酵母定点双DSB同步诱导模型的构建方法
文献发布时间:2023-06-19 09:36:59
技术领域
本发明属于基因工程技术,尤其是一种酵母定点双DSB同步诱导模型的构建方法。
背景技术
DSB是一种非常严重的DNA损伤,若不及时修复,则会导致基因突变、诱发癌症等。DSB主要通过同源重组(HR)和非同源末端连接(NHEJ)来进行修复,构建可化学或物理诱导的DSB是研究上述DNA损伤途径必不可少的工具。传统的DNA损伤模型多用电离辐射、化学诱变剂等来诱导,并不能很好地将损伤类型限制在单一DSB损伤,还常常伴随DNA单链断裂(SSB)、碱基突变以及DNA双链交联等。此外,自然条件下细胞内DSB的产生通常不止一个,研究多个DSB同步产生时的毒理作用,以及机体的修复过程,具有重要的生物医学基础理论意义和使用价值。
如图1所示,电离辐射、化学诱变剂等外界因素诱导的DNA损伤类型复杂,且位点随机,无法有效研究HR、NHEJ等特定修复DSB的分子信号通路。本文构建了一个能稳定诱导产生双DSB定点产生的模型,两个DSB的间隔距离可根据需要改变,因而不仅可以排除其他损伤类型的干扰,更重要的是可研究同步产生的“聚集型”DSB相对于单个DSB,对机体的特殊危害,以及细胞修复应答的协同过程。鉴于“聚集型”DNA损伤是癌症放疗及众多化疗药物所产生的特征损伤类型,这一模型是研究细胞信号通路及药物毒理的强大工具。
通过检索,尚未发现与本发明专利申请相关的专利公开文献。
发明内容
本发明的目的在于克服现有技术的不足之处,提供一种定点同步双DSB模型的构建方法。
本发明解决其技术问题所采用的技术方案是:
一种酵母定点双DSB同步诱导模型的构建方法,步骤如下:
⑴设计上、下游引物,引物两端加上I-SceI识别位点,用上、下游引物将酿酒酵母诱导型启动子控制的I-SceI基因的表达盒及KanMX筛选基因GAL-I-SceI-KanMX从质粒pGSKU扩增下来;
⑵设计第二段引物进行第二次PCR扩增,使上、下游引物的同源臂为70-80bp;
⑶使用转化方法,将PCR扩增得到的GAL-I-SceI基因序列整合到酿酒酵母基因组上,利用筛选标记基因获得成功整合的菌株,并设计验证引物验证是否构建成功;
⑷含有GAL或其他启动子所控制表达I-SceI基因的酵母,激活启动子时,就能够表达I-SceI内切酶,进而识别两端预先放置的I-SceI识别位点并进行切割,从而产生双DSB。
而且,所述步骤⑴中上游引物为SEQNO.1,下游引物为SEQNO.2;
DNA聚合酶反应体系:ddH
DNA聚合酶程序设定:预变性96℃2min设置1个循环,变性94℃30s、退火56℃60s、延伸72℃1500bp/min设置32个循环,后延伸72℃7min设置1个循环。
而且,所述步骤⑵中上游引物为SEQNO.3,下游引物为SEQNO.4;
DNA聚合酶反应体系:ddH
DNA聚合酶程序设定:预变性96℃2min设置1个循环,变性94℃30s、退火56℃60s、延伸72℃1500bp/min设置32个循环,后延伸72℃7min设置1个循环。
而且,所述步骤⑶中上游引物为SEQNO.5,下游引物为SEQNO.6。
而且,所述步骤⑴中酿酒酵母诱导型启动子为半乳糖启动子GAL、己糖转运载体或乙醇脱氢酶Ⅱ。
而且,所述步骤⑴中能够根据需要通过重叠延伸PCR引入一段无用序列,从而调节两个DSB的间隔距离。
而且,所述步骤⑴中I-SceI基因的表达盒包括半乳糖启动子、I-SceI内切酶、KanMX/潮霉素/诺尔斯菌素筛选标记。
本发明取得的优点和积极效果为:
1、本发明建立了一种在酵母基因组的任何位点稳定诱导、同步产生两个DSB的方法,并可根据需要设置两个DSB的间隔距离。这一方法稍作延伸,即可在基因组的任何位置,同步产生多个DSB,是一个强大的DNA修复应答研究模型。特别适用于研究同源重组修复(HR)和非同源末端连接修复(NHEJ),以及不同修复途径间的协同及互补机制,可以用于人体或其他生物的细胞。
2、多个DSB同步产生的“聚集型”DNA损伤是癌症放疗及众多化疗药物的特征损伤类型,本发明所建立的双DSB定点诱导模型,与传统的电离辐射和化学诱变剂等诱导的随机DNA损伤相比,只诱导单纯的DSB损伤,不仅可排除其他类型DNA损伤的干扰,更重要的是可在基因组的任何位置诱导“聚集型”DSB的产生,从而可精准获取多个DSB同时产生时,对机体的特殊危害,以及细胞修复应答的协同过程,是研究抗癌药物毒理及精准医疗的强有利工具。
3、本发明构建了一个能稳定诱导产生双DSB定点产生的模型,两个DSB的间隔距离可根据需要改变,因而不仅可以排除其他损伤类型的干扰,更重要的是可研究同步产生的“聚集型”DSB相对于单个DSB,对机体的特殊危害,以及细胞修复应答的协同过程。鉴于“聚集型”DNA损伤是癌症放疗及众多化疗药物所产生的特征损伤类型,这一模型是研究细胞信号通路及药物毒理的强大工具。
附图说明
图1为现有技术中产生DSB损伤的主要方法示意图;
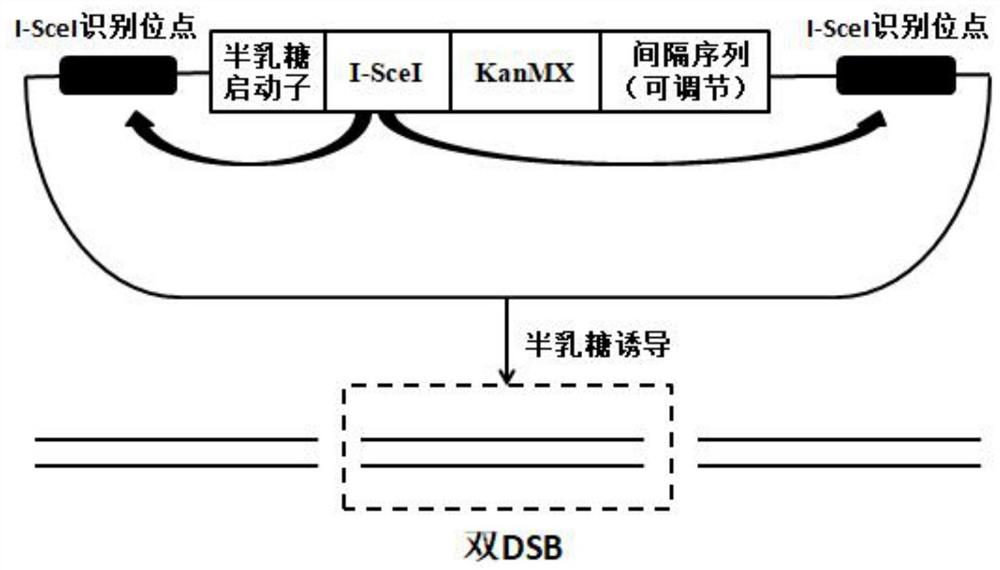
图2为本发明双DSB同步诱导模型的一种构建示意图;可通过调节间隔序列(基因之间没有遗传效应的DNA片段)的长短来产生不同间隔长度的DSB;
图3为本发明中验证双DSB定点损伤模型构建是否成功示意图;实验组1-5显示扩增出3620bp的条带,表明间隔序列为3620bp的双DSB损伤模型构建成功。
具体实施方式
下面结合实施例,对本发明进一步说明,下属实施例是叙述性的,不是限定性的,不能以下述实施例来限定本发明的保护范围。
本发明中所使用的原料,如无特殊说明,均为常规市售产品,本发明中所使用的方法,如无特殊说明,均为本领域常规方法,本发明所用各物质质量均为常规使用质量。
一种酵母定点双DSB同步诱导模型的构建方法,步骤如下:
⑴设计上、下游引物,引物两端加上I-SceI识别位点,用上、下游引物将酿酒酵母诱导型启动子控制的I-SceI基因的表达盒及KanMX筛选基因GAL-I-SceI-KanMX从质粒pGSKU扩增下来;
⑵设计第二段引物进行第二次PCR扩增,使上、下游引物的同源臂为70-80bp;
⑶使用转化方法,将PCR扩增得到的GAL-I-SceI基因序列整合到酿酒酵母基因组上,利用筛选标记基因获得成功整合的菌株,并设计验证引物验证是否构建成功;
⑷含有GAL或其他启动子所控制表达SceI基因的酵母,激活启动子时,就能够表达I-SceI内切酶,进而识别两端预先放置的I-SceI识别位点并进行切割,从而产生双DSB。可以如图2所示。
较优地,所述步骤⑴中上游引物为SEQNO.1,下游引物为SEQNO.2;
DNA聚合酶反应体系:ddH
DNA聚合酶程序设定:预变性96℃2min设置1个循环,变性94℃30s、退火56℃60s、延伸72℃1500bp/min设置32个循环,后延伸72℃7min设置1个循环。
较优地,所述步骤⑵中上游引物为SEQNO.3,下游引物为SEQNO.4;
DNA聚合酶反应体系:ddH
DNA聚合酶程序设定:预变性96℃2min设置1个循环,变性94℃30s、退火56℃60s、延伸72℃1500bp/min设置32个循环,后延伸72℃7min设置1个循环。
较优地,所述步骤⑶中上游引物为SEQNO.5,下游引物为SEQNO.6。
较优地,所述步骤⑴中酿酒酵母诱导型启动子为半乳糖启动子GAL、己糖转运载体或乙醇脱氢酶Ⅱ。
较优地,所述步骤⑴中能够根据需要通过重叠延伸PCR引入一段无用序列,从而调节两个DSB的间隔距离。
较优地,所述步骤⑴中I-SceI基因的表达盒包括半乳糖启动子、I-SceI内切酶、KanMX/潮霉素/诺尔斯菌素筛选标记。
具体地:
一种酵母中双DSB模型的构建方法,步骤如下:
⑴在质粒中构建由半乳糖启动子(GAL)控制的I-SceI基因的表达盒(GAL-I-SceI)及添加必要的抗性筛选基因(HYG,KanMX等)。I-SceI是一类由内含子编码的归位内切酶,含235个氨基酸。此内切酶能特异性地识别含18个非对称核苷酸的DNA序列(TAGGGATAACAGGGTAAT),在含有此碱基序列的位点处将DNA切割,形成DSB。类似的内切酶还有多种,可根据需要灵活选用。
⑵通过PCR扩增GAL-I-SceI基因序列,设计的PCR上、下游引物两端分别添加I-SceI酶所识别的DNA序列(TAGGGATAACAGGGTAAT),以及基因组整合所需要的同源臂。根据需要调整两个I-SceI酶识别序列的插入位置,以便产生不同距离间隔的双DSB。为有效的将GAL-I-SceI整合到目标生物基因组的特定位点处,同源臂的长度应大于80bp。
⑶使用化学转化法或其他转化方法,将PCR扩增得到的GAL-I-SceI基因序列整合到酿酒酵母基因组上,利用筛选标记基因获得成功整合的菌株,并设计验证引物验证是否构建成功。
⑷含有GAL或其他启动子所控制表达I-SceI基因的酵母,激活启动子时,即可大量表达I-SceI内切酶,进而识别两端预先放置的I-SceI识别位点并进行切割,从而产生双DSB。
更为具体地,相关制备及检测如下:
一种酵母定点双DSB同步诱导模型的构建方法,具体步骤如下:
⑴设计上、下游引物F1、F2,引物两端加上I-SceI识别位点,用F1、F2将半乳糖启动子(GAL)控制的I-SceI基因的表达盒及KanMX筛选基因(GAL-I-SceI-KanMX)从质粒pGSKU扩增下来。该引物设计的两个DSB的间隔距离为3620,可根据实验需要通过重叠延伸PCR引入一段间隔序列(基因之间没有遗传效应的DNA片段),从而调节两个DSB的间隔距离。F1序列:TTTCAGAGGTCGCCTGACGCTAGGGATAACAGGGTAATATACGCAAACCGCCTCTCCC,F2序列:TCCCAATTTTTCAGTTGAAAATAGGGATAACAGGGTAATGTCACCAAAGAACCAAGGGG;
pGSKU是可公开获取的质粒,I-SceI基因的表达盒是从pGSKU质粒上直接扩增出来的目的片段。pGSKU图谱连接:https://www.addgene.org/72243/。
FastpfuDNA聚合酶反应体系:ddH
FastpfuDNA聚合酶程序设定:预变性96℃2min设置1个循环,变性94℃30s、退火56℃60s、延伸72℃1500bp/min设置32个循环,后延伸72℃7min设置1个循环。
⑵设计第二段引物F2、R2进行第二次PCR扩增,使上、下游引物的同源臂为70-80bp。F2:ATCAAATTCGATGACTGGAAATTTTTTGTTAATTTCAGAGGTCGCCTGACGC;R2:ATGAAAAGCCGGTTCCGGCGCTCTCACCTTTCCTTTTTCTCCCAATTTTTCAGTTGAAA。该引物设计的插入位点为Ⅲ号染色体91055-91143处,插入位点可根据不同的实验需求更改。
FastpfuDNA聚合酶反应体系:ddH
FastpfuDNA聚合酶程序设定:预变性96℃2min设置1个循环,变性94℃30s、退火56℃60s、延伸72℃1500bp/min设置32个循环,后延伸72℃7min设置1个循环。
⑶使用化学转化法或其他转化方法,将PCR扩增得到的GAL-I-SceI基因序列整合到酿酒酵母基因组上,利用筛选标记基因获得成功整合的菌株,并设计验证引物验证是否构建成功。上游引物:CAAGTAATTGGTTGTTTGGC;下游引物:TAGCCAGTTTGTTGAAAGCTTGGT。其中,对照组(W):不能用验证引物扩增出条带;实验组(1-5):能用引物扩增出3620bp的片段;结果如图3所示;
⑷含有GAL或其他启动子所控制表达I-SceI基因的酵母,激活启动子时,即可大量表达I-SceI内切酶,进而识别两端预先放置的I-SceI识别位点并进行切割,从而产生双DSB。
尽管为说明目的公开了本发明的实施例,但是本领域的技术人员可以理解:在不脱离本发明及所附权利要求的精神和范围内,各种替换、变化和修改都是可能的,因此,本发明的范围不局限于实施例所公开的内容。
序列表
<110> 天津科技大学,齐鲁理工学院
<120> 酵母定点双DSB同步诱导模型的构建方法
<160> 7
<170> SIPOSequenceListing 1.0
<210> 1
<211> 58
<212> DNA/RNA
<213> F1序列(Unknown)
<400> 1
tttcagaggt cgcctgacgc tagggataac agggtaatat acgcaaaccg cctctccc 58
<210> 2
<211> 59
<212> DNA/RNA
<213> F2序列(Unknown)
<400> 2
tcccaatttt tcagttgaaa atagggataa cagggtaatg tcaccaaaga accaagggg 59
<210> 3
<211> 52
<212> DNA/RNA
<213> 第二段引物F2(Unknown)
<400> 3
atcaaattcg atgactggaa attttttgtt aatttcagag gtcgcctgac gc 52
<210> 4
<211> 59
<212> DNA/RNA
<213> 第二段引物R2(Unknown)
<400> 4
atgaaaagcc ggttccggcg ctctcacctt tcctttttct cccaattttt cagttgaaa 59
<210> 5
<211> 20
<212> DNA/RNA
<213> 上游引物(Unknown)
<400> 5
caagtaattg gttgtttggc 20
<210> 6
<211> 24
<212> DNA/RNA
<213> 下游引物(Unknown)
<400> 6
tagccagttt gttgaaagct tggt 24
<210> 7
<211> 18
<212> DNA/RNA
<213> 18个非对称核苷酸的DNA序列(Unknown)
<400> 7
tagggataac agggtaat 18
- 酵母定点双DSB同步诱导模型的构建方法
- 表达外源蛋白的毕赤酵母、其构建方法及其诱导表达方法