用于局部麻醉剂的乳剂
文献发布时间:2024-04-18 20:01:30
相关申请的交叉引用
本申请要求于2021年3月31日提交的美国临时专利申请第63/169,121号以及于2021年7月7日提交的美国专利申请第17/369,369号的优先权,出于所有目的,它们的内容以其整体援引加入本文。
公开领域
本公开涉及含有局部麻醉剂的药理学活性、安全且长效的乳剂及其用途。
背景
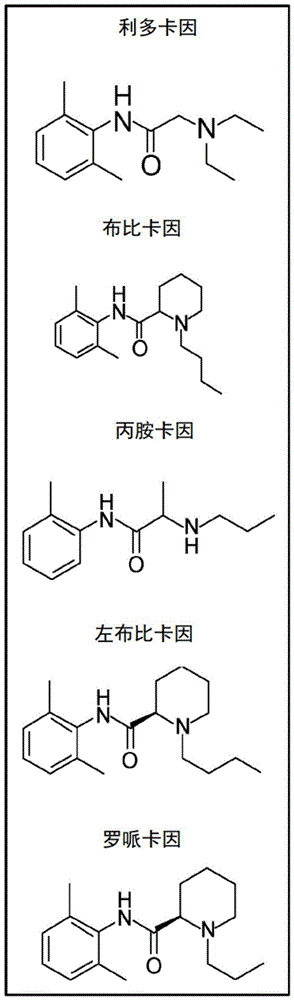
局部麻醉药(LA)如利多卡因、布比卡因、丙胺卡因、左布比卡因和罗哌卡因是化学相关的酰胺局部麻醉剂,并且通过如下方式享有相同的作用机制:抑制电压门控钠通道,这减缓了膜去极化和复极化并且导致膜稳定化(参见Daniel E.Becker和Kenneth L.Reed,Local Anesthetics:Pharmacological Considerations,Anesth Prog 59:90,102 2012)。
已表明局部麻醉剂是有效的,并且被广泛用于预防或减少小手术、切口、活组织检查、牙科和产科手术的疼痛以及伤口疼痛。
局部麻醉剂通常以局部单次注射到病变或切口区域中给予,并且由于其半衰期短(至多约4小时),其麻醉活性持续约数小时。为了更长的作用,向受影响的组织的缓慢且连续的输注可以在数天内通过硬膜外或伤口递送导管来施用。这种较久输注不便于给药,并且单次注射缓释或长效制剂以提供LA的延长作用是高度期望的。
已开发出多种方法来延长LA的作用持续时间,包括:
1.将LA嵌入脂质体囊泡中以减缓其释放(参见美国专利第8,182,835和10,206,876号)
2.将LA掺入聚合物基质中以实现缓慢释放(参见美国专利第10,898,575号)。
3.LA与血管限制剂肾上腺素共同给药(参见Lixtraxen注射液(盐酸利多卡因和肾上腺素注射液,溶液剂)
乳剂历来用于使LA失活或者作为LA过量的解毒剂。有许多文献证明,不含药物的乳剂(如英脱利匹特)通常用于降低LA的心血管效应或毒性。多位研究人员已清楚地展示,乳剂可以使LA失活,从而导致LA的药理效应和毒性丧失(参见Zausig,York A.等人,LipidEmulsion Improves Recovery from Bupivacaine-Induced Cardiac Arrest,but Notfrom Ropivacaine-or Mepivacaine-Induced Cardiac Arrest,Anesthesia&Analgesia:2009年10月-第109卷-第4期-第1323-1326页;E.Litonius等人,Effect of intravenouslipid emulsion on bupivacaine plasma concentration in humans,Anaesthesia2012,67,600–605;Lotte C.G.等人,Systematic review of the effect of intravenouslipid emulsion therapy for local anesthetic toxicity,Clinical Toxicology,2016,第54卷,第3期,167-193,http://dx.doi.org/10.3109/15563650.2015.1121270)。使用乳剂来递送药理学活性LA违背了常识和医学实践。
本申请涉及一项令人惊讶的发现,该发现与关于使用乳剂使LA失活的当前理解和医学实践相反。本申请公开了乳剂不仅维持LA所期望的药理效应,而且延长其作用持续时间并降低其毒性的用途。本发明的乳剂特别可用于管理手术或创伤相关的疼痛,因为本公开的乳剂可以在单次注射后将期望的LA活性延长1-5天,从而提供延长的疼痛缓解益处。
发明概述
本发明涉及含有LA的乳剂组合物的用途,所述含有LA的乳剂组合物:
a.维持并延长所述LA的药理活性(实施例2)
b.比其常规溶液制剂中的LA更安全(实施例3)
c.与相同LA的常规溶液剂相比,提供延长的药代动力学(PK)特性(实施例1),以及
d.提供多至约1-3天的延长麻醉作用(实施例1&2)
因此,本发明教导了一种乳剂组合物,所述乳剂组合物包含、基本上由以下组成、或由以下组成:
a)局部麻醉剂,
b)油相,和
c)水相,
其中
1.所述局部麻醉剂的浓度为按所述乳剂的重量计多至4%,
2.所述油相包含重量比为2:1至1:2的卵磷脂和植物油,
3.所述油相的浓度为按所述乳剂的重量计约33%至99.5%,以及
4.所述乳剂含有直径为约30至约2500纳米的颗粒或液滴。
在一个实施方案中,本发明的乳剂含有选自利多卡因、布比卡因以及罗哌卡因的LA(实施例2&21)。
在一个实施方案中,本发明的乳剂含有呈游离碱形式(或非离子化形式)、作为药学上可接受的盐如盐酸盐(呈离子化形式)(实施例18)、或它们的组合的LA。
在一个实施方案中,本发明的乳剂是可注射液体或可用注射器通过皮下注射针手动排出的液体(实施例11)。
在一个实施方案中,本发明的乳剂具有直径尺寸为约30至约2500纳米的颗粒或油滴。该粒度范围是优选的,因为小于30nm可由于LA从乳剂中快速释放而导致作用缩短,并且大于2500nm可导致难以注射的非常粘稠的液体(实施例9、10)。
在一个实施方案中,本发明的乳剂具有平均直径为约200nm至2500nm的颗粒或油滴(实施例9)。
在一个实施方案中,本发明的乳剂具有直径小于100nm的颗粒以及直径大于100nm的颗粒。
在一个实施方案中,本发明的乳剂具有尺寸多分散的颗粒,其多分散性指数(PDI)>0.7(实施例17)。
在一个实施方案中,本发明的乳剂具有PDI高于0.7,优选地高于0.8并且最优选高于0.9的颗粒(实施例17)。
在一个实施方案中,本发明的乳剂具有含实心的非脂质体颗粒,并且所述颗粒周围不存在可检测的脂质双层膜(实施例10)。
在一个实施方案中,本发明的乳剂在用水稀释或与水混合之后形成(实施例9)。
在一个实施方案中,所述油相以所述乳剂的约33重量%至99.5重量%的浓度存在于所述乳剂中(实施例12&13)。
在一个实施方案中,所述油相包含卵磷脂、油以及LA。
在一个实施方案中,卵磷脂以油相的约55重量%至67重量%的浓度存在于油相中。
在一个实施方案中,油以油相的约33重量%至44重量%的浓度存在于油相中。
在一个实施方案中,卵磷脂和油以2:1至5:4的重量比存在于油相中。该范围内的比例是优选的,因为过多卵磷脂可使乳剂太粘而无法通过针头注射,而过多油不能充分溶解LA,因为油本身不是用于LA的良好溶剂,也无法提供LA的延长释放。
在一个实施方案中,本发明的乳剂含有按所述乳剂的重量计多至4%的LA。
在一个实施方案中,超过90重量%的LA分布在乳剂的油相中或者与乳剂的油相非共价结合。
在一个实施方案中,本发明的乳剂为粘度在约300至600cps范围内的半透明或不透明液体(实施例8)。
在一个实施方案中,无论LA的疏水性如何,本发明的乳剂中不少于90%的LA与乳剂油相结合(实施例3)。
在一个实施方案中,本发明的乳剂中不少于90%的LA与乳剂油相结合,即使LA不疏水或者log P值小于1.5也是如此。
在一个实施方案中,本发明的乳剂使用LA游离碱(即非离子化形式)或LA盐(即离子化形式,例如HCl盐)作为起始材料来制备(实施例3)。
在一个实施方案中,本发明的乳剂含有疏水性且水不溶性LA,如利多卡因、布比卡因或罗哌卡因的游离碱(非离子化或未离子化)形式。
在一个优选的实施方案中,本发明的乳剂含有离子化形式的非疏水性或水溶性LA,如罗哌卡因、布比卡因或利多卡因的盐酸盐。
在一个实施方案中,本发明的乳剂含有离子化、非离子化形式或这两者的LA(实施例18)。
在一个实施方案中,本发明的乳剂含有约5摩尔%至25摩尔%非离子化LA以及约75摩尔%至95摩尔%离子化LA(实施例18)。
在一个实施方案中,本发明的乳剂是液晶,其像液体一样流动,但其分子以晶体状方式取向,其可通过其独特的光学或光谱特性(如小角X射线散射或傅里叶变换红外光谱或FTIR)来表征和区别于其它液体制剂(实施例19&实施例20)。
在一个实施方案中,本发明的乳剂是溶致液晶,其表现出作为所添加的水相或水的量的函数的相变。在一个实施方案中,本发明的乳剂具有在约1%水含量下的相变点(实施例15)。
在一个实施方案中,本发明的乳剂的油滴基本上不含脂质双层膜,所述脂质双层膜是脂质体的基本结构特征(实施例10)。
在一个实施方案中,本发明的乳剂不含聚合物。
在一个实施方案中,本发明的乳剂缓慢地释放LA,如超过1、2、3或4天的释放和功效(实施例7、13)。
在一个实施方案中,本发明的乳剂以与含有相同LA的前脂质体或脂质体组合物不同的速率释放LA(实施例7)。
在一个实施方案中,本发明的乳剂在结构上不同于含有相同LA的前脂质体或脂质体组合物。
在一个实施方案中,本发明的乳剂不具有任何有机溶剂,如醇。
在另一个实施方案中,本发明的乳剂可以具有可与水混溶的有机溶剂。
在一个实施方案中,本发明的乳剂可以用10mL注射器通过18-27G针头或22G导管来注射(实施例11)。
在一个实施方案中,本发明的乳剂包含水相和油相,其中油相包含LA、植物油,以及衍生自卵或大豆的卵磷脂。
在另一个实施方案中,本发明的乳剂具有pH 3至pH 8的pH。
在一个实施方案中,本发明的乳剂组合物不含有任何合成表面活性剂如聚山梨醇酯80、聚合物如PLGA、或生物聚合物如胶原蛋白。
在一个实施方案中,本发明的乳剂是即用型或即注射型。
在另一个实施方案中,本发明的乳剂在注射前与水或水溶液(如盐水)混合。
在一个实施方案中,所述乳剂是油包水乳剂。在另一个实施方案中,所述乳剂是水包油乳剂。在另一个实施方案中,用稀释剂(如水、盐水)稀释油包水乳剂使乳剂转化为水包油型。在某些其它实施方案中,通过例如真空干燥从水包油乳剂移除过量的水,使水包油乳剂转化为油包水乳剂。
在一个实施方案中,本发明的乳剂基本上为水含量>10%的水包油乳剂、水含量介于3%与10%之间的水包油与油包水型两者的混合物、主要为水含量介于1%与3%之间的油包水乳剂,以及基本上为水含量不超过1%的油包水乳剂。这些乳剂类型中的任一者均可用于提供长效且安全的LA药物。可以在该步骤中通过控制水含量来实现优选的乳剂类型的选择。
在一个实施方案中,本发明的乳剂具有相变点为0.3%(重量/重量)至3%(重量/重量)水,优选地0.5%(重量/重量)至2%(重量/重量)水,并且更优选地约1%(重量/重量)水的液晶结构。
在一个实施方案中,本发明的乳剂在物理上稳定,并且在25℃下储存2年后不形成LA沉淀、经历相分离或外观变化(实施例8)。
在一个实施方案中,本发明的乳剂是化学稳定的,并且在25℃下储存2年后保留不少于95%的完整LA(实施例8)。
在一个实施方案中,本发明的乳剂在25℃下储存2年后含有少于0.2%的LA的N-氧化物(实施例8)。
在另一个实施方案中,本发明的乳剂可以任选地含有选自甲硫氨酸、半胱氨酸、右旋糖、果糖、乳糖,以及依地酸盐(EDTA)的抗氧化剂。
在一个实施方案中,本发明的乳剂含有EDTA钠和半胱氨酸的组合(实施例16)。
在一个实施方案中,本发明的乳剂用于经由浸润或注射到伤口表面、手术切口上或软组织中来给药。
在一个实施方案中,在注射到软组织中后,本发明的乳剂与相同LA的溶液制剂相比提供了对LA延长的PK特性(实施例1)。
在一个实施方案中,本发明的乳剂提供了长期抑制疼痛的作用(实施例2)。
在一个实施方案中,本发明的乳剂比相同LA的溶液制剂更安全(实施例4)。
在一个实施方案中,本发明提供了用于预防或治疗疼痛的方法,所述方法包括向患者给药本发明的乳剂。
在一些实施方案中,将本发明的乳剂作为单次剂量(即,整个治疗内仅一次)给药。
在一些实施方案中,将本发明的乳剂多次(即,在治疗过程期间超过一次)给药。
在一些实施方案中,将本发明的乳剂按原样(即,未经稀释)给药。
在一些实施方案中,将本发明的乳剂在用水或对人类给药安全的另一种液体稀释后给药。
在一些实施方案中,将本发明的乳剂给药到疼痛部位附近的组织中,如手术切口或创伤伤口。
在一些实施方案中,将本发明的乳剂给药于手术切口,包括但不限于软组织手术的切口,包括但不限于经由切开技术或腹腔镜技术进行的普外科手术;腹部外科手术,包括但不限于胃切除术、直肠结肠切除术、阑尾切除术、胰切除术、胆囊切除术、疝修补术;结直肠外科手术,如结肠切除术、回肠造口术、APR,以及痔切除术;胸外科手术,如肺切除术、食管切除术;肝脏外科手术,如肝癌手术;整形外科手术,如腹壁整形术、隆乳术或乳房固定术;泌尿外科手术,如前列腺切除术或膀胱切除术;妇产科外科手术,包括肌瘤切除术、绝育术、子宫切除术,以及经腹子宫全切和双侧输卵管卵巢切除术(total abdominalhysterectomy bilateral salpingo oophorectomy,TAHBSO);以及耳鼻喉外科手术。在一些实施方案中,也将本发明的乳剂给药于骨组织外科手术的切口,包括但不限于骨科外科手术,如髋膝关节置换术、骨折和关节的切开复位内固定、椎板切除术以及脊柱融合术;胸外科手术,如胸骨切口和漏斗胸修复术;足外科手术,包括拇囊炎切除术;以及普通骨外科手术,包括髂嵴移植。
在一些实施方案中,将本发明的乳剂给药于创伤伤口,包括但不限于擦伤、撕裂伤、皮肤撕裂、咬伤、烧伤和穿透性创伤伤口。
在一些实施方案中,本发明的乳剂以0.5%至4%,优选地1%至3%并且最优选地1.5%至2.5%的LA浓度给药于手术切口或创伤伤口。
在一些实施方案中,本发明的乳剂通过浸润或注射到手术部位、疼痛部位或神经处或附近的软组织中来给药。
在一些实施方案中,本发明的乳剂通过浸润或注射到软组织或骨组织外科手术中的切口部位中来给药。
在一些实施方案中,本发明的乳剂通过滴注或局部施用到手术切口或伤口部位上来给药。
在一些实施方案中,本发明的乳剂通过滴注或局部施用到软组织或骨组织上来给药。
在一些实施方案中,本发明的乳剂通过浸润和滴注两者来给药。
在一些实施方案中,本发明的乳剂通过滴注以每个手术切口或伤口部位多至5、10、15、20、25、30、40或50mL的体积来给药。
在一些实施方案中,本发明的乳剂通过滴注以每个手术切口或伤口部位多至30mL,优选地25mL或更优选地20mL的体积来给药。
在一些实施方案中,本发明的乳剂通过浸润以每个手术切口或伤口部位多至5、10、15、20、25、30、40或50mL的体积来给药。
在一些实施方案中,本发明的乳剂通过浸润以每个手术切口或伤口部位多至1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、25、30、40和50mL的体积来给药。
在一些实施方案中,本发明的乳剂通过滴注和浸润两者以每个手术切口或伤口部位总共多至10、15、20、25、30、40和50mL来给药。
在一些实施方案中,本发明的乳剂通过滴注和浸润以1:20至20:1的体积比来给药。
在一些实施方案中,本发明的乳剂在外科手术之前、期间和之后通过滴注和浸润到手术切口来给药。
在一些实施方案中,本发明的乳剂在通过缝合闭合切口之前,通过滴注和浸润到手术切口来给药。
在一些实施方案中,本发明的乳剂在通过缝合闭合切口之前,通过注射或浸润到切口中的软组织或骨组织(包括但不限于骨、关节、筋膜、肌肉、脂肪、皮下组织、皮肤组织)之中、之上或周围来给药。
在一些实施方案中,本发明的乳剂在通过缝合闭合切口之前,通过滴注到切口中的软组织或骨组织(包括但不限于骨、关节、筋膜、肌肉、脂肪、皮下组织、皮肤组织)之中、之上或周围来给药。
在一些实施方案中,本发明的乳剂通过肌内、关节内、关节周围、皮下注射或经皮注射到闭合切口后切口处或周围的软组织或骨组织(包括但不限于骨、关节、筋膜、肌肉、脂肪、皮下组织、皮肤组织)之中、之上或周围来给药。
在一些实施方案中,本发明的乳剂通过局部施用到闭合的切口或伤口上来给药。
在一些实施方案中,使用附有针头的注射器通过浸润来给药本发明的乳剂。
在一些实施方案中,使用附有或不附有针头的注射器通过滴注来给药本发明的乳剂。
在一些实施方案中,本发明的乳剂通过给药到从受侵袭疼痛部位(如手术切口部位或创伤伤口)传递疼痛信号的神经附近的组织中来给药以阻滞神经。
在一些实施方案中,将本发明的乳剂给药到神经以用于区域阻滞和外周神经阻滞两者,包括但不限于TAP阻滞、PEC阻滞、椎旁阻滞、脊椎旁阻滞、臂丛神经阻滞、颈丛阻滞、腹腔神经丛阻滞、小关节阻滞、或特定周围神经阻滞(例如:三叉神经、眼、上颌、肌间沟、锁骨下、腋、髂腹股沟、阴茎、股、坐骨、腿弯部或隐神经)。
在一些实施方案中,将本发明的乳剂以多至1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、25、30、40和50mL的体积给药以用于神经阻滞。
在一些实施方案中,将本发明的乳剂以0.1%至3%,优选地0.2%至2%,并且最优选地0.4%至2%的LA浓度给药以用于神经阻滞。
在一些实施方案中,本发明的乳剂在用氯化钠溶液、右旋糖溶液、水、或可安全用于神经阻滞的液体稀释之后给药以用于神经阻滞。
在优选的实施方案中,本发明的乳剂在用生理盐水稀释至0.1%至2%,优选地0.2%至2%,并且最优选地0.4至2%的LA浓度之后给药以用于神经阻滞。范围优选的原因是为了在稀释后实现乳剂的优选粘度、可注射性以及延长释放特性。过低稀释度导致粘度较高且难以注射,过高稀释度导致延长释放特性降低和受损(实施例11、12、13)。
在一些实施方案中,本发明的乳剂在用氯化钠溶液、右旋糖溶液、或可安全用于神经阻滞的液体稀释至不超过约40K,优选地400并且最优选地4厘泊的粘度之后给药以用于神经阻滞(实施例11、12)。
在一些实施方案中,将本发明的乳剂给药到软组织中,其中它在软组织中提供比在血液中更高的LA浓度。
在一些实施方案中,将本发明的乳剂给药到软组织中,其中软组织中的LA浓度是血液中的1、2、3、5和10倍以上。
在一些实施方案中,将本发明的乳剂给药到软组织中以提供LA的延长释放,其中1、2、3、4、5、6或7天后LA血浆浓度超过5ng/mL(实施例1)。
在一些实施方案中,将本发明的乳剂给药到软组织中以提供无峰或低C
在一些实施方案中,将本发明的乳剂给药到软组织中以提供C
在一些实施方案中,将本发明的乳剂以2-800mg罗哌卡因的剂量给药到软组织中,以提供不大于3300ng/mL的C
在一些实施方案中,将本发明的乳剂以2-800mg罗哌卡因的剂量给药到软组织中以提供0.5至48小时的T
在一些实施方案中,将本发明的乳剂以2-800mg罗哌卡因的剂量给药到人个体的软组织中,以提供200至251,000ng/mL*小时的AUC(实施例1)。AUC的范围是优选的,因为较低的AUC产生低治疗效应,而较高AUC导致毒性。
在一些实施方案中,本发明的乳剂在给药到软组织中之后不影响软组织的伤口愈合过程(实施例2、5)。
在一些实施方案中,本发明的乳剂不影响缝合线的物理完整性(实施例6)。
在一些实施方案中,本发明的乳剂不影响外科网的物理完整性(实施例6)。
在一些实施方案中,本发明的乳剂提供疼痛缓解至少约24小时。
在一些实施方案中,本发明的乳剂提供疼痛缓解24至48小时。
在一些实施方案中,本发明的乳剂提供疼痛缓解至少48小时。
在一些实施方案中,本发明的乳剂提供疼痛缓解48至72小时。
在一些实施方案中,本发明的乳剂提供疼痛缓解至少72小时。
在优选的实施方案中,本发明的乳剂用于提供延长的疼痛缓解或神经阻滞。
在优选的实施方案中,本发明的乳剂用于术后疼痛管理。
在另一个实施方案中,本发明的乳剂用于围术期神经阻滞,包括但不限于术前、术中和术后神经阻滞,以实现外科手术之前、期间和之后延长的疼痛抑制。
当阅读以下具体描述和附图时,这些和其它方面、目标和实施方案会变得明显。
附图简述
图1示出了酰胺局部麻醉剂的示意性结构。
图2示出了罗哌卡因N-氧化物的结构。
图3示出了脂质体双层膜的示意图。
图4A-C示出了电子显微照片,它们显示本发明乳剂即未稀释乳剂(上图,图4A)和稀释的乳剂(下图,图4B)中的颗粒或液滴以及充分表征且FDA批准的脂质体药物(Ambisome
图5示出了与根据美国专利第10,206,876B2号中的实施例1&2制备的罗哌卡因脂质体组合物相比,本发明的罗哌卡因乳剂的体外释放。
图6示出了与罗哌卡因溶液剂
图7示出了小角X射线散射(SAXS)测试结果,其显示出本发明的罗哌卡因乳剂制剂与前脂质体制剂(“前体脂质体”)之间的结构差异。
图8A-B显示通过FTIR得到的本发明的罗哌卡因乳剂制剂与前脂质体制剂(“前体脂质体”)之间的光谱差异。
详细描述
I.定义
如本文所用,各种术语应具有以下定义。
如本文所用,“约”描述了范围覆盖从目标值两侧扩展10%的量。例如,“约100”意指介于90与110之间的任何值,包括90至110。
如本文所用,“酸”是指药学上可接受的酸,如盐酸、乙酸、柠檬酸、甲磺酸和硫酸等等,其中盐酸是优选的酸。
如本文所用,“碱性物(alkaline)”或“碱”是指药学上可接受的碱,如氢氧化钠、氢氧化钾、氢氧化铵、赖氨酸、精氨酸等等,其中氢氧化钠和氢氧化钾是优选的碱。
“抗氧化剂”是可添加到组合物中以防止活性药物或非活性组分氧化的药物添加剂。抗氧化剂包括还原剂、金属离子螯合剂以及惰性气体。用于本发明的乳剂的优选的抗氧化剂是EDTA钠、半胱氨酸、CO
术语“水相”是指使油滴悬浮在乳剂中的水溶液。在本发明的乳剂中,可以通过超滤法使水相与油相分离,这使得能够对水相和油相中的各组分进行单独定量。除了水之外,水相还可含有其它水溶性或亲水性成分。本发明的乳剂的水相可含有pH缓冲剂、防腐剂、抗氧化剂、金属离子螯合剂或螯合剂以及活性药物(LA)的一小部分。在一个实施方案中,水相是生物流体,如血液和体液。在一个实施方案中,本发明乳剂的水相包含还原剂、螯合剂以及不超过10重量%的添加的LA。在一个实施方案中,本发明乳剂的水相的浓度为按所述乳剂的重量计0.1%至90%,优选地0.2%至80%,更优选地0.2%至70%,并且最优选地0.2%至66%。
如本文所用,“化学稳定性”或“化学稳定的”意指能够使其活性成分维持在不少于其初始量的90%且某些杂质低于可接受限度的药物制剂的状态。如果本发明的乳剂能够维持在不少于其初始LA量的90%且LA N-氧化物不超过按LA重量计0.2%,则认为本发明的乳剂是化学稳定的。在一个实施方案中,本发明的乳剂是化学稳定的。在一个优选的实施方案中,本发明的乳剂在25℃下储存1.5年或优选地2年后使LA维持在不少于其初始量的90%且LA N-氧化物不超过按LA重量计0.2%。
如本文所用,“乳剂”是不可混溶油相和水相的混合物。乳剂可为水包油或油包水型,通常是光学不透明的并且具有有限的物理稳定性。在某些情况下,本发明的组合物是在连续油相中悬浮有离散水或水相的光学半透或半透明且物理稳定的油包水乳剂。在一个实施方案中,本发明的乳剂是水包油型,其中油相由悬浮在连续水相中的离散油滴构成。在某些其它实施方案中,本发明的乳剂是水包油乳剂,但可在移除水后转化为油包水。在另一个实施方案中,本发明的乳剂是油包水型,但可在添加水或用生物流体如血液稀释之后转化为水包油。在另一个实施方案中,本发明的乳剂是水包油型,并且在用水或生物流体稀释后保持为水包油。在一个实施方案中,本发明的乳剂为油包水与水包油型两者的混合物。在另一个实施方案中,本发明的乳剂是光学半透或半透明且物理稳定的。在另一个实施方案中,本发明的乳剂是白色和不透明的。
如本文所用,术语“油滴”是指作为由水相包围的离散颗粒存在的本发明的乳剂油相。油滴由乳剂中的水不溶性组分构成,包括卵磷脂和油。油滴不是脂质体颗粒,因为它们不具有限定脂质体结构的脂质双层。油滴也不同于胶束或胶束颗粒。胶束存在于表面活性剂水溶液中并且尺寸小得多(<100nm,与本发明的油滴200-2500nm相比)。可形成胶束的表面活性剂是水溶性表面活性剂,如聚山梨醇酯、氢化蓖麻油等,由于它们的毒性而不可取,并且在本发明的乳剂中被排除在外。由于存在高含量的油(按油相的重量计30-70%),油滴也不同于固体脂质纳米颗粒或SLN,后者的油通常比本发明乳剂中使用的少得多。物理上讲,油滴为柔软的柔性液体,并且可易于过滤通过具有小孔(0.2-0.45微米)的膜或者可注射通过细针头,而脂质体和SLN为固体的不可弯曲刚性颗粒,并且难以推送通过细针头或过滤器。
如本文所用,术语“疼痛缓解”意指使用药物或其它方法来预防、减少或摆脱疼痛;预防、减少或摆脱疼痛的行为。
如本文所用,“粒度”是指本发明的乳剂中或其用水或生物流体稀释后的颗粒或油滴的直径。油滴尺寸可对本发明的乳剂的LA的体外释放、药代动力学特性和可注射性具有深远影响。如US 9,517,202中所教导的极小乳剂液滴(<100nm)更难以制备、不太稳定并且往往快速释放LA,并且产生短药代动力学特性或短作用持续时间。另一方面,较大乳剂液滴(>2500nm)可导致难以通过针头排出或不可注射的非常粘稠的乳剂。在某些实施方案中,本发明的乳剂中的油相以大于100nm的粒度存在。在另一个实施方案中,本发明的乳剂中的粒度为200nm至2500nm,优选地200nm至500nm并且最优选地300nm至400nm。
如本文所用,术语“药物掺入能力”定义了可装载到制剂中且同时仍然维持制剂的所有期望特性的药物的最大量。在本发明乳剂的乳剂中,LA的药物掺入能力为按所述乳剂的重量计约4%。如果LA添加到超过4%,则乳剂将变得浑浊,形成沉淀,和/或丧失所期望的体外释放特性。在一个实施方案中,本发明的乳剂含有不超过4重量%的LA,优选不超过3.5重量%的LA,并且最优选地0.5重量%、1重量%、1.5重量%、2重量%、2.5重量%或3重量%的LA。
如本文所用,“可过滤”意指本发明的乳剂通过特定孔径(如0.2-5微米)的过滤膜而没有吸附到膜上的LA的显著(>10%)损失的能力。在一个实施方案中,本发明的乳剂可过滤通过孔径为0.2至5微米的过滤器。在另一个实施方案中,本发明的乳剂可过滤通过孔径为5微米、1.2微米、0.8微米、0.45微米或0.2微米的过滤器。在一个实施方案中,本发明的乳剂可在不采用任何有机溶剂的情况下过滤。在一个优选的实施方案中,本发明的乳剂可采用有机溶剂、水相或它们的混合物过滤。
如本文所用,术语“细针头”或“针头”包括附接到注射器或导管以用于注射和浸润的小口径的中空皮下注射针头。针头的外径由针规系统指示。根据Stubs Needle Gauge系统,常用医疗用途中的皮下注射针头的范围为7(最大)到33号(G)(最小)。在一个实施方案中,本发明的乳剂可使用注射器通过针或导管挤出或注射。在一个优选的实施方案中,本发明的乳剂可使用1mL至50mL,优选地10mL范围内尺寸的注射器,通过18至27G,优选地18G至25G,并且最优选地18G至22G范围内的针头挤出。在另一个优选的实施方案中,本发明的乳剂可使用1mL至50mL范围内尺寸的注射器,通过附接到针头(16-25G)的导管(6”-24”长)挤出。这种针头-导管-注射器组合对于使用本发明的LA乳剂进行神经阻滞特别有用。
如本文所用,术语“疏水性”或“疏水的”意指所关注的LA分子具有1.5或更大的logP,其中P是辛醇/水分配系数。LA的游离碱或未离子化形式通常不溶于水,并且具有高log P或者是疏水的。相比之下,相同LA的离子化或盐形式通常可溶于水,具有低于1.5的log P或者是非疏水的。例如,呈游离碱的罗哌卡因的log P为2.9(P=辛醇:水分配系数=794)并且不溶于水,而罗哌卡因盐酸盐是非疏水的,并且log P为1.1(P=14)。在一个实施方案中,本发明的乳剂可以掺入呈其游离碱或盐形式的LA,即,无论LA疏水性如何。在另一个实施方案中,本发明的乳剂提供与LA游离碱或其盐类似的药理活性(实施例3)。
如本文所用,术语“浸润”意指注射到组织中的一个或多个部位处。例如,可以将LA注射到手术伤口组织中超过一个点处以便浸润一个区域,从而为整个区域提供镇痛效应或疼痛缓解。
如本文所用,术语“可注射”意指当10mL的液体可在小于10分钟内用10mL注射器以25牛顿力通过细针头排出时(实施例8)。
如本文所用,术语“滴注”意指将药品直接或局部给药或施用(而非注射)到组织上。例如,可以将LA以滴注局部给药到手术伤口,从而为整个伤口提供镇痛效应或疼痛缓解。
如本文所用,术语“体外释放”是指LA从制剂(例如,本发明的乳剂)中解离成变得在水或所关注的水溶液(例如,盐水或生物流体)中未结合或游离的速率和程度,如在试管或容器中所研究。因此,体外释放研究揭示乳剂释放LA的起效、速率以及持续时间。在一组体外释放测试条件下,起效通过达到最大LA浓度的20%的时间、达到最大LA浓度的50%的时间的速率,以及达到最大LA浓度的时间的持续时间进行量化。如果两种LA制剂不享有相同的体外释放特性,则它们的生物活性不被认为是等效的,即,非生物等效的。体外释放特征的差异也表明这两种制剂在结构上不同,即使它们可能用相同或相似的材料制成也是如此(实施例7)。此外,用于制备本发明的乳剂的工艺条件可对体外释放具有深远影响。所添加的LA的类型和量、水相和油相中使用的材料、它们的净和相对(比率)质量、它们添加的次序、液滴尺寸,以及任选地成分(如本发明的乳剂中的有机溶剂)均可以改变体外释放特性。因此,在一个实施方案中,本发明的乳剂提供了比相同LA的溶液剂更缓慢和更长并且不同于基于脂质体的LA制剂的体外释放特性(实施例7)。
如本文所用,“离子化”是指分子如LA分子如下通过失去电子而获取正电荷:
LA-电子=LA
其中LA
在一个实施方案中,本发明的乳剂是用100%离子化LA如罗哌卡因HCl盐作为起始材料来制备的,其中最终乳剂被制备成含有约5至25摩尔%(或5摩尔%至25摩尔%)的非离子化LA形式,优选地10至20摩尔%非离子化LA形式,并且更优选地12至18摩尔%非离子化LA。
这种离子化和非离子化LA的混合物可通过如下方式获得:添加离子化和非离子化LA起始材料的混合物,通过添加计算量的酸如盐酸使非离子化LA起始材料离子化,或者通过在乳剂制备过程期间添加计算量的碱性物或碱例如氢氧化钠和氢氧化钾来使离子化LA起始材料去离子化(实施例18)。
如本文所用,“局部麻醉剂”是指具有如图1所显示的化学结构之一的酰胺局部麻醉药物分子。
如本文所用,“LA-油滴结合”意指本发明的乳剂中LA与油滴之间的非共价结合。非共价结合通过使未结合或游离LA分子与水或所关注的水性介质(例如,血液或生物流体)中的油滴所结合的LA分离,然后测定它们相应的浓度进行测量。可以通过超滤获得游离与结合的LA的分离,并且可以通过HPLC测量LA浓度。LA-油滴结合比的程度以浓度比表示(LA-油滴结合(%)=结合LA的浓度/总LA浓度×100)。在一个实施方案中,本发明的乳剂的LA-油滴结合(%)不少于LA的90%。在一个优选的实施方案中,本发明的乳剂的LA-油滴结合(%)不少于LA的90%,无论其疏水性或log P(即,大于或小于1.5)如何。
如本文所用,“卵磷脂”是源自天然来源的磷脂的混合物。可注射卵磷脂包括源自卵或大豆的卵磷脂,其已经过纯化并且基本上不含刺激剂、致敏剂、致炎因子或引起其它有害生物反应的物质。对于本发明,优选的卵磷脂包括含有超过75重量/重量%磷脂酰胆碱(PC)、不溶于水且基本上不含溶血卵磷脂(即,含有不超过1-4重量%的溶血卵磷脂)的那些。高PC含量使得卵磷脂相对柔软,并且产生相对更丝滑且更易注射的乳剂。溶血卵磷脂具溶血性,并且出于安全原因是不可取的。优选的卵磷脂的实例包括但不限于商品名为LIPOID S 75、LIPOID S100、LIPOID E 80和Phospholipon 90G的卵磷脂产品。卵磷脂分子上的一些脂肪酸链是不饱和的,并且可以经历氧化形成过氧化物,过氧化物继而可使LA氧化形成LA的N-氧化物。因此,在一个实施方案中,本发明的乳剂含有抗氧化剂和/或金属离子螯合剂或它们的组合以抑制卵磷脂氧化。一些卵磷脂氢化成仅含有饱和脂肪酸。虽然这些氢化卵磷脂对氧化不太敏感,但它们具有较高熔点并且产生坚硬(乳脂状)且难以注射的乳剂。在本发明的乳剂中,需要卵磷脂以形成油滴,结合LA并提供LA的缓慢释放。如果卵磷脂以低于乳剂的20重量%使用,则乳剂丧失其提供LA的缓慢释放或延长的作用特性的能力。另一方面,如果卵磷脂超过乳剂的70重量%,则乳剂变硬且难以注射。因此,本发明的乳剂中使用的卵磷脂的优选量为乳剂重量的20重量%至70重量%。在一个实施方案中,用于本发明的乳剂的优选卵磷脂具有不少于75重量%的PC含量。在另一个实施方案中,用于本发明的乳剂的优选卵磷脂具有不超过1-4重量%的溶血卵磷脂。在另一个实施方案中,优选的卵磷脂源自大豆或卵黄。在另一个实施方案中,优选的卵磷脂选自LIPOID S 75、LIPOIDS100、LIPOID E 80,以及Phospholipon 90G或它们的混合物。在另一个实施方案中,本发明的乳剂中使用的卵磷脂含有不超过25重量%的具有完全饱和脂肪酸侧链的卵磷脂或氢化卵磷脂。在一个实施方案中,本发明乳剂中使用的卵磷脂的量为按乳剂的重量(重量/重量)计20%至70%,优选地40%至60%并且最优选地50%至60%。
如本文所用,“脂质双层”是指围绕脂质体颗粒或囊泡的特征性结构膜。脂质体是由一个或多个磷脂双层构成的球形囊泡,其与细胞膜的双层结构极为相似。脂质体包封亲水性或亲脂性药物的能力使这些囊泡能够变为可用的药物递送系统(例如
如本文所用,“脂质体”是指由一个或多个磷脂双层构成的球形囊泡(图3),其与细胞膜的结构极为相似。脂质体包封亲水性或亲脂性药物并可注射的能力使这些囊泡能够变为可用的药物递送系统。
如本文所用,“液晶”是指具有介于常规液体的那些与固体晶体的那些之间的特性的物质状态。例如,液晶可像液体一样流动,但其分子能够以类似晶体的方式取向。存在着许多不同类型的液晶相,它们可以通过其不同的光学(如小角X射线散射)或光谱(例如FTIR)特性来区分。液晶结构通常被称为其中液晶分子沿不同方向取向的域。然而,在一个域内,分子是良好有序的。小角X射线散射可以测量域之间的平均间距(“晶格间距”)。另一方面,FTIR可以检测液晶中分子的红外吸收带的变化,这是由于其有序的结构或分子间相互作用。在一个实施方案中,本发明的乳剂为具有独特的光学和光谱特性的液晶。在一个实施方案中,本发明的乳剂的晶格间距大于2.8nm,优选地3nm至5nm,并且更优选地约3.8nm。
如本文所用,“溶致液晶”是指主要由有机分子(如本发明的乳剂中的那些)和溶剂(例如水)组成的液晶类型。溶致液晶表现出作为溶剂含量或浓度的函数的相变。在一个实施方案中,本发明的乳剂为溶致液晶型,当水浓度从低于1%增加到高于约1%时具有从油包水型向水包油型乳剂的明显相变。在一个实施方案中,本发明的乳剂在约1%水下具有相变点。
术语“金属离子螯合剂或螯合剂”包括安全用于可注射产品中的金属离子螯合剂。金属离子螯合剂通过与金属离子结合而起作用,并由此降低该金属离子对氧化、水解或其它降解反应的催化效应。可用于本发明的金属螯合剂可包括乙二胺四乙酸(EDTA,依地酸盐)、甘氨酸和柠檬酸以及它们相应的盐或混合物。在一个实施方案中,本发明的乳剂含有抗氧化剂和/或金属离子螯合剂或它们的组合。
如本文所用,术语“神经阻滞”意指其中将麻醉剂或镇痛剂注射到神经周围或直接靠近神经的区域中以阻滞身体特定部分的疼痛、感觉或运动的过程。神经阻滞可为局部或周围神经阻滞,并且为局部麻醉或疼痛缓解的形式。
如本文所用,“N-氧化物”是指LA的氧化产物,其中氧共价连接至LA结构上的氮原子(N)。例如,罗哌卡因N-氧化物示出于图2中。氮原子的氧化是含有卵磷脂或油的制剂中LA的主要降解产物,因此必须保持在0.2%以下(基于LA的重量计)。根据FDA的行业指南Q3B(R2)新药品中的杂质(Guidance for Industry Q3B(R2)Impurities in New DrugProducts),0.2%限值是杂质的鉴定和质控限度(Identification and QualificationThresholds)。在一个实施方案中,本发明的乳剂含有抗氧化剂和/或金属离子螯合剂或它们的组合以抑制N-氧化物的形成。在优选的实施方案中,本发明的乳剂含有不超过0.2%(按LA的重量计)的N-氧化物。
如本文所用,“中性pH”在4至8的范围内,优选地5.2至7.2。
如本文所用,“水包油乳剂”是其中油相呈悬浮或分散在水相(连续相)中的小液滴(分散相)形式的乳剂,并且“油包水乳剂”是指其中小水滴悬浮或分散在油相中的形式。在一个优选的实施方案中,本发明的乳剂是水包油乳剂。在另一个优选的实施方案中,本发明的乳剂是油包水乳剂,其在用水或生物流体如血液稀释后转化为水包油乳剂。在另一个实施方案中,本发明的乳剂是水包油与油包水乳剂的混合物。
如本文所用,“油相”是指包含油和磷脂的乳剂(o/w)的水不混溶相。油相也可含有其它亲脂性添加剂,包括抗氧化剂和抗微生物防腐剂等,以及如本发明的乳剂中的LA。在一个实施方案中,本发明的乳剂含有浓度为乳剂重量的约20%至99.5%的油相。当油相低于20重量%时,乳剂就丧失其提供期望的LA延长释放和延长作用的能力。另一方面,油相可以高达乳剂重量的99.8%,优选99.7%,并且更优选99.5%。油相大于99.8%的乳剂可能难以制备,因为残余的水(<0.2%)难以使用本发明的乳剂的当前乳剂制备操作移除(实施例18)。当含水量超过乳剂重量的约50%时,乳剂主要为水包油型,或者当含水量小于乳剂重量的约5%时主要为油包水型。当含水量为乳剂重量的约5%至约50%时,乳剂包含水包油和油包水型两者。乳剂在注射或滴注到人体中时以水包油和油包水型两者存在。
如本文所用,术语“有机溶剂”是指安全注射到人体中的水不混溶性溶剂。有机溶剂的实例包括但不限于甘油、乙醇、醇、苄醇、丙二醇、聚乙二醇、或它们的组合。向本发明的乳剂中添加有机溶剂的主要目的是降低乳剂的粘度。有机溶剂也可充当抗微生物防腐剂,以使乳剂能够在延长的时间段内使用。然而,有机溶剂的量必须控制在按所述乳剂的重量计约2%至12%的范围内。如果有机溶剂添加到4%以下,则可能无法实现降粘效果,但有机溶剂超过乳剂重量的12%,本发明的乳剂将破坏,且油滴尺寸大大增加(例如>1-5微米,由于液滴聚集),并且乳剂继而变成较稠的糊状物,使其难以注射。在一个实施方案中,本发明的乳剂含有0%有机溶剂。在另一个实施方案中,本发明的乳剂含有按所述乳剂的重量计2%至12%,优选按所述乳剂的重量计4%至10%,或更优选按所述乳剂的重量计6%至8%的有机溶剂。
如本文所用,术语“疼痛强度量表”或“疼痛强度评分”、或“疼痛量表”或“疼痛评分”是帮助评估人员的疼痛水平的工具。人员通常使用专门设计的量表自我报告他们的疼痛,有时在医生、父母或监护人的帮助下进行。常用的疼痛量表包括但不限于数字等级量表(NRS)、直观模拟量表(VAS)、分类量表、Wong-Baker面部表情疼痛量表;“静息疼痛评分或NRS-R和活动疼痛评分(NRS-A)”意指针对疼痛缓解的数字等级量表(NRS),其要求患者按照限定的量表对他们的疼痛进行评分。例如,0-10,其中0是无痛,并且10是可想象的最严重疼痛。常用的NRS为11个点(0-10)、21个点(0-20)和101个点(0-100)。NRS-R是个体静息时报告的疼痛评分。NRS-A是个体执行某些活动或运动之时或之后报告的疼痛评分。
如本文所用,术语“pH缓冲剂”或“pH缓冲盐”包括具有抗衡离子如铵、钠或钾等的可离子化pH缓冲盐,如磷酸盐、乙酸盐、柠檬酸盐、碳酸氢盐等等。
在优选的方面,本发明乳剂处于约4至约8的pH,更优选地约pH 4至约7,如约4.0、4.2、4.4、4.6、4.8、5.0、5.2、5.4、5.6、5.8、6.0、6.2、6.4、6.6、6.8、7.0或7.2。乳剂的pH通过使根据本发明的乳剂组合物的所有组分合并、添加酸(例如,HCl、乙酸、磷酸)和/或碱(例如,NaOH、KOH、精氨酸、赖氨酸)以调节至目标pH来实现。当pH低于pH 4时,乳剂有刺激性,并且可引起注射部位反应。当pH高于7时,LA降解加速,并且乳剂容易形成沉淀。
如本文所用,LA的“药理学活性”或“药理活性”是指LA的预期生物活性的体内或体外测量。与游离或未结合的LA相比,本发明的乳剂中配制的LA的药理活性不受到损害(实施例2)。这与乳剂降低LA的生物活性的常识形成鲜明对比,表明本发明的乳剂在功能上不同于其它常用于使LA失活或作为LA的解毒剂的乳剂,如英脱利匹特(Intralipid)。在一个实施方案中,本发明的乳剂在相同剂量下使其LA药理活性维持在游离LA活性的50%至150%范围内。
如本文所用,术语“药代动力学特性”或“PK特性”是指药物在给药于动物或人个体后随时间推移的血液浓度特性。对于本发明乳剂,PK特性以随时间推移的LA血浆浓度表示,具有以下四个关键参数:
C
T
AUC:测量LA的生物利用度的曲线下面积
T
在一个实施方案中,本发明的乳剂表现出如实施例1中所列的PK参数。在另一个实施方案中,在将2重量%本发明的罗哌卡因乳剂在人类中以200mg剂量注射并浸润到手术切口中后,本发明的乳剂表现出C
如本文所用,“磷脂”是指具有两个脂肪酸和一个磷酸根离子的任意甘油三酯,所述磷酸根离子共价连接至有机小分子(如胆碱、乙醇胺、甘油、丝氨酸、或无任何物质)。因此,示例性磷脂包括磷脂酰胆碱(PC)、磷脂酰乙醇胺(PE)、磷脂酰甘油(PG)、磷脂酰丝氨酸(PS)以及磷脂酸(PA)。脂肪酸通常具有约10至约18个具有不同饱和度的碳原子。这些磷脂可以从天然来源获得或者合成制备。天然源的磷脂被称为卵磷脂。合成磷脂的实例为1,2-二肉豆蔻酰基-sn-甘油-3-磷酸胆碱(DMPC)、1,2-二硬脂酰基-sn-甘油-3-磷酸乙醇胺(DSPE)、1,2-二肉豆蔻酰基-sn-甘油-3-磷酸甘油钠盐(DMPG,Na)以及1,2-二棕榈酰基-sn-甘油-3-磷酸-L-丝氨酸钠盐(DPPS,Na)。合成制备的磷脂通常难以掺入油相中,具有有限的安全性且昂贵。在某些实施方案中,本发明的乳剂基本上不含合成磷脂。在另一个实施方案中,卵磷脂是本发明的乳剂的优选磷脂。
如本文所用,在提及到本发明的乳剂时,“物理稳定性”或“物理稳定”能够在25℃下2年后维持特定期望的物理特性,如液滴尺寸、视觉均匀度、不存在LA沉淀、粘度以及体外释放特性。在某些实施方案中,本发明的乳剂在25℃下约2年后保持视觉上均匀、基本上不含任何LA沉淀以及相同的粘度(初始值的约90%至约110%)。在另一个实施方案中,本发明的乳剂通过使起效、速率以及持续时间均维持在其初始值的约50%至约150%而保留相同的体外释放特性。
如本文所用,“还原剂”包括但不限于抗坏血酸、抗坏血酸盐、棕榈酸抗坏血酸酯、偏亚硫酸氢盐、没食子酸丙酯、丁基化羟基苯甲醚、丁基化羟基甲苯、生育酚、半胱氨酸、甲硫氨酸、柠檬酸、柠檬酸盐、还原糖,如葡萄糖、果糖、甘油醛、半乳糖、乳糖、麦芽糖、它们的盐或混合物。在某些实施方案中,本发明的乳剂含有还原剂。在优选的实施方案中,本发明的乳剂含有选自抗坏血酸、棕榈酸抗坏血酸酯、生育酚、半胱氨酸、甲硫氨酸的还原剂。在另一个实施方案中,所用还原剂为乳剂重量的0.1%至3%。
如本文所用,术语“半透”或“半透明”是指液体的部分透明度或澄清度。半透或半透明的液体药物使得能够视觉检查液体中的异物颗粒和微生物污染。因此,半透或半透明比不透明(如乳)更优选。液体的透明度可以通过使用UV-vis分光光度计在固定波长(例如700nm)下测量透光率(T,%)进行定量。使用10mm光程长度石英比色皿,本发明的乳剂典型地具有在700nm下不小于50%的T值。在相同的条件下,水的T值为约100%,并且普通牛乳的T值小于10%。在一个实施方案中,本发明的乳剂是半透或半透明的(油包水型乳剂)。在另一个实施方案中,本发明的乳剂的T值(使用10mm光程长度石英比色皿在700nm下测量)大于25%,优选地大于40%,并且更优选地50%(油包水与水包油乳剂的混合物或水包油型)。
如本文所用,术语“小角X射线散射”或“SAXS”是指测量由样品散射的X射线强度作为散射角的函数的分析技术。测量以典型地在0.1度至5度范围内的极小角度进行。布拉格定律表明,越来越小的散射角对应于越来越大的结构特征。每当材料含有纳米长度尺度(典型地在1-100nm范围内)的结构特征时,就会观察到SAXS信号。SAXS方法是用于纳米材料如液晶的结构表征的最通用技术之一。SAXS探针用于结构特征如纳米颗粒尺寸分布、颗粒形状、颗粒结构,以及液晶相等。
如本文所用,术语“软组织”意指体内未通过骨化或钙化(如骨和牙齿)过程硬化的所有组织。软组织连接、包围或支撑内脏和骨,并且包括但不限于肌肉、腱、韧带、脂肪、纤维组织、淋巴和血管、筋膜、滑膜或皮肤。
如本文所用,术语“基本上不含”意指小于组合物的总重量的1%。
如本文所用,“溶液”是指仅由一个相构成的澄清、均匀的混合物。本发明的乳剂不是溶液,因为它具有油滴(一个相),其悬浮在水相(第二相)中。
如本文所用,“表面活性剂”是指降低液体的表面张力或介于油与水之间的界面张力的水溶性化合物。表面活性剂的实例是聚山梨醇酯、司盘类、氢化蓖麻油、维生素E TPGS、月桂基硫酸钠、泊洛沙姆以及泰洛沙泊等。本发明的乳剂基本上不含表面活性剂,优选地基本上不含常用于固体脂质纳米颗粒(SLN)的可离子化阳离子脂质、聚乙二醇化脂质或胆固醇。
如本文所用,术语“外科网”意指可在外科手术期间植入以支撑各种组织或器官的塑料、有机、或生物材料的一类网状片材。外科网可以由无机、有机、聚合物和生物材料制成。
如本文所用,术语“缝合线”意指用于在受伤或外科手术后使身体组织固定在一起的医疗装置。应用通常涉及使用附带有一定长度线的针头。
如本文所用,“超滤”(UF)是指其中液体静压迫使液体抵靠半透膜的多种膜过滤技术。悬浮的固体、油滴或高分子量或尺寸的溶质得以保留,而水和低分子量溶质(例如游离药物)通过膜。超滤(例如,采用30K道尔顿MW截止膜)用于分离本发明乳剂的油相(油滴被留在膜上)和水相(通过膜)。然后,对于各相中的LA浓度,可以分析分离的油和水相。
如本文所用,“植物油”是指源自植物种子或坚果的油。示例性植物油包括但不限于杏仁油、琉璃苣油、黑加仑籽油、玉米油、红花油、大豆油、芝麻油、棉籽油、花生油、橄榄油、菜籽油、椰子油、棕榈油、芥花油、蓖麻油等。植物油典型地含有长链甘油三酯,该长链甘油三酯是当三个脂肪酸(长度通常为约14至约22个碳且具有含有不同数量和位置的不饱和键的链,这取决于油的来源)与甘油上的三个羟基形成酯键时形成的。在某些实施方案中,高度纯化级(也称为“超精制(super refined)”)的植物油通常用于确保药物级水包油乳剂的安全性和稳定性。对于本发明,优选的油是大豆油、玉米油和芝麻油。与其它油相比,蓖麻油更具亲水性,因为它在脂肪酸侧链上具有羟基。在一个实施方案中,不使用蓖麻油,因为使用蓖麻油时乳剂物理稳定性较差。因为油分子上的一些脂肪酸链是不饱和的,并且可经历氧化形成过氧化物,该过氧化物可继而氧化同一制剂中的LA。因此,在一个实施方案中,本发明的乳剂含有抗氧化剂和/或金属离子螯合剂或它们的组合以抑制油氧化。在一个实施方案中,本发明的乳剂含有按所述乳剂的重量计约10%至约50%,优选地约20%至约40%或更优选地约30%至约40%的植物油。当油低于10%时,乳剂极硬,难以过滤和注射,另一方面,当油超过50%时,所形成的乳剂是白色、不透明、乳脂状和粘稠的,并且也难以过滤和注射。在另一个实施方案中,本发明的乳剂中卵磷脂与油的重量比为7:1至2:5,优选地3:1至1:1,并且更优选地2:1至5:4。
如本文所用,“水溶性”描述了能够以每十重量份水中不少于一重量份溶质的程度溶解在水中形成均匀溶液的固体或液体溶质。
如本文所用,“水溶性表面活性剂”是通过降低介于水与另一种液体之间或介于水与固体之间的界面表面而帮助溶解化合物以形成澄清且单相水溶液的化合物。水溶性表面活性剂包括亲水-亲脂平衡(HLB)大于7的任何表面活性剂或表面活性物质。水溶性表面活性剂的实例包括但不限于聚山梨醇酯、司盘、溶血卵磷脂、辛酸癸酸聚乙二醇甘油酯(labrasol)、氢化蓖麻油、solutol、月桂酸聚乙二醇甘油酯(gelucire)、SDS以及TPGS等。相比之下,卵磷脂不是水溶性表面活性剂,因为它不溶于水。水溶性表面活性剂通常用于乳剂中,但对于本发明的乳剂而言是不可取的,因为它们中的大多数可引起溶血(红细胞膜的破坏)以及注射部位的刺激或疼痛。在某些实施方案中,本发明的乳剂基本上不含水溶性表面活性剂。在一个优选的实施方案中,本发明的乳剂基本上不含溶血卵磷脂。
如本文所用,“伤口愈合过程”或“伤口愈合”意指活体生物被破坏或受损的组织被新产生的组织替换。它典型地涉及四个阶段:止血、炎症、增殖以及成熟。良好的伤口愈合过程不导致疤痕形成以及密封组织的物理强度弱化。
II.实施方案
本发明提供了一种乳剂组合物,所述乳剂组合物包含、基本上由以下组成、或由以下组成:
a)局部麻醉剂,
b)油相,和
c)水相,
其中
i.所述局部麻醉剂的浓度为按所述乳剂的重量计多至4%,
ii.所述油相的浓度为按所述乳剂的重量计约20%至99.8%,
iii.所述油相包含重量比为7:1至2:5的卵磷脂和植物油,以及
iv.所述乳剂含有平均直径为约200-2500纳米的颗粒或液滴。
本发明提供了一种乳剂,如水包油组合物,所述乳剂包含、基本上由以下组成、或由以下组成:
a)罗哌卡因、其盐或它们的混合物,
b)油相,和
c)水相,
其中
i.罗哌卡因的浓度为按所述乳剂的重量计0.5%至4%,
ii.所述油相的浓度为按所述乳剂的重量计约20%至99.8%,
iii.所述油相包含重量比为2:1至5:4的卵磷脂和植物油,以及
iv.所述乳剂含有平均直径为约200-2500纳米的颗粒或液滴。
v.所述乳剂包含还原剂和金属离子螯合剂,以及
vi.所述乳剂的pH为4至7。
本发明提供了一种乳剂,如水包油组合物,所述乳剂包含、基本上由以下组成、或由以下组成:
a)盐酸罗哌卡因,
b)由大豆卵磷脂和芝麻油组成的油相,以及
c)由EDTA钠、半胱氨酸和水组成的水相,
其中
i.罗哌卡因的浓度为按所述乳剂的重量计0.5%至4%,
ii.碱性物或碱,其量足以使约5至25摩尔%的添加的盐酸罗哌卡因转化为非离子化形式,以及将所述乳剂pH调节至pH目标,
iii.大豆卵磷脂和植物油的重量比为2:1至5:4,
iv.所述乳剂含有平均直径为约200-2500纳米的颗粒或液滴,
v.油滴的浓度为所述乳剂的重量的约33%至99.5%,以及
vi.所述乳剂的pH为4至7。
本发明提供了一种乳剂,如水包油组合物,所述乳剂包含、基本上由以下组成、或由以下组成:
d)罗哌卡因游离碱,
e)由大豆卵磷脂和芝麻油组成的油相,以及
f)由EDTA钠、半胱氨酸和水组成的水相,
其中
vii.罗哌卡因的浓度为按所述乳剂的重量计0.5%至4%,
viii.酸,其量足以使约75至95摩尔%的添加的罗哌卡因游离碱转化为离子化形式,以及将所述乳剂pH调节至pH目标,
ix.大豆卵磷脂和植物油的重量比为2:1至5:4,
x.所述乳剂含有平均直径为约200-2500纳米的颗粒或液滴,
xi.油滴的浓度为所述乳剂的重量的约33%至99.5%,以及
xii.乳剂的pH为4至7。
III.制备乳剂的方法
在一个实施方案中,本发明提供了用于制备本发明的乳剂组合物的方法,所述方法包括、基本上由以下组成、或由以下组成:
步骤1.使油相组分(包括卵磷脂和植物油)合并。
步骤2.均质化以形成平滑的半固体糊状物(“油相”)。
步骤3.使LA、还原剂、金属螯合剂和水合并。混合以溶解所有的固体,以形成水溶液(“水相”)。
步骤4.使水相和油相合并,并通过均化混合,以形成水包油乳剂。
步骤5.添加足以使5至25摩尔%的步骤3中添加的LA(呈盐形式)转化为非离子化形式的量的碱,或者添加足以使75至95摩尔%的步骤3中添加的LA(呈游离碱形式)转化为离子化形式的量的酸。然后,调节乳剂pH到4至7。
步骤6.将水包油乳剂均质化为油滴,以使平均直径达到200至2500nm。
步骤7.根据需要,通过添加更多的水或经真空干燥移除一些水,将含水量调节到20%至99.8%。
步骤8.任选地,添加可混溶有机溶剂(例如乙醇)。
步骤9.过滤乳剂以对其灭菌。
IV.使用方法
易于用局部麻醉剂管理的疼痛
本发明的LA乳剂的给药可用于提供与各种各样的病症、病况或疾病中的任一种相关联的疼痛缓解。疼痛的原因可能是可鉴定或不可鉴定的。在可鉴定的情况下,疼痛的起源可为例如恶性、非恶性、感染性、非感染性或自身免疫性来源。
当前未患有疾病或病况但易受这些影响的个体也可以受益于本发明的预防性疼痛缓解方法,例如在创伤外科手术之前。适合根据本发明的疗法的疼痛可以涉及与无痛间隔交替的延长疼痛发作、或严重程度有所不同的基本上不间断的疼痛。
一般来说,疼痛可为躯体源性、神经源性或心因性。躯体源性疼痛可为肌肉或骨骼的(即,骨关节炎、腰骶背痛、创伤后、肌筋膜)、内脏的(即,慢性胰腺炎、溃疡、肠易激)、缺血性(即,闭塞性动脉硬化)、或与癌症进展有关(例如,恶性或非恶性)。神经源性疼痛可能是由于创伤后和术后神经痛,可与神经病(即糖尿病、毒性等)有关,并且可与神经卡压、面神经痛、会阴神经痛、截肢术后、丘脑、灼痛,以及反射性交感神经营养不良有关。每种可能性均是本发明的单独实施方案。
适合管理的病况、疾病、病症和疼痛起源的具体实例包括但不限于术后疼痛(也称为手术后疼痛)、癌症疼痛(例如,转移性或非转移性癌症)、慢性炎性疾病疼痛、神经性疼痛、医源性疼痛(例如,在侵入性手术或高剂量放射疗法后的疼痛,例如,涉及瘢痕组织形成,导致运动自由度妨碍性损害和显著的慢性疼痛)、复杂性局部疼痛综合症、腰椎手术失败性疼痛(failed-back pain)(慢性背痛)、软组织疼痛、关节和骨疼痛、中枢性疼痛、损伤(例如,致残性伤害,例如截瘫、四肢瘫痪等,以及非致残性伤害(例如,背部、颈部、脊柱、关节、腿、臂、手、足等)、关节炎疼痛(例如,类风湿性关节炎、骨关节炎、病因不明的关节炎症状等)、遗传性疾病(例如镰状细胞性贫血)、感染性疾病以及所得的综合症(例如,莱姆病、AIDS等)、慢性头痛(例如,移行性)、灼痛、感觉过敏、交感神经营养不良、幻肢综合症、失神经支配等等。疼痛可以与身体的任何部分相关联,例如肌肉骨骼系统、内脏器官、皮肤、神经系统等。每种可能性均是本发明的单独实施方案。
癌症疼痛是可使用局部麻醉剂的乳剂缓解的一大类疼痛的实例。癌症疼痛的潜在原因之一是肿瘤病变对组织的严重局部拉伸。例如,当癌细胞以不受限制方式增殖时,癌细胞增殖的局部区域中的组织经受机械应力,该机械应力是移位组织并适应肿瘤块所占据的增加的体积所需的。当肿瘤负荷局限于小封闭区室(如骨髓)时,所得的压力可导致严重疼痛。疼痛的另一个原因可能是由用于对抗患者的癌症的激进疗法(例如放射疗法、化学疗法等)引起的。这样的癌症疗法可涉及局部或广泛的组织损伤,从而导致疼痛。
与任何类型的恶性或非恶性癌症相关联的疼痛可适合根据本文所述的方法来缓解。可与疼痛相关联的癌症的具体实例(由于癌症本身的性质或治疗癌症的疗法)包括但不一定限于肺癌、膀胱癌、黑素瘤、骨癌、多发性骨髓瘤、脑癌、非霍奇金淋巴瘤、乳腺癌、口腔癌、宫颈癌、卵巢癌、结肠癌、直肠癌、胰腺癌、发育异常痣、内分泌癌、前列腺癌、头颈癌、肉瘤、霍奇金病、皮肤癌、肾癌、胃癌、白血病、睾丸癌、肝癌、子宫癌以及再生障碍性贫血。某些类型的神经性疼痛也适合根据本发明治疗。
也可适合使用本文所述的方法管理的慢性背痛是另一大类疼痛。慢性背痛通常是由于以下六种原因中的一种或多种:(i)由滑移、关节炎、楔形变或脊柱侧凸引起的椎间小关节压力;(ii)神经根病变,即由椎间盘突出或肿瘤所致的神经根的机械性压迫;(iii)肌腱炎或筋扭;(iv)肌肉痉挛或肌肉扭伤;(v)缺血,即循环流局部不足;以及(vi)神经病,即代谢性病因或由脊髓肿瘤或中枢神经系统疾病引起的神经组织损伤。
本文所述的乳剂可用于需要LA的长效或缓释制剂的多种治疗目的。
作为常见的医疗实践,乳剂一直被用来使LA失活或者作为LA过量的解毒剂。有许多文献证明,乳剂(不含药物)通常用于降低LA的心血管效应或毒性。多位研究人员清楚地展示,乳剂可以使LA失活,从而导致LA的药理效应和其毒性丧失(参见Zausig,York A.等人,Lipid Emulsion Improves Recovery from Bupivacaine-Induced Cardiac Arrest,but Not from Ropivacaine-or Mepivacaine-Induced Cardiac Arrest,Anesthesia&Analgesia:2009年10月-第109卷-第4期-第1323-1326页;E.Litonius等人,Effect ofintravenous lipid emulsion on bupivacaine plasma concentration in humans,Anaesthesia 2012,67,600-605;Lotte C.G.等人,Systematic review of the effect ofintravenous lipid emulsion therapy for local anesthetic toxicity,ClinicalToxicology,2016,第54卷,第3期,167-193,http://dx.doi.org/10.3109/15563650.2015.1121270)。
因此,使用乳剂来递送药理学活性LA与常识和医学实践相悖。本发明涉及一项令人惊奇的发现,与使用乳剂使LA失活的常识和医学实践相反,本申请公开了乳剂的使用不仅维持LA的药理效应,而且延长其作用持续时间并降低其毒性。
通过注射、浸润和/或滴注相对高剂量的LA一次性施用以在施用的局部组织中提供高浓度的LA,使得本发明的乳剂的这种延长作用成为可能。
患有或易受疼痛影响的个体可以受益于根据本文所述的方法的疼痛缓解12小时至24小时、24小时至48小时、48小时至72小时、或更多。如果期望更长时段的疼痛缓解,可以重复给药局部麻醉剂乳剂。典型地,乳剂的给药可以在约1周或数个月内重复两次、三次或更多次。
用于治疗或预防疼痛的本发明方法可进一步包括共同给药另一种预防剂或治疗剂,其包括但不限于抗感染剂、抗炎剂、抗肾衰竭剂,以及抗心血管疾病剂、镇吐剂、抗焦虑剂和镇痛剂、或用于减少局部麻醉剂的任何潜在副作用的解毒剂。这样的潜在副作用包括但不限于恶心、呕吐、头痛、白细胞计数低、红细胞计数低、血小板计数低、头痛、发烧、嗜睡、肌肉酸痛、全身痛、骨痛、注射部位疼痛、痢疾、神经病、瘙痒、口腔溃疡、脱发、焦虑或抑郁。
预期使用本发明的乳剂缓解疼痛的治疗益处之一是减少全身麻醉剂或患者自控镇痛(尤其是阿片剂或阿片类药物)的使用。
尽管在一次性施用中给出相对高的剂量,但本发明的乳剂不产生LA的爆发释放或者超过药物的心血管或CNS毒性水平的高LA C
LA剂量、乳剂体积或施用频率(一次或多次)可以通过药物功效来确定,如通过疼痛评分(如NRS-R NRS-A)来量度,并且全身麻醉药物使用的减少(这可能导致心血管和CNS毒性较小)可以通过局部和全身药代动力学特性和/或体外释放特性的研究所确定。
类似地,施用方法(注射、浸润或滴注或它们的组合)可以通过药物功效来确定,如通过疼痛评分(如NRS-R NRS-A)来量度,或者全身麻醉药物使用的减少(这可能导致心血管和CNS毒性较小)可以通过局部和全身药代动力学特性和/或体外释放特性的研究所确定。
优选的方法是给药高体积的本发明的乳剂以用于更好的功效,只要LA C
本发明的乳剂中LA的剂量也取决于手术或伤口的类型、疼痛的严重性以及其它麻醉剂的共同给药。
作用持续时间也取决于给药途径,但通常预计相对于多次注射较小体积(例如浸润),将较大体积的乳剂单次注射到软组织中会提供更长的作用持续时间。类似地,注射或浸润到深层组织中通常会提供比局部施用或滴注更长的镇痛效果。此外,未经稀释的乳剂往往比稀释的乳剂提供更长的作用。
对于手术切口或伤口,浸润和滴注的组合是常见的给药方法。对于给定的切口/伤口,通常期望通过浸润施用最大乳剂体积以覆盖最大组织面积。还期望浸润切口/伤口并且滴注伤口可容纳的任何剩余体积。对于神经阻滞,注射或浸润是优选的给药途径。
预期本发明的乳剂的疼痛缓解可以通过酰胺LA如但不限于罗哌卡因来递送,因为所有的酰胺LA享有相似的物理、化学和药理学特性。
一旦确定乳剂剂量体积,就可以将其在多种时间段内给药,包括但不限于约每12小时、约每24小时、约每36小时、约每48小时、约每72小时、约每周、约每两周、约每三周、约每月、以及约每两个月。
乳剂的剂量和给药频率可以根据多种因素进行调节,包括个体的类型、年龄、体重、性别和医疗状况;待治疗病况的严重程度;给药途径;个体的肾或肝功能;以及所采用的特定局部麻醉剂。本领域技术人员可以容易地确定可用于治疗疼痛(包括待治疗的特定类型的疼痛)的局部麻醉剂的有效量。
本发明的给定乳剂的体内释放特性可以通过进行体外释放测试来评价,这对于确保本发明的乳剂在修改后保留其缓释特性尤其有用。例如,体外释放测试可以用于确定可用于稀释乳剂而不丧失其缓慢释放特性的稀释度。
本发明的乳剂的粘度也是选择给药或稀释途径的考虑因素。对于滴注或局部施用,乳剂粘度可相对较高,因为乳剂可使用无针头注射器通过挤出来容易地给药。对于浸润或注射,乳剂需要较低的粘度,因为它必须使用注射器通过针头注射。此外,对于神经阻滞应用,乳剂必须为甚至更低的粘度,因为这种应用通常需要较长的针头或导管。本发明的乳剂的粘度随含水量或稀释比而变化。低比率稀释(用较少水)可导致粘度太高而无法注射。另一方面,极高的稀释度(用过多水)将致使乳剂丧失其缓释特性。因此,可以平衡乳剂粘度和体外释放特性,以限定本发明的乳剂可接受的稀释剂和稀释方法。
本发明的乳剂可以在阿片样物质或非阿片类镇痛剂之前、同时、或之后、或在同一天、或在彼此1小时、2小时、12小时、24小时、48小时或72小时内给药。
在一个实施方案中,本发明的乳剂是填充到玻璃或塑料容器如小瓶、瓶、安瓿、注射器和袋中的半透明均匀液体。每个容器的典型体积为5mL至50mL,优选地10mL至25mL。将液体在使用前吸入注射器中。
在一个优选的实施方案中,本发明的乳剂被提供在附有皮下注射针头的预填充注射器中并且准备好用于注射。该特征对于外科手术期间的施用是特别可取的。
在一个实施方案中,预填充注射器也可包含适用于乳剂的注射、滴注(installation)和浸润以及局部施用的针头。
在一个实施方案中,用于乳剂的针头是18-25G针头,优选地21G针头。
在某个实施方案中,本发明的乳剂经由使用附接到注射器的针头或附接到导管然后注射器的针头而注射或浸润到软组织中或神经附近来给药。
在某个实施方案中,将本发明的乳剂局部地或用注射器注射而浸润或施用到开放性手术伤口。在一些实施方案中,制剂通过皮下、皮内、肌内、或经皮注射来给药。
在一个实施方案中,乳剂经由伤口浸润给药到手术部位中。
在一个实施方案中,乳剂经由伤口滴注给药到手术部位中。
在一个实施方案中,乳剂经由伤口浸润和滴注的组合给药到手术部位中。给药体积的范围取决于手术切口尺寸和手术切口位置。典型的体积为5mL至50mL。优选的给药体积为10mL至40mL。浸润与滴注之间的典型体积比为1:20至20:1。典型的注射器和针头组合用于进行伤口浸润。注射器的典型尺寸为1mL至50mL。注射器的优选尺寸为10mL至30mL。针头的典型尺寸为18-30G,3/8至3-1/2英寸。针头的优选尺寸为19-22G,1至1-1/2英寸。
在另一个实施方案中,乳剂经由特定神经或神经束附近的局部神经阻滞、区域阻滞、平面阻滞、周围神经阻滞来给药,以阻滞来自身体特定区域的疼痛感。
在一个优选的实施方案中,乳剂经由超声引导的局部神经阻滞来给药。典型的体积为5mL至50mL。优选的给药体积为10mL至40mL。在另一个优选的实施方案中,乳剂使用具有或不具有管道和延长装置的神经阻滞针头来给药。典型的神经阻滞针头为20-22G,具有1-3/8至6英寸。在一个实施方案中,乳剂经由硬膜外针头来给药。典型的硬膜外针头为17-22G。
稀释对用于手术疼痛控制的药物而言总是期望的,以能够调节剂量体积、浓度和释放特性,因为外科手术的切口尺寸和疼痛程度有所不同。此外,神经阻滞应用通常需要较低的LA浓度。已知一些基于聚合物的延长释放制剂不可稀释,因此限制了它们在手术中的适用性。作为水包油乳剂,本发明的组合物与水相容,因此可以稀释。在优选的实施方案中,在给药到患者中之前,用水、盐水、右旋糖溶液或其它安全使用的可注射稀释剂来稀释本发明的乳剂。
本发明的乳剂的稀释可以改变其粘度和药物释放特性。因此,稀释仅可在限定的范围内进行。高粘度(例如,>400厘泊)使通过针头注射变得困难。本发明的乳剂的粘度对稀释剂和稀释比敏感。用于本发明的乳剂的稀释剂可为水、盐水、右旋糖溶液、IV输注流体如林格乳酸盐溶液、或其它安全使用的可注射稀释剂如乙醇或丙二醇,或它们的组合。稀释比定义为稀释剂的体积相对于乳剂的体积。令人惊奇地,用水稀释初始会增加本发明的乳剂的粘度,之后在过度稀释后随后降低粘度。例如,用盐水1:1稀释和用水2:1稀释分别将乳剂粘度从约500增加至17000-28000以及至大约40000厘泊。另一方面,过度稀释(例如,稀释比>10:1)致使乳剂改变缓慢LA释放特性,并且损害疼痛缓解功效或作用持续时间。为了平衡粘度和释放特性,将本发明的乳剂用稀释剂以10:1至1:1的稀释比稀释。本发明的乳剂与盐水或水的优选稀释比为4:1至2:1。例如,用生理盐水2:1稀释导致约21厘泊的乳剂粘度,同时维持可接受的体外释放特性(实施例12、13)。由于不同的稀释剂也对粘度具有不同的影响,并且可能需要调节至稀释比,新稀释剂的优选稀释比是导致乳剂粘度不超过400厘泊和可接受的LA体外释放特性的比率。
在一个实施方案中,将乳剂用盐水稀释,然后通过物理混合动作(包括但不限于在开放容器中使用注射器重复抽出和排出动作,以及摇动密闭容器(例如瓶、小瓶、袋或注射器)中的乳剂和稀释剂)混合至均匀稠度。用于混合的优选容器是用于手术床边混合的开放无菌容器和无菌注射器。3-20次的抽出/排出循环将生成均匀的混合物。优选的重复8-12个循环也生成均匀的混合物。
在一个实施方案中,乳剂及其稀释乳剂具有不同的体外释放特性。例如,未经稀释乳剂具有12小时19-46%、24小时30-60%、48小时45-77%、72小时56-84%的LA释放范围。使用如实施例13中所述的体外释放方法,2:1盐水稀释的乳剂具有12小时49-90%、24小时67-100%、48小时83-100%、72小时89-100%的LA释放范围。在一个实施方案中,本发明的乳剂具有12小时19-90%、24小时30-100%、48小时45-100%、72小时56-100%的LA释放范围,优选地12小时19-49%、24小时30-67%、48小时45-83%、72小时56-89%的LA释放范围。
一些液体可注射药物或植入物与缝合线或其它手术材料(如网片)不相容,因此不能用于需要缝合线或网片的外科手术。在一个实施方案中,本发明的乳剂与外科手术材料如网片和缝合线相容,并且可安全地用于其中使用网片和缝合线的外科手术。典型的网片材料包括聚丙烯(PP)、膨体聚四氟乙烯(e-PTFE)、PP/PCG-25(聚卡普隆25)。典型的缝合线材料包括尼龙、聚二噁烷酮、聚丙烯、聚葡糖酸酯、羟乙酸乳酸聚酯(polyglactin)910。
在一个实施方案中,外科网在本发明的乳剂中于37℃浸泡至少7天后保留其原始抗拉强度的90-110%(实施例6)。
在一个实施方案中,外科用缝合线在本发明的乳剂中于37℃浸泡至少7天后保留其原始抗拉强度的90-110%(实施例6)。
用于给药至手术或伤口部位的良好药物不应影响切口或伤口的愈合过程。已知一些药物改变伤口愈合过程,因此改变施用后的愈合率或疤痕形成。本发明的乳剂显示出对伤口愈合过程没有任何有害效应,如由来自手术伤口的愈合组织的不变抗拉强度和无疤痕外观所表明(实施例2、5)。在一个实施方案中,本发明的乳剂对软组织的伤口愈合不具有不利影响。在另一个实施方案中,本发明的乳剂对骨愈合不具有不利影响。在另一个实施方案中,本发明的乳剂对神经组织(包括但不限于坐骨神经、脊神经前根/分支、以及伤口部位附近组织中的外周神经)不具有不利影响。
手术疼痛可持续多至3-5天,因此期望止痛药的效果持续多至3-5天。本发明的乳剂被设计成在注射部位具有延长的停留并缓慢地释放LA,以产生对切口/伤口部位的局部麻醉剂效果或神经阻滞。与常规溶液制剂(例如
在一个实施方案中,与常规溶液制剂(如
在一个实施方案中,与相同LA的溶液制剂所产生的相比,本发明的乳剂的药代动力学特性具有更低的C
在一个实施方案中,本发明的乳剂提供的药代动力学特性的C
在一个实施方案中,本发明的乳剂提供的药代动力学特性的T
在一个实施方案中,本发明的乳剂提供的药代动力学特性的T
在动物和人类中,与溶液LA制剂(例如
在一个实施方案中,与
在一个实施方案中,在人类中,与
在另一个实施方案中,当经由伤口浸润给药时,在0-24小时、0-48小时、0-72小时、24-48小时、48-72小时和24-72小时的给药后窗口期间,在人类中,本发明的乳剂提供的静息疼痛评分(NRS-R)或活动疼痛评分(NRS-A)低于
在另一个实施方案中,经由伤口浸润/滴注或神经阻滞给药本发明的乳剂减少了外科手术后患者的阿片样物质消耗。在一个优选的实施方案中,经由伤口浸润/滴注或神经阻滞给药乳剂使患者的阿片样物质消耗下降不少于5%、10%、20%、30%、40%或50%。在另一个优选的实施方案中,与给药
在一个实施方案中,当经由伤口浸润或神经阻滞给药时,本发明的乳剂不引起局部麻醉剂全身毒性(LAST)。在另一个优选的实施方案中,当与
乳剂组合物用于疼痛缓解的用途,所述乳剂组合物包含:
a.局部麻醉剂,
b.油相,和
c.水相,其中
i.所述局部麻醉剂的浓度为按所述乳剂的重量计多至4%,
ii.所述油相的浓度为按所述乳剂的重量计约20%至99.8%,
iii.所述油相包含重量比为7:1至2:5的卵磷脂和植物油,以及
iv.所述乳剂含有约30至2500纳米的颗粒或液滴。
本公开提供了用于在需要其的个体中缓解疼痛的方法,所述方法包括:
向个体给药有效量的乳剂组合物,所述乳剂组合物包含:
酰胺局部麻醉剂,其中酰胺局部麻醉剂为按所述乳剂的重量计不超过约4%;
油相,所述油相包含重量比为2:1至5:4的卵磷脂和植物油,并且其中所述油相的浓度为按所述乳剂的重量计约20%至99.8%;以及
水相,其中所述乳剂含有直径为约30nm至约2500nm的颗粒或液滴。
在某些方面,所述疼痛是选自躯体源性、神经源性,以及心因性疼痛的成员。
在某些方面,所述疼痛是术后疼痛或癌症疼痛。
在某些方面,所述组合物的给药在个体中提供至少24小时的疼痛缓解。
在某些方面,所述组合物通过选自伤口浸润、滴注以及神经阻滞的成员给药于所述个体。
在某些方面,所述酰胺局部麻醉剂是选自布比卡因、罗哌卡因以及它们的药学上可接受的盐的成员。
在某些方面,所述酰胺局部麻醉剂是罗哌卡因。
在某些方面,通过伤口浸润或滴注给药罗哌卡因组合物使罗哌卡因血浆水平维持在其心脏毒性水平以下。
在某些方面,所述组合物的给药不引起任何可检测的局部麻醉剂全身毒性(LAST)。
在某些方面,所述植物油是选自芝麻油、大豆油、橄榄油以及它们的组合的成员。
在某些方面,所述植物油是芝麻油。
在某些方面,其中所述组合物进一步包含选自乙醇、丙二醇、甘油以及液体聚乙二醇的水混溶性有机溶剂。
在某些方面,所述组合物具有约2至600厘泊的粘度。
在某些方面,所述组合物具有约4至约7的pH。
在某些方面,其中所述组合物是半透明或白色不透明液体,并且可通过0.2微米过滤器过滤。
在某些方面,所述卵磷脂含有不少于75重量%的磷脂酰胆碱。
在某些方面,所述组合物在给药前用盐水稀释至2:1比率或更大,或者用水稀释至4:1比率或更大。
在某些方面,超过90重量%的酰胺局部麻醉剂与油滴非共价结合。
在某些方面,所述组合物使用注射器、带针头注射器或带导管注射器给药。
在某些方面,与溶液制剂中的相同局部麻醉剂相比,所述组合物的给药提供了具有更低C
在某些方面,所述组合物通过使如由疼痛强度量表所测量的疼痛强度在多至72小时内下降不少于10%而提供疼痛缓解。
在某些方面,所述组合物的给药在至少60分钟内提供快速缓解起效,并且在给药后持续约48-72小时。
在某些方面,所述组合物提供了个体对阿片样物质使用的延迟。
在某些方面,所述组合物提供于小瓶或注射器中,准备好用于注射或准备好用于给药。
在某些方面,所述组合物经由带针头注射器或带导管注射器给药。
在某些方面,所述油滴是非脂质体的,并且基本上不含脂质体双层膜结构。
将通过参考以下非限制性实施例进一步理解本发明。
实施例1
罗哌卡因乳剂与罗哌卡因溶液剂
在该实施例中,对代码为F-53的组合物中的本发明的罗哌卡因乳剂进行了人药代动力学(PK)研究。F-53包含约2.6%罗哌卡因HCl、约52%大豆卵磷脂以及约35%芝麻油、约0.02% EDTA钠、约0.1%半胱氨酸、乙醇以及水。将F-53以200mg的剂量在12名人个体的手术切口部位处注射并浸润。收集血样,并且通过LC-MS分析测定血液中的罗哌卡因浓度。将PK结果与报告的罗哌卡因溶液剂(
结论:与溶液制剂
实施例2
人功效和安全性研究
在该实施例中,在小切口腹壁整形术后的人个体中进行了罗哌卡因乳剂的人临床安全性和功效研究。这是一项随机化、双盲、单中心研究,以评价F-53在≥18且≤70岁的男性和女性中用于管理小切口腹壁整形手术之后的术后疼痛的安全性、PK特性以及镇痛作用持续时间。在小切口腹壁整形术后闭合之前,每个随机分配的患者接受了通过伤口浸润和滴注给药到软组织中的F-53(200mg罗哌卡因)或安慰剂(0.9% NaCl)。该研究揭示了以下关键发现:
1.在多至72小时内,F-53在每12-24小时的时间段有效地将患者的疼痛强度(NRS-R)下降10-25%
2.F-53镇痛效应出现迅速(60分钟内起效),并在药物给药后0-72小时期间均匀地持续。
3.F-53治疗延迟了患者首次使用阿片样物质的时间:F-53中16.5小时vs安慰剂中12.3小时。
4.F-53耐受性良好,并且未显示出局部组织反应或伤口愈合受损的迹象。没有个体被研究人员评估为具有LAST。研究中未观察到严重不良事件(SAE)。临床实验室数据和生命体征值并未揭示出任何表明F-53负面影响的异常情况。F-53组中没有个体经历临床上显著的ECG变化。
实施例3
罗哌卡因乳剂与
本研究的目的是比较本发明的罗哌卡因乳剂、
使用针刺模型,在豚鼠中评价了皮下注射后这些制剂的局部麻醉剂功效。该模型涉及在豚鼠的皮肤下注射制剂以形成风团(小肿胀),用针刺破风团区域并观察动物的反应。如果动物对针刺没有反应,则确认局部麻醉剂活性。
F-10、F-11和F-13是根据本发明制备的乳剂制剂,它们各自含有罗哌卡因(1%作为HCl盐于F-10中、2%作为HCl盐于F-11中,或2%作为游离碱于F-13中)、约52%大豆卵磷脂以及约35%芝麻油、约0.02% EDTA、约0.1%半胱氨酸、乙醇和水。罗哌卡因HCl和罗哌卡因在疏水性方面与罗哌卡因游离碱差别很大,其中HCl盐的log P为1.1,而游离碱的log P为2.9。
为了比较,还测试了含有1%罗哌卡因HCl的
除V-10外,所有制剂均表现出显著的局部麻醉剂作用。然而,局部麻醉剂作用的持续时间(维持50%无反应的时间)按以下次序非常不同:
结论:本发明的含有局部麻醉剂药物的盐或游离碱的乳剂不仅能够维持局部麻醉剂活性,而且还提供期望的延长局部麻醉剂功效。本发明的乳剂表现出比溶液制剂
实施例4
罗哌卡因乳剂的安全性评估
本研究的目的是测量在Sprague-Dawley(SD)大鼠中罗哌卡因乳剂(F-32,其组成与实施例1中的F-53相似)通过单次皮下注射的注射部位处的最大耐受剂量(MTD)和局部毒性。为了比较,还测试了
/>
在第1天以44mg/kg或88mg/kg剂量给药
结论:F-32是比
实施例5
乳剂对伤口愈合的影响
在该实施例中,在伤口浸润/滴注到小型猪的背部皮肤上所成的手术切口中之后,评价了罗哌卡因乳剂制剂(F-53)对伤口愈合的效果。使切口愈合,并在给药后28天收集切口部位的猪皮肤,并切成小样品,以根据ASTM D638进行拉伸测试。以一致的速率牵拉开样品,直到发生断裂或分离,并记录抗拉强度[MPa],并在乳剂处理的皮肤与盐水处理的皮肤之间比较。第28天,比较盐水浸润的皮肤(2.3MPa+/-0.5[雄性]、4.8+/-1.3[雌性])与乳剂浸润的皮肤(3.0MPa+/-0.7[雄性]、3.5+/-0.5[雌性]),拉伸断裂强度也是相似的,且任一组均未观察到疤痕。
总之,罗哌卡因乳剂对伤口愈合不具有不利影响。
实施例6
乳剂与缝合线和网片的相容性
在该实施例中,在体外评价了罗哌卡因乳剂制剂(F-53)与缝合线和网片的相容性。
本研究中测试的每条缝合线均被切成13”长,并在F-53或生理盐水中于37℃浸泡7天。根据ASTM D2256通过拉伸测试测量断裂力。测试了四种类型的缝合线(PDS II、Prolene、Maxon、Vicryl)。对于所有测试的缝合线,在浸泡多至7天后,F-53浸泡的样品表现出与可比较的生理盐水浸泡的对照相同的断裂力,表明F-53对缝合线具有与生理盐水相同的机械效应,并且所有暴露于F-53多至7天的缝合线在延长暴露于F-53后均未展示出抗拉强度的显著下降。
对于网片测试,每个网片样品沿其长度被切成0.5”x1.5”的条,其中1.5”尺寸为纵向。将条在F-53或生理盐水中于37℃浸没(浸泡)7天。测试了三种类型的网片(Bard Dulex、Prolene、Ultrapro)。每个条样品根据ASTM D5035通过拉伸测试进行测量。Bard Dulex和Prolene的断裂力在用F-53或生理盐水浸泡的情况下似乎未改变,而UltraPro网片表现出在与F-53或生理盐水接触后断裂力略有下降。F-53和生理盐水显示出对测试网片的机械完整性的类似效果。
总之,本发明的乳剂与缝合线或网片相容。
实施例7
罗哌卡因乳剂的体外释放
在该实施例中,测量了罗哌卡因乳剂制剂的体外释放特性,并与根据美国专利第9,849,088B2号的实施例1和2中描述的组合物和方法所制备的前脂质体制剂进行了比较。罗哌卡因乳剂制剂使用如实施例18中所述的乳剂方法制备,而前脂质体使用美国专利第9,849,088B2号的实施例1和2中描述的方法制备。最终组成汇总于下表中:
使用USP 4型溶出仪测量了从乳剂(代码为F-5-2)和前脂质体制剂中罗哌卡因的体外释放。图5提供了体外释放特性比较。
结论:本发明的乳剂表现出与前脂质体制剂极为不同的罗哌卡因释放特性,尽管前脂质体制剂具有相似的组分。与前脂质体制剂相比,乳剂在前2小时内释放速率较快,之后速率较慢。显然,尽管前脂质体制剂由相似的成分制成,但乳剂与前脂质体制剂并不是生物等效的。
实施例8
罗哌卡因乳剂的稳定性
在该实施例中,如实施例1中所述的组合物中的罗哌卡因乳剂的长期稳定性数据提供在下表中:2-8℃下的稳定性
25℃下的稳定性
结论:本发明的乳剂在2-8℃和25℃下24个月内是稳定的。具体地,在2-8℃和25℃下24个月后,乳剂能够使罗哌卡因测定保持在标签宣称的90%至110%之间,N-氧化物在0.2%以下,罗哌卡因相关化合物A在0.01%以下,且总杂质在0.2%以下。
实施例9
罗哌卡因乳剂的粒度分析
在该实施例中,在不同的条件下测量与实施例1中所述相同的组合物(F-53)中的罗哌卡因乳剂的粒度。使用动态光散射光谱仪(Malvern Zetasizer Model Nano)测量用水稀释之前和之后的乳剂液滴的平均粒度(报告为直径的Z平均值)。粒度随水稀释比和制备方法有所不同,然而,所测量的粒度通过相同的样品制备方法保持一致。
结论:本发明的F-53乳剂的平均粒度为209nm。当用水稀释时,其粒度范围为约213nm至2504nm。
实施例10
罗哌卡因乳剂的电子显微镜成像
在该实施例中,使用Talos 120L TEM和负染色法,使用透射电子显微镜(TEM)检查了与如实施例1中所述的相同组合物中罗哌卡因乳剂的微观结构。为了比较,还对公知的脂质体药物
TEM图像显示于图4A-B中。本发明的乳剂中存在的各颗粒/液滴具有实心(即,没有空的内部空隙)并且不具有独特的膜。本发明的乳剂中存在的颗粒/液滴在结构上不同于脂质体颗粒,所述脂质体颗粒具有空心并且由独特的双层脂质膜围绕,参见图4C。
结论:本发明的乳剂含有约30nm至500nm的颗粒,并且水稀释的乳剂含有30-2500nm的可见颗粒,与实施例9中的动态光散射发现一致。本发明的乳剂在用水稀释之前或之后不含有脂质体颗粒。
实施例11
罗哌卡因乳剂的可注射性
在该实施例中,测量了与实施例1中所述的相同组合物中的罗哌卡因乳剂的可注射性。可注射性被定义为使用10mL塑料注射器通过皮下注射针头排出10mL乳剂所需的时间。
注射时间测量为以约或不大于25牛顿的力通过附接的针头或导管挤出10mL注射器中填充的10mL液体所需的时间。25牛顿的注射力被大多数医疗从业者认为是可接受的。典型的注射力测量为20-30N(http://www.sciencedirect.com/science/article/pii/S1098733903005741?via%3Dihub),并且注射时间为5-10分钟,并且18-25G针头被认为是可接受的(“腹腔镜袖状胃切除术中的TAP(TAP in Laparoscopic Sleeve Gastrectomy)”。https://videos.exparel.com/video-playlist/19)。下表提供了罗哌卡因乳剂在未经稀释或用水或乙醇稀释时记录的注射时间。
/>
结论:可以在少于7分钟内使用25牛顿力通过18-27G针头或22G导管从10mL塑料注射器注射十(10)毫升的罗哌卡因乳剂。
实施例12
罗哌卡因乳剂稀释后的粘度
在该实施例中,罗哌卡因乳剂(F-53)用水或生理盐水以不同的比率稀释,并测量经稀释乳剂的粘度:
结论:本发明的乳剂能够以2:1或更大的比率用盐水或者以4:1或更大的比率用水稀释,并且仍然维持其可注射性。
实施例13
稀释乳剂的体外释放
在该实施例中,将罗哌卡因乳剂(F-53)用盐水以2:1的体积比稀释(粘度=21厘泊),并测定其体外释放特性。体外释放方法涉及具有透析膜的USP溶出仪。
结论:本发明的未经稀释和盐水稀释的乳剂均在约3天或72小时内维持缓慢释放特性。盐水稀释的乳剂表现出相对低的粘度并且易于通过小针头注射。
实施例14
pH对罗哌卡因乳剂稳定性的效果
在该实施例中,检查了与实施例1所述的相同组合物中的乳剂中罗哌卡因的稳定性作为乳剂pH的函数。具有不同pH水平的多个样品以相同的碱罗哌卡因乳剂组合物制备。每个样品在121°下热应激60分钟。加热后的杂质生长汇总于下表中:
结论:在介于pH 4与pH 7之间的乳剂pH下观察到最佳的化学稳定性。然而,在pH 7下,乳剂似乎物理稳定较差。
实施例15
水对罗哌卡因乳剂稳定性的效果
在该实施例中,检查了与实施例1所述的相同组合物中的乳剂中罗哌卡因的物理稳定性作为乳剂中水含量的函数。使用相同的碱罗哌卡因乳剂组合物制备了多个具有不同含水量的样品。各个样品储存于2-8℃、25℃和40℃,并观察随时间推移的外观。发现汇总于下表中:
结论:本发明的乳剂以1%水作为转变点随着水含量的增加经历从油包水型到水包油型的相转变。低于该转变点,乳剂是半透明的并且基本上为油包水型。高于转变点,乳剂开始转变成水包油型。多至约3%水,乳剂大部分仍为油包水。在3%与10%之间,乳剂含有油包水和水包油两者,并且呈乳脂状。在10%以上,乳剂再次变成液体,但呈白色且不透明。水含量低于1%是优选的,因为这样的乳剂是半透明的(能够进行视觉检查)、稀液体(易于注射),并且在所有3个温度(2-8、25和40℃)下1.5年内是物理稳定的。
实施例16
一种或多种用于罗哌卡因乳剂的最佳抗氧化剂
在该实施例中,研究了EDTA和半胱氨酸对与实施例1相同的组合物中的罗哌卡因乳剂的稳定性的效果。用不同浓度的EDTA和L-半胱氨酸制备了相同碱组合物的多批乳剂。将乳剂储存于25℃和40℃以研究杂质(特别是罗哌卡因N-氧化物)的生长。下表汇总了发现:
结论:在不采用抗氧化剂的情况下,罗哌卡因乳剂形成了包括N-氧化物在内的多种杂质。当仅将EDTA钠(0.02%或0.04%)或仅将半胱氨酸(0.1%或0.2%)添加到罗哌卡因乳剂中时,N-氧化物停止形成,但其它杂质继续生长。当添加EDTA钠(0.02%或0.04%)和半胱氨酸(0.1%或0.2%)两者时,包括N-氧化物在内的所有杂质的生长均受到抑制。因此,EDTA钠(0.02%或0.04%)和半胱氨酸(0.1%或0.2%)的组合是本发明的乳剂的优选抗氧化剂。
在后续研究中,还向含有EDTA钠和半胱氨酸组合(0.02%+0.1%)的相同罗哌卡因乳剂中添加了如下额外的抗氧化剂:
每个样品均密封在玻璃小瓶中。在密封之前,将一个F-11小瓶的小瓶顶部空间用二氧化碳气体填充。所有小瓶均在121℃下加热2小时。通过HPLC测量加热之前和之后的杂质含量。N-氧化物的水平显示于下表中:
结论:额外的抗氧化剂(包括CO
实施例17
未经稀释乳剂中的粒度和分布
在该实施例中,使用Malvern Zetasizer,通过动态光散射(DLS)测量乳剂(F-53)以确定粒度和多分散指数(PDI)。
Z平均值以纳米(nm)报告,并定义为“通过DLS测量的颗粒集合的强度加权平均流体动力学尺寸(intensity weighted mean hydrodynamic size of the ensemblecollection of particles measured by DLS)”。多分散指数(PDI)是检测到的颗粒的尺寸非均匀性的量度。大于0.7的值表明样品具有非常广泛的尺寸分布。D(v.0.5)是体积中值直径D(v,0.5),即50%的分布高于该直径且50%低于该直径的直径。类似地,D(v,0.9)是90%的体积分布低于该值的直径。
结论:本发明的乳剂具有大于100nm的平均粒度以及如由高PDI值所表明的宽尺寸分布。
实施例18
用于制备乳剂的方法
步骤1.油相和水相的制备
将油和卵磷脂添加到不锈钢配混容器中。混合直至形成平滑糊状物(“油相”)。
将LA(罗哌卡因HCl)和其它赋形剂(如抗氧化剂)溶解在水中。混合以溶解固体,以获得澄清溶液(“水相”)。
步骤2.水包油乳剂的制备
使油相和水相以体积或重量比>9:1合并(即,最终含水量大于10%)。用匀化器混合形成平均油滴大于1微米的水包油(“粗乳剂”)。
步骤3.部分离子化/去离子化和pH调节
添加碱性物或碱如氢氧化钠,其量等同于步骤1中添加的LA(作为HCl盐)的约5-25摩尔%,以使离子化LA起始材料(罗哌卡因HCl)部分转化以形成非离子化罗哌卡因。如果使用非离子化起始材料如罗哌卡因游离碱,则添加足够的酸(如HCl)以使非离子化LA起始材料(罗哌卡因)部分转化,以形成75-95摩尔%的离子化罗哌卡因并调节乳剂的pH。在该步骤后,乳剂含有离子化和非离子化LA或罗哌卡因两者。为了调节乳剂pH,需要极少量(少于LA部分离子化/去离子化所需的15%)的酸或碱。
步骤4.继续均化直至水包油乳剂的平均液滴尺寸为100nm至2500nm,优选地200nm至1000nm并且最优选地200nm至400nm。对于本发明的乳剂,不期望使平均液滴尺寸低于100nm,因为平均液滴尺寸<100nm的乳剂随着尺寸快速增长而在物理上不稳定,具有快速释放LA的倾向(爆发释放),并且需要极长的均化时间(多天)来实现这样小的液滴。另一方面,大于2500nm的平均液滴尺寸也不是优选的,因为平均液滴尺寸超过2500nm的乳剂可能容易在步骤5期间分离成两相(例如,乳脂状出来),相对较粘稠(难以注射),并且不能通过0.2微米过滤器(步骤7)。平均尺寸为200nm至400nm的乳剂在物理上最稳定,提供了期望的LA释放速率,并且需要较短的处理时间(数小时内)。
步骤5.水移除
通过真空干燥移除步骤4乳剂包油中的水,以初始获得水含量>10%的水包油乳剂,然后是水含量介于3%与10%之间的水包油与油包水型两者的混合物、主要为水含量介于1%与3%之间的油包水,最终是水含量不超过1%的油包水乳剂。这些乳剂类型中的任一者均可用于提供长效且安全的LA药物。可以在该步骤中通过控制水含量来实现优选的乳剂类型的选择。
步骤6.稀释或粘度调节
根据需要,调节含水量和/或添加降粘剂(例如醇)以调节步骤5乳剂的粘度。
步骤7.无菌过滤和填充
将步骤6乳剂通过无菌0.2微米孔径过滤器以对乳剂灭菌,并将过滤的乳剂无菌填充并密封到玻璃小瓶或注射器中。
实施例19
在该实施例中,使用小角X射线散射(SAXS)来光学比较使用实施例18中描述的方法制备的罗哌卡因乳剂制剂与根据美国专利第9,849,088B2号的实施例1和2中描述的组合物和方法制备的前脂质体制剂(“前体脂质体”)的结构。测试样品的组成汇总于实施例7中。
SAXS使用具有转动阳极发生器和Highstar多线检测器的Bruker SAXS仪器进行。将样品安装在自动测角仪上的He室中。为了防止从空气散射,在收集每个样品之前将He气体吹扫到室中。数据在0.8至4.7°20的360°χ圆上以0.02度的宽度进行平滑处理和积分。
表示每个测试样品中重复和有序结构之间的平均间距或距离(以纳米或nm计)的d值或“晶格间距”根据布拉格定律计算并显示于图7中。
结论:本发明的乳剂的d值为3.76nm,而前脂质体制剂的d值为2.8nm,表明本发明的乳剂的平均颗粒间距离显著高于前体脂质体制剂。图7中的SAXS数据显示本发明的乳剂在结构上不同于前体脂质体制剂。
实施例20
在该实施例中,使用FTIR来光谱学比较使用实施例18中描述的方法制备的罗哌卡因乳剂制剂与根据美国专利第9,849,088B2号的实施例1和2中描述的组合物和方法制备的前脂质体制剂(“前体脂质体”)。
配备有DialPath传输液体取样器和Microlab PC软件的Agilent Cary 630FTIR用于数据采集。检测范围设定为4000至650(全范围),扫描为140,分辨率为4(cm^-1),且路径长度为50μm。每个样品均按原样或未稀释测量。
如图8A-B中所显示,FTIR检测到两个谱带;一个在3300cm
结论:本发明的乳剂在光谱学上与前体脂质体制剂不同。
实施例21
该实施例描述了本发明的乳剂组合物中其它酰胺局部麻醉剂的预言性组合物。每种组合物均可以使用与实施例18中描述的相同或相似的方法制备。
预期以上每种制剂均具有延长的LA药理活性、优异的安全性,以及如在本发明的罗哌卡因乳剂中观察到的期望的物理和化学特性(实施例1-20)。
根据前面的具体描述,本发明的修改和变化对于本领域技术人员会是明显的,并且旨在落入以下权利要求书的范围内。本文引用的所有参考文献的教导均明确援引加入。