抗基质金属蛋白酶7 (MMP-7)抑制性抗体及其用途
文献发布时间:2023-06-19 13:26:15
相关申请
本申请要求于2019年2月10日提交的以色列专利申请号264768的优先权利益,所述以色列专利申请的内容整体引入本文作为参考。
序列表声明
与本申请的提交同时递交,包含4,599字节,于2020年2月6日创建,名称为81393Sequence Listing.txt的ASCII文件引入本文作为参考。
技术领域和背景技术
在其一些实施方案中,本发明涉及与MMP-7的催化位点结合的抗体。该抗体可以用于治疗癌症,并且更具体地但非排他地,用于治疗胰腺癌。
由于其与多重肿瘤类型的临床行为的相关,基质金属蛋白酶7(MMP-7,基质溶解因子)已成为用于开发下一代癌症治疗学的重要靶。它是晚期结肠直肠癌的患者的总体存活的独立预后因素。MMP-7的除去显著减少了多发性肠瘤形成小鼠模型中的肿瘤形成。以类似的方式,发现MMP-7的过表达加速MMTV-neu小鼠乳腺癌模型中的肿瘤发展。MMP-7通过其促成早期肿瘤生长的机制尚未明确。MMP-7裸(null)小鼠已显示在胰管结扎后易于丧失化生性病变,并且用广谱MMP抑制剂抑制MMP-7减少肠息肉的数目。
MMP-7在MMP中是独特的,因为它缺乏血液结合素样结构域,并且其主要结构由信号肽、前肽和含锌催化结构域组成。MMP-7是分泌型MMP,并且如同其它MMP,在前肽的切割后被活化。导致其活化的蛋白酶尚未完全确定。MMP-7已牵涉大分子如IV型胶原、明胶、层粘连蛋白、内功素/巢蛋白和腱生蛋白C的分解。除这些经典的酶促作用之外,MMP-7还已牵涉修饰信号传导途径和调控细胞因子的活性。在MMP-7的非经典信号传导相关活性中,最佳表征的底物包括Fas-L、Fas- R/CD-95、TNF-α、VEGF、纤溶酶原、E-钙粘蛋白和整联蛋白β-4。MMP-7涉及ErbB4活性的调控, IL-17介导的上皮间充质转变的诱导,并且经由Wnt/β-连环蛋白途径充当促侵袭效应分子。通过这些效应,MMP-7作用于肿瘤细胞以及基质细胞,致使其成为感兴趣的靶,在蛋白酶网络和信号传导级联两者中具有多重下游和上游牵涉。在胰腺癌中,血清MMP-7最近显示为术前预后标记物,因为其的表达增加与不可切除的疾病相关联。先前的证据指示了,血清、血浆和胰液中的MMP-7连同有关的疾病指标,可用作诊断标记物。
胰腺癌是致命的、高度侵袭性的恶性肿瘤,估计到2030年将成为美国癌症相关死亡的第二大原因。虽然其发病率急剧增加,但用于胰腺癌的治疗选项仍然很少,并且迫切需要新型治疗学策略。
背景技术包括WO2012/056455,其公开了针对MMP-7生成的抗体。
发明内容
根据本发明的一些实施方案的一个方面,提供了包含结合MMP-7的催化位点的抗原识别区的抗体,其具有如以下中所示的互补决定区(CDR)氨基酸序列:在抗体的轻链上从N到C序贯排列的SEQ ID NO: 3(CDR1)、4(CDR2)和5(CDR3);以及在抗体的重链上从N到C序贯排列的SEQ ID NO: 6(CDR1)、7(CDR2)和8(CDR3)。
根据本发明的一些实施方案的一个方面,提供了治疗有此需要的受试者中与MMP-7的不平衡或异常活性相关的疾病的方法,该方法包括向受试者施用治疗有效量的权利要求1的抗体,从而治疗受试者中与MMP-7的不平衡或异常活性相关的疾病。
根据本发明的一些实施方案的一个方面,提供了诊断受试者中与MMP-7的不平衡或异常活性相关的疾病的方法,该方法包括使受试者的样品与本文所述的抗体接触,以便分析MMP-7的表达,其中所述MMP-7表达的上调指示与MMP-7的不平衡或异常活性相关的疾病。
根据本发明的一些实施方案的一个方面,提供了治疗有此需要的受试者中的癌症的方法,其包括:
(a)在受试者的样品中分析MMP-7的量;和
(b)在确认MMP-7的量高于预定水平后,向受试者施用治疗有效量的本文所述的抗体,从而治疗癌症。
根据本发明的一些实施方案的一个方面,提供了治疗有此需要的受试者中的癌症的方法,其包括:
(a)使用本文所述的抗体在受试者的样品中分析MMP-7的量;和
(b)在确认MMP-7的量高于预定水平后,向受试者施用治疗有效量的药剂,所述药剂下调MMP-7的量,从而治疗癌症。
根据本发明的一些实施方案的一个方面,提供了治疗有此需要的受试者中的胰腺癌的方法,其包括向受试者施用治疗有效量的抗体,所述抗体包含结合MMP-7的催化位点的抗原识别区,其中所述抗体抑制MMP-7的活性,并且其中抗体针对MMP-7的Ki是抗体针对MMP2或MMP9的Ki的至多1/5,从而治疗胰腺癌。
根据本发明的一些实施方案的一个方面,提供了包含本文所述的抗体的药物组合物。
根据本发明的一些实施方案的一个方面,提供了编码至少一个CDR氨基酸序列的分离的多核苷酸,所述CDR氨基酸序列选自SEQ ID NO: 3、4、5、6、7和8。
根据本发明的一些实施方案的一个方面,提供了包含本文所述的抗体和化学治疗剂的制造物品。
根据本发明的一些实施方案,抗体具有如SEQ ID NO: 1中所示的VL氨基酸序列。
根据本发明的一些实施方案,抗体具有如SEQ ID NO: 2中所示的VH氨基酸序列。
根据本发明的一些实施方案,抗体附着至可检测部分或治疗部分。
根据本发明的一些实施方案,疾病是癌症。
根据本发明的一些实施方案,癌症是胰腺癌。
根据本发明的一些实施方案,使用抗体实现分析。
根据本发明的一些实施方案,抗体是本文所述的抗体。
根据本发明的一些实施方案,多核苷酸编码如SEQ ID NO: 3-5中所示的CDR氨基酸序列。
根据本发明的一些实施方案,多核苷酸编码如SEQ ID NO: 6-8中所示的CDR氨基酸序列。
根据本发明的一些实施方案,多核苷酸编码如SEQ ID NO: 3-8中所示的CDR氨基酸序列。
根据本发明的一些实施方案,该方法进一步包括向受试者施用化学治疗剂。
根据本发明的一些实施方案,化学治疗剂是核苷类似物。
根据本发明的一些实施方案,核苷酸类似物包含吉西他滨。
根据本发明的一些实施方案,化学治疗剂是奥沙利铂。
根据本发明的一些实施方案,当作为单一药剂使用时,化学治疗剂的剂量小于黄金标准剂量。
根据本发明的一些实施方案,化学治疗剂是核苷类似物。
根据本发明的一些实施方案,核苷酸类似物包含吉西他滨。
根据本发明的一些实施方案,化学治疗剂是奥沙利铂。
根据本发明的一些实施方案,将抗体和化学治疗剂配制在单一组合物中。
根据本发明的一些实施方案,制造物品用于治疗癌症。
根据本发明的一些实施方案,癌症是胰腺癌。
除非另有定义,否则本文使用的所有技术和/或科学术语具有与本发明所属领域的普通技术人员通常理解相同的含义。尽管与本文描述的那些类似或等价的方法和材料,可以用于本发明的实施方案的实践或测试中,但下文描述了示例性的方法和/或材料。在冲突的情况下,以专利说明书包括定义为准。另外,材料、方法和实施例仅是说明性的,并不预期必然是限制性的。
附图说明
参考附图,仅作为示例,在本文中描述了本发明的一些实施方案。现在详细地具体参考附图,要强调的是,所示出的细节是作为示例并且用于本发明的实施方案的说明性讨论的目的。在这方面,结合附图进行的描述对于本领域技术人员而言显而易见的是,可以如何实践本发明的实施方案。
在附图中:
图1A-B. 用单独的合成Zn Tripod或人MMP-7催化酶免疫并不产生特异性结合活化酶的抗体候选物。A,在来自用单独的Zn Tripod免疫的小鼠的出血中,使用ELISA在血清中检查针对抗原Zn-Tripod-KLH(Zn Tripod)、MMP-7活化形式、MMP-7酶原/前体形式和BSA对照的免疫应答。该图显示了与各种抗原稀释度的结合。ELISA并未显示结合MMP-7活化构象或酶原形式的抗体的存在。观察到针对Zn Tripod的反应性。B,还使用来自用单独的重组人MMP-7活化形式免疫的小鼠的出血,在ELISA中检查免疫应答。弱应答的特征在于针对MMP-7前体形式和MMP-7活化形式两者的结合。值得注意的是,在Zn Tripod包被的孔中没有观察到结合。
图2A-F. 交替免疫产生针对酶的活化形式具有高结合亲和力和选择性的抗MMP-7抗体。A,雌性BALB/c小鼠以3周间隔用由完全弗氏佐剂乳化的合成Zn Tripod或活化形式的重组人MMP-7进行免疫。使用ELISA观察到根据重复注射的渐进性免疫应答。在第18周选择来自具有高结合亲和力的候选物的B细胞用于杂交瘤生成。B,当使用ELISA检查时,所选小鼠在融合之前的血清免疫应答显示针对Zn Tripod和MMP-7的双特异性,并且显示在各种稀释度下与MMP-14可忽略不计的结合。C,纯化的杂交瘤单克隆抗体GSM-192与MMP-7活性酶的结合曲线(Kd= 43.1±1.43 nM)。该抗体并未与其它MMP的实验对象组有效结合,指示了其针对MMP-7的高特异性。D,使用短荧光肽在体外检查GSM-192 Fab对人MMP-7的酶促活性的作用。GSM-192 Fab以Ki =131.98±10.23 nM抑制MMP-7活性。相关的MMP,如MMP-14、-9和-12,在GSM-192的存在下显示持续的活性,具有可忽略不计的效应或没有效应。E,显示了模拟高度保守的MMP活性位点的合成Zn Tripod的结构。在这种结构中,与3个组氨酸(以红色突出显示)的锌配位与所有MMP活性位点一样是保守的。F,斑点印迹显示了抗MMP-7 Ab在不同浓度下以高亲和力与酶的活化形式的选择性结合。GSM-192并不与酶原形式结合。
图3A-D. GSM-192结构和对接显示了抗体对结合位点的独特亲和力,所述结合位点接近于酶的活化形式内的保守活性位点。A,GSM-192的Fab片段的晶体结构(2.3 Å)作为带状图表示。显示了重链(浅绿色)和轻链(深绿色)(PDB:6FBJ)。B,GSM-192与活化的人MMP-7的对接模型揭示了与酶活性位点的边缘(外位点(exosite))的直接结合。MMP-7的表面以米色显示,并且制备为半透明的。抗体以深绿色勾勒,显示了三条侧链:位于Leu锚定点(紫色)附近的L100H、R101H和Y33L。后者在锚定外位点的活性位点处与MMP-7相互作用。乙酰氧肟酸(AHA)显示为球棍模型,其中碳原子为黄色,氧为红色且氮为蓝色。C,MMP-7(米色)、GSM-192(绿色)复合物的对接结构显示了极佳的表面互补性和小“隧道”,小抑制剂AHA可以通过所述“隧道”在界面处插入并且与Zn2+离子结合。D,球棍对接模型显示了在活性位点锌(灰色)附近的重要接触。
图4A-C. 通过基因沉默和抑制性抗体GSM-192两者的MMP-7抑制导致体外肿瘤细胞死亡。A,在基于FACS的细胞周期分析中,MMP-7慢病毒沉默导致subG1峰值增加,指示了AsPC-1细胞死亡的猛增。两种载体,慢病毒1(LV-1)和慢病毒2(LV-2)显示了亚G1峰值的类似增加。B,MMP-7抑制经由AsPC-1细胞中的细胞凋亡导致肿瘤细胞死亡。FACS分析显示了在用渐增剂量的GSM-192 Fab处理后,增加的膜联蛋白V有关的细胞凋亡(Q1+Q2)。此处FACS数据输出指示了作为Q1的早期凋亡细胞和作为Q2的晚期凋亡细胞。使用平均值± s.e.m(灰色条形图)绘制经历细胞凋亡的细胞百分比增加。使用相同管道生成的LOXL-2 Fab用作处理对照。C,蛋白质印迹显示了在具有或没有GSM-192 Fab处理的细胞裂解物中的FasL表达。观察到抗MMP-7 Fab处理的细胞中的FasL水平的总体增加。以1.5 μM浓度的抗LOXL-2 Fab和星形孢菌素(STS)(促凋亡药物)用作处理对照。α微管蛋白用作样品制备对照。图中的数据代表α微管蛋白标准化的平均值± s.e.m。
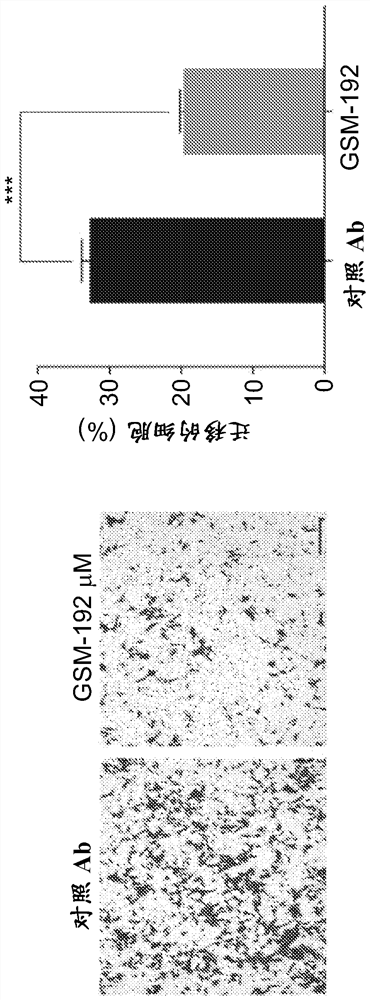
图5A-B. GSM-192处理分别在划痕测定和transwell测定中延缓或减少细胞迁移。A,在标准划痕试验中,用GSM-192处理减缓了细胞迁移。处理后7小时,对照抗体GST(非特异性的合并的IgG)处理的孔使划痕闭合,但GSM-192处理的孔则不。数据代表平均值±s.e.m,并且用双尾t检验评估显著性。比例尺,20 μm。**P ≤ 0.01。B,GSM-192在处理后15小时显著减少跨越transwell膜的细胞迁移。数据代表了平均值± s.e.m,并且用双尾t检验评估显著性。比例尺,20 μm。***P ≤ 0.001。
图6A-B. GSM-192处理后的MMP-7表达和细胞存活。A,细胞培养基的蛋白质印迹分析显示了在各种胰腺癌细胞系中的MMP-7表达。B,MTT细胞存活测定用于生成剂量响应曲线拟合分析,并且获得了不同浓度的GSM-192处理对AsPC-1和CFPAC-1胰腺导管腺癌细胞的IC50值。对于AsPC-1为2.33μM且对于CFPAC-1为4.34μM的这些IC
图7A-B. 人PDAC组织用GSM-192的免疫组织化学染色。A,商业抗MMP-7 Ab染色显示了与人PDAC肿瘤的外围相比,在核心中MMP-7蛋白的表达增加。B,GSM-192和抗MMP-7商业抗体染色的比较显示了不同的定位。GSM-192占优势地染色细胞表面上的活性酶,并且商业抗体染色核和细胞质区室中的酶原和催化酶两者。比例 = 20 μm。
图8. 抗MMP-7mAb并不通过RIP1-Tag2小鼠血清中的循环MMP变得隔离。与纯催化MMP-7相比,抗体并不与衍生自荷有晚期胰岛素瘤(13.5周)的小鼠的大范围总血清蛋白结合。它指示了血液中缺乏催化形式的MMP-7以及抗MMP-7抗体对催化酶的高特异性。因此,抗体不易去除循环酶或由于结合而被去除。
图9A-C. A,早期治疗总体上减少了RIP1-Tag2胰岛素瘤(PanNET)模型中经历血管生成转换的胰岛数目。GSM-192治疗的小鼠中血管生成性胰岛的数目是对照组小鼠中可见的总数的一半。B,在体内,抗MMP-7 mAb没有显示Ki-67和半胱天冬酶-3阳性细胞的数目的显著差异,但它减少了经历血管生成的胰岛中由CD34+细胞覆盖的面积。C,晚期治疗显示了对RIP1-Tag2小鼠模型中的肿瘤体积的持续作用。如所示的,总肿瘤体积是所有肿瘤基因座的合计体积。相对于对照小鼠,它在治疗的小鼠中显著减少。**P ≤ 0.01,*P ≤ 0.05。
图10A-B. A,当用1μM GSM-192治疗时,通过HUVEC细胞的管形成被破坏。与GST对照抗体治疗的组相比,闭环和分支点的总数目在治疗组中显著减少。B,主动脉环出芽测定显示了,用1μM GSM-192显著减少新血管出芽。在离体温育后7天,出芽在GSM-192治疗的组中显著减少。这些结果证实了用GSM-192治疗后对血管生成的直接影响。
图11A-B. A,根据吉西他滨(GEM)浓度的对数绘制并且在可变斜率S形回归模型上拟合的MTT细胞死亡测定吸光度数据,显示了与GEM+PBS组相比,GEM+GSM-192组的IC
具体实施方式
在其一些实施方案中,本发明涉及与MMP-7的催化位点结合的抗体。该抗体可以用于治疗癌症,并且更具体地但非排他地,用于治疗胰腺癌。
在详细解释本发明的至少一个实施方案之前,应理解,本发明在其应用中并不一定限于下述描述中阐述的或由实施例例示的细节。本发明能够具有其它实施方案,或者能够以各种方式进行实践或执行。
基质金属蛋白酶参与许多生物过程,其范围从细胞增殖、分化和细胞外基质(ECM)的重塑到血管形成和细胞迁移。这些过程需要在基质金属蛋白酶(MMP)与其天然组织抑制剂(TIMP)的功能之间的微妙平衡。这种平衡的丧失是众多病理学状况的标志,所述病理学状况包括转移性肿瘤、神经退行性疾病和骨关节炎。
众多MMP抑制剂是本领域已知的,包括小肽抑制剂例如异羟肟酸盐/酯(hydroxomate)、非微生物四环素和单克隆抗体。
本发明人先前发现了识别金属酶的催化位点的电子和结构决定簇两者的抗体可以用作其有力的抑制剂。使用模拟金属酶的金属结合的催化位点的半抗原作为免疫原致使能够生成高度有效的治疗性抗体,其显示能够治疗特征在于金属蛋白活性升高的临床状况(参见WO2004/087042和WO2008/102359)。
本发明人现在已理解另外的筛选步骤对于发现高度特异性的MMP-7抗体是必不可少的。仅能够结合活化形式的MMP-7而不是酶原的抗体应该被选择用于进一步开发(即,选择用于融合且开发以生成单克隆抗体)。本发明人通过进行斑点印迹分析来执行这个步骤,所述斑点印迹分析使用分别涂布有不同浓度的活性MMP-7及其酶原的硝酸纤维素膜。通过进行这种另外的测定,本发明人发现了针对MMP-7的高度特异性的单克隆抗体,在本文中称为GSM-192,其结合酶的催化结构域而不是酶的酶原形式。
GSM-192的Fv结构域与可用的MMP-7结构的对接提示了GSM-192有效结合催化裂隙区域(图3A-D),产生阻断底物分子进入的立体位阻(stearic hindrance)。
在分析该抗体的特异性的同时,本发明人发现与有关的MMP相比,GSM-192以令人印象深刻的选择性和亲和力结合MMP-7(图2A-F)。使用GSM-192 Fab抑制MMP-7以浓度依赖性方式诱导胰腺癌细胞的细胞死亡。用GSM-192处理PDAC人细胞导致主要外在死亡途径配体 - Fas配体的浓度依赖性稳定。因此,活性MMP-7的抑制在体外导致细胞死亡,具有处理组中的细胞表面Fas配体的伴随增加(图4A-C)。不受理论束缚,可以得出,通过GSM-192抑制MMP-7可能是由于MMP-7的下游功能之一,即保护癌细胞免于FasL诱导的细胞凋亡,的阻断。此外,PDAC细胞(CFPAC-1)的GSM-192处理削弱了其迁移通过transwell孔且在划痕测定中闭合人工伤口的能力(图5A-B)。总之,使用高亲和力新型抗体特异性抑制MMP-7进一步确证了其作为胰腺癌进展中的关键靶的作用。
本发明人提出抗体如GSM-192可能对治疗、诊断和研究应用具有显著且直接的影响。具体而言,对于诊断应用,识别MMP的活性构象的抗体是有价值的,因为患病组织显示了活化形式超过其酶原的显著差异表达。
因此,根据本发明的第一个方面,提供了包含结合MMP-7的催化位点的抗原识别区的抗体,其具有如以下中所示的互补决定区(CDR)氨基酸序列:在抗体的轻链上从N到C序贯排列的SEQ ID NO: 3(CDR1)、4(CDR2)和5(CDR3);以及在抗体的重链上从N到C序贯排列的SEQ ID NO: 7(CDR1)、8(CDR2)和9(CDR3)。
本发明的这个方面的抗体可以包含如SEQ ID NO: 2中所示的VH氨基酸序列、以及如SEQ ID NO: 1中所示的VL氨基酸序列。抗体的CDR序列在SEQ ID NO. 3-8中提供。
根据一个特定的实施方案,抗体包含与如SEQ ID NO: 1和2中所示的序列至少90%同源、至少91%同源、至少92%同源、至少93%同源、至少94%同源、至少95%同源、至少96%同源、至少97%同源、至少98%同源、至少99%同源或甚至100%同源的氨基酸序列(其中抗体的CDR序列始终与本文上文提供的那些序列100%同源)。
如本文使用的,术语“抗体”指完整的抗体分子,并且短语“抗体片段”指其功能片段,例如能够与巨噬细胞结合的Fab、F(ab')
如本文使用的,术语“互补决定区”或“CDR”可互换使用,以指在重链和轻链多肽的可变区内发现的抗原结合区。一般地,抗体包含在每个VH中的三个CDR(CDRH1或H1;CDRH2或H2;和CDRH3或H3)、以及在每个VL中的三个CDR(CDRL1或L1;CDRL2或L2;和CDRL3或L3)。
特定抗体中构成可变区或CDR的氨基酸残基的身份(identity)可以使用本领域众所周知的方法进行确定,并且包括方法例如Kabat等人定义的序列变异性(参见例如,Kabat等人,1992,Sequences of Proteins of Immunological Interest,第5版,Public HealthService,NIH,Washington D.C.)、Chothia等人定义的结构环区域的定位(参见例如,Chothia等人,Nature 342:877-883,1989.)、使用Oxford Molecular的AbM抗体建模软件(现在的Accelrys®,参见Martin等人,1989,Proc. Natl Acad Sci USA. 86:9268;以及万维网网站www(dot)bioinf-org(dot)uk/abs)在Kabat和Chothia之间的妥协、如通过接触定义(参见MacCallum等人,J. Mol. Biol. 262:732-745,1996)和“构象定义”(参见例如,Makabe等人,Journal of Biological Chemistry,283:1156-1166,2008)限定的可用的复杂晶体结构。
如本文使用的,“可变区”和“CDR”可以指通过本领域已知的任何方法(包括方法的组合)定义的可变区和CDR。
生成抗体(即,单克隆的和多克隆的)的方法是本领域众所周知的。抗体可以经由本领域已知的几种方法中的任何一种生成,所述方法可以采用抗体分子的体内产生的诱导、筛选免疫球蛋白文库或如公开的高度特异性结合试剂的实验对象组[Orlandi D.R.等人(1989)Proc. Natl. Acad. Sci. 86:3833-3837,Winter G.等人(1991)Nature 349:293-299]、或通过培养中的连续细胞系生成单克隆抗体分子。这些包括但不限于杂交瘤技术、人B细胞杂交瘤技术和EB病毒(EBV)-杂交瘤技术[Kohler G.等人(1975)Nature 256:495-497,Kozbor D.等人(1985)J. Immunol. Methods 81:31-42,Cote R.J.等人(1983)Proc. Natl. Acad. Sci. 80:2026-2030,Cole S.P.等人(1984)Mol. Cell. Biol. 62:109-120]。
在其中本发明的化合物太小而不能引发强免疫原性应答的情况下,此类抗原(半抗原)可以偶联至抗原中性的载体,例如匙孔血蓝蛋白(KLH)或血清白蛋白[例如牛血清白蛋白(BSA)]载体(参见美国专利号5,189,178和5,239,078)。可以使用本领域众所周知的方法来实现与载体的偶联;例如,可以实现与氨基的直接偶联,并且任选地随后为所形成的亚氨基键合的还原。可替代地,载体可以使用缩合剂如二环己基碳二亚胺或其它碳二亚胺脱水剂进行偶联。接头化合物也可以用于实现偶联;同双功能和异双功能接头两者均可从Pierce Chemical Company,Rockford,Ill获得。然后可以将所得到的免疫原性复合物注射到合适的哺乳动物受试者,例如小鼠、兔等等内。合适的方案涉及根据时间表在佐剂的存在下重复注射免疫原,其加强血清中的抗体产生。免疫血清的滴度可以使用本领域众所周知的免疫测定程序容易地测量。
所获得的抗血清可以直接使用,或者可以如上文所述获得单克隆抗体。
可以使用本领域众所周知的方法获得抗体片段。(参见例如,引入本文作为参考的Harlow和Lane,Antibodies: A Laboratory Manual,Cold Spring Harbor Laboratory,New York,1988)。例如,可以通过抗体的蛋白酶解水解、或者通过编码该片段的DNA在大肠杆菌或哺乳动物细胞(例如中国仓鼠卵巢细胞培养物或其它蛋白质表达系统)中的表达,来制备根据本发明的抗体片段。
可替代地,可以通过常规方法,通过完整抗体的胃蛋白酶或木瓜蛋白酶消化来获得抗体片段。例如,抗体片段可以通过用胃蛋白酶酶促切割抗体来产生,以提供表示为F(ab')2的5S片段。这种片段可以使用硫醇还原剂和任选的阻断基进一步切割,所述阻断基用于起因于二硫键的切割的巯基,以产生3.5S Fab'单价片段。可替代地,使用胃蛋白酶的酶促切割直接产生两个单价Fab'片段和一个Fc片段。这些方法例如由Goldenberg,美国专利号4,036,945和4,331,647以及其中包含的参考文献进行描述,所述专利在处整体引入作为参考。还参见Porter,R. R.,Biochem. J.,73: 119-126,1959。也可以使用切割抗体的其它方法,例如重链的分离以形成单价轻链-重链片段,片段的进一步切割,或者其它酶促、化学或遗传技术,只要片段与由完整抗体识别的抗原结合。
Fv片段包含V
CDR肽(“最小识别单元”)可以通过构建编码目的抗体的CDR的基因来获得。例如,通过使用聚合酶链反应从抗体产生细胞的RNA合成可变区,来制备此类基因。参见例如,Larrick和Fry,Methods,2: 106-10,1991。
应了解,对于人治疗或诊断,优选使用人源化抗体。非人(例如鼠)抗体的人源化形式是免疫球蛋白、免疫球蛋白链或其片段(例如Fv、Fab、Fab'、F(ab')
因此,根据本发明的另一个方面,提供了分离的多核苷酸,其编码选自SEQ ID NO:3、4、5、6、7和8的至少一个CDR氨基酸序列。多核苷酸可以编码轻链的CDR(例如SEQ ID NO:3-5)和/或重链的CDR(例如SEQ ID NO: 6-8)。任选地,多核苷酸可以编码抗体的每个CDR。多核苷酸可以进一步编码其编码抗体主链的序列(例如IgG1、2、3或4)。主链可以包含人序列。
优选克隆到本发明的一些实施方案的核酸构建体内,本发明的一些实施方案的多核苷酸可以用于在遗传上指导在本发明的一些实施方案的转化宿主细胞中的抗体或抗体链产生。
本发明的一些实施方案的多核苷酸可以通过本领域内的各种已知方法中的任何一种引入细胞内。此类方法一般可以在以下中描述:Sambrook等人,[Molecular Cloning:A Laboratory Manual,Cold Springs Harbor Laboratory,New York(1989,1992)];Ausubel等人,[Current Protocols in Molecular Biology,John Wiley and Sons,Baltimore,Maryland(1989)];Chang等人,[Somatic Gene Therapy,CRC Press,AnnArbor,MI(1995)];Vega等人,[Gene Targeting,CRC Press,Ann Arbor MI(1995)];Vectors [A Survey of Molecular Cloning Vectors and Their Uses,Butterworths,Boston MA(1988)]以及Gilboa等人[Biotechniques 4(6): 504-512(1986)],并且包括例如稳定或瞬时转染、脂质转染、电穿孔和用重组病毒载体的感染。
本发明还考虑的是表达本文所述的多核苷酸的细胞。
用于使非人抗体人源化的方法是本领域众所周知的。一般地,人源化抗体具有从其为非人的来源引入的一个或多个氨基酸残基。这些非人氨基酸残基经常被称为输入残基,其通常取自输入可变结构域。人源化可以基本上按照Winter及同事的方法[Jones等人,Nature,321:522-525(1986);Riechmann等人,Nature 332:323-327(1988);Verhoeyen等人,Science,239:1534-1536(1988)],通过用啮齿类动物CDR或CDR序列取代人抗体的相应序列来执行。相应地,此类人源化抗体是嵌合抗体(美国专利号4,816,567),其中基本上少于完整的人可变结构域已被来自非人物种的相应序列取代。在实践中,人源化抗体通常是人抗体,其中一些CDR残基和可能的某些FR残基被来自啮齿类动物抗体中的类似位点的残基取代。
还可以使用本领域已知的各种技术产生人抗体,所述技术包括噬菌体展示文库[Hoogenboom和Winter,J. Mol. Biol.,227:381(1991);Marks等人,J. Mol. Biol.,222:581(1991)]。Cole等人和Boerner等人的技术也可用于制备人单克隆抗体(Cole等人,Monoclonal Antibodies and Cancer Therapy,Alan R. Liss,第77页(1985),以及Boerner等人,J. Immunol.,147(1):86-95(1991)]。类似地,可以通过将人免疫球蛋白基因座引入转基因动物内来制备人,所述转基因动物例如其中内源性免疫球蛋白基因已部分或完全失活的小鼠。在攻击后,观察到人抗体产生,其在所有方面都密切类似在人中可见的,包括基因重排、组装和抗体储库。这种方法例如在美国专利号5,545,807;5,545,806;5,569,825;5,625,126;5,633,425;5,661,016和下述科学出版物中进行描述:Marks等人,Bio/Technology 10,779-783(1992);Lonberg等人,Nature 368 856-859(1994);Morrison,Nature 368 812-13(1994);Fishwild等人,Nature Biotechnology 14,845-51(1996);Neuberger,Nature Biotechnology 14,826(1996);Lonberg和Huszar,Intern.Rev. Immunol. 13 65-93(1995)。
一旦获得抗体,就可以测试它们的金属酶抑制活性和亲和力。用于金属蛋白抑制活性的适当测定条件在以下中进行描述:Knight等人,FEBS Letters 296(3):263-266(1992),Cawston等人,Anal. Biochem,99:340-345(1979),Cawston等人,Methods inEnzymology 80:771 et seq.(1981);Cawston等人,Biochem. J.,195:159-165(1981),Weingarten等人,Biochem. Biophys. Res. Comm.,139:1184-1187(1984),以及美国专利号4,743,587和5,240,958。
本发明的这个方面的抑制性抗体通常针对MMP-7具有非常高的结合亲和力和特异性,具有40-50 nM,更通常为约40-45 nM的K
抗体可以与催化位点的金属配位离子结合而不是与金属离子本身结合,或者可替代地,抗体可以与存在于活性位点袋中的金属离子及其配位离子两者结合。应了解,除与催化位点结合之外,抗体还可以与MMP-7表面上的位点结合。
本发明还提供了包含这些抗体的至少一个、两个、三个、四个、五个或六个CDR序列以及其同源物和片段的任何(多)肽序列,只要其金属酶抑制活性得到保留(金属蛋白的催化活性的特异性抑制)。
根据本发明的一些实施方案,抗体可以与功能性部分(也称为“免疫缀合物”)例如可检测部分或治疗部分缀合。免疫缀合物分子可以是分离的分子,例如可溶性和/或合成分子。
各种类型的可检测部分或报告部分可以与本发明的抗体缀合。这些包括但不限于放射性同位素(例如
合适荧光团的实例包括但不限于藻红蛋白(PE)、异硫氰酸荧光素(FITC)、细胞色素(Cy-chrome)、罗丹明、绿色荧光蛋白(GFP)、蓝色荧光蛋白(BFP)、德克萨斯红、PE-Cy5等等。关于荧光团选择、将荧光团连接到各种类型分子的方法的另外指导,参见Richard P.Haugland,“Molecular Probes: Handbook of Fluorescent Probes and ResearchChemicals 1992–1994”,第5版,Molecular Probes,Inc.(1994);授予Oncoimmunin Inc.的美国专利号6,037,137;Hermanson,“Bioconjugate Techniques”,Academic Press NewYork,N.Y.(1995);Kay M.等人,1995. Biochemistry 34:293;Stubbs等人,1996.Biochemistry 35:937;Gakamsky D.等人,“Evaluating Receptor Stoichiometry byFluorescence Resonance Energy Transfer,” in “Receptors: A PracticalApproach,” 第2版,Stanford C.和Horton R.(编辑),Oxford University Press,UK.(2001);授予Targesome,Inc.的美国专利号6,350,466]。可以用于检测与荧光可检测部分缀合的抗体的荧光检测方法包括例如,荧光活化流式细胞术(FACS)、免疫荧光共聚焦显微镜检查、荧光原位杂交(FISH)和荧光共振能量转移(FRET))。
众多类型的酶可以附着至本发明的抗体[例如辣根过氧化物酶(HPR)、β-半乳糖苷酶和碱性磷酸酶(AP)],并且可以使用ELISA(例如,在溶液中)、酶联免疫组织化学测定(例如,在固定组织中)、酶联化学发光测定(例如,在电泳分离的蛋白质混合物中)或本领域已知的其它方法[参见例如,Khatkhatay MI.和Desai M.,1999. J Immunoassay 20:151-83;Wisdom GB.,1994. Methods Mol Biol. 32:433-40;Ishikawa E.等人,1983. JImmunoassay 4:209-327;Oellerich M.,1980. J Clin Chem Clin Biochem. 18:197-208;Schuurs AH.和van Weemen BK.,1980. J Immunoassay 1:229-49),来执行酶缀合的抗体的检测。
亲和标签(或结合对的成员)可以是可由相应抗体鉴定的抗原[例如,由抗DIG抗体鉴定的地高辛配基(DIG)]或针对标签具有高亲和力的分子[例如,链霉抗生物素蛋白和生物素]。如上所述,抗体或结合亲和标签的分子可以是荧光标记的或缀合至酶的。
本领域广泛实践的各种方法可以用于将链霉抗生物素蛋白或生物素分子附着至本发明的抗体。例如,生物素分子可以经由生物素蛋白连接酶(例如,BirA)的识别序列附着至本发明的抗体,如下述实施例节段和Denkberg,G.等人,2000. Eur. J. Immunol. 30:3522-3532中所述。可替代地,链霉抗生物素蛋白分子可以附着至抗体片段例如单链Fv,基本上如以下中所述:Cloutier SM.等人,2000. Molecular Immunology 37:1067-1077;Dubel S.等人,1995. J Immunol Methods 178:201;Huston JS.等人,1991. Methods inEnzymology 203:46;Kipriyanov SM.等人,1995. Hum Antibodies Hybridomas 6:93;Kipriyanov SM.等人,1996. Protein Engineering 9:203;Pearce LA.等人,1997.Biochem Molec Biol Intl 42:1179-1188)。
与链霉抗生物素蛋白缀合的功能性部分例如荧光团,从免疫荧光流式细胞术试剂的基本上所有主要供应商(例如,Pharmingen或Becton-Dickinson)商购可得。
根据本发明的一些实施方案,生物素缀合的抗体与链霉抗生物素蛋白分子结合,以形成多价组合物(例如,抗体的二聚体或四聚体形式)。
表1提供了可以与本发明的抗体缀合的可鉴定部分的非限制性实例。
如提到的,抗体可以与治疗部分缀合。治疗部分可以是例如细胞毒性部分、毒性部分、细胞因子部分、以及包含与本发明的抗体不同特异性的第二抗体部分。
可以与本发明的抗体缀合的治疗部分的非限制性实例在下表2中提供。
取决于上下文、应用和目的,功能性部分(本发明的可检测部分或治疗部分)可以以各种方式附着或缀合至本发明的抗体。
当功能性部分是多肽时,免疫缀合物可以通过重组手段产生。例如,编码毒素(例如,PE38KDEL)或荧光蛋白[例如,绿色荧光蛋白(GFP)、红色荧光蛋白(RFP)或黄色荧光蛋白(YFP)]的核酸序列可以与编码本发明的抗体的核酸序列在框内连接,并且在宿主细胞中表达以产生重组缀合抗体。可替代地,功能性部分可以通过例如以限定次序逐步添加一个或多个氨基酸残基,例如固相肽合成技术来化学合成。
也可以使用本领域广泛实践的标准化学合成技术,将功能性部分附着至本发明的抗体[参见例如,hypertexttransferprotocol://worldwideweb(dot)chemistry(dot)org/portal/Chemistry)],例如使用任何合适的直接或间接的化学键合,如经由肽键(当功能性部分是多肽时),或经由与介入接头元件例如接头肽或其它化学部分例如有机聚合物的共价键合。嵌合肽可以经由在肽的羧基(C)或氨基(N)末端处的键合,或者经由与内部化学基团例如线性、分支或环状侧链,内部碳或氮原子等等的键合进行连接。抗体的荧光标记的描述在美国专利号3,940,475、4,289,747和4,376,110中详细提供。
用于将肽部分(治疗部分或可检测部分)与本发明的抗体缀合的示例性方法在本文下文进行描述:
SPDP
如上文提到的,针对MMP-7的抗体的一种具体用途是预防或治疗与MMP-7的不平衡或异常活性相关的疾病。
此类疾病的实例包括但不限于关节炎疾病,例如骨关节炎(OA)、类风湿性关节炎(RA)、脓毒性关节炎、软组织风湿病、多软骨炎和肌腱炎;转移性肿瘤,牙周病;角膜溃疡,例如由碱或其它烧伤、辐射、维生素E或类视黄醇缺乏诱导的;肾小球疾病,例如蛋白尿、营养不良性大疱性表皮松解症;骨吸收疾病,例如骨质疏松症、佩吉特氏病、甲状旁腺功能亢进和胆脂瘤;通过预防排卵或植入的生育控制;涉及肿瘤生长或者与糖尿病视网膜病变和黄斑变性相关的新血管形成的血管生成;与动脉粥样硬化斑块破裂相关的冠状动脉血栓形成;肺气肿、伤口愈合和HIV感染。
根据一个实施方案,疾病是癌症。示例性癌症包括胰腺癌、卵巢癌、肾细胞癌、结肠癌、乳腺癌、胃癌、直肠癌和前列腺癌。
治疗的癌症可能处于早期或晚期。在一个实施方案中,癌症已转移。
另外考虑的癌症包括但不限于肾上腺皮质癌,遗传性;膀胱癌;乳腺癌;乳腺癌,导管型;乳腺癌,浸润性导管内;乳腺癌,散发性;乳腺癌,易感性;乳腺癌,4型;乳腺癌,4型;乳腺癌-1;乳腺癌-3;乳腺-卵巢癌;伯基特氏淋巴瘤;宫颈癌;结肠直肠腺瘤;结肠直肠癌;结肠直肠癌,遗传性非息肉病性,1型;结肠直肠癌,遗传性非息肉病性,2型;结肠直肠癌,遗传性非息肉病性,3型;结肠直肠癌,遗传性非息肉病性,6型;结肠直肠癌,遗传性非息肉病性,7型;隆突性皮肤纤维肉瘤;子宫内膜癌;食道癌;胃癌,纤维肉瘤,多形性胶质母细胞瘤;血管球瘤,多发性;肝母细胞瘤;肝细胞癌;肝细胞癌;白血病,急性成淋巴细胞性;白血病,急性髓样;白血病,急性髓样,伴嗜酸性粒细胞增多症;白血病,急性非淋巴细胞性;白血病,慢性髓样;李-佛美尼综合征(Li-Fraumeni syndrome);脂肪肉瘤,肺癌;肺癌,小细胞;淋巴瘤,非霍奇金氏;林奇癌症家族综合征II;男性生殖细胞肿瘤;肥大细胞白血病;甲状腺髓质;髓母细胞瘤;黑色素瘤,脑膜瘤;多内分泌性腺瘤形成;髓样恶性肿瘤,易感性;粘液肉瘤,神经母细胞瘤;骨肉瘤;卵巢癌;卵巢癌,浆液性;卵巢癌;卵巢性索肿瘤;胰腺癌;胰腺内分泌肿瘤;副神经节瘤,家族性非嗜铬性;毛母质瘤;垂体瘤,侵袭性;前列腺腺癌;前列腺癌;肾细胞癌,乳头状,家族性和散发性;视网膜母细胞瘤;横纹样瘤易感综合征,家族性;横纹肌样瘤;横纹肌肉瘤;肺小细胞癌;软组织肉瘤,鳞状细胞癌,头颈部;T细胞急性成淋巴细胞性白血病;Turcot综合征伴胶质母细胞瘤;胼胝形成伴食道癌;子宫颈癌,肾母细胞瘤,2型;和肾母细胞瘤,1型等。
根据一个特定的实施方案,癌症是胰腺癌。
根据一个特定的实施方案,胰腺癌是胰腺腺癌、神经内分泌肿瘤或胰腺腺泡癌。
因此,本说明书提供了治疗有此需要的受试者中的胰腺癌的方法,其包括向受试者施用治疗有效量的抗体,所述抗体包含结合MMP-7的催化位点的抗原识别区,其中所述抗体抑制所述MMP-7的活性,并且其中抗体针对所述MMP-7的Ki是抗体针对MMP2或MMP9的Ki的至多1/5,从而治疗胰腺癌。
此类抗体的实例在本文以及WO2012/056455和WO2010/012167中提供,所述专利的内容引入本文作为参考。
可以使用本领域已知的测定来确认抗体的特异性。用于金属蛋白抑制活性的适当测定条件在以下中进行描述:Knight等人,FEBS Letters 296(3):263-266(1992),Cawston等人,Anal. Biochem,99:340-345(1979),Cawston等人,Methods in Enzymology 80:771et seq.(1981);Cawston等人,Biochem. J.,195:159-165(1981),Weingarten等人,Biochem. Biophys. Res. Comm.,139:1184-1187(1984),以及美国专利号4,743,587和5,240,958。
根据另一个实施方案,该疾病是炎性肠病(IBD),其是特征在于肠道炎症和组织重塑的严重的胃肠道病症,在频率中增加并且可能证明对于患者的致残。IBD、溃疡性结肠炎(UC)和克罗恩氏病的主要形式是慢性复发性状况,其在临床上的特征在于腹痛、腹泻、直肠出血和发烧。
可以治疗的受试者包括哺乳动物受试者,例如人。
针对MMP-7的抗体的另一个用途是诊断与MMP-7的表达上调相关的疾病。
因此,根据本发明的另一个方面,提供了诊断受试者中与MMP-7的不平衡或异常活性相关的疾病的方法,该方法包括使受试者的样品与本文所述的抗体(例如GSM-192)接触,以便分析MMP-7的表达,其中所述MMP-7表达的上调指示与MMP-7的不平衡或异常活性相关的疾病。
使用所公开的抗体分析MMP-7表达的方法包括但不限于蛋白质分析、免疫沉淀和免疫组织化学。
样品可以是液体,例如尿、唾液、脑脊髓液、血液、血清等等;固体或半固体,例如组织、粪便等等;或者可替代地,实体组织,例如组织学诊断中常用的那些实体组织。
通常,将MMP-7的量与对照(来自健康受试者的相应样品)或对应于健康受试者的已知量的MMP-7进行比较。
在诊断之后,可以将结果告知受试者。在使用本文公开的MMP-7抗体的测试结果的基础上,可以进行进一步的另外的诊断测试。
应了解,除了在体外(即,对受试者的样品)执行诊断外,还可以在体内实现诊断。
可以诊断的疾病包括上文对于可以治疗的疾病列出的那些疾病。
如果诊断结果为阴性 - 即不存在MMP-7表达的显著增加,并且确证患者患有癌症,则可以根据癌症的类型开出化学治疗剂(其并非MMP-7抗体)。
如果诊断结果为阳性 - 即MMP-7表达的增加,则本发明人考虑用下调MMP-7的量/活性的药剂治疗疾病。在一个实施方案中,用于治疗疾病的药剂是例如在本文以及WO2012/056455和WO2010/012167中公开的抑制性MMP-7抗体,所述专利的内容引入本文作为参考。
生成MMP-7抗体的方法是本领域已知的。在一个实施方案中,该方法包括用以下免疫受试者(例如动物受试者):
(i)合成的锌模拟化合物,其具有类似于MMP-7的催化结构域的结构和电子特性;和
(ii)MMP-7。
具有类似于金属酶的催化结构域的结构和电子特性的合成的锌模拟化合物通常是包含螯合金属离子的化合物。金属离子通常是锌或者其类似离子钴或镉。
根据一个实施方案,螯合剂是卟啉。
可以基于靶多肽中的实际催化结构域的结构和电子特性来选择锌模拟化合物。通常,靶多肽包括提供过渡金属配位所需的三个接触点的3个氨基酸。根据过渡金属离子,代表性配位络合物几何形状可以是四面体、正方形平面或三角形。一般而言,本发明的模拟组合物基于氨基酸侧链结构和配位的几何形状来选择。通常,可以配位过渡金属结合的氨基酸是组氨酸、精氨酸、谷氨酸、半胱氨酸、甲硫氨酸、色氨酸、丝氨酸、苏氨酸和酪氨酸,其中前两种是优选的。
示例性的合成的锌模拟化合物公开于WO2004/087042和WO2008/102359中,其引入本文作为参考。
根据一个实施方案,合成的锌模拟化合物具有通式(I):
其中:
m和n各自独立地为1至6的整数;
X
R
R是O-(CH
然而:
x和y各自独立地为1至6的整数;和
R'和R''各自独立地选自氢、烷基和环烷基。
根据本发明的这个方面的一个特定实施方案,该化合物是[2-(2-氨基乙基羰基(minoethylcarbomoyl))-乙氧基甲基]-三-[2-(N-(3-咪唑-1-基-丙基))-乙氧基甲基]甲烷,称为Imisdp,具有通式(II):
其中R是O-CH
如提到的,本发明的方法通过用本文上文所述的锌模拟化合物和MMP-7金属酶本身两者免疫来进行。
本发明考虑了用全长MMP-7或其一部分的免疫。通常,该部分应该含有对MMP-7特异性的抗原决定簇(即表位)。
根据一个实施方案,抗原决定簇在MMP-7的表面上。
用于免疫的MMP-7可以从其体内环境进行纯化,或者可替代地可以通过重组手段生成。
根据一个实施方案,免疫程序包括用锌模拟化合物的初始免疫和用MMP-7的后续(例如三至十二周后)免疫。可以通过检查来查看免疫动物(例如小鼠)中是否存在免疫应答来确定免疫的确切时间。例如,可以分析血清中的抗体滴度。
根据另一个实施方案,免疫程序包括用MMP-7的初始免疫和用锌模拟化合物的后续(例如三至十二周后)免疫。
根据又一个实施方案,免疫程序包括用MMP-7和锌模拟化合物两者的共免疫(即同时)。
本发明的抗体可以本身或作为药物组合物的部分施用于受试者。
如本文使用的,“药物组合物”指本文所述的一种或多种活性成分与其它化学组分,如生理学上合适的载体和赋形剂的制剂。药物组合物的目的是促进化合物对生物的施用。
在本文中,术语“活性成分”指可负责生物效应的抗体。
在下文中,可以互换使用的短语“生理学上可接受的载体”和“药学上可接受的载体”指这样的载体或稀释剂,其不引起对生物的明显刺激,并且不消除所施用化合物的生物活性和特性。佐剂包括在这些短语下。
在本文中,术语“赋形剂”指加入药物组合物中,以进一步促进活性成分的施用的惰性物质。赋形剂的实例包括但不限于碳酸钙、磷酸钙、各种类型的糖和淀粉、纤维素衍生物、明胶、植物油和聚乙二醇。
关于药物配制和施用的技术可以在“Remington’s Pharmaceutical Sciences,”Mack Publishing Co.,Easton,PA,最新版本中找到,所述参考文献引入本文作为参考。
合适的施用途径可以例如包括经口,直肠,经粘膜,尤其是经鼻、肠或肠胃外递送,包括肌内、皮下和髓内注射,以及鞘内、直接心室内、心内例如在右心室腔或左心室腔内、在普通冠状动脉内、静脉内、腹膜内、鼻内或眼内注射。
用于将药物递送到CNS的常规方法包括:神经外科策略(例如,脑内注射或脑室内输注);在开发BBB的内源性转运途径之一的尝试中,试剂的分子操纵(例如,产生嵌合融合蛋白,其包含与自身无法穿越BBB的试剂组合的,对于内皮细胞表面分子具有亲和力的转运肽);设计为增加试剂的脂质溶解度的药理学策略(例如,水溶性试剂与脂质或胆固醇载体的缀合);以及BBB的完整性通过高渗破坏(起因于甘露糖醇溶液输注到颈动脉内、或使用生物活性剂如血管紧张素肽)的瞬时破坏。然而,这些策略各自具有局限性,例如与侵入性手术操作相关的固有风险,由内源性转运系统固有的局限性施加的大小局限性,与嵌合分子的全身施用相关的潜在不期望的生物副作用,所述嵌合分子包含可能在CNS外部活跃的载体基序,以及在其中BBB被破坏的脑区域内的脑损伤的可能风险,这致使其成为次优的递送方法。
可替代地,可以例如经由将药物组合物直接注射到患者的组织区域内,以局部而非全身方式施用药物组合物。
术语“组织”指由具有相似结构和/或共同功能的细胞聚集体组成的生物的部分。实例包括但不限于脑组织、视网膜、皮肤组织、肝组织、胰腺组织、骨骼、软骨、结缔组织、血液组织、肌肉组织、心脏组织、脑组织、血管组织、肾组织、肺组织、性腺组织、造血组织。
本发明的药物组合物可以通过本领域众所周知的工艺来制造,例如,借助于常规混合、溶解、制粒、糖衣丸制备、水飞、乳化、包囊、包埋或冻干工艺。
因此,用于依照本发明使用的药物组合物可以以常规方式进行配制,使用包含赋形剂和助剂的一种或多种生理学上可接受的载体,其促进活性成分加工成可以在药学上使用的制剂。适当的制剂取决于选择的施用途径。
对于注射,药物组合物的活性成分可以在水溶液中进行配制,优选在生理学相容的缓冲液中,例如汉克氏溶液、林格氏溶液或生理盐缓冲液。对于经粘膜施用,在制剂中使用对于待渗透的屏障适当的渗透剂。此类渗透剂是本领域一般已知的。
对于经口施用,可以通过将活性化合物与本领域众所周知的药学上可接受的载体组合,来容易地配制药物组合物。此类载体使得药物组合物能够被配制为片剂、丸剂、糖衣丸、胶囊、液体、凝胶、糖浆剂、浆料、悬浮液等等,用于通过患者的经口摄取。用于经口使用的药物制剂可以通过以下进行制备:使用固体赋形剂,任选地研磨所得到的混合物,并且在需要时加入合适的助剂后,加工颗粒的混合物,以获得片剂或糖衣丸芯。合适的赋形剂特别是填料,例如糖,包括乳糖、蔗糖、甘露糖醇或山梨糖醇;纤维素制剂,例如玉蜀黍淀粉、小麦淀粉、大米淀粉、马铃薯淀粉、明胶、黄蓍树胶、甲基纤维素、羟丙基甲基纤维素、羧甲基纤维素钠;和/或生理学上可接受的聚合物,例如聚乙烯吡咯烷酮(PVP)。需要时,可以加入崩解剂,例如交联的聚乙烯吡咯烷酮、琼脂或海藻酸或其盐例如海藻酸钠。
糖衣丸芯提供有合适的包衣。为此,可以使用浓缩的糖溶液,其可以任选地含有阿拉伯树胶、滑石、聚乙烯吡咯烷酮、卡波姆凝胶、聚乙二醇、二氧化钛、漆溶液和合适的有机溶剂或溶剂混合物。可以将染料或色素加入片剂或糖衣丸包衣中,用于鉴定或表征活性化合物剂量的不同组合。
可以经口使用的药物组合物包括由明胶制成的推入配合胶囊,以及由明胶和增塑剂如甘油或山梨糖醇制成的密封软胶囊。推入配合胶囊可以含有与填料如乳糖、粘合剂如淀粉、润滑剂如滑石或硬脂酸镁以及任选的稳定剂混合的活性成分。在软胶囊中,活性成分可以溶解或悬浮于合适的液体中,所述液体例如脂肪油、液体石蜡或液体聚乙二醇。另外,可以添加稳定剂。用于经口施用的所有制剂应该为适合于选择的施用途径的剂量。
对于颊部施用,组合物可以采取以常规方式配制的片剂或锭剂的形式。
对于通过鼻吸入的施用,用于根据本发明使用的活性成分,以来自加压包装或喷雾器的气溶胶喷雾呈现的形式方便地递送,其中使用合适的推进剂,例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷或二氧化碳。在加压气溶胶的情况下,剂量单位可以通过提供阀门以递送计量的量来确定。可以配制用于分配器中的例如明胶的胶囊和药液筒,其含有化合物和合适的粉末基质例如乳糖或淀粉的粉末混合物。
本文所述的药物组合物可以配制用于肠胃外施用,例如通过推注或连续输注。用于注射的制剂可以以单位剂型存在于例如安瓿或具有任选添加的防腐剂的多剂量容器中。组合物可以是在油性或水性媒介物中的悬浮液、溶液或乳剂,并且可以含有配制试剂例如悬浮剂、稳定剂和/或分散剂。
用于肠胃外施用的药物组合物包括水溶性形式的活性制剂的水溶液。另外,可以将活性成分的悬浮液制备为适当地基于油或水的注射悬浮液。合适的亲脂性溶剂或媒介物包括脂肪油例如芝麻油,或者合成脂肪酸酯例如油酸乙酯、甘油三酯或脂质体。水性注射悬浮液可以含有增加悬浮液的粘度的物质,例如羧甲基纤维素钠、山梨糖醇或葡聚糖。任选地,悬浮液还可以含有合适的稳定剂或增加活性成分的溶解度的试剂,以允许制备高度浓缩的溶液。
可替代地,活性成分可以是粉末形式,用于在使用前由合适的媒介物例如无菌、无热原的水基溶液构造。
本发明的药物组合物也可以使用例如常规的栓剂基质,例如可可脂或其它甘油酯配制成直肠组合物,例如栓剂或保留灌肠剂。
适用于本发明的背景下的药物组合物包括这样的组合物,其中有效成分以有效达到预期目的的量包含。更具体而言,治疗有效量意指这样的活性成分(抗体)的量,其有效预防、减轻或改善病症(例如,癌症/炭疽感染)的症状,或者延长待治疗受试者的存活。
治疗有效量的确定完全在本领域技术人员的能力内,尤其是根据本文提供的详细公开内容。
对于本发明的方法中使用的任何制剂,治疗有效量或剂量可以最初从体外和细胞培养测定进行估计。例如,可以在动物模型中制定剂量,以达到所需的浓度或滴度。此类信息可以用于更准确地确定在人中的有用剂量。
本文所述的活性成分的毒性和治疗功效可以通过在体外、细胞培养或实验动物中的标准药学程序来确定。从这些体外和细胞培养测定以及动物研究中获得的数据可以用于制定用于在人中使用的剂量范围。剂量可以根据采用的剂型和利用的施用途径而变。确切的制剂、施用途径和剂量可以由各个医生考虑到患者的状况加以选择。(参见例如,Fingl等人,1975,于"The Pharmacological Basis of Therapeutics",第1章,第1页)。
剂量的量和间隔可以个别地调整,以提供活性成分的组织或血液水平,其足以诱导或压制生物效应(最小有效浓度,MEC)。MEC对于每种制剂不同,但可以从体外数据进行估计。达到MEC必需的剂量取决于个体特征和施用途径。检测测定可以用于确定血浆浓度。
取决于待治疗状况的严重性和响应性,给药可以是单次施用或多次施用,其中治疗过程持续数天至数周,或者直至实现治愈或达到疾病状态的减轻。
当然,待施用的组合物的量取决于待治疗的受试者、痛苦的严重性、施用方式、开处方医生的判断等。
本发明人已显示了,本发明的这个方面的抗体增强了化学治疗剂的敏感性。因此,本发明人考虑将抗体与化学治疗剂一起施用。
化学治疗剂的实例包括但不限于阿西维辛;阿柔比星;盐酸阿考达唑;阿克罗宁;阿霉素;阿多来新;阿地白介素;六甲蜜胺;安波霉素;乙酸阿美蒽醌;氨鲁米特;氨茶碱;阿那曲唑;安曲霉素;天冬酰胺酶;曲林菌素;阿扎胞苷;阿扎替派;阿佐霉素;巴马司他;苯佐替派;比卡鲁胺;盐酸比生群;甲磺酸双奈法德;比折来新;硫酸博来霉素;布喹那钠;溴匹立明;白消安;放线菌素C;卡芦睾酮;卡醋胺;卡贝替姆;卡铂;卡莫司汀;盐酸卡柔比星;卡折来新;西地芬戈;苯丁酸氮芥;西罗霉素;顺铂;克拉屈滨;甲磺酸克立那托;环磷酰胺;阿糖胞苷;达卡巴嗪;更生霉素;盐酸柔红霉素;地西他滨;右奥马铂;地扎胍宁;甲磺酸地扎胍宁;地吖醌;多西他赛;多柔比星;盐酸多柔比星;屈洛昔芬;柠檬酸屈洛昔芬;丙酸屈他雄酮;达佐霉素;依达曲沙;盐酸依氟鸟氨酸;依沙芦星;恩洛铂;恩普氨酯;依匹哌啶;盐酸表柔比星;厄布洛唑;盐酸依索比星;雌莫司汀;雌莫司汀磷酸钠;依替硝唑;依托泊苷;磷酸依托泊苷;氯苯乙嘧胺;盐酸法倔唑;法扎拉滨;芬维A胺;氟尿苷;磷酸氟达拉滨;氟尿嘧啶;氟西他滨;磷喹酮;福司曲星钠;吉西他滨;盐酸吉西他滨;羟基脲;盐酸依达比星;异环磷酰胺;伊莫福新;干扰素α-2a;干扰素α-2b;干扰素α-n1;干扰素α-n3;干扰素β-I a;干扰素γ-I b;异丙基铂;盐酸伊立替康;乙酸兰瑞肽;来曲唑;乙酸亮丙瑞林;盐酸利拉唑;洛美曲索钠;洛莫司汀;盐酸洛索蒽醌;马索罗酚;美登素;盐酸二氯甲基二乙胺;乙酸甲地孕酮;乙酸美伦孕酮;美法仑;美诺立尔;巯嘌呤;氨甲蝶呤;氨甲蝶呤钠;氯苯氨啶;美妥替哌;米丁度胺;米托卡星;丝裂红素;米托洁林;米托马星;丝裂霉素;米托司培;米托坦;盐酸米托蒽醌;霉酚酸;诺考达唑;诺拉霉素;奥马铂;奥昔舒仑;紫杉醇;奥沙利铂;培门冬酶;培利霉素;奈莫司汀;硫酸培洛霉素;培磷酰胺;哌泊溴烷;哌泊舒凡;盐酸吡罗蒽醌;普卡霉素;普洛美坦;卟吩姆钠;泊非霉素;泼尼氮芥;盐酸丙卡巴肼;嘌呤霉素;盐酸嘌呤霉素;吡唑霉素;利波腺苷;罗谷亚胺;沙芬戈;盐酸沙芬戈;司莫司汀;辛曲秦;磷乙酰天冬氨酸钠;司帕霉素;盐酸螺旋锗;螺莫司汀;螺铂;链黑霉素;链脲佐菌素;磺氯苯脲;他利霉素;泰素;替可加兰钠;替加氟;盐酸替洛蒽醌;替莫泊芬;替尼泊苷;替罗昔隆;睾内酯;硫咪嘌呤;硫鸟嘌呤;噻替哌;噻唑呋林(Tiazofuirin);替拉扎明;盐酸拓扑替康;柠檬酸托瑞米芬;乙酸曲托龙;磷酸曲西立滨;三甲曲沙;葡萄糖醛酸三甲曲沙;曲普瑞林;盐酸妥布氯唑;尿嘧啶氮芥;乌瑞替哌;伐普肽;维替泊芬;硫酸长春碱;硫酸长春新碱;长春地辛;硫酸长春地辛;硫酸长春匹定;硫酸长春甘酯;硫酸环氧长春碱;酒石酸长春瑞滨;硫酸异长春碱;硫酸长春利定;伏氯唑;折尼铂;净司他丁 ;盐酸佐柔比星。另外的抗肿瘤剂包括以下中公开的那些抗肿瘤剂:Goodman和Gilman的"The Pharmacological Basis of Therapeutics",第八版,1990,McGraw-Hill,Inc.(Health Professions Division)的第52章,Antineoplastic Agents(Paul Calabresi and Bruce A. Chabner),以及其引言,1202-1263。
在一个特定的实施方案中,化学治疗剂是核苷类似物,其实例包括但不限于吉西他滨、氨甲蝶呤、5-氟尿嘧啶、阿糖胞苷、山嵛酰阿糖胞苷、替加氟、UFT等等。
根据一个特定的实施方案,核苷酸药剂是吉西他滨。
根据一个特定的实施方案,化学治疗剂是奥沙利铂。
在组合疗法的背景下,化学治疗剂可以通过与施用抗体相同的施用途径(例如肺内、经口、肠内等)进行施用。在替代方案中,化学治疗剂可以通过与抗体不同的施用途径进行施用。
化学治疗剂可以在抗体之前(或之后)、与抗体在同一天、之前(或之后)一天、之前(或之后)一周、之前(或之后)一个月、或之前(或之后)两个月等等进行施用。
化学治疗剂和抗体可以伴随施用,即,其中这些试剂各自的施用可以以彼此部分或完全重叠的时间间隔发生。化学治疗剂和抗体可以在彼此不重叠的时间间隔过程中施用。例如,化学治疗剂可以在t=0到1小时的时帧内施用,而抗体可以在t=1到2小时的时帧内施用。另外,化学治疗剂可以在t=0到1小时的时帧内施用,而抗体可以在t=2-3小时、t=3-4小时、t=4-5小时、t=5-6小时、t=6-7小时、t=7-8小时、t=8-9小时、t=9-10小时等等的时帧内的某处施用。此外,抗体可以在t=负2-3小时、t=负3-4小时、t=负4-5小时、t=负5-6小时、t=负6-7小时、t=负7-8小时、t=负8-9小时、t=负9-10小时的时帧内的某处施用。
本发明的抗体和化学治疗剂通常以组合的量提供,以实现治疗、预防和/或疼痛缓解效果。该量显然取决于选择使用的特定化合物,其它治疗模态的性质和数目,待治疗、预防和/或缓解的状况,受试者的物种、年龄、性别、重量、健康和预后,施用模式,靶向的有效性,停留时间,清除模式,药物组合物的副作用的类型和严重程度,以及对于本领域技术人员显而易见的许多其它因素。抗体将在其下观察到与化学治疗剂组合的治疗、预防和/或疼痛缓解效果的水平下使用。
化学治疗剂可以作为单一药剂以黄金标准计量、作为单一药剂低于黄金标准计量、或作为单一药剂高于黄金标准计量(连同抗体一起)施用。
根据具体实施方案,化学治疗剂作为单一药剂低于黄金标准计量施用。
如本文使用的,术语“黄金标准计量”指对于处于给定阶段的给定肿瘤,通过监管机构(例如,FDA)推荐的计量。
根据其它具体实施方案,化学治疗剂以并不发挥与黄金标准计量相关的至少一种副作用的剂量施用。化学治疗剂治疗的副作用的非限制性实例包括皮疹、腹泻、口疮、甲沟炎、疲劳、高血糖、肝毒性、肾衰竭、心血管效应、电解质异常和GI穿孔。
因此,在一个实施方案中,当作为单一疗法使用时,化学治疗剂的量低于治疗、预防和/或疼痛缓解效果所需的最小剂量(例如,该最小剂量的10-99%,优选25至75%)。这允许减少由化学治疗剂引起的副作用,但疗法因为与抗体组合而致使有效,该组合总体上是有效的。
在本发明的一个方面,抗体和化学治疗剂就其剂量而言是协同的。也就是说,由本发明的抗体提供的效应大于由分开使用时化学治疗剂和抗体的累加效应将预料的效应。在一个替代实施方案中,本发明的化学治疗剂和抗体就其副作用而言是协同的。也就是说,由抗体与化学治疗剂组合引起的副作用小于由分开使用时的化学治疗剂或抗体提供的等价治疗效应时将预料的副作用。
需要时,本发明的组合物可以存在于包装或分配器装置,例如FDA批准的试剂盒中,其可以包含含有活性成分的一个或多个单位剂型。包装可以例如包含金属箔或塑料箔,例如泡罩包装。包装或分配器装置可能伴随有施用说明书。包装或分配器也可以由与容器结合的通告来容纳,所述通告为由管理药品的制造、使用或销售的政府机构规定的形式,所述通告反映了组合物的形式或者人或兽医学施用通过机构的批准。例如,此类通告可以是由美国食品药品监督管理局(U.S. Food and Drug Administration)批准用于处方药的标记,或者批准的产品插页。如上文进一步详述的,还可以制备包含在相容的药物载体中配制的本发明的制剂的组合物,置于适当的容器中,并且标记用于治疗指示的状况。
术语“治疗”指抑制、预防或停止病理状态(疾病、病症或状况)的发展和/或引起病理状态的减轻、缓解或消退。本领域技术人员将理解,各种方法和测定可以用于评价病理状态的发展,并且类似地,各种方法和测定可以用于评价病理状态的减轻、缓解或消退。
如本文使用的,术语“预防”指阻止疾病、病症或状况在受试者中发生,所述受试者可能处于疾病的风险中,但尚未被诊断为患有该疾病。
如本文使用的,术语“受试者”包括患有该病理状态的哺乳动物,优选处于任何年龄的人类。优选地,该术语包括处于发展病理状态的风险中的个体。
术语“包含(comprises)”、“包含(comprising)”、“包括(includes)”、“包括(including)”、“具有”及其词形变化意指“包括但不限于”。
如本文使用的,术语“方法”指用于完成给定任务的方式、手段、技术和程序,包括但不限于化学、药理学、生物学、生物化学和医学领域的从业人员已知的,或者从已知的方式、手段、技术和程序容易开发的那些方式、手段、技术和程序。
应了解,为了清楚起见,在分开的实施方案的上下文中描述的本发明的某些特点,也可以在单个实施方案中组合提供。相反,为了简洁起见,在单个实施方案的上下文中描述的本发明的各种特点,也可以分开地或以任何合适的子组合或如本发明的任何其它所述实施方案中合适地提供。在各个实施方案的上下文中描述的某些特点,不应被视为那些实施方案的必要特点,除非该实施方案没有那些要素是不可操作的。
如上文描绘的以及如下文权利要求节段中请求保护的,本发明的各个实施方案和方面在下述实施例中找到实验支持。
实施例
现在参考下述实施例,其连同上文说明书一起,以非限制性方式示出了本发明的一些实施方案。
一般地,本文使用的命名法和本发明中利用的实验室程序包括分子、生物化学、微生物学和重组DNA技术。此类技术在文献中得到详尽解释。参见例如,"Molecular Cloning:A laboratory Manual" Sambrook等人,(1989);"Current Protocols in MolecularBiology"第I-III卷Ausubel,R. M.编辑(1994);Ausubel等人,"Current Protocols inMolecular Biology",John Wiley and Sons,Baltimore,Maryland(1989);Perbal,"APractical Guide to Molecular Cloning",John Wiley & Sons,New York(1988);Watson等人,"Recombinant DNA",Scientific American Books,New York;Birren等人(编辑)"Genome Analysis: A Laboratory Manual Series",第1-4卷,Cold Spring HarborLaboratory Press,New York(1998);如美国专利号4,666,828;4,683,202;4,801,531;5,192,659和5,272,057中所示的方法学;"Cell Biology: A Laboratory Handbook",第I-III卷Cellis,J. E.编辑(1994);"Culture of Animal Cells - A Manual of BasicTechnique" by Freshney,Wiley-Liss,N. Y.(1994),第三版;"Current Protocols inImmunology"第I-III卷Coligan J. E.编辑(1994);Stites等人(编辑),"Basic andClinical Immunology"(第8版),Appleton & Lange,Norwalk,CT(1994);Mishell和Shiigi(编辑),"Selected Methods in Cellular Immunology",W. H. Freeman and Co.,NewYork(1980);可用的免疫测定在专利和科学文献中得到广泛描述,参见例如美国专利号3,791,932;3,839,153;3,850,752;3,850,578;3,853,987;3,867,517;3,879,262;3,901,654;3,935,074;3,984,533;3,996,345;4,034,074;4,098,876;4,879,219;5,011,771和5,281,521;"Oligonucleotide Synthesis" Gait,M. J.编辑(1984);“Nucleic AcidHybridization" Hames,B. D.和Higgins S. J.编辑(1985);"Transcription andTranslation" Hames,B. D.和Higgins S. J.编辑(1984);"Animal Cell Culture"Freshney,R. I.编辑(1986);"Immobilized Cells and Enzymes" IRL Press,(1986);"APractical Guide to Molecular Cloning" Perbal,B.,(1984),以及"Methods inEnzymology"第1-317卷,Academic Press;"PCR Protocols: A Guide To Methods AndApplications",Academic Press,San Diego,CA(1990);Marshak等人,"Strategies forProtein Purification and Characterization - A Laboratory Course Manual" CSHLPress(1996);所有这些都引入作为参考,如同它在本文中充分阐述一样。本文件自始至终提供了其它一般参考文献。其中的程序被认为是本领域众所周知的,并且为了读者的方便而提供。其中包含的所有信息都引入本文作为参考。
材料和方法
使用基于真空的歧管96孔斑点印迹系统(GE 10447900 Minifold斑点印迹),通过将1、1.5和2 µg酶原和活化的MMP-7重组酶固定到用TBST预先润湿的硝酸纤维素膜上执行斑点印迹。封闭在室温下用3% BSA的TBST溶液执行1小时。然后膜通过温育过夜探测通过在较早的步骤中洗脱的抗体的结合。所使用的抗MMP-7 mAb为在TBST中2 ug/ml的浓度。使在TBST中洗涤3次后的膜与山羊抗小鼠HRP抗体一起温育,并且使用ECL显色以确定仅针对催化形式结合的选择性。
MMP-7 Elisa
MMP-7
测量初始反应速率,并且通过将数据拟合到方程来评估抑制常数,其中Vi是在抑制剂的存在下的初始速度,V0是在抑制剂的不存在下的初始速度,且I是抑制剂浓度。为了确定抑制的类型,在Fab GSM-192的几个固定浓度(0-500 nM)下,根据底物浓度(0-30 mM)测量MMP-7的初始速度。通过线性化导出表观KM和Vmax的值。
GSM-192
D I VT Q S P A S L A V S L G Q R A T I S C R A S E S F D S Y G N T FV H W Y Q Q K P G Q P P K L L I Y L V S N L E S G V P A G F R G R G S R T D FT L T I D P V E A D D A A T Y Y C Q Q N N E D P Y T F G G G T K L E I K R A
GSM-192
E V Q L Q Q S G P E L V K P G A S V K I P C K A S G Y T F T D Y N M DW M K Q S H G K S L E W I G H I N P N N G G T F Y N Q K F K D K A T F I V D KS S N T A Y M E L R S L T S E D T A V Y F C A R G G G L R R G P F A Y W G Q GT L V T V S
GSM-192 Fab
将过滤的模型进一步筛选,以仅包括其中相互作用涉及抗体CDR中的暴露残基的模型。这种筛选计数了暴露的CDR残基和靶分子之间的原子间接触数目(≤5Å距离)。MolFit的GEH评分对在分子的相对取向上的小变化很敏感,并且先前发现局部刚体精化对于鉴定真正的高评分对接模型是非常有效的。因此,通过允许以2°步长的小局部旋转,对来自几次扫描的模型进行精化。精化突出显示了对接结果中的一个模型。这个模型在采用与结构2y6a非常类似的正常模式构象异构体的对接扫描中排名第1,并且其精化评分比下一个模型高3.1σ,且比平均评分高9σ(平均评分和σ通过将极端值分布函数拟合到GEH评分的分布
锚定点用于鉴定在蛋白质的表面上的单条氨基酸侧链的优选结合位置。映射用ANCHORSmap
MMP-7
MTT
FACS
Trans-well
结果
仅能够结合活化形式的MMP-7而不是酶原的抗体被选择用于进一步开发(即,选择用于融合且开发以生成单克隆抗体)。这通过斑点印迹分析来进行,所述斑点印迹分析使用分别涂布有不同浓度的活性MMP-7及其酶原的硝酸纤维素膜。显示了GSM-192结合酶的催化结构域而不是酶的酶原形式(图2F)。
MMP-7
值得注意的是,考虑到免疫加强还包括常见的锌基序,该抗体对于MMP-7是完全选择性的。根据标准方案,使用短荧光肽,在体外检查GSM-192 Fab片段对人MMP-7的酶促活性的作用。在浓度渐增的GSM-192的Fab片段的存在下,测量重组活性MMP-7的酶促活性。发现GSM-192 Fab抑制MMP-7活性,具有131.98±10.23 nM的Ki。同时测量抗体对有关MMP的催化活性的作用。MMP-9、-14和-12的体外催化活性或者不受影响,或者在MMP-14的情况下仅受抗MMP-7 Fab处理可忽略不计地影响(图2D)。
蛋白质数据库(PDB)包括MMP-7的催化结构域的几种结构,当叠加时所述结构显示了环在活性位点附近的运动性。在正常模式分析中可见类似的迁移率,并且因此抗体GSM-192(Fv结构域L2-L112和Q0-Q116)与展示活性位点的不同开口的几种MMP-7构象异构体对接。如结构2y6c31中可见的,与具有开放活性位点的MMP-7构象异构体对接,产生了图3B中所示的统计上突出的模型,具有高于下一个模型> 3σ的互补性评分。
对接模型暗示了结合通过由氨基酸L181、A216和Y241分隔,在MMP-7活性位点内的口袋中L100H的强疏水性锚定得到稳定。计算锚定点
值得注意的是,该模型显示了乙酰氧肟酸(AHA)是可逆的锌结合异羟肟酸盐,其直接在催化裂隙中结合,可以连同抗体一起容纳在结合位点中。图3C呈现了MMP-7/GSM-192复合物的预测结构的视图,一方面突出显示了极佳的表面互补性,以及另一方面在GSM-192的存在下,AHA可以通过其插入并结合MMP-7的小开口。
总之,所提议的抗体的结合模式由疏水性锚定和众多其它接触组成;它并不涉及Zn2+并允许AHA与Zn2+的同时结合。
在类似管道中产生的抗LOXL-2(赖氨酰氧化酶样2)mAb33用作同种型对照。GSM-192 Fab显示经由细胞凋亡诱导细胞死亡,并且对应于抗MMP-7治疗的浓度增加,经历细胞凋亡的细胞群体的比例增加(图4B)。为了测试细胞凋亡的分子标记物,通过蛋白质印迹分析来分析Fas配体的表达。AsPC-1细胞用GSM-192 Fab在亚致死浓度下处理24小时。事实上,与未处理的对照相比,这些细胞显示了Fas配体(FasL)表达的增加(图4C)。
GSM-192 Fab
GSM-192
参考文献
1.White,R.J.,Margolis,P.S.,Trias,J. & Yuan,Z. Targetingmetalloenzymes: a strategy that works. Curr Opin Pharmacol 3,502-7 (2003).
2.Aharoni,A.等人The 'evolvability' of promiscuous protein functions.Nat Genet 37,73-6 (2005).
3.Bonnans,C.,Chou,J. & Werb,Z. Remodelling the extracellular matrixin development and disease. Nat Rev Mol Cell Biol 15,786-801 (2014).
4.Shiomi,T. & Okada,Y. MT1-MMP and MMP-7 in invasion and metastasisof human cancers. Cancer Metastasis Rev 22,145-52 (2003).
5.Jackson,H.W.,Defamie,V.,Waterhouse,P. & Khokha,R. TIMPs: versatileextracellular regulators in cancer. Nat Rev Cancer 17,38-53 (2017).
6.Murphy,G. Tissue inhibitors of metalloproteinases. Genome Biol 12,233 (2011).
7.Jung,Y.S.等人TIMP-1 induces an EMT-like phenotypic conversion inMDCK cells independent of its MMP-inhibitory domain. PLoS One 7,e38773(2012).
8.Drews,J. Drug discovery: a historical perspective. Science 287,1960-4 (2000).
9.Overall,C.M. & Kleifeld,O. Tumour microenvironment - opinion:validating matrix metalloproteinases as drug targets and anti-targets forcancer therapy. Nat Rev Cancer 6,227-39 (2006).
10.Maurel,J.等人Serum matrix metalloproteinase 7 levels identifiespoor prognosis advanced colorectal cancer patients. Int J Cancer 121,1066-71(2007).
11.Wilson,C.L.,Heppner,K.J.,Labosky,P.A.,Hogan,B.L. & Matrisian,L.M.Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinasematrilysin. Proc Natl Acad Sci U S A 94,1402-7 (1997).
12.Rudolph-Owen,L.A.,Chan,R.,Muller,W.J. & Matrisian,L.M. The matrixmetalloproteinase matrilysin influences early-stage mammary tumorigenesis.Cancer Res 58,5500-6 (1998).
13.Crawford,H.C.,Scoggins,C.R.,Washington,M.K.,Matrisian,L.M. &Leach,S.D. Matrix metalloproteinase-7 is expressed by pancreatic cancerprecursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. JClin Invest 109,1437-44 (2002).
14.Wagenaar-Miller,R.A.等人Cooperative effects of matrixmetalloproteinase and cyclooxygenase-2 inhibition on intestinal adenomareduction. Br J Cancer 88,1445-52 (2003).
15.Dufour,A. & Overall,C.M. Missing the target: matrixmetalloproteinase antitargets in inflammation and cancer. Trends PharmacolSci 34,233-42 (2013).
16.Strand,S.等人Cleavage of CD95 by matrix metalloproteinase-7induces apoptosis resistance in tumour cells. Oncogene 23,3732-6 (2004).
17.Ito,T.K.,Ishii,G.,Chiba,H. & Ochiai,A. The VEGF angiogenic switchof fibroblasts is regulated by MMP-7 from cancer cells. Oncogene 26,7194-203(2007).
18.Patterson,B.C. & Sang,Q.A. Angiostatin-converting enzymeactivities of human matrilysin (MMP-7) and gelatinase B/type IV collagenase(MMP-9). J Biol Chem 272,28823-5 (1997).
19.McGuire,J.K.,Li,Q. & Parks,W.C. Matrilysin (matrixmetalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lungepithelium. Am J Pathol 162,1831-43 (2003).
20.McCawley,L.J. & Matrisian,L.M. Matrix metalloproteinases: they'renot just for matrix anymore! Curr Opin Cell Biol 13,534-40 (2001).
21.Lynch,C.C.等人Matrix metalloproteinase 7 mediates mammaryepithelial cell tumorigenesis through the ErbB4 receptor. Cancer Res 67,6760-7 (2007).
22.Zhang,Q.等人Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene (2016).
23.Brabletz,T.,Jung,A.,Dag,S.,Hlubek,F. & Kirchner,T. beta-cateninregulates the expression of the matrix metalloproteinase-7 in humancolorectal cancer. Am J Pathol 155,1033-8 (1999).
24.Wang,S.C.等人A Pilot Study Evaluating Serum MMP7 as a PreoperativePrognostic Marker for Pancreatic Ductal Adenocarcinoma Patients. JGastrointest Surg 20,899-904 (2016).
25.Park,H.D.等人Serum CA19-9,cathepsin D,and matrixmetalloproteinase-7 as a diagnostic panel for pancreatic ductaladenocarcinoma. Proteomics 12,3590-7 (2012).
26.Kuhlmann,K.F.等人Evaluation of matrix metalloproteinase 7 inplasma and pancreatic juice as a biomarker for pancreatic cancer. CancerEpidemiol Biomarkers Prev 16,886-91 (2007).
27.Tamburrino,D.等人Selection criteria in resectable pancreaticcancer: a biological and morphological approach. World J Gastroenterol 20,11210-5 (2014).
28.Chari,S.T.等人Early detection of sporadic pancreatic cancer:summative review. Pancreas 44,693-712 (2015).
29.Sela-Passwell,N.等人Antibodies targeting the catalytic zinccomplex of activated matrix metalloproteinases show therapeutic potential.Nat Med 18,143-7 (2011).
30.Knight,C.G.,Willenbrock,F. & Murphy,G. A novel coumarin-labelledpeptide for sensitive continuous assays of the matrix metalloproteinases.FEBS Lett 296,263-6 (1992).
31.Otwinowski,Z. & Minor,W. Processing of X-ray diffraction datacollected in oscillation mode. Methods Enzymol 276,307-26 (1997).
32.Ben-Shimon,A. & Eisenstein,M. Computational mapping of anchoringspots on protein surfaces. J Mol Biol 402,259-77 (2010).
33.Grossman,M.等人Tumor Cell Invasion Can Be Blocked by Modulators ofCollagen Fibril Alignment That Control Assembly of the Extracellular Matrix.Cancer Res 76,4249-58 (2016).
34.Page-McCaw,A.,Ewald,A.J. & Werb,Z. Matrix metalloproteinases andthe regulation of tissue remodelling. Nat Rev Mol Cell Biol 8,221-33 (2007).
35.Egeblad,M. & Werb,Z. New functions for the matrixmetalloproteinases in cancer progression. Nat Rev Cancer 2,161-74 (2002).
36.Fukuda,A.等人Stat3 and MMP7 contribute to pancreatic ductaladenocarcinoma initiation and progression. Cancer Cell 19,441-55 (2011).
37.Almendro,V.等人The role of MMP7 and its cross-talk with the FAS/FASL system during the acquisition of chemoresistance to oxaliplatin. PLoSOne 4,e4728 (2009).
38.Ametller,E.等人Differential regulation of MMP7 in colon cancercells resistant and sensitive to oxaliplatin-induced cell death. Cancer BiolTher 11,4-13 (2011).
39.Terwilliger,T.C. Maximum-likelihood density modification. ActaCrystallogr D Biol Crystallogr 56,965-72 (2000).
40.French,S. & Wilson,K. On the treatment of negative intensityobservations. Acta Crystallographica Section A: Crystal Physics,Diffraction,Theoretical and General Crystallography 34,517-525 (1978).
41.McCoy,A.J.等人Phaser crystallographic software. J Appl Crystallogr40,658-674 (2007).
42.Terwilliger,T.C. & Berendzen,J. Automated MAD and MIR structuresolution. Acta Crystallogr D Biol Crystallogr 55,849-61 (1999).
43.Emsley,P. & Cowtan,K. Coot: model-building tools for moleculargraphics. Acta Crystallogr D Biol Crystallogr 60,2126-32 (2004).
44.Laskowski,R.A.,MacArthur,M.W.,Moss,D.S. & Thornton,J.M. PROCHECK:a program to check the stereochemical quality of protein structures. Journalof applied crystallography 26,283-291 (1993).
45.Suhre,K. & Sanejouand,Y.H. ElNemo: a normal mode web server forprotein movement analysis and the generation of templates for molecularreplacement. Nucleic Acids Res 32,W610-4 (2004).
46.Katchalski-Katzir,E.等人Molecular surface recognition:determination of geometric fit between proteins and their ligands bycorrelation techniques. Proc Natl Acad Sci U S A 89,2195-9 (1992).
47.Heifetz,A.,Katchalski-Katzir,E. & Eisenstein,M. Electrostatics inprotein-protein docking. Protein Sci 11,571-87 (2002).
48.Berchanski,A.,Shapira,B. & Eisenstein,M. Hydrophobiccomplementarity in protein-protein docking. Proteins 56,130-42 (2004).
49.Kowalsman,N. & Eisenstein,M. Combining interface core and wholeinterface descriptors in postscan processing of protein-protein dockingmodels. Proteins 77,297-318 (2009).
50.Kowalsman,N. & Eisenstein,M. Inherent limitations in protein-protein docking procedures. Bioinformatics 23,421-6 (2007).
51.Pettersen,E.F.等人UCSF Chimera--a visualization system forexploratory research and analysis. J Comput Chem 25,1605-12 (2004).
52.Yee,J.K.,Friedmann,T. & Burns,J.C. Generation of high-titerpseudotyped retroviral vectors with very broad host range. Methods Cell Biol43 Pt A,99-112 (1994).
53.Vermes,I.,Haanen,C.,Steffens-Nakken,H. & Reutelingsperger,C. Anovel assay for apoptosis. Flow cytometric detection of phosphatidylserineexpression on early apoptotic cells using fluorescein labelled Annexin V. JImmunol Methods 184,39-51 (1995).
尽管本发明已与其具体实施方案结合进行描述,但很明显,许多替代、修改和变化对于本领域技术人员将是显而易见的。相应地,它预期涵盖落入所附权利要求的精神和广泛范围内的所有此类替代、修改和变化。
本说明书中提到的所有出版物、专利和专利申请都在本文中整体引入说明书内作为参考,其程度与每个个别出版物、专利或专利申请具体地且个别地指示引入本文作为参考相同。另外,在本申请中的任何参考文献的引用或鉴定,不应解释为承认此类参考文献可用作本发明的现有技术。就使用节段标题而言,它们不应被解释为必然的限制。
另外,本申请的任何优先权文件在此整体引入本文作为参考。