小麦耐碱生长素响应蛋白基因TaSAUR215及其应用
文献发布时间:2023-06-19 19:27:02
技术领域
本发明属于生物基因工程技术领域,尤其涉及耐碱基因——小麦耐碱生长素响应蛋白基因TaSAUR215及其应用。
背景技术
土壤盐碱化严重影响农作物产量。特别是随着工业的发展,土壤盐碱化愈来愈严重,已成为一个全球关注的社会问题。我国人口众多,而土壤盐碱化更为严重,已经成为制约我国经济和社会发展的重要因素。因此,除了缓解土壤盐碱化以外,培育耐盐碱农作物新品种已成为当前一个十分迫切的任务。
利用转基因改良植物技术将新的性状转入高生物量植物中,以此开发高效的转基因植物新品种并用于在盐碱地种植,是一项具有广阔应用前景的技术。
利用基因工程技术开展植物耐碱方面研究已取得了较大的进展,克隆了大量相关基因,并将这些基因转入植物中,用于耐碱机制研究。一些实验表明,将植物本身以及其它生物中与耐碱相关的基因转入植物中,其异源转录和翻译产物可以使转基因植物的抗碱能力得到提高。
目前,已发现了一些能显著提高植物耐碱能力的基因,但检索发现小麦耐碱生长素响应蛋白基因TaSAUR215及其在作物中调控小麦耐碱性未见报道
发明内容
针对现有技术的不足,本发明的目的是提供一种耐碱基因——小麦耐碱生长素响应蛋白基因TaSAUR215及其应用。
本发明所述的小麦耐碱生长素响应蛋白基因TaSAUR215,其特征在于:所述基因cDNA的核苷酸序列如SEQ ID No.1所示。
本发明还提供了含上述基因TaSAUR215的植物表达载体,其特征在于:所述植物表达载体是pGA3426-TaSAUR215或pTCK303-TaSAUR215。
本发明的技术方案在于从小麦中分离得到小麦基因TaSAUR215,然后将该基因转化到普通小麦YM20中以实现研究TaSAUR215基因的功能以及植物的耐碱机理。
本发明所述小麦耐碱生长素响应蛋白基因TaSAUR215在培育抗碱植物中的应用。
本发明所述植物表达载体pGA3426-TaSAUR215或pTCK303-TaSAUR215在培育抗碱植物中的应用。
其中:所述植物优选是普通小麦。
将本发明所述基因小麦耐碱生长素响应蛋白基因TaSAUR215导入植物细胞,植物就可以获得碱胁迫的耐受能力。为了便于对转基因植物或细胞系进行筛选,可以对含有所述基因TaSAUR215的植物表达载体(pGA3426-TaSAUR215或pTCK303-TaSAUR215)进行加工,如可以加入选择标记(GUS等)或者具有抗性的抗生素标记物(潮霉素,卡那霉素,庆大霉素等)等。
事实上,任何一种可以将外源基因导入植物中表达的载体都可以应用,本发明优选的载体是pGA3426。
本发明提供了一种小麦耐碱生长素响应蛋白基因TaSAUR215,所述基因可广泛用于培育耐碱农作物品种。本发明的有益效果是:利用现有的植物基因工程技术,本发明克隆得到了小麦碱胁迫应答基因TaSAUR215,并通过根瘤农杆菌介导的方法将该基因过量表达于小麦,经过实验比较分析,TaSAUR215过表达促进了小麦对碱胁迫的抗性,证明转基因植株的耐碱能力明显提高。
附图说明
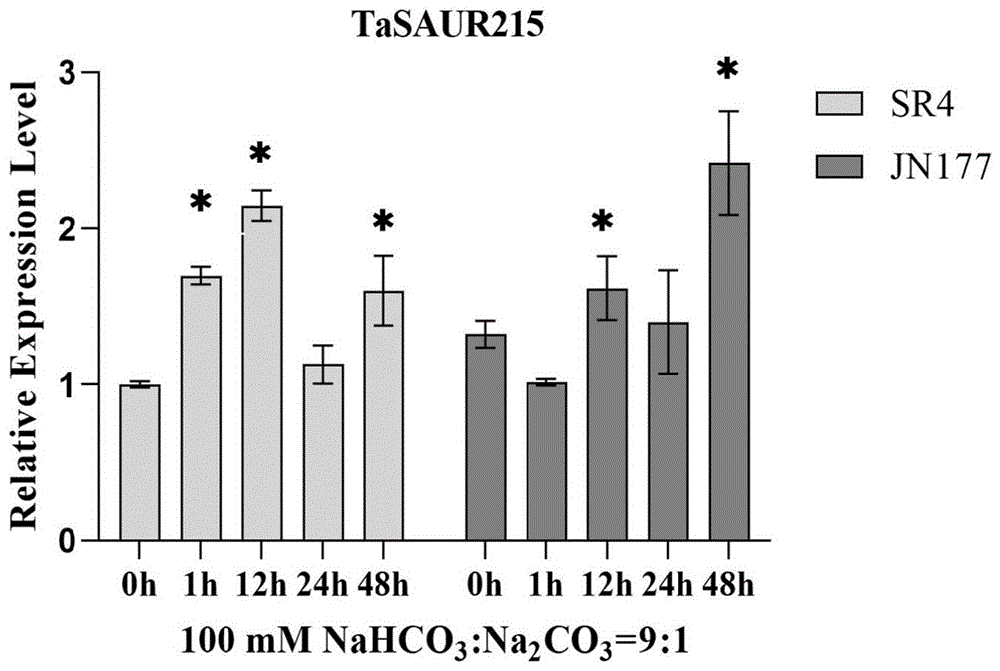
图1山融4号和济南177碱胁迫TaSAUR215的RT-PCR分析。
图2TaSAUR215转基因小麦表达量鉴定。
图3TaSAUR215转基因小麦碱胁迫下的表型。
其中:(A)TaSAUR215转基因小麦碱胁迫下的表型;(B)TaSAUR215-OE碱胁迫下的鲜重统计;(C)TaSAUR215-OE碱胁迫下与对照的相对鲜重;(D)TaSAUR215-RNAi碱胁迫下的鲜重统计;(E)TaSAUR215-RNAi碱胁迫下与对照的相对鲜重。
具体实施方式
下面结合附图和具体实施例对本发明内容进行详细说明。如下所述例子仅是本发明的较佳实施方式而已,应该说明的是,下述说明仅仅是为了解释本发明,并非对本发明作任何形式上的限制,凡是依据本发明的技术实质对实施方式所做的任何简单修改,等同变化与修饰,均属于本发明技术方案的范围内。
下述实施例中,所使用的材料、试剂、载体、菌株等,如无特殊说明,均从商业途径得到。
实施例1、TaSAUR215的克隆
1.1提取小麦Total RNA
1.将组织材料与钢株放入2.0的EP管中,液氮速冻并用组织研磨机研磨成粉末;
2.待液氮挥发干,每100mg材料约加入1ml的Invitrogen公司的TRIzol提取液,融化后,用加样枪反复吸吹,剧烈振荡混匀样品,使样品充分裂解,室温放置5min;
3.加入0.2ml氯仿(chloroform),剧烈振荡混匀15sec,室温放置10min;
4.4℃,12000rpm离心15min;
5.用移液器小心吸出上层水相,加入新的1.5ml的离心管中,加入500μl的异丙醇(1:1体积),充分混匀,-20℃2沉淀30min或过夜;
6.4℃,12000rpm离心10min,小心弃去上清液;
7.RNA沉淀用1ml的75%乙醇洗涤。4℃,12000rpm离心10min收集沉淀;
8.重复用75%乙醇洗涤一次RNA沉淀;
9.去上清,RNA沉淀于无菌操作台上晾干约10-15分钟,RNA略显透明,加入适当体积(30-50μl)的RNase-free水充分溶解(可放于-80℃长期保存);
10.紫外分光光度计及1%Agrose凝胶电泳检测RNA浓度及质量。
注:a)用紫外分光光度计检测RNA的产量,在260nm处的吸光度,1OD=40μg/ml。根据在260nm和280nm处的吸光值,检测RNA的纯度2纯RNA的OD
b)用1%Agrose凝胶电泳检侧RNA的质量及大小。吸取1μl RNA加入3μl的RNase-free水,加1μl上样缓冲液65℃变性5min。电泳后用EB染色,另取3μl的1kb DNAMarker作为对照。
1.2cDNA反转录
反转录酶:M-MLV Reverse Transcriptase(Invitrogen)。
1.12μl体系:
2.65℃变性5min,迅速插入冰中,然后依次加入:
5×First-Strand Buffer 4μl
0.1M DTT 2μl
RNaseOUT(Invitrogen) 1μl
3.轻轻混匀,37℃反应2min;
4.加入1μl M-MLV RT,混匀,37℃反应50min;
5.70℃温育15min使M-MLV RT失活;
6.加入1μl RNase H(Invitrogen),37℃反应20min;
7.用超纯水稀释至合适浓度。作为PCR模板。
1.3开放阅读框的克隆和序列测定
1.引物序列:根据测序结果,设计基因上下游引物(TaSAUR215-F、TaSAUR215-R),扩增基因的开放阅读框。
TaSAUR215-F:5’-ATGGGGGAGCAAGGCAGGGC-3’
TaSAUR215-R:5’-TCATGTGCACAAGATCCCATG-3’
2.PCR反应体系(50μl):
3.PCR反应程序为:94℃预变性5min;94℃变性45sec,55℃复性45sec,72℃延伸1.5min,循环35次;72℃延伸7min。
4.扩增片段回收后与pEASY-T1载体连接并转化大肠杆菌Trans1 T1,测序由青岛擎科公司完成。
1.4基因表达分析(RT-PCR和real-time PCR)
a.胁迫下RNA的提取
山融4号和济南177种子正常萌发,Hangload培养液培养至株高约10cm时(约2周时间)
开始施加碱性盐胁迫100mM NaHCO
b.反转录(RT)产生cDNA
反转录产生cDNA,方法同上。
c.PCR反应及电泳
1.以cDNA为模板,进行PCR反应。引物如下
TaSAUR215-RT-F:5’-GAGATGAGGAGGTTCGTCATC-3’
TaSAUR215-RT-R:5’-CTCCGAAGAGGAGTAGGAC-3’
2.PCR体系:
3.PCR程序:
95℃5min2 25~30cycles 95℃20sec2 57℃60sec2 72℃60sec;72℃7min.
根据内参Actin的扩增情况确定PCR的循环数,调整cDNA模板的加入量。
结果见图1。
实施例2、植物表达载体的构建(Ubi启动子)
2.1Ubi启动子植物表达载体的构建
利用植物表达载体pGA3426,选用KKKⅠ和HHKHⅢ分别对pGA3426和含有目的基因的pEASY-T1载体进行双酶切,分别回收载体大片段和目的基因小片段,用T
(1)质粒pGA3426空载体和pEASY-T1的KKKⅠ和HHKHⅢ双酶切
碱裂解法提取pGA3426空载体和pEASY-T1质粒,各取10μg酶切,酶切体系如下:
于30℃恒温水浴锅酶切2小时以上。双酶切后以1×TAE为电泳缓冲液,将酶切产物进行1%琼脂糖凝胶电泳。在紫外透射仪下用洁净刀片切下pGA3426中11kb的载体大片段和pEASY-T1中大约0.4kb的目的基因条带,回收该条带。
(2)经酶切和脱磷的pGA3426载体片段(约11kb)和pEASY-T1双酶切回收片段(约0.4kb)以摩尔比1:4的比例进行16℃连接过夜。
(3)连接产物热激法转化大肠杆菌Trans1 T1感受态细胞,转化菌在含Kan 50μg/ml的LB固体平板上37℃培养16小时左右。
(4)重组子的鉴定
①质粒的PCR验证
挑取单菌落分别接种于5ml含Kan的LB液体培养基中37℃振荡培养过夜,碱变性法提取质粒,用基因特异引物(TaSAUR215-F,TaSAUR215-R)进行PCR扩增,引物如下:
TaSAUR215-F:5’-ATGGGGGAGCAAGGCAGGGC-3’
TaSAUR215-R:5’-TCATGTGCACAAGATCCCATG-3’
体系如下:
PCR反应条件如下:预变性94℃3min,35个循环为:94℃30sec,55℃30sec,72℃1min,最后,72℃延伸10min。PCR产物用1.0%琼脂糖凝胶电泳鉴定。
②质粒酶切鉴定
提质粒进行KKKⅠ和HHKHⅢ双酶切,酶切体系同上。
1%琼脂糖凝胶电泳,检测是否含有预期分子量大小的片段,验证载体的正确构建。
实施例3、农杆菌感受态的制备与转化
3.1农杆菌EHA105感受态的制备
(1)从YEP平板(含50μg/ml利福平)上挑取根癌农杆菌单菌落,接种于含50μg/ml利福平的YEP液体培养基中,200rpm/min,28℃培养过夜。
(2)取2ml过夜培养液接种于50ml含相同抗生素的YEP液体培养基中在相同条件下培养至OD
(3)菌液冰浴30min,4℃,5000rpm离心10min,收集菌体。
(4)将菌体重悬于冰浴的10ml 0.15mol/L的NaCl中,离心收集菌体。
(5)再悬浮于1ml 20mmol/L冰预冷的CaCl
3.2冻融法转化根癌农杆菌EHA105
(1)在室温下融化农杆菌感受态细胞,加入1μg表达载体质粒DNA,混匀后冰浴30min。
(2)置液氮速冻1min,迅速移至37℃保温3min。
(3)加入无抗生素的YEP 800μl,28℃震荡培养3小时。
(4)7000rpm离心30s收集菌体,涂于含50μg/ml利福平、50μg/ml Kan的YEP平板上,28℃倒置暗培养2-3天。
3.3菌体PCR鉴定。
实施例4、转基因功能验证-小麦转化及筛选
小麦转化
(1)挑选饱满的YM20种子,70%乙醇中浸泡5min,然后清洁剂(20%漂水(白猫,上海),0.1% Triton)洗涤10-15min,无菌水冲洗4次,并浸泡于无菌水中过夜,将泡发的YM20种子转移至垫有浸湿滤纸的无菌培养皿中暗培养3天。
(2)转化前一天,取2ml活化的农杆菌EHA105加到含相应抗生素的200ml YEP培养基中,过夜培养至OD
(3)离心收集菌体,并重悬于浸染液中,使OD
(4)采用茎尖法进行小麦转化,用解剖刀切开小麦嫩芽,暴露其生长点并浸泡在侵染液中抽真空15min并保持压力15min。
(5)待抽真空完毕后倒掉侵染液并沥干侵染液,将小麦幼苗平铺于垫有浸湿滤纸的无菌培养皿中暗培养3天。
(6)暗处理完成后将小麦幼芽转移到培养土中正常生长。
(7)将正常生长的小麦幼苗取叶片于填装钢株的2.0EP管中,采用CTAB法提取小麦全基因组DNA,并采用TaSAUR215特异性引物(UBI-F2TaSAUR215-R)鉴定小麦转基因事件。
UBI-F:5’-GCCCTGCCTTCATACGCT-3’
TaSAUR215-R:5’-TCATGTGCACAAGATCCCATG-3’
(8)提取小麦Total RNA并用TaSAUR215引物(TaSAUR215-RT-F、TaSAUR215-RT-R)鉴定转基因小麦植株表达量变化。
TaSAUR215-RT-F:5’-GAGATGAGGAGGTTCGTCATC-3’
TaSAUR215-RT-R:5’-CTCCGAAGAGGAGTAGGAC-3’
(9)将转基因小麦种子与对照YM20种子泡发过夜,平铺于浸湿的滤纸上,待种子萌发后转移到锥形瓶或花盆中。
(10)待部分锥形瓶中的TaSAUR215-OE、TaSAUR215-RNAi及YM20生长至一叶一心时施加100mM NaHCO
(11)将鲜重作为表型标准,正常生长时TaSAUR215-OE较YM20鲜重更高,TaSAUR215-RNAi鲜重低于YM20,而在100mM NaHCO
结果见图2、图3。