施方案,用于集成的单细胞数字PCR的微流控装置的示意图。
图29为图28的微流控装置的示意图,描绘了通过分叉旋转混合器将PCR溶液与模板分子混合。
图30为根据本发明一个实施方案,用于通过将细胞产物递送至更大的室中以对该其进行连续稀释的微流控装置的示意图。
图31为根据本发明一个实施方案,用于多路复用单细胞RT-qPCR的微流控装置的示意图。
详细描述
为了分离和测量单细胞,已经开发了各种技术。这些技术在它们能够分析的细胞数目和能够进行的平行或多路测量的数目方面差异都很大。
定义
如本文所用,“流式细胞术”是基于测量单个粒子的荧光性、光散射和吸光度,用于快速、定量地检测和分选细胞群、细胞器和其他组分的常用技术(18)。流式细胞术使用光学多路复用技术来测量不同属性,每分钟能够处理和测量多达成百上千个细胞。该技术已经用于测量单细胞的物理特性和化学组成(19)。通过将转录报告基因如GFP与关注的基因或miRNA结合并记录每个细胞的荧光,已经使用流式细胞术来测量mRNA(20)和miRNA(21)表达。此前也有报道通过流式细胞术分选单细胞,以通过RT-qPCR用于进一步遗传分析(22)。
如本文所用,“激光捕获显微切割”(LCM)是可将来自异质样品的纯群体在直接可见下分离用于进一步分析的技术(23)。该技术可应用于组织(23)、细胞学制备(24)和活细胞培养(25)。在LCM中,关注的细胞在显微镜下可见且使用者限定该区域以与样品的其他部分分离。然后,通过去除周围区域或从样品切离期望的部分,可将这些选定的区域与样品的其余部分分离。由于该方法对待分离的样品为非破环性的,通过该方法分离的细胞可使用任何传统分子生物学技术来测量。
如本文所用,“微流控装置”是指允许对
如本文所用,“细胞捕获室”是指在微流控装置内的封闭空间,其中使细胞流经所述装置时,一个或多个细胞可从更大的细胞群中分离出来。每个细胞捕获室将具有至少一个入口和至少一个出口,所述入口允许流体包括含有细胞的流体进入所述室中,所述出口允许流体流出所述室。本领域技术人员应理解,入口或出口可在结构和尺寸方面有很大改变,从最普遍意义上可表征为能够在开启位置和关闭位置之间可逆地切换的孔口,以便在开启位置时允许流体流入或流出所述细胞捕获室,在关闭位置时封闭所述细胞捕获室,从而分离和保留其内容物,由此所述孔口也可介于开启位置和关闭位置之间以允许一些流体流动。
流体流经细胞捕获室的方向指示了所述细胞捕获室的“上游”和“下游”方向。因此,入口将位于所述室的上游位置,而出口通常将位于所述室的下游位置。然而,本领域技术人员应理解,单独的孔口也可即作为入口又作为出口。
如本文所用,“入口”或“出口”可包括任何口或孔口,由此流体流动限制于流经所述入口、出口或孔口。可有阀门来控制流量,或通过用阻止液流(例如,油)的层分隔所述通道来控制流量。
“细胞捕获室”可进一步包括通常位于所述“细胞漏斗”下游的“细胞捕获器”,以此所述细胞捕获器被定位以接收(和保留)从所述细胞漏斗向下游流动的细胞。
如本文所用,“细胞漏斗”是指一种设备,其被设计为用于将来自细胞处于分散状态的上游位置的细胞流会聚到细胞捕获室中一个或多个期望的下游位置,所述细胞捕获室具有细胞流的更小横截面。所述细胞漏斗施加力以引导细胞流向细胞捕获室中的一个或多个期望位置。为清楚的目的,本文将“力”定义为导致自由体(例如,细胞)经历速度上变化的任何支配力。漏斗可跨越所述细胞捕获室的整个高和/或宽,或者部分跨越所述高和/或宽。
图1a-l提供了根据本发明的各种实施方案的示例性机械漏斗的图示。如图1b和e中所示,可通过制作更长的颈缩部12对图1a的基本机械漏斗10进行改进。可以有多个漏斗,如图1d、i和j中的嵌套漏斗10a和10b以及图1f中的10c。所述漏斗的角θ可变化,如图1k中所示例。所述漏斗的颈缩部12可如图1d、e、f、g、k所示为直的,或者如图1a、b、h、i、j、k、l所示为斜的。还可通过组合机械力和水力法来额外实现汇集,如图1c所示(即不同流速的不同或相同的流体,可经所述外部通道流入)。漏斗还可包括上游通道如凹槽的横截面变化,其导致细胞在遇到细胞捕获器之前沿着所述通道的宽被定位于特定部分。
如本文所用,“细胞捕获器”通常指随着时间在预定位置接收和保留细胞的装置。细胞捕获器可包括定位的表面修饰用于化学固定细胞。或者,所述细胞捕获器可为机械力捕获器、水力捕获器(10,26-28)、水力平衡捕获器(29,30)、活动阀调捕获器(2,10,31,32)、介电泳力捕获器(33)、DNA固定捕获器(17)、凝胶封装捕获器(34)、磁力捕获器、声学力捕获器或光学力捕获器(35)。在本发明的各种实施方案中,细胞捕获器通常将直接定位于由漏斗产生的细胞流较小横截面的通道上。根据本发明的各种实施方案,当使用如图1所示的机械漏斗时,捕获器可直接定位于所述漏斗下游开口后方。
如本文所用,“机械力捕获器”是指诸如笼的细胞捕获器。
如本文所用,“水力捕获器”是指其中运动流体的力在保留所捕获细胞于其位置上起作用的细胞捕获器。水力捕获器也可由机械力捕获器组成,细胞被捕获并保留于其中。在利用水力捕获器的本发明一些实施方案中,可取的是具有3个或更多进入细胞捕获室的入口,从而可对流体进行调整以便引导细胞到所述捕获器。
如本文所用,“介电泳力捕获器”是指其中作为介电物体的细胞通过不均匀电场产生的力被保留的细胞捕获器。
如本文所用,“磁力捕获器”是指利用磁场来保留细胞的细胞捕获器。通常,用磁性粒子标记细胞,然后通过磁场进行定位和保留。然而,磁力捕获器也可用于捕获在合适缓冲液中的非磁性细胞。
如本文所用,“声学力捕获器”是指这样的细胞捕获器,其中将超声驻波用于产生施加定位和保留细胞的力的静压梯度。
如本文所用,“光学力捕获器”是指这样的细胞捕获器,其中深聚焦激光束,通常为近红外激光束,被用于将细胞拉向所述光束的方向。
细胞捕获器的大小可根据所期望捕获细胞的大小、类型、机械特性或数目来变化。根据本发明的各种实施方案,微流控装置还可包括设计用于捕获多种细胞类型的捕获器组合。而且,每个细胞捕获室可包括多个捕获器。在这样的实施方案中,在每个大小上被捕获细胞的频率可用作诊断结论。或者,可对单个捕获器中捕获的细胞群内容物进行处理和分析。
图1m-x提供了根据本发明的各种实施方案用于水力捕获器中的示例性物理笼的图示。如图1m-x中的笼20所示,所述笼的内部形状可为杯形以容纳球形细胞,或可设计为各种几何形状以容纳独特形状和/或大小的细胞。所述笼可形成基本上跨越所述室的整个高或宽,或者部分跨越所述室的高或宽的堰,或二者兼有。如图1m所示,所述笼的外形可被改变以调节流向捕获器周围的流体流动。如图1o、p、q、r、s和x所示,可增加孔或筛元件22以促进和调节流体流经所述杯。如图1x所示,孔的大小可减小或增大。所述孔可为所述室的整个高或宽,或者部分跨越所述室的高或宽的堰,或二者兼有。笼的大小可制作得如图1t所示更长,如图1u所示更宽,如图1v所示更长且更宽,或者如图1w所示更短和更窄。
形成所述漏斗的物理结构可为两侧均为斜的,即在漏斗表面的相对侧也为斜的,以便于从所述捕获器中去除细胞,具有如图1k和l所示的ψ角。其他特征,除捕获器外,可定位于漏斗的下游以进一步调节流体阻抗,如图1h和1l中的元件14。
如本文所用,“流体注入通道”是指流体通过其被引入至所述装置的室中的任何管道。流体注入通道可用于将包括细胞悬浮液、洗涤缓冲液、反应混合物等的任何流体递送至室中。
如本文所用,“辅助室”是指辅助细胞捕获室的任何室。辅助室可用于处理或分析所捕获的细胞,或其分离的内容物。处理可包括细胞制备步骤,包括培养、洗涤、裂解和分级。分析可包括DNA和RNA扩增和检测,包括线粒体PCR、基因组PCR、数字PCR、RT-PCR、RTq-PCR、多重置换扩增(DNA)、滚环扩增测序、简并PCR、分子倒置探针、分子信标,以及其他DNA/RNA扩增和检测方法、体外转录、连接、免疫化学、报告基因表达分析、杂交研究等。可将几个辅助室以串联和/或并联的方式连接至单个细胞捕获室,从而可对单个细胞捕获室中的内容物进行多重处理。阀门可定位于辅助室和细胞捕获室之间,或位于辅助室之间,以调节室之间的流体流动。
如本文所用,“可膨胀的”细胞捕获室是指在进行操作以容纳不同体积的流体期间可膨胀或收缩的细胞捕获室。如此,单个室可用于细胞捕获和随后的处理和分析必须在不同体积的流体中进行的目的,从而避免了对多个分离的室的需要。
可以用几种方式实现可膨胀的细胞捕获室的膨胀。例如,所述室可包括注射器,其中通过使用活塞来调节室的体积。或者,所述室可用能够随着流体添加而膨胀或当流体体积减少时收缩的弹性材料构建。还或者,所述室可由不相混合的液体(如空气或油)来部分限定,从而通过所述液体边界的移动可容纳更大体积的流体。
本领域技术人员应理解,辅助室可为可膨胀的。因此,微流控装置可由不可膨胀的和/或可膨胀的细胞捕获室和辅助室组合构成。
如本文所用,“污染物”是指干扰对所述细胞或细胞内容物进行准确和/或精确分析的任何物质。污染物包括但不限于,蛋白、小分子、盐、缓冲剂、RNA、DNA、其他细胞、粒子等。
如本文所用,“微小RNA”(miRNA)为短的(19-23个核苷酸长度)非编码核糖核酸(RNA)聚合物,其涉及将互补mRNA翻译为蛋白的转录后调控(36)。它们表达为形成自身互补的“发卡”RNA的更大转录本。已经表明miRNA对很多生物途径非常重要,包括增殖(37)、分化(38)和死亡(39)、发育时间(40)和方式(41)、神经系统发育模式(39,42)和抗病毒抗性(43,44)。各种功能作用,结合miRNA表达谱是细胞状态的独特信号的知识(45),暗示通过研究miRNA表达可深入探究很多疾病如癌症(45-47)和心脏病(48-51)的复杂生化性质,。
“逆转录定量聚合酶链式反应”(RT-qPCR)为通常用于测量基因表达的分子生物学技术(52-54)。RT-qPCR扩大了传统聚合酶链式反应(PCR)的功能,所述PCR为特异性并以指数方式原位扩增单个或多个拷贝DNA的方法(54)。在RT-qPCR中,通过称为逆转录的过程从RNA模板合成第一链DNA(RT;54)。通过利用RT酶需要具有DNA-RNA双链,将特异性增加到逆转录步骤。然后使用设计为与期望靶标互补且被称为引物的寡核苷酸来特异性地将一段RNA转录为DNA。合成第一链以后,在荧光报告基因和设计用于扩增此前步骤所转录DNA的引物存在下,进行传统的PCR(54)。然后,每个循坏后均监测该反应,记录荧光,产生了特征性的S形曲线。反应完成后,就可能确定基因的相对起始丰度(54)。利用RT-qPCR测量单细胞mRNA(55,56)和miRNA(57,58)表达水平为大家所认可。本领域技术人员应理解,利用光学多路复用技术策略可以在单个反应中进行多于一个序列的分析。本领域技术人员还应理解,除了所限定序列的定量,可使用RT-PCR来扩增一个或多个基因,用于随后的回收和/或分析。
尽管本文所述本发明的实施方案通常涉及捕获和分离细胞并随后处理,应理解,根据本发明的各种方面,所述微流控装置可用于捕获和分析除细胞以外的预定数目的其他实体,包括细胞器、病毒、微粒、液滴等。
实施例1
实施例1.1材料和方法
核酸检测和定量
通过微阵列分析进行DNA定量是一种分析样品中上千个不同的靶标的多路复用的公认方法。将固定在固体表面上的特异性DNA序列用作溶液中靶分子的探针。使含有这些靶分子的溶液流经微阵列的表面,靶分子通过标准DNA碱基配对结合至固定的探针。通过检测荧光团或银色的或化学发光的靶标来检测和定量探针-靶标杂交。尽管微阵列分析已经用于单细胞,但是为了产生满足分析检测限的靶分子量,其需要扩增步骤(59)。
单分子成像技术包括荧光原位杂交(FISH;60,61),单个荧光团成像(62)已经用于直接计数单细胞中的转录本数目。尽管这些方法通过直接观察在其定量转录本丰度的能力上是无与伦比的,但是它们仅能进行光学多路复用并需要高度特化的仪器。
已经开发了特异性检测和定量miRNA的方法,包括northern印迹杂交(64)、原位杂交(60,61)、单分子成像(62)、微阵列(65,66)、新一代测序(67)和具有茎环RT引物和TaqMan探针的RT-qPCR(68)。由于大的动态范围、高度的特异性和许多单细胞转录本定量方法需要PCR扩增这一事实,为了测量miRNA丰度而选择使用茎环引物和TaqMan探针的RT-qPCR。茎环引物含有自身互补区,因而其自身的“回折”产生了发卡结构。该结构防止引物结合至除分子最末端以外的RNA分子,其防止miRNA前体被扩增(68)。TaqMan探针是设计用于增加qPCR特异性的水解探针(69)。水解探针是在一端标记荧光基团而另一端标记淬灭基团的寡核苷酸。当完整的探针游离于溶液中时,所述荧光基团足够贴近所述淬灭基团,从而所述荧光基团发出的任何荧光均通过荧光共振能量转移(FRET)被所述淬灭基团所淬灭(69)。在PCR期间,该探针结合至其互补序列,通过Taq聚合酶的外切酶活性而被切割,由此分离所述荧光基团和所述淬灭基团(69)。因此,每个循环以后,理想状态是会有两倍的荧光性增加。通过在每个循环后或“实时”测量荧光,就可能确定相对起始丰度。实验中(称为标准)系列稀释已知浓度的内含物产生校正曲线,使得能够计算出分子的最初起始数目。
制造
多层软蚀刻技术(MSL)被用于制造装置(63,70)。该制造过程利用了已确立的光蚀刻技术和微电子制造技术上的进展。MSL的第一个步骤是利用计算机绘图软件绘制设计图,然后将其印刷在高分辨率掩膜上。使覆盖有光刻胶的硅晶片暴露于紫外光,其在某些区域通过所述掩膜滤出。根据所述光刻胶是否为负性或正性的,暴露的(负性)或未暴露的(正性)面积将进行交联且所述光刻胶将聚合。未聚合的光刻胶在显影液中是可溶的并随后被冲洗掉。通过组合不同的光刻胶并以不同的速度旋转涂覆,可使晶片形成各种不同形状和高度的图案。然后将该晶片用作模具把所述图案转移至聚二甲硅氧烷(PDMS)。在MSL中,在彼此顶部堆叠以不同模具模制的不同的PDMS层,是用于在重叠的“流动层”和“控制层”产生通道(63,70)。通过以互补的化学计量比率混合密封的预聚合组分和硬化剂组分将两层(或更多层)结合在一起以实现硬化作用。为了制造简易的微流控芯片,“厚”层由含有流动层的模具模制,而“薄”层由含有控制层的模具模制。在两层均部分硬化以后,把流动层从所述模具上剥离,并手动与控制层对准。使这些层结合,然后将该双层片从所述控制模具上剥离,然后打出入口和出口的孔,再将该双层片结合至PDMS的空白层上。在更多的结合时间之后,把完成的元件安装至载玻片上。
使用芯片外可计算机编程的螺线管控制所述装置中的流体流,其启动压力施加于控制层中的流体。当压力施加于这些管线时,位于重叠的正交控制和流体管线之间的弹性膜向流体通道中偏转,有效地阀调所述流体。这些阀门的不同组合可用于产生蠕动泵、多路复用控制,并分隔所述芯片的不同区域。
实施例1.2:细胞捕获
为了提高捕获效率,降低捕获器设计的大小选择性,并试图表征捕获器尺寸如何影响对不同细胞类型的捕获效率,设计了各种不同的捕获器几何体并对两种不同细胞系测试:K562细胞,平均直径18微米的人红白血病细胞系;以及nBAF3,平均直径12微米的鼠pro-B-细胞系。
制造了96种不同捕获器几何体,并在6种独立的装置中测试。细胞捕获测试装置的示意图如图2a所示。单个装置能够分析16种不同的捕获器几何体,在单个柱中各自重复83次,各个捕获器分开150微米。使用二元多路分用器(装置的顶部)来选择所需的捕获器几何体,且使用微流体蠕动泵(装置的底部)来泵送细胞流经所述装置。比例尺为500微米。该插图显示了包括两个细胞捕获器的放大区域。筛选几何体的测试参数为:“a”流体聚集器和捕获堰之间的距离,“b”从所述堰移除部分(即,筛元件)的宽度,“c”所移除部分(即,筛元件)的数目,和“d”捕获堰的尺寸。该插图的比例尺为100微米。图2b和2c分别显示了原始的和优化的捕获器几何体。图2d显示了接近图2c所示捕获器几何体的单个K562的光学显微图。图2e显示了被捕获后的同一细胞。在B-E中的比例尺为100微米。
优化了制造方案,从而能够可重复地制造出贯穿晶片的高的长宽比(7:2至14:1的范围)。在所有情况下,所述通道包含捕获器,在这些实施方案中,形成堰的捕获笼高为14微米,且所述漏斗由两个10微米宽的障碍物成角度组成。
细胞捕获器和漏斗之间的距离在22.5至8.5微米之间变化,对应于K562细胞平均直径大约1.25-0.5x范围,以及nBAF3细胞平均直径大约1.9-0.7x范围。对细胞进入细胞捕获室的观察表明,具有捕获细胞距离漏斗8.5微米的几何体使得单细胞填充系数最大化。然而,虽然该距离使填充系数最大化,还观察到阻碍数目的增加,因而选择10.5微米的距离在最终的装置中集成。
捕获笼的尺寸在12x12微米至20x36微米之间变化。如所预期,随着杯尺寸增加,观察到含有多于一个单细胞的捕获器数目也增加了。
还测试了去除1、2、3和4个部分以产生宽度在1-6微米(1,2,4,6)范围变化的筛元件。对细胞进入捕获器的观察表明,宽6微米的筛元件允许一些细胞挤压通过所述捕获器。在从捕获器中去除一个或两个部分(即在捕获器中产生一个或两个筛元件)之间没有任何可观测的差异。为了确定是否有足够的流体流经具有筛元件的捕获器,从而所述漏斗能够从该设计中去除,选择具有不同数目筛元件的捕获器进行了无漏斗的测试。在所有情况下,在不是直接位于漏斗下游的捕获器中没有观察到细胞被捕获。
因此,测试96种不同的捕获器几何体后,位于距离流体聚集器10.5微米处的12x12微米捕获杯,且从捕获堰中去除两个4微米片段,针对K562细胞观察到满意的捕获效率。
实施例1.3:细胞裂解
因其易于整合至微流控装置而选择了两种细胞裂解方法:涉及把样品加热至85℃达7min的热裂解,或由Invitrogen
该测试的结果可见于图3中。化学裂解的效率更接近类似于每10-倍稀释约-3.3个循环的预期值。然而,化学裂解所增加的益处,由于使用其需要在最终装置上添加额外的室从而牺牲重要的密度这一事实而被降低。
实施例1.4:细胞上样和芯片上细胞洗涤
如图4中所示的初步结果表明,不可能区分背景信号和来自单细胞的信号。然而,排除了该信号仅来自试剂污染的可能性,因为在芯片上细胞没有流经的区域不同于其余背景。该结果显示,在悬浮细胞的介质中存在不可忽视量的游离漂浮miRNA。该问题的解决方案是,在细胞裂解之前用干净的培养介质洗涤捕获器中所捕获的细胞,由此洗涤任何游离漂浮的miRNA、过量的细胞和来自所述芯片的任何其他碎片。
实施例1.5:RT-qPCR的细胞裂解物抑制作用
高浓度的细胞裂解物抑制分子生物学反应。为了确定最佳的细胞裂解物稀释度,以便能对样品进行随后的RT和qPCR反应,进行了下述实验。将芯片外制备的10x系列稀释的K562细胞裂解物(每个捕获室10个细胞至1/1000细胞等同物)上样至常规微流控装置,其含有测试各种细胞裂解物至RT体积稀释比率的一系列室。所有反应均在技术上一式三份进行。将人工合成的来自秀丽隐杆线虫(Caenorhabditis elegans)(cel-mir-2)的miRNA种类以0.2ng/nL的恒定浓度加入至逆转录混合物中。进行标准的miRNA脉冲逆转录38,39,将来自该反应的产物稀释5倍,进行芯片上qPCR,分析合成的秀丽隐杆线虫miRNA。
由于在K562细胞中没有发现分析的miRNA,可推测由于不同浓度的细胞裂解物对RT-qPCR的抑制效果。该实验的结果如图5所示。1nL、2nL、4nL和8nL RT室体积时,在每个捕获室10个细胞的浓度下观测到循环阈值(C
实施例1.6:RT-PCR稀释
测试了1:2.1至1:2 1稀释比率的RT-PCR,以确定该稀释度对qPCR性能的影响。将K562裂解物于试管中处理,逆转录,并以不同的稀释度加入至PCR混合物中。
图6显示了不同的RT-PCR稀释度对qPCR性能的影响。从左至右的点表示稀释系数2.1、3、4、5、6、7、8、10、16和21。将所有结果归一化至1:5,因其在由ASDF提出的方案中使用。由该图可知,在低(1:2.1,1:3)稀释系数下可看到反应抑制。模板的总浓度,以及因此分析的灵敏度随更高的稀释系数而降低。因此5x的稀释系数被用作所述最终装置中的稀释系数。误差线表示两个样品的标准偏差。
实施例1.7:预扩增验证和洗脱效率
设计、制造并测试了包括上述指出的设计考虑的装置,以便验证细胞捕获、洗涤和裂解的综合功能,然后进行逆转录和预扩增,并测试样品洗脱策略。该装置的示意图如图7所示。该装置特征在于能够平行捕获10个单细胞,1nL细胞处理室,15nL逆转录反应室,75nLPCR预扩增反应室,用于单独分配每个样品的因子多路分用器,以及蠕动泵。
10个K562单细胞被捕获和裂解。然后将人miRNA 16(hsa-mir-16)逆转录,通过qPCR确定相对拷贝数。该实验的实时PCR曲线如图8所示。所有细胞均表达miRNA 16,且所有曲线均出现平均循环阈值26,标准偏差在平均值的1C
实施例1.8:洗脱方法
如上所述,用于验证所述生物化学分析的综合功能的设计如图7所示,其包括因子多路分用器以便能够独立地分配每个室,以及微流体蠕动泵。整合了这两个设计特征以测试洗脱方法。由于qPCR测量溶液中DNA的相对起始浓度,用同样的体积洗脱每个室很重要。测试了多种洗脱方法:40 000微流体蠕动泵循环,压力驱动流体一段时间,洗脱至Whatman滤纸,然后将DNA重悬于TE缓冲液中,洗脱然后干燥样品并以已知体积重悬,用HarvardApparatus公司注射泵(PHD 2000)将流体注射至所述装置中。各测试结果总结于表1中。所有提供的数据均为10个重复的平均值,误差测量为测量值的标准差。
表1.测试洗脱方法测量结果的总结
尽管洗脱至Whatman滤纸上,且洗脱然后干燥样品产生了最具重复性的结果,这些方法增加了工作流程的大量时间。确定了用注射泵和高精度玻璃注射器(Hamilton,500uL)洗脱均产生了一致的洗脱结果而没有明显妨碍所述实验方案。由于注射泵可由计算机控制,该洗脱方法也可完全自动化。
为了评估洗脱效率,使上部分提出的qPCR反应达到饱和(40个循环,参见图8),通过用注射泵注入20uL将所得产物从所述装置中洗脱出来,以此稀释产物266x。来自这些室的实时曲线可参见图9。然后将这些室中的三个第二次洗脱,以评估洗脱效率。相对丰度平均降低约813±5倍,对应于99.877%±0.0008%的洗脱效率。
实施例1.9:最终的集成装置设计
使用适合高度多路复用单细胞基因分析所需的所有元件,设计了能够完全平行处理40个单细胞,7个标准物和NTC的装置。所述装置的设计与用于终端定量的Fluidigm48.48动态阵列(DA)的处理能力匹配。所述装置的示意图如图10所示。该装置的特征在于能够完全平行处理来自多达4种不同群体(可测试多达4种独立的细胞群,每种含有10个样品)的40个单细胞,7个标准物和无模板对照(NTC)。所述插图显示了阵列中一个单元的放大图。每个单元连接至试剂注入管线a和细胞上样管线f。控制管线b用于将每个单元与试剂注入管线分隔。多个控制管线c用于单独地递送每个样品。控制管线d、e和g用于将细胞处理区与随后的反应室和邻近的处理单元分隔。逆转录反应室位于细胞处理区下游的h处,紧接着是在j处的PCR预扩增室。控制管线i用于分隔各个反应室。控制管线k用于在热循环中使PCR室进行水合反应,而控制管线l用于分隔所述PCR反应室。通过出口m洗脱预扩增产物。该比例尺为1cm,且该插图的比例尺为1mm。
该过程的基本示意图如图11所示。简而言之,使用实施例1.1(图11A)中所述优化的捕获堰捕获细胞。通过使空白培养基流经所捕获的细胞对其进行洗涤后(将未捕获的细胞、碎片和细胞外miRNA洗涤出所述装置),阀门将上样通道分隔为1nL线性排列的裂解室(图11B)。然后,通过将整个装置放置于平板式热循环仪上并加热至85℃达7min来裂解细胞。裂解后,通过试剂注入管线注入逆转录混合物,将每个裂解室的所有内容物均推送至逆转录室(图11C)。用水冲洗试剂注入管线(图11D),然后将所述装置放置于平板式热循环仪上,运行脉冲RT方案。然后将所述RT产物推送至含有PCR混合物的预扩增室(图11E)。将所述装置于室温下放置1小时以使RT产物和PCR混合物扩散混合,并再次冲洗试剂注入管线,如图11D所示。再次将所述装置放置于平板式热循环仪上,并运行低循环预扩增PCR方案。预扩增后,将毛细管移液器吸头插入至每个出口,并连接注射泵中的注射器。通过将10uL洗脱混合物注入至每个室中系列地洗脱每个样品,稀释所述产物100x(图11F)。洗脱后将每个样品上样至Fluidigm 48.48DA芯片上单独的孔内。
整个过程耗费约16小时,并使用了不到5μL的RT混合物、10μL的预扩增混合物和500μL的qPCR混合物。总共约210个移液步骤。在试管中进行等同的实验需要多于240μL的RT混合物、960μL的预扩增混合物和多于46mL的qPCR混合物。该基于试管的实验将需要多于4700个移液步骤。因此,所提出的工作带来了改善的方案,大大降低了进行高度平行、高度多路复用单细胞分析所需的成本和时间。
实施例2
如图12A所示的集成微流控装置,可进行300个平行RT-qPCR分析,并执行单细胞捕获、裂解、逆转录和qPCR的所有步骤。为便于不同样品和细胞类型的精确比较,所述装置由6个独立的样品上样通道组成,各自含有50个细胞处理单元,使用约20μL的试剂进行总共300个RT-qPCR反应。设计要素包括1)允许有效分配单细胞而不产生机械损伤,2)使来自培养基中游离RNA或细胞碎片的背景信号最小化,和3)避免在nL体积中由细胞裂解物引起的反应抑制。
为了降低该装置的复杂性和避免RNA纯化的需要,将所述装置优化为与使用仅需向单个反应容器中连续加入试剂的“一步法”RT-qPCR方案的可商业提供的分析兼容。图12A中的矩形框显示了图12B中描绘的区域。每个细胞处理单元由复合的室组成,所述室由细胞捕获室顺序连接至两个用于RT和qPCR的较大辅助室构成(图12B)。每个单元的组成如下:(i)试剂注入管线,(ii)具有集成细胞捕获器的0.6nL细胞捕获室,(iii)10nL的RT室,和(iv)50nL的PCR室(比例尺:400μm)。图12C为具有黑色箭头所标示的所捕获单细胞的两个细胞捕获室的光学显微图。每个捕获器包括细胞漏斗,其包含引导细胞进入捕获区的导向板(比例尺:400μm)。图12D-I为所述装置操作的示意图(D-I的比例尺:400μm)。在图12D中,将单细胞悬浮液注入所述装置中。在图12E中,细胞捕获器将单细胞从液流中分离,并允许对细胞进行洗涤以去除细胞外RNA。在图12F中,启动气动阀导致在热裂解之前单细胞分离。在图12G中,通过试剂注入管线注入用于逆转录(RT)反应(10nL)的试剂。在图12H中,用随后用于PCR的试剂冲洗所述试剂注入管线。在图12I中,将用于qPCR的试剂与RT产物在50nL的qPCR室中混合。
该简单的流控结构允许在热裂解后进行两步法RT-qPCR(图12D-I),或者化学裂解后进行一步法RT-qPCR。所有通道均连接至一个共同的供给通道,在每个反应步骤完成后其用于通过上游的室来注入下一步反应的预混液(master mix),从而稀释该中间产物(细胞裂解物或cDNA)并汇集下一步反应的混合物。该以微流控形式汇集的平行反应,确保了所有反应步骤相同的时间,且大大降低了与以μL体积移液和混合步骤相关的技术不定性。进行荧光测量以确保每个步骤反应物高效和可重复的转移,表明样品转移中的损失小于5%。为使装置成本和复杂性最小化,使用外围硬件包括安装在平板式热循环仪上的CCD检测器进行温度控制和荧光检测。
实施例2.1:材料和方法
装置制造和操作
如上所述,对单细胞进行mRNA和miRNA丰度测量,发现与通过数字PCR对细胞裂解物测量的平均拷贝数一致(图20c)。通过多层软蚀刻技术制造微流控装置(70,71)。使用用CAD软件(AutoCAD,Autodesk公司)设计的光掩膜,通过光蚀刻限定平面硅模具,并以20,000点/英寸的分辨率(CAD/Art services公司)印刷在透明膜上。使用SU8-2025光刻胶(Microchem,美国)制造“对照”模具,以沉积特征为24μm高的阀。用3步光刻制造“流动”模具。首先,使用高13μm的SPR220-7光刻胶(Shipley,美国)制造用于试剂注入的通道以及室之间的连接。所述SPR通道为圆形,以便于通过在115℃孵育15min阀门关闭。于190℃硬烤2小时,以防止在加入随后层期间SPR光刻胶的侵蚀。其次,所述细胞捕获器的特征为以14μmSU8-2010光刻胶(Microchem,美国)来限定。最后,用高150μm的SU8-100光刻胶构建较大的室和流体总线。根据生产商的说明书来进行所有光刻胶处理。
由这些聚二甲硅氧烷(PDMS,RTV615,General Electric,美国)模具浇铸微流控装置。每个装置由3层弹性结构组成,具有空白底层,中间“控制”层,和上面“流动”层,其中所述“控制”层具有通道,其通过向上推和缩小上面“流动”层中的通道来起着阀门的作用。所述模具首先用三甲基氯硅烷(TMCS,Aldrich)蒸汽处理2min以防止PDMS结合至光刻胶结构。所述液流层通过以下制备:将PDMS(5份RTV615A:1份RTV615B)混合物倒入所述流动模具中,排气,并在80℃烘烤60min。所述薄的控制层通过以下制备:用PDMS(20份RTV615A:1份RTV615B)以1800rpm旋转涂覆所述控制模具,并在80℃烘烤45min。烘烤后,将流动层的PDMS从流动模具上剥离下来,并与所述控制层对准。在80℃烘烤60min后,将该粘结的两层结构与所述控制模具分开,进行通道进入孔打孔。所述空白层(无通道)通过以下制备:在空白晶片上旋转(2000rpm)涂覆PDMS(20份RTV615A:1份RTV615B),并在80℃烘烤45min。将粘结的流动和控制结构安装至空白层上,并在80℃烘烤3小时。最后,将该3层粘结的模具结构从所述空白模具上移除,切分为单个装置,且通过在80℃烘烤过夜将这些单个装置各自粘结至干净的载玻片。
所述装置操作需要控制9个气动阀门,并可用简单的各种手动阀门操作。对于当前的研究,使用了半自动的操作,其中微流控阀门通过螺线管制动器来控制(Fluidigm公司,旧金山),所述制动器通过使用LabView驱动器(National Instruments)操作的数字化输入输出卡(NI-DAQ,DIO-32H,National Instruments)来控制。通过放入控制管线孔口的20gauge不锈钢测针(Small Parts公司),聚乙烯管将所述螺线管连接至所述微流控装置。使用Krytox(DuPont)油作为控制管线中的流体,以30psi的压力启动阀门。
微流控单细胞RT-qPCR
所述装置设计为与可商业获得的RT-qPCR产物相兼容。将热裂解后进行两步法RT-qPCR的方案用于miRNA和OCT4 mRNA分析。可选择地,将化学裂解后进行一步法RT-qPCR的方案用于SNVs和GAPDH的mRNA测量。
通过热裂解和两步法RT-qPCR进行单细胞转录测量
通过使含有0.5mg/mL牛血清蛋白(BSA)和0.5U/μL RNA酶抑制剂的PBS流入所有通道来灌注所述装置,同时使RT和PCR室保持为空的并通过阀门分隔。BSA有助于防止细胞粘附至通道壁上。在灌注后细胞上样前,所有阀门均为关闭的。通过向插入至样品入口处的微量移液管吸头施加压力(~2-3psi),将单细胞悬浮液注入至所述装置。样品入口首先针对入口阀进行死端灌注以防止起泡进入所述装置。样品入口阀、细胞室阀和出口阀均打开以允许细胞悬浮液流经所述样品通道。将细胞上样至该装置,所述装置悬浮于培养基中(直接来自培养)。细胞上样浓度保持在5x10
在注入细胞悬浮液和捕获单细胞后,关闭细胞样品入口阀,通过用于灌注该装置的PBS溶液冲洗该管线来洗涤所述细胞。这去除了未捕获的单细胞、胞外RNA和碎片。芯片上洗涤后,关闭细胞室阀门以分隔细胞上样通道,并隔离单独的细胞反应器。使用显微镜目测细胞捕获室来确认和计数各个室中的细胞数目。通过将微流控装置放置在平板式热循环仪上并于85℃加热7min(然后冷却至4℃)来裂解细胞。
通过使用ABI高容量逆转录试剂盒(68),并加入表面活性剂以防止和端和蛋白吸附至PDMS表面,在装置中进行逆转录(RT)(2μL 10×逆转录缓冲液,来自ABI的4μL 5×RT茎环miRNA引物,1μL 100mMdNTPs,1.34μL的50U/μL Multiscribe逆转录酶,0.26μL的20U/μLRNA酶抑制剂,2μL 1%吐温20,9.4μL PCR级水)。将所述RT混合物上样至所述装置并流经所述试剂注入通道冲洗。通过打开连通细胞室与RT室的阀门,并打开连通细胞室与试剂注入管线的阀门,将RT试剂注入至该反应。在关闭至试剂注入管线的连接之前,对所述RT室进行死端灌注。通过将微流控装置于平板式热循环仪上实施脉冲温度RT方案(16℃下2min,然后20℃下30s,42℃下30s,50℃下1s共60个循环)。于85℃(5min)下使RT酶失活,然后将装置冷却至4℃。
用25μL的2×TaqMman通用预混液(ABI)、2.5μL 20×Real-TimemiRNA分析试剂(引物和探针,ABI)、5μL的1%吐温20和7.5μL的PCR级水制备PCR试剂。使该PCR试剂流经所述试剂注入通道以冲走所述RT试剂。打开阀门并将PCR试剂注入以稀释该RT产物至PCR反应室中。完全灌注PCR反应室后,启动关闭PCR室的阀门,将所述装置转移至封闭空间进行实时PCR(Biomark
通过化学细胞裂解和1-步法RT-qPCR进行单细胞转录测量
使用Cells Direct试剂盒(Invitrogen,美国)进行mRNA转录测量(SP1,GAPDH)。除了几点区别以外,用于化学裂解和一步法RT-qPCR的微流控装置的操作,类似于上述用于热裂解和两步法RT-qPCR的方法。灌注所述装置并用含有0.5mg/mL BSA的PBS洗涤细胞。由于化学裂解缓冲液(10μL裂解重悬缓冲液,1μL裂解促进液,Invitrogen,美国)含有RNA稳定剂,无需再使用RNase抑制剂。细胞上样与热裂解和两步法RT-qPCR的方案相同。通过注入化学裂解缓冲液流经细胞捕获室并装满10nL室(用于两步法方案中的RT试剂注入)来裂解单细胞。于室温下孵育该裂解反应10min,然后通过将该装置放置于平板式热循环仪并于70℃孵育10min来使裂解试剂热失活。然后将一步法RT-qPCR混合液(1μL的SuperScript IIIRT/铂Taq混合液,25μL的2×反应混合液(含有ROX参比染料),2.5μL的20×Taqman分析试剂(引物和探针,ABI),1μL的50mM MgSO
数字PCR实验
为了进行mRNA数字PCR分析,将细胞用含有0.5mg/mL BSA的PBS洗涤,溶解于化学裂解缓冲液中,根据以上描述的方案在试管中进行逆转录,将所得cDNA产物上样至数字PCR阵列。用于miRNA研究,将细胞裂解于含有0.5mg/mL BSA和0.5U/μLRNase抑制剂的PBS中。使用miRNA茎环引物(Applied Biosystems,美国)和高容量cDNA逆转录试剂盒(AppliedBiosystems,美国)以10μL体积进行逆转录。在注入微流控数字PCR阵列之前,如以上描述的芯片上两步法RT-qPCR方案,将RT产物加入至PCR试剂。也使用与以上描述相同的方案进行数字PCR阵列的热循环。通过多层软光刻技术制造PDMS数字PCR阵列,其包括765个2nL独立的PCR室,类似于Warren等人描述的设计72)。热循环后,计数阳性室,基于二项分布推断出实际分子数目。
实时PCR系统
BioMark
数据采集的基本规格包括:
图像分辨率和位深度:4百万像素,16位
滤光片:FAM:Ex 485/20Em 525/25;
VIC:Ex 530/20Em 570/30;
ROX:Ex 580/25Em 610/15;
QAS:Ex 580/25Em 680/25,
光源:175W氙弧灯泡
RT-qPCR分析
在基因组DNA存在下测量mRNA需要设计特异性靶向成熟mRNA序列的引物。在许多情况下,这可通过设计内含子跨越引物来实现。将特别设计的茎环RT引物系统(AppliedBiosystems)用于特异性靶向成熟miRNA。
用于GAPDH和miRNA的TaqMan分析仪(Applied Biosystems,分析仪IDHs99999905_m1)获自Applied Biosystems公司。对于GAPDH,以微升体积的大块细胞裂解物(以芯片上单细胞的相等浓度,10
OCT4(POU5F1)引物序列获自RTPrimerDB
将具有增强的双链稳定性的BHQ-Plus探针(Biosearch Technologies公司)用于SNV检测以允许序列长度更短并增强特异性。对于SP1位点,SNV位置选自Shah等人(73)中的表2。将hg18序列上该位置侧翼的200bp用于使用引物3的分析设计。所得引物和探针序列如下(所述SNV有下划线):
SP1突变体探针:FAM-AGGCCAGCAAAAA
5'修饰:FAM,3'修饰:BHQ-1Plus。Tm=62.7℃
SP1 WT探针:Cal Fluor-CAGGCCAGCAAAAA
5'修饰:CAL Fluor Orange 560,3'修饰:BHQ-1plus。Tm=62.1℃
SP1正向引物:CCAGACATCTGGAGGCTCATTG Tm=65.8℃
SP1反向引物:TGAACTAGCTGAGGCTGGATATm=66.0℃
无逆转录酶的对照实验显示阳性扩增。因此,通过RT-qPCR在单细胞中测量SP1突变体和野生型丰度,在成熟mRNA转录本和基因组DNA之间没有区别。
图像分析
每个PCR循环后,拍摄整个装置至少两种不同颜色(一种被动参比染料和一种或多种报告基团染料)的荧光图像,使用MATLAB(MathWorks)软件记录的常规数据进行分析以产生实时扩增曲线。将反应室与使用被动参比染料的第一图像的其余图像分割。人工旋转该图像以便所有反应室图像为方形边缘。其次,计算每行和列的平均图像密度,并人工设定阈值以将亮区与背景区分。含有发光行和发光列的区域被分配给反应室。
通过使该原始图像和待分析图像的平均行和列强度之间的绝对距离最小化,将所有后面的图像均自动与该原始图像比对。对于每个图像,针对每个反应室记录了报告基团染料和被动染料的强度。通过将每个报告基团染料的强度归一化至所述被动染料的强度来产生实时扩增曲线。通过将一条线的等式与指数前区域拟合,并将来自整个曲线的结果进行外推和扣除来从这些曲线中去除线性成分。用于确定CT值的阈值被自动确定为所有扩增曲线的最大二阶导数的归一化荧光值中位数。
mRNA FISH
发现在单细胞中Oct4表达的微流控测量结果与通过mRNAFISH获得的OCT4测量结果一致。对于mRNAFISH的测量,用PBS洗涤生长于LABTEK腔室盖玻片上的细胞,于室温下4%甲醛中固定10min,于40℃下70%乙醇中过夜渗透。第二天用洗涤缓冲液(含15%甲醛胺的2×SSC)洗涤细胞,然后于30℃与适当稀释的对OCT4特异的mRNAFISH探针(参见表2)在杂交溶液(含硫酸葡聚糖,酵母tRNA,NEB,BSA,15%甲醛胺的2×SSC)中杂交过夜。第二天早上抽吸OCT4杂交溶液,连续冲洗细胞并于30℃用洗涤缓冲液孵育30min,然后用2×SSC洗涤。将一滴(25μL)含有DAPI的Slowfade GOLD抗荧光淬灭试剂加入至所述细胞,立即用盖片封盖并成像。使用带有100×物镜(N.A.1.3)的Leica DMI6000B倒置显微镜,在DAPI和Texas红滤光片光谱中,获得了每个细胞32-64张堆叠的mRNA杂交图像(间隔0.5μm)。
人工评估每个图像栈中对应于单个mRNA分子的荧光点,因为发现使用此前报道算法的自动阈值设定是不可靠的。该过程中自动化的困难在于使用已报道方案的信噪比不一致,且可能涉及hESC细胞的厚度(~15μm)。此外,需要人工干预来确定邻近细胞的边界。为了优化信噪比,我们有系统地改变探针浓度、孵育时间、孵育温度,以及杂交缓冲溶液中的甲醛胺浓度。
表2.对OCT4特异的mRNAFISH探针序列
细胞培养
将K562细胞培养于补充有10%胎牛血清(FBS)(Gibco)的杜尔伯克氏改良伊格尔培养基(DMEM)(Gibco)中。使用RNA MiniPrep(Qiagen,美国)试剂盒从K562细胞提取纯化的RNA。
在额外补充有抗生素-抗真菌素(100U/mL青霉素,100mg/mL链霉素和0.25mg/mL两性霉素B)(Invitrogen,Carlsbad,CA,美国)的mTeSR(76)基础培养基(STEMCELLTechnologies公司,温哥华BC,加拿大)中增殖CA1S hESCs(74,75)。细胞转代时,用磷酸盐缓冲液(PBS)洗涤hESC,然后用TrypLE Express(Invitrogen,Carlsbad,CA,美国)于37℃孵育10min,以基于融合将单个hESC从4-8天的培养物中分离。用补充有抗生素-抗真菌素的mTeSR来中和TrypLE Express,然后将悬浮液转移至含有1:30稀释的Matrigel基质胶(Becton Dickinson,San Jose,CA,美国)和补充有抗生素-抗真菌素的mTeSR预涂层的新的组织培养皿中。为了分化,接种细胞1天后,用含有10%胎牛血清(FBS)的杜尔伯克氏改良伊格尔培养基更换mTeSR。
当收获hESC用于qRT-PCR时,用TrypLE Express(Invitrogen,Carlsbad,CA,美国)于37℃孵育细胞20min,以便从4-8天的培养物中产生更均匀的单细胞悬浮液。
分离自小叶乳腺癌转移灶的原代细胞的冻存管,由BC癌症研究中心根据英属哥伦比亚大学伦理指导提供。为增加活力,将细胞转移至新鲜培养基,在于所述微流控装置中分析之前孵育2天。
转移效率测量
将含有10μM FAM-标记的40-mer poly-A寡核苷酸(IDT,美国)、0.1%吐温20和100×稀释的ROX被动参比染料(来自CellsDirect试剂盒,Invitrogen,P/N 54880)的溶液上样至细胞捕获室,依次推送至10nL和50nL室,所述室中含有0.1%吐温20和100×稀释的ROX参比染料的水溶液。将获得的FAM和ROX荧光图像用于测量寡核苷酸从一个室至下一个室的转移。每个室的转移效率计算为(初始信号–最终信号)/(初始信号),其中,信号=(FAM强度–FAM背景)/(ROX强度–ROX背景)。转移效率较低限的保守估计值采用转移效率的平均测量值的一个标准偏差。
细胞捕获测量
使用以上描述的方案制造了具有细胞捕获器几何体的线性排列的常规微流控装置。将所述装置安装至倒置显微镜(Leica DM IRE2),使用CCD照相机(Hamamatsu ORCA-ER)在光亮区域成像。用含0.05%牛血清蛋白(BSA)(Gibco)的磷酸盐缓冲液(PBS)(Gibco)灌注所述装置。在该装置中上样之前,在新鲜培养基(补充有10%胎牛血清(Gibco)的杜尔伯克氏改良伊格尔培养基(DMEM)(Gibco))中洗涤细胞两次。之后将最终的洗涤细胞重悬为1百万/mL浓度。用Cedex自动化细胞计数仪(Roche innovatis AG)测量输入样品的活力。
为了测量捕获效率,用下流微流体蠕动泵以约1nL/秒的速率泵送细胞流经所述阵列,记录了在成功捕获事件之前流经每个捕获器的细胞数目。使用几何分布的最大似然估计值,将这些计数与R(2.11.1版)的fitdistr函数(MASS软件包,7.3-6版)拟合。将效率记录为成功捕获每个细胞的可能性。
为了测量上样后的细胞活力,如上所述使用压力驱动流体将细胞上样至所述阵列中直到观察到高的捕获占有率为止。使含0.2%台盼蓝(Gibco)的PBS溶液流经所捕获的细胞。用未染色细胞除以细胞总数计算出细胞活力。
使用ImageJ(1.43u版)从Cedex图像和微流控装置中所捕获细胞的图像测量细胞的直径。使用双样本t-检测来测试所得大小分布差异很大的假设。使用F检测测试同方差性假设。为了优化细胞捕获器几何体,通过使所述杯在聚集器的一个细胞直径之内,并通过包括穿过所述杯的小旁路分流,类似于Skelley等人(10)中所示的杯几何体,将细胞捕获效率提高至87%。
通过扩散混合
通过扩散混合溶液在所述微流控装置中表征为将荧光标记的40bppoly-A寡核苷酸上样至10nL室,并推送所述室中的内含物至邻近的50nL室。使用延时成像来测量在PCR室中荧光标记的寡核苷酸分布随时间的演变(图S7)。每个室中随时间的像素强度标准偏差被用作混合的度量。将所有分析室所得的曲线(N=200)采用最小二乘回归各自进行指数衰减拟合,以确定特征性混合时间常数。这导致15.2±1min的平均混合时间。
使用Stokes-Einstein关系式并假设无规卷曲,我们估计40bp寡核苷酸的分散常数为:
其中K
实施例2.2:微流控RT-qPCR的精确度和灵敏度
将室体积设计为确保每个处理步骤之间足够的稀释,以避免反应抑制并同时维持高模板浓度和分析灵敏度。
图13A为整个装置的荧光图像,其显示了6个通道中的300个反应,系列稀释的K562细胞的纯化总RNA经40个PCR循环后所拍摄。从左至右的样品为40pg/室、5pg/室、625fg/室、78fg/室、10fg/室和无模板对照(NTC)。有限稀释的单分子扩增导致了10fg和78fg通道的数字扩增模式。在NTC通道(N=50)没有观测到扩增。图13B显示了从类似于图13A图像的处理序列产生的300个实时扩增曲线。确定CT值的阈值由虚线表示。图13C描绘了来自8×系列稀释的纯化总RNA针对GAPDH的芯片上(下方)和芯片外(上方)RT-qPCR,其显示了在nL体积的反应中提高的灵敏度。在所述微流控系统中,10fg样品的CT值对应于50个室的19个中检测的单分子扩增。单细胞测量结果的平均值和标准偏差对于芯片上和芯片外分析均显示为绿色。获自芯片上的CT值对应于每个细胞20pg RNA的平均值。由于上清液中碎片和游离RNA的残留信号,在PBS中洗涤两次并通过玻璃毛细管分离的K562单细胞在芯片外的测量结果表现出人为增加的水平。在约2μL上清液中转移细胞,测量该上清液含有~20pg细胞外RNA。误差线代表所有扩增反应所测量CT值的标准偏差。图13D显示了在K562细胞裂解物稀释液中GAPDH的实时扩增曲线。图13D显示了在系列稀释的细胞裂解物中所测量的GAPDH的CT值。单细胞裂解物中没有发生抑制作用。
细胞裂解物稀释显示,在浓度大于0.2个细胞/nL,或每50nL反应中10个细胞时,反应抑制变得明显(图13D)。进行试管中实验以确定,至少5:1的稀释率(PCR混合物:RT产物)是PCR效率的优选。因此,我们设计组合的反应器,其具有总体积60.6nL,由0.6nL细胞捕获室、10nLRT室和50nL qPCR室组成。这些体积允许单分子的可靠扩增(图13A),当从单细胞等量的RNA(20pg)起始时,其产生330ng/mL的最终模板浓度。使用较大体积的RT和PCR室还具有降低其表面积与体积比率的额外优点,从而使试剂通过透气性装置材料(聚二甲硅氧烷)的蒸发最小化。
通过进行范围为40pg(~2个细胞等同物)至10fg(~1/2000个细胞等同物)的8-倍稀释系列的总RNA的GAPDH表达测量,测定所述装置的RT-qPCR灵敏度和精确度。RNA纯化自K562细胞——来自慢性骨髓白血病患者的BCR-ABL阳性人细胞系(77)(图13A-C)。通过4个最高模板浓度(40pg、5pg、625fg、78.125fg)确定扩增效率,作为log2(C)对CT的线性最小二乘拟合的斜率,并发现其为0.988±.055。在3个最高浓度(对于40pg、5pg和625fg样品分别为s.d.=0.08,0.10,0.14)时,CT值的标准偏差小于0.15,表明整个阵列均匀扩增且技术误差小于绝对浓度的10%,接近qPCR精确度限度。在78fg样品中观测到最高的测量结果变异性,其散粒噪声(泊松样品噪声)最明显,并占测量结果变异的约50%。模板量小于625fg导致单分子扩增的数字模式特征(78fg为49/50,10fg为19/50),并与通过二项式分布(2)确定的预期的室占有率一致。基于在10fg样品中的单分子检测频率,我们测量了GAPDH的平均拷贝数为每个单细胞等同物(20pg)有979±240个转录本拷贝(图13)。来自芯片上qPCR和芯片外合成的cDNA的CT值比较表明,在同样的RNA浓度下工作时,芯片上RT效率等于芯片外的效率。图14显示了来自K562细胞裂解物在微流控装置中进行RT或在所述装置中进行qPCR之前在试管中进行RT的GAPDH测量结果比较。获得的CT值(插图)显示无明显的效率差异。最后,对于同一系列稀释的RNA,在芯片上和在试管中(20μL体积)的GAPDH分析比较(图13C)表明,芯片上分析提供了提高的灵敏度。
实施例2.3:细胞捕获
并入漏斗(导向板)的细胞捕获室位于距离捕获器22.5μm处,从而将细胞会聚于捕获效率最高的中心流线(图12C)。使用这些结构实现了阵列位置的高单细胞占有率(图15A,B)。在8个单独的实验中,~60秒的上样方案(106个细胞/mL,20nL/s每个通道)导致成功地分离了1518/1700个室中的单细胞(89.3%),细胞捕获效率为5.0±0.5%。上样后用TrypanBlueTM染色来评估细胞活力,并确定为相当于输入样品的活力(97.4%活力vs.输入时96.8%)。最后,在上样之前和之后对细胞直径分布的测量结果表明,在选择不同大小的细胞时细胞捕获没有带来明显的偏差(p=0.67,双样本t-检测)。图16提供的柱状图显示,通过Cedex测量的原始样品中的细胞大小分布(图16A)与通过微流控捕获器分离的细胞的大小分布(图16B)一致,假定捕获细胞的球形细胞直径分布对应于平均体积4.2pL,标准偏差为2.0pL。将该细胞捕获器几何体和上样方案用于下述的所有后续qPCR测量。对捕获器和漏斗(即导向板)几何体的进一步改善发现,达到填充系数>99%(分析的100个捕获器中捕获了100个单细胞)和细胞捕获效率为87.0±4.5%,细胞活力再次与输入样品相当(>98%),且细胞大小无明显偏差(p=0.35,双样本t-检测),使得该方法可用于分析有限量的样品,如较少的干细胞或临床样品。
图15A显示了沿着装置的所有6个通道的每个室中细胞的位置,如通过亮视野显微镜所确定,其表示为白色圆圈并覆盖于通过K562细胞中GAPDH的RT-qPCR测量获得的CT值热点图上。深色圆圈显示为NTC。图15B为显示图15A所示实验的CT测量结果的散点图。插图中的柱形图显示了93.2%的单细胞占有率。图15C显示了K562单细胞(N=233)中测量的GAPDH转录本数目分布。
实施例2.4
将细胞捕获室的捕获器内细胞的固定同样用于在裂解之前在芯片上洗涤细胞,以去除游离RNA、细胞碎片和未捕获的细胞,否则它们会产生背景信号或导致单细胞占有率低。图17A提供了注入微流控装置之前,在芯片外PBS中洗涤的细胞的GAPDH测量结果,没有芯片上洗涤,该测量结果含有来自上清液中模板的背景信号。没有芯片上洗涤,未捕获的细胞保留在捕获室中,导致了更少的单细胞测量结果(插入的柱形图)。如图17B所示,发现芯片上洗涤降低了来自上清液中游离RNA的背景信号,并显著增加了所分析的单细胞数目。如图18所示,由于小体积处理,通过与芯片外的结果比较,洗涤后残余RNA的检测显著降低。图18A描绘了沿着装置的所有6个通道的每个室中的细胞位置,如通过亮视野显微镜所确定,其表示为白色圆圈并覆盖在通过RT-qPCR测量K562细胞中的miR27a获得的CT值热点图上。深色圆圈显示为NTC。图18B为对应于图18A所示的实验在30个PCR循环后整个装置的荧光图像。由于断续的荧光点邻近反应室,热裂解后保留了细胞尸体并为可见的。
图17C提供了来自上样的纯化RNA洗涤或未洗涤所述细胞捕获室的GAPDH测量结果比较。通过上样纯化的RNA模板(36.5ng/mL),然后洗涤并进行RT-qPCR分析,确定室洗涤效率为>99.99%(未洗涤测量到1.1′104拷贝,洗涤后检测为0拷贝)。此外,检测不同细胞上样和洗涤方案的RT-qPCR测量结果表明,相比芯片外洗涤步骤后在μL体积中分析,芯片上洗涤允许从培养基中直接上样具有低的背景(图17C)。重要的是,芯片上洗涤允许在洗涤的几秒钟内裂解,从而最小化可能由于连续的培养基更换和旋转(spin)步骤引起的假转录响应。
实施例2.4
通过比较纯化RNA的GAPDH测量结果和直接对K562单细胞进行的测量结果,评估了芯片上细胞处理的效率和可靠性(图13C,图15C)。上样直接来自培养基的K562细胞,然后洗涤,并用化学裂解和一步法RT-qPCR方案(Cells Direct
实施例2.5:单细胞miRNA表达测量的应用
接着将该技术应用于单细胞miRNA表达研究。miRNA的长度短(~22个核苷酸)使其难以通过杂交方法来检测,从而RT-qPCR是其主要的定量策略。为了证明该技术的稳定性和生产能力,进行了总计1672个单细胞测量,以检测在跨越宽范围丰度的9种miRNA表达(>16000拷贝/细胞至<0.2平均拷贝/细胞)中的单细胞活力。再次将K562细胞选定为该研究的异质群体,因为已知其表现出红细胞、粒细胞和单核细胞的综合特征(77,78)。将评估miR-16(在许多种组织中发现高度表达的微小RNA(79))的表达作为归一化的内部标准(57)。miR-16在K562细胞中为对数正态分布,但相比GAPDH具有略低的表达和明显更严格的调控,具有平均804(s.d.=261)拷贝/细胞和标准偏差30%(CT平均值=21.4,s.d.=0.4)。对通过微量移液管分离至20μL体积小管中的单细胞的比较实验,显示测量结果变异性增加至~90%(CT平均值=29.5,s.d.=0.9),证明在nL体积中的平行微流控细胞处理提高了精确度(图20A)。在芯片上和芯片外测量结果之间所观测的CT平均值的变化是由于较低的模板浓度,因而增加了在芯片外样品中所需的PCR循环。
(C)(D)在K562细胞中表达的9种miRNA的2072个单细胞测量结果。间接得到的柱形图表示了每种miRNA的表达分布。
为了证明该装置在单细胞中测量差异表达的效用,测量了miR-223(涉及髓样分化的miRNA)的表达。相比miR-16,发现K562细胞的miR-223表达高度可变(CT平均值=22.2,s.d.=1.6,拷贝数=513,s.d.=406),且不是对数正态分布(图20B;最右边的柱表示未检测到miR-223的细胞(ND)),与已知的K562细胞功能异质性一致。这些测量结果突出了单细胞miRNA表达分析在评估细胞群异质性和鉴定作为细胞状态有用生物标记物的miRNA中的效用。为进一步探索该可能性,测量了其他7种miRNA(共9种)的表达,对K562细胞群中单细胞表达的模式绘图。图20C显示了相比来自大块细胞裂解物的数字PCR测量结果,在微流控装置中通过RT-qPCR测量的平均单细胞miRNA拷贝数。误差线表示每种miRNA的单细胞测量结果的标准偏差。按照以上描述的过程,将通过数字PCR获得的单分子CT值用于将所测量的CT值转换为绝对拷贝数。假设100%的扩增效率,观测到计算为2(CT(单细胞)—CT(单分子))的拷贝数,与通过由大块裂解物制备的cDNA的数字PCR获得的平均拷贝数具有很好的相关性(系数为0.9932)。图20D显示了在K562细胞中表达的9种miRNA的2072个单细胞测量结果。间接得到的柱形图表示了每种miRNA的表达分布
单细胞测量结果揭示了miRNA表达的不同模式,其中miR-16、miR-92和miR-17-5p分别显示出单峰和严格调控的分布,而miR-223、miR-196a和miR-145显示出多峰分布和高水平的细胞异质性。对于最低丰度的miRNA即miR-200a,仅在一小部分细胞中检测到其表达,且表达水平低于每个细胞~5拷贝。所有细胞的平均miR-200a拷贝数在通过数字PCR获得的因子2之内(每个细胞0.2拷贝)。反之,发现miR-92为最高丰度的miRNA,在每个细胞中存在约60,000拷贝。这些测量结果确立了具有大于104的动态范围和在单分子灵敏度上的单细胞miRNA定量。
为了说明单细胞测量结果在精确评估在两种不同细胞群之间平均表达和异质性两方面的差异的效用,将K562细胞中miR-16和miR-223的表达水平与在CA1S细胞(人胚胎干细胞系(hESC))中的表达水平进行比较。尽管发现miR-16在hESC中的表达水平类似于其在K562中的表达水平(ΔCT=0.6),观测到约2倍大的表达变异性(平均CT=22.0,s.d.=0.7)(图20A)。相反,相比K562,miR-223的CA1S单细胞测量结果显示了巨大下调,其中仅在3.6%的细胞中检测到miR-223。
实施例2.6:单细胞中miR-145和OCT4的共调控
单细胞中多种转录的测量允许定量测量基因共调控,否则其可能会被细胞的异质性所掩蔽。为了证明这种能力,在hESC分化期间进行光学上多路复用的单细胞RT-qPCR分析以研究miR-145和OCT4(已知的miR-145靶标)的共调控(图21A-C)。图21A为在分化的hESC中OCT4和miR145共表达的多路复用分析的散点图。彩色编码的点表示每个时间点的单细胞测量结果(N=547)。十字交叉表示群体的平均拷贝数。图21B和21C是显示表示在坐标轴上投射的每个转录本分布的柱形图,虚线表示平均拷贝数。在分化的第0、4、6和8天进行了总计1094个单细胞的测量。用每个时间点的细胞分布绘图显示出这些转录本的演化,并显示8天后平均miR-145水平增加了约20倍(拷贝数:D0:平均值=18.9,s.d.=25.5,D8:平均值=380.3,s.d.=259.4)。miR-145的增加还伴随着OCT4逐渐下调,8天后最终达到平均30-倍抑制(拷贝数:D0:平均值=755.7,s.d.=306.4,D8:平均值=27.8,s.d.=124.5)。这通过mRNA-FISH得以单独地证明,如图22所示在CA1S细胞中用DAPI复染OCT4的mRNA-FISH图像。图22A为FBS分化7天后,CA1S细胞中OCT4的mRNA-FISH代表性图像。通过人工检查图像堆栈来估计OCT4 mRNA的平均拷贝数,确定为42(s.d.=41,N=6)。图22B为未分化CA1S细胞的代表性图像。通过人工检查图像堆栈来估计平均拷贝数,确定为988(s.d.=368,N=6)。比例尺=10am。
尤其是,第6天的单细胞分析显示OCT4和miR-145的双峰分布,揭示了细胞状态的转换。不意欲受假说束缚,认为这很可能反映了细胞亚群的自发分化。所观测到的miR-145和OCT4共调控的单细胞动力学在群体测量中并不明显,突出了可扩展的单细胞转录分析在将分子标签与细胞抉择机制相关联中的应用。
实施例2.7:原代细胞中的SNV检测
为了确定所述方法的特异性,将mRNA单核苷酸变体(SNV)的多路复用测量用于评估原代肿瘤样品中的基因组异质性。分析了分离自转移性乳腺癌胸膜积液的总计117个单细胞的转录因子SP1的SNV突变体表达,此前通过深度测序确定(73)(图21D)。使用在SNV位置侧翼的序列设计引物,且所述引物在基因组DNA和mRNA转录本之间没有区别。在分析的117个原代细胞中,对于突变体和野生型等位基因,22个(18.8%)为杂合性的,85个(72.6%)为纯合子野生型,1个(0.9%)为纯合子突变体,有9个(7.7%)未检测到转录本。在37个对照K562细胞中未检测到SP1突变,且在这些细胞中仅有2个未检测到野生型转录本。在原代样品中没有拷贝数改变的情况下,这些观测到的频率表明突变体与野生型SP1比率为11.2%(18.8×1+0.9×2=20.6突变体比18.8×1+72.6×2=164野生型)。然而,对来自原代样品的纯化DNA使用数字PCR,发现突变体比野生型SP1等位基因的比率为18.7±2.3%,与此前报道的通过深度测序获得的比率21.9%一致(73)。鉴于原始样品中肿瘤细胞的频率为约89%(73),DNA分子计数和单细胞RNA表达测量结果均显示,该肿瘤的癌症转移源自多种癌细胞谱系。
实施例3:文库构建
如此前所描述,将K52细胞悬浮液直接上样至微流控装置。用PBS溶液于3psi下洗涤固定在捕获器中的捕获细胞10min。通过启动阀门来分隔包含单细胞的室,热裂解(85度5min),并通过第二入口用第一反应混合物装满位于细胞捕获器上游的细胞捕获室,所述第一反应混合物包括用于连接的3'miRNA接头。
接着,通过所述第二入口加入将5’DNA接头连接至miRNA的5'端的连接混合物。将包含裂解细胞和第一反应混合物的细胞捕获室与较大的第一辅助室之间的接口阀打开,以使细胞捕获室的内含物和连接混合物在5psi压力下一起冲入第一辅助室。
在第一辅助室中进行连接后,通过所述第二入口加入cDNA合成混合物,以由5'接头-miRNA-3'接头分子合成cDNA。将包含裂解细胞和第一反应混合物及连接混合物的第一辅助室与较大的第二辅助室之间的接口阀打开,以使第二辅助室的内含物和cDNA合成混合物在5psi压力下一起冲入第二辅助室。合成cDNA后,用5uL的新鲜PCR混合液通过洗脱口洗脱反应产物,以用于cDNA扩增,随后在Illumina测序仪上进行高通量测序。
对多种RNA种类包括高百分比的miRNA进行鉴别测序(图23)。在一个单细胞中发现的miRNA种类列举于表中。在单个微流控装置中连同阳性(纯化的RNA)和阴性(无细胞)对照一起总共处理了11个单细胞。
实施例4
设计了由两个流体通路组成的微流控芯片,用于测序和表征独立的转录组(图24)。第一通路从入口经包含8个水力细胞捕获器的水平通道通向出口。该水平通道可被分隔以将8个捕获器分离成8个单独的细胞捕获室和一个等量体积的空室用作对照。第二流体通路从位于芯片顶部的第二“试剂”入口引导,垂直往下流进连续的9个反应柱中。每个柱与分隔的细胞捕获室或对照体积相交,然后流经筛孔阀至单独的出口。筛孔阀为“渗漏”阀,其允许流体流过,但阻止较大物体如RNA捕获珠通过。
该芯片能够进行单细胞捕获、裂解、mRNA纯化和逆转录。简而言之,通过试剂入口将mRNA捕获珠载入并堆积于该筛孔阀。然后关闭一组阀门以分隔和保护所述堆积的柱。接着,引导细胞流经水平通道并流到装满每个细胞捕获器,此后可用流经所述水平通道的干净缓冲液洗涤所述细胞。然后将细胞捕获室分隔以分离所述细胞,并将芯片转移至平板式热循环仪以进行热裂解。然后打开垂直通道,该细胞裂解物流经珠子柱以捕获mRNA。当裂解物从细胞捕获室流向所述柱时,其流经裂解物稀释室。该室非常重要,因为纯的裂解物含有蛋白和其他会聚集并阻塞所述珠子柱的物质。该稀释步骤有助于避免如此。一旦完成捕获,再次将芯片置于热循环仪上孵育,通过反应入口引入逆转录混合物并流经所述柱。最后,打开筛孔阀,使现已结合cDNA的珠子流至其各自的出口。然后可将所述cDNA用于PCR扩增和随后的测序。此外,可将随机序列结合至所述捕获珠子上,致使每个捕获的mRNA分子被给予独特的条码。因此,测序数据可用作转录本丰度的数字计数。
以这种方式对4种细胞表征的结果如表3和4所示。
实施例5
图25显示了具有下游筛孔阀的细胞捕获室。在流体压力下功能化的珠子(例如用寡聚-dTs功能化)堆积于所述筛孔阀(图25B),然后通过确定性的细胞上样将单细胞(或预定数目的细胞)添加至所述室中,其中通过使用阀门或如上所述通过使用细胞捕获器(图25A),流体路径可用于从大块样品中分离单细胞。通过水力细胞捕获器或通过渗漏“筛孔”阀可将细胞固定于每个指定的位置。通过使化学裂解试剂流经所述室来裂解细胞,且在所述珠子上捕获并纯化所述底物(例如,mRNA)。后面的试剂可流经反应室,同时通过筛孔阀使结合底物的珠子保持被固定。所述室可用(无渗漏的)阀门进行封闭以进行每个反应步骤。
/>
实施例6
图26说明了用于磁性固定功能化珠子以便于在单个室中进行多步反应的微流控装置。在邻近较大反应室的捕获室中分离细胞。将例如用poly-T寡核苷酸功能化的磁珠悬浮于裂解试剂中,然后经细胞捕获室注入至反应室中,在该过程中裂解细胞。珠子结合底物(例如mRNA)后,通过磁场被固定于室的顶部(或所述室的任何合适的壁),从而允许反应室的流体内含物被更换。这允许在单个室中进行底物纯化和多步处理结合的底物。
实施例7
图27为根据本发明一个实施方案,将单细胞处理与微阵列点样集成以将不同试剂(即引物/探针)递送至不同反应的微流控装置示意图。在该方案中,所捕获的细胞被热裂解,然后该细胞裂解物和每一种后面的试剂一起被推送至邻近室中(例如,用于逆转录(RT)和PCR)。通过该室的出口可回收所述反应产物。在该设计中,可使用激光溶蚀流体通孔(互相连接)向下至在排列的点样试剂上对准的室中。使用自动微阵列点样器,这些点可用于为每个反应提供不同的引物或探针。引物可用特有的DNA碱基识别序列来点样。该“条码”被并入每个单细胞反应中,并有助于许多平行单细胞的DNA和/或RNA转录本测序,所述单细胞具有每条序列编码于该条码的细胞来源。
实施例8
根据本发明一个实施方案,集成的单细胞数字PCR的微流控装置如图28所示。该装置可进行用于平行的单分子扩增和计数来自单细胞的RNA转录本的所有流体处理步骤。所述装置组合了实施例2所示的细胞处理和用于数字PCR的2000x1nL室的阵列。将细胞捕获,然后洗涤和裂解。将其内含物递送至邻近较大的室中进行逆转录。最后,将DNA模板(RT产物)与PCR试剂一起推送至数字PCR室的阵列中。为了混合PCR液和模板分子,将数字阵列设计为大的分叉旋转混合器,其中通过使用蠕动泵使流体围绕所述阵列循环(图29)。混合后,阀门(红色)将所述通道分隔为2000个反应室。进行数字PCR,并计数扩增孔的数目以直接计数来自样品即单细胞的单个分子。与高密度数字PCR阵列组合时,该策略能够通过数字PCR高通量分析单细胞,非常高效地将来自细胞的核苷酸转移至数字阵列;如果细胞在小管中处理后再转移至用于数字PCR分析的装置中,这是很难实现的。应理解,该几何结构可与化学法或酶法DNA断裂步骤一起使用,以对基因组DNA进行数字PCR分析,且发现其可应用于分析单细胞的拷贝数目变化。
实施例9
根据本发明一个实施方案,用于将系列稀释的细胞产物递送至较大的室中的微流控装置如图30所示,其可包括或不包括主动混合。显示了7个连续的室。通过用随后的试剂稀释,将试剂从一个室中推送至下一个室。前面的室也可被绕过或冲洗。主动混合用于增加在较大的室中混合速度。右边的图为主动混合的荧光图像,通过使用阀门作为室外部周围通道中的蠕动泵来循环溶液。将该设计优化用于多步反应,如Tang等所描述的从单细胞扩增RNA以产生足够的物质用于测序(80)。例如,多个步骤,包括细胞捕获、细胞裂解、cDNA合成、引物的EXO1降解、poly-A加尾、第二链合成和PCR,可通过在室之间连续转移细胞内含物来完成。应注意,本文中每个后面的室提供3:1的稀释,以实现高效转移每一步的内含物,这需要在每个步骤合适地选择试剂的浓度。而且,应理解,可能有且可能是所期望的不同稀释比例。
实施例10:空间多路复用
在本发明又一个实施方案中,可将细胞裂解物分开到多个室中平行进行不同的分析。图31显示了根据本发明一个实施方案用于(低)特异性多路复用的微流控装置。空间多路复用允许从空间上限定多个分析,并去除光学限制。所述装置的特征在于如实施例2所示的细胞处理单元,其中单细胞被水力捕获、洗涤和裂解。通过推送将细胞裂解物(DNA、RNA或模板)和逆转录(RT)试剂一起转移至邻近室中。进行RT后,所述产物(cDNA)可被继续转移和处理至多个邻近室中。在图31(下方)中,描绘了单个较大的室,其可用于通过扩散混合模板和PCR液,和/或用于通过PCR预扩增模板。
为了多路复用,将所述模板(与PCR试剂一起)向下推送至具有多个(例如10个)室的通道。该通道自身环接,且模板和试剂可通过旋转混合器进行混合。混合后,所述通道被分隔为单独的室。将不同的分析物(即引物/探针)上样至邻近室中(通过激光溶蚀层间连接促进流体路径选择)。打开分隔组合的PCR/模板溶液和引物/探针分析物的阀门,允许其内含物通过扩散进行混合(可选择的设计可通过水平对流使这两种溶液组合)。以这种方式,单细胞的内含物可被查询用于多个靶标(即不同的转录本)。
实施例11
在本发明又一个实施方案中,微流控装置可用于在分析之前处理单细胞(或预定数目的细胞)。例如,可将固定的细胞以任何方式(例如,通过化学制剂、病原体、电流等)进行刺激并裂解,将细胞裂解物转移至反应室用于分析。例如,位于捕获器中的细胞在洗涤步骤期间可接受所包括的信号转导分子的刺激。在一定量的时间后,所述细胞可能被裂解,并且其内容物被转移至辅助室用于通过RT-qPCR、数字PCR、测序、微阵列分析等进行核酸扩增和/或定量,所述时间可能针对不同细胞而改变。这可用于研究由于刺激的早期转录事件,具有单细胞精确性、瞬时分辨力和通量,不可能使用其他替代方法。
本领域技术人员应进一步理解,所述方法不必是破坏性的。例如,可对固定的细胞进行刺激,然后可将随后的细胞分泌物洗涤至下游的反应室用于分析。可将完整的细胞进一步处理或回收用于培养。这类分泌物可包括代谢物、激素、细胞因子、其他蛋白、miRNA、mRNA等。作为使用荧光夹心分析进行定量的来自细胞的这类蛋白的实例,其中抗体用于捕获和检测固体表面如通道壁或固定的微珠上的目标蛋白。或者,使用免疫PCR方法在下游的室中进行qPCR可检测分泌蛋白。或者,使用酶法扩增和通过荧光底物裂解可进行数字化蛋白计数,或通过进行数字免疫PCR,其中少于一个蛋白存在于下游反应器的每个阵列中。本领域技术人员应理解,有很多可能的分析方法能够针对单细胞分泌的DNA、RNA、蛋白和代谢物进行分析。
实施例12
在本发明另一个实施方案中,微流控装置可用于在流动下分离用于培养分析的细胞。具体地,如果捕获器为高效率的且仅能容纳单细胞,那么通道中的捕获器阵列可用于连续地捕获细胞的子代(从而允许进行谱系分析)。这样的实施方案在研究干细胞不对称分裂中具有效用,其中细胞的子代可被编程为处于不同于原始细胞的分化状态。例如,通过细胞分裂,捕获器阵列的起始位置处的单细胞可用于占据该阵列的其他捕获器中。然后所述细胞可被单独处理以扩增和定量mRNA或miRNA表达,从而获得有关干细胞及其子代在限定条件下的转录状态信息。
实施例13
在本发明又一个实施方案中,微流控装置可用于捕获和固定细胞以改善转染。在该实施方案中,极少量的细胞类型能被固定并暴露于病毒溶液流,以增加细胞-病毒的相互作用效率。在一种情形下,细胞可被定位于产生关注病毒的其他细胞的下游。
实施例14
在本发明又一个实施方案中,细胞捕获器可用于在细胞裂解之前或之后固定和染色细胞,包括抗体染色、mRNAFISH、FISH或化学染色。
实施例15
在本发明又一个实施方案中,细胞裂解后的细胞尸体可保留在捕获器内,同时细胞内容物被递送至下游的室中,从而能够分别处理DNA和其他内容物(例如RNA或蛋白)。例如,已知使用去污剂NP40的化学裂解可使细胞核保持完整。可选择地,可使用热裂解来破坏细胞膜,释放细胞溶质组分,同时保留基因组DNA的定位。
操作
尽管已经描述和说明了本发明的具体实施方案,这些实施方案应看作是仅用于说明本发明的目的,而不限制根据所附权利要求所解释的本发明。
参考文献
1.Taniguchi K,Kajiyama T,&Kambara H(2009)Quantitative analysis ofgene expression in a single cell by qPCR.Nat Methods 6(7):503-506.
2.Warren L,Bryder D,Weissman IL,&Quake SR(2006)Transcription factorprofiling in individual hematopoietic progenitors by digital RT-PCR.Proc NatlAcad Sci U S A 103(47):17807-17812.
3.Raj A,van den Bogaard P,Rifkin SA,van Oudenaarden A,&Tyagi S(2008)Imaging individual mRNA molecules using multiple singly labeled probes.NatureMethods 5(10):877-879.
4.Larsson C,Grundberg I,Soderberg O,&Nilsson M(2010)In situ detectionand genotyping of individual mRNA molecules.Nature Methods 7(5):395-U381.
5.Tang F,et al.(2009)mRNA-Seq whole-transcriptome analysis of asingle cell.Nat Methods 6(5):377-382.
6.Bengtsson M,Hemberg M,Rorsman P,&Stahlberg A(2008)Quantification ofmRNA in single cells and modelling of RT-qPCR induced noise.BMC Mol Biol 9:63.
7.Zare RN&Kim S(2010)Microfluidic platforms for single-cellanalysis.Annu Rev Biomed Eng 12:187-201.
8.Sims CE&Allbritton NL(2007)Analysis of single mammalian cells on-chip.Lab Chip 7(4):423-440.
9.Wheeler AR,et al.(2003)Microfluidic device for single-cellanalysis.Anal Chem 75(14):3581-3586.
10.Skelley AM,Kirak O,Suh H,Jaenisch R,&Voldman J(2009)Microfluidiccontrol of cell pairing and fusion.Nat Methods 6(2):147-152.
11.Marcus JS,Anderson WF,&Quake SR(2006)Microfluidic single-cell mRNAisolation and analysis.Anal Chem 78(9):3084-3089.
12.Bontoux N,et al.(2008)Integrating whole transcriptome assays on alab-on-a-chip for single cell gene profiling.Lab on a Chip 8(3):443-450.
13.Zhong JF,et al.(2008)A microfluidic processor for gene expressionprofiling of single human embryonic stem cells.Lab Chip 8(1):68-74.
14.Guo GJ,et al.(2010)Resolution of Cell Fate Decisions Revealed bySingle-Cell Gene Expression Analysis from Zygote to Blastocyst.DevelopmentalCell 18(4):675-685.
15.Petriv OI,et al.(2010)Comprehensive microRNA expression profilingof the hematopoietic hierarchy.Proc Natl Acad Sci USA 107(35):15443-15448.
16.Diehn M,et al.(2009)Association of reactive oxygen species levelsand radioresistance in cancer stem cells.Nature 458(7239):780-783.
17.Toriello NM,et al.(2008)Integrated microfluidic bioprocessor forsingle-cell gene expression analysis.Proc Natl Acad Sci USA 105(51):20173-20178.
18.Rieseberg,M.,Kasper,C.,Reardon,K.&T,S.Flow cytometry inbiotechnology.Applied Microbiology and Biotechnology 56,350-360(2001).
19.Cohen,D.et al.Chemical Cytometry:Fluorescence-Based Single-CellAnalysis.Annual Review of Analytical Chemistry 1,165-190,doi:doi:10.1146/annurev.anchem.1.031207.113104(2008).
20.Veening,J.-W.,Smits,W.K.,Hamoen,L.W.&Kuipers,O.P.Single cellanalysis of gene expression patterns of competence development and initiationof sporulation inBacillus subtilisgrown on chemically definedmedia.Journal of Applied Microbiology 101,531-541(2006).
21.Kato,Y.et al.Real-time functional imaging for monitoring miR-133during myogenic differentiation.The International Journal of Biochemistry&Cell Biology 41,2225-2231,doi:DOI:10.1016/j.biocel.2009.04.018(2009).
22.Zhang,L.et al.Whole genome amplification from a single cell:implications for genetic analysis.Proc.Natl.Acad.Sci.U.S.A.89,5847-5851(1992).
23.Emmert-Buck,M.R.et al.Laser Capture Microdissection.SCIENCE 274,998-1001,doi:10.1126/science.274.5289.998(1996).
24.Orba,Y.et al.Application of laser capture microdissection tocytologic specimens for the detection of immunoglobulin heavy chain generearrangement in patients with malignant lymphoma.Cancer Cytopathology 99,198-204(2003).
25.Stich,M.et al.Live Cell Catapulting and Recultivation.Pathology-Research and Practice 199,405-409,doi:Doi:10.1078/0344-0338-00437(2003).
26.Li,P.C.H.,Camprieu,L.d.,Cai,J.&Sangar,M.Transport,retention andfluorescent measurement of single biological cells studied in microfluidicchips.Lab on a Chip 4,174-180(2004).
27.Li,X.&Li,P.C.H.Microfluidic Selection and Retention of a SingleCardiac Myocyte,On-Chip Dye Loading,Cell Contraction by Chemical Stimulation,and Quantitative Fluorescent Analysis of Intracellular Calcium.Anal.Chem.77,4315-4322,doi:10.1021/ac048240a(2005).
28.Di Carlo,D.,Aghdam,N.&Lee,L.P.Single-Cell Enzyme Concentrations,Kinetics,and Inhibition Analysis Using High-Density Hydrodynamic CellIsolation Arrays.Anal.Chem.78,4925-4930,doi:10.1021/ac060541s(2006).
29.Rowat,A.C.,Bird,J.C.,Agresti,J.J.,Rando,O.J.&Weitz,D.A.Trackinglineages of single cells in lines using a microfluidic device.Proceedings ofthe National Academy of Sciences 106,18149-18154,doi:10.1073/pnas.0903163106(2009).
30.Kobel,S.,Valero,A.,Latt,J.,Renaud,P.&Lutolf,M.Optimization ofmicrofluidic single cell trapping for long-term on-chip culture.Lab on a Chip10,857-863(2010).
31.King,K.R.et al.A high-throughput microfluidic real-time geneexpression living cell array.Lab on a Chip 7,77-85(2007).
32.Marcy,Y.et al.Dissecting biological"dark matter"with single-cellgenetic analysis of rare and uncultivated TM7 microbes from the human mouth.Proc.Natl.Acad.Sci.U.S.A.104,11889-11894(2007).
33.Voldman,J.,Gray,M.L.,Toner,M.&Schmidt,M.A.A microfabrication-baseddynamic array cytometer.Anal.Chem.74,3984-3990,doi:10.1021/ac0256235(2002).
34.Braschler,T.,Johann,R.,Heule,M.,Metref,L.&Renaud,P.Gentle celltrapping and release on a microfluidic chip by in situ alginate hydrogelformation.Lab on a Chip 5,553-559(2005).
35.Neuman,K.C.,Chadd,E.H.,Liou,G.F.,Bergman,K.&Block,S.M.Characterization of photodamage to Escherichia coli in opticaltraps.Biophys.J.77,2856-2863(1999).
36.Bartel,D.P.MicroRNAs:Genomics,Biogenesis,Mechanism,andFunction.Cell 116,281-297,doi:Doi:10.1016/s0092-8674(04)00045-5(2004).
37.O'Donnell,K.A.,Wentzel,E.A.,Zeller,K.I.,Dang,C.V.&Mendell,J.T.c-Myc-regulated microRNAs modulate E2F1 expression.Nature 435,839-843,doi:http://www.nature.com/nature/journal/v435/n7043/suppinfo/nature03677_S1.html(2005).
38.Chen,C.-Z.,Li,L.,Lodish,H.F.&Bartel,D.P.MicroRNAs ModulateHematopoietic Lineage Differentiation.SCIENCE 303,83-86,doi:10.1126/science.1091903(2004).
39.Miska,E.et al.Microarray analysis of microRNA expression in thedeveloping mammalian brain.Genome Biology 5,R68(2004).
40.Sindelka,R.,Jonak,J.,Hands,R.,Bustin,S.A.&Kubista,M.Intracellularexpression profiles measured by real-time PCR tomography in the Xenopuslaevis oocyte.Nucleic Acids Research 36,387-392(2008).
41.Chang,S.,Johnston,R.J.&Hobert,O.A transcriptional regulatorycascade that controls left/right asymmetry in chemosensory neurons ofC.elegans.Genes&Development 17,2123-2137,doi:10.1101/gad.1117903(2003).
42.Krichevsky,A.M.,King,K.S.,Donahue,C.P.,Khrapko,K.&Kosik,K.S.AmicroRNA array reveals extensive regulation of microRNAs during braindevelopment.RNA 9,1274-1281,doi:10.1261/rna.5980303(2003).
43.Niu,Q.-W.et al.Expression of artificial microRNAs in transgenicArabidopsis thaliana confers virus resistance.Nat Biotech 24,1420-1428,doi:http://www.nature.com/nbt/journal/v24/n11/suppinfo/nbt1255_S1.html(2006).
44.Scaria,V.,Hariharan,M.,Maiti,S.,Pillai,B.&Brahmachari,S.Host-virusinteraction:a new role for microRNAs.Retrovirology 3,68(2006).
45.Calin,G.A.et al.MicroRNA profiling reveals distinct signatures inB cell chronic lymphocytic leukemias.Proc.Natl.Acad.Sci.U.S.A.101,11755-11760(2004).
46.Cui,J.-W.et al.Retroviral insertional activation of the Fli-3locus in erythroleukemias encoding a cluster of microRNAs that convert Epo-induced differentiation to proliferation.Blood 110,2631-2640,doi:10.1182/blood-2006-10-053850(2007).
47.Lu,J.et al.MicroRNA expression profiles classify humancancers.Nature 435,834-838,doi:http://www.nature.com/nature/journal/v435/n7043/suppinfo/nature03702_S1.html(2005).
48.Ikeda,S.et al.Altered microRNA expression in human heartdisease.Physiol.Genomics 31,367-373,doi:10.1152/physiolgenomics.00144.2007(2007).
49.Thum,T.,Catalucci,D.&Bauersachs,J.MicroRNAs:novel regulators incardiac development and disease.Cardiovasc Res 79,562-570,doi:10.1093/cvr/cvn137(2008).
50.Thum,T.et al.MicroRNAs in the Human Heart:A Clue to Fetal GeneReprogramming in Heart Failure.Circulation 116,258-267,doi:10.1161/circulationaha.107.687947(2007).
51.Wang,N.,Zhou,Z.,Liao,X.&Zhang,T.Role of microRNAs in cardiachypertrophy and heart failure.IUBMB Life 61,566-571(2009).
52.Gibson,U.E.,Heid,C.A.&Williams,P.M.A novel method for real timequantitative RT-PCR.Genome Research 6,995-1001,doi:10.1101/gr.6.10.995(1996).
53.Ginzinger,D.G.Gene quantification using real-time quantitativePCR:An emerging technology hits the mainstream.Experimental Hematology 30,503-512,doi:Doi:10.1016/s0301-472x(02)00806-8(2002).
54.Nolan,T.,Hands,R.E.&Bustin,S.A.Quantification of mRNA using real-time RT-PCR.Nat.Protocols 1,1559-15382(2006).
55.Bengtsson,M.,Hemberg,M.,Rorsman,P.&Stahlberg,A.Quantification ofmRNA in single cells and modelling of RT-qPCR induced noise.Bmc MolecularBiology 9(2008).
56.Taniguchi,K.,Kajiyama,T.&Kambara,H.Quantitative analysis of geneexpression in a single cell by qPCR.Nature Methods 6,503-U550(2009).
57.Tang,F.C.,Hajkova,P.,Barton,S.C.,Lao,K.Q.&Surani,M.A.MicroRNAexpression profiling of single whole embryonic stem cells.Nucleic AcidsResearch 34(2006).
58.Tang,F.C.et al.220-plex microRNA expression profile of a singlecell.Nature Protocols 1,1154-1159(2006).
59.Kamme,F.et al.Single-Cell Microarray Analysis in Hippocampus CA1:Demonstration and Validation of Cellular Heterogeneity.J.Neurosci.23,3607-3615(2003).
60.Kloosterman,W.P.,Wienholds,E.,de Bruijn,E.,Kauppinen,S.&Plasterk,R.H.A.In situ detection of miRNAs in animal embryos using LNA-modifiedoligonucleotide probes.Nat Meth 3,27-29,doi:http://www.nature.com/nmeth/journal/v3/n1/suppinfo/nmeth843_S1.html(2006).
61.Lu,J.&Tsourkas,A.Imaging individual microRNAs in single mammaliancells in situ.Nucleic Acids Research 37(2009).
62.Neely,L.A.et al.A single-molecule method for the quantitation ofmicroRNA gene expression.Nat Meth 3,41-46,
63.Unger,M.A.et al.Microfabricated Elastomeric Valve and Pump SystemsUSA patent(2005).
64.Lee,R.C.,Feinbaum,R.L.&Ambros,V.The C.elegans heterochronic genelin-4 encodes small RNAs with antisense complementarity to lin-14.Cell 75,843-854,doi:Doi:10.1016/0092-8674(93)90529-y(1993).
65.Nelson,P.T.et al.Microarray-based,high-throughput gene expressionprofiling of microRNAs.Nat Meth 1,155-161,doi:http://www.nature.com/nmeth/journal/v1/n2/suppinfo/nmeth717_S1.html(2004).
66.Thomson,J.M.,Parker,J.,Perou,C.M.&Hammond,S.M.A custom microarrayplatform for analysis of microRNA gene expression.Nat Meth 1,47-53,doi:http://www.nature.com/nmeth/journal/v1/n1/suppinfo/nmeth704_S1.html(2004).
67.Willenbrock,H.et al.Quantitative miRNA expression analysis:comparing microarrays with next-generation sequencing.RNA 15,2028-2034,doi:doi:10.1261/rna.1699809(2009).
68.Chen,C.et al.Real-time quantification of microRNAs by stem-loopRT-PCR.Nucl.Acids Res.33,e179-,doi:10.1093/nar/gni178(2005).
69.Livak,K.J.,Flood,S.J.,Marmaro,J.,Giusti,W.&Deetz,K.Oligonucleotides with fluorescent dyes at opposite ends provide a quenchedprobe system useful for detecting PCR product and nucleic acidhybridization.Genome Research 4,357-362(1995).
70.Unger,M.A.,Chou,H.P.,Thorsen,T.,Scherer,A.&Quake,S.R.Monolithicmicrofabricated valves and pumps by multilayer soft lithography.SCIENCE 288,113-116(2000).
71.Thorsen T,Maerkl SJ,&Quake SR(2002)Microfluidic large-scaleintegration.Science 298(5593):580-584.
72.Warren L,Bryder D,Weissman IL,&Quake SR(2006)Transcription factorprofiling in individual hematopoietic progenitors by digital RT-PCR.Proc NatlAcad Sci U S A 103(47):17807-17812.
73.Shah SP,et al.(2009)Mutational evolution in a lobular breasttumour profiled at single nucleotide resolution.Nature 461(7265):809-813.
74.Schulze HG,et al.(2010)Assessing Differentiation Status of HumanEmbryonic Stem Cells Noninvasively Using Raman Microspectroscopy.AnalyticalChemistry 82(12):5020-5027.
75.Adewumi O,et al.(2007)Characterization of human embryonic stemcell lines by the International Stem Cell Initiative.Nature Biotechnology 25(7):803-816.
76.Ludwig TE,et al.(2006)Feeder-independent culture of humanembryonic stem cells.Nat Methods 3(8):637-646.
77.Lozzio BB,Lozzio CB,Bamberger EG,&Feliu AS(1981)A multipotentialleukemia cell line(K-562)of human origin.Proc Soc Exp Biol Med 166(4):546-550.
78.Yuan JY,et al.(2009)MicroRNA-223 reversibly regulates erythroidand megakaryocytic differentiation of K562 cells.J Cell Mol Med 13(11-12):4551-4559.
79.Landgraf P,et al.(2007)A mammalian microRNA expression atlas basedon small RNA library sequencing.Cell 129(7):1401-1414.
80.Tang FC,Barbacioru C,Nordman E,Li B,Xu NL,Bashkirov VI,Lao KQ,Surani MA:RNA-Seq analysis to capture the transcriptome landscape of a singlecell.Nature Protocols 5:516-535.