一种鲸蜡基-PG羟乙基棕榈酰胺的合成方法
文献发布时间:2024-04-18 20:01:30
技术领域
本发明属于有机合成领域,具体涉及一种鲸蜡基-PG羟乙基棕榈酰胺的合成方法。
背景技术
鲸蜡基-PG羟乙基棕榈酰胺(简称“神经酰胺E”)是人工合成类神经酰胺,其具有显著的敏感肌屏障修护功效,能够改善皮肤屏障;还能增强表皮细胞内聚力和促进表皮的水合,提高皮肤的保水能力,适合应用于各类护肤化妆品中。
关于神经酰胺E的制备,已有部分文献报道,CN112321446A公开了一种酰胺衍生物的合成方法,其采用丙酮缩甘油制备丙酮缩甘油醚随后水解加入TsCl反应得到对甲苯磺酸酯衍生物,衍生物在脱去对甲苯磺酸得到缩水甘油醚,缩水甘油醚再与乙醇胺反应得到仲胺衍生物,仲胺衍生物再与烷基羧酸甲酯反应生成目标产品,但此方法合成出的神经酰胺总产率低,且纯度不高,操作繁琐,整体成本高;CN112441937A公开了一种使用烯丙基溴与十六醇先反应生成烯丙基烷基醚,再通过间氯过氧苯甲酸氧化得到缩水甘油醚,然后与乙醇胺、烷基羧酸甲酯反应得到产物的方法,但此方法在反应过程中使用氢化钠、间氯过氧苯甲酸等危险等级高,且腐蚀性强的反应助剂,反应过程较为危险,操作难度大,且副产品较多。CN112321445A是采用烷基醇与环氧氯丙烷反应制备缩水甘油醚,再与乙醇胺反应得到仲胺衍生物,仲胺衍生物与烷基羧酸甲酯反应生成目标产品;CN112441937A采用烯丙烯溴与烷基醇在强碱和催化剂作用下制备缩水甘油醚,缩水甘油醚随之胺解得仲胺衍生物,再与烷基羧酸甲酯反应得到酰胺衍生物。
上述合成神经酰胺E的路线均为先通过合成缩水甘油醚,缩水甘油醚再通过与乙醇胺缩合得到仲胺衍生物,仲胺衍生物再与烷基羧酸甲酯反应得到目标产品,但是,制备缩水甘油醚的工艺较为复杂,使用此方法合成出的神经酰胺总产率低、副产物较多、纯度低、且成本较高,不适合工业规模化生产。
因此,若能提供一种合成方法简单、神经酰胺总产率高、纯度高且成本低的神经酰胺E的合成方法,将会更适于工业化生产,对于本领域来说将是一种显著的进步。
发明内容
为了解决上述的技术问题,本发明提供了一种鲸蜡基-PG羟乙基棕榈酰胺的合成方法,第一步合成出的中间体与乙醇胺直接反应得到仲胺衍生物,仲胺衍生物再与酰氯化合物反应制备目标产物,避免了缩水甘油醚的制备,使用此方法合成的神经酰胺E的总产率高、纯度高且成本低,适于工业规模化生产。
本发明的技术方案如下:
一种鲸蜡基-PG羟乙基棕榈酰胺的合成方法,包括以下步骤:
(1)制备SN1中间体:将十六醇和氯丙二醇溶于有机溶剂,添加质子酸,反应得到SN1中间体;
(2)制备SN2仲胺衍生物:将SN1中间体与乙醇胺反应,得到SN2仲胺衍生物;
(3)制备鲸蜡基-PG羟乙基棕榈酰胺:SN2仲胺衍生物与棕榈酰氯进行反应,制得鲸蜡基-PG羟乙基棕榈酰胺。
优选的,所述合成方法具体包括以下步骤:
(1)制备SN1中间体:将十六醇和氯丙二醇溶于有机溶剂,添加质子酸,反应得到SN1中间体,如下反应所示:
(2)制备SN2仲胺衍生物:将SN1中间体与乙醇胺反应,得到SN2仲胺衍生物,SN2仲胺衍生物通过减压蒸馏将产品中剩余的乙醇胺脱除,得提纯后的SN2仲胺衍生物,如下反应所示:
(3)制备鲸蜡基-PG羟乙基棕榈酰胺:溶解提纯后的SN2仲胺衍生物,先添加少量的缚酸剂,然后滴加棕榈酰氯进行反应,将反应产物用有机溶剂重结晶2~3次,脱除杂质,制得鲸蜡基-PG羟乙基棕榈酰胺,如下反应所示:
优选的,步骤(1)所述溶剂为石油醚、苯、甲苯、二甲苯、吡啶等中的一种,所述质子酸为浓硫酸、磷酸、硝酸的至少一种;
优选的,步骤(1)中,所述质子酸的用量为十六醇摩尔量的0.3%~2%;十六醇与氯丙二醇的摩尔比为1:1~1:3;
优选的,步骤(1)中,反应温度为100~120℃,反应时间为4-6h;
优选的,步骤(2)乙醇胺与SN1中间体摩尔比为1:1~2:1;
优选的,步骤(2)中的反应溶剂为乙醇、二氯甲烷、二氯乙烷、石油醚中的一种;
优选的,步骤(2)中,反应温度为30~50℃,反应时间为10-12h;
优选的,步骤(3)中,所述缚酸剂为碳酸氢钠、碳酸钠、三乙胺、吡啶中的一种;溶解SN2仲胺衍生物所用溶剂为乙酸乙酯、二氯甲烷、二氯乙烷、正己烷中的一种;重结晶所用有机溶剂为乙酸乙酯、乙醇、正己烷、甲醇中的一种;
优选的,步骤(3)中,棕榈酰氯与SN2仲胺衍生物摩尔比为1:1~2:1;缚酸剂与棕榈酰氯摩尔比为0.5:1~1:1;
优选的,步骤(3)中,反应温度为10℃,反应时间为1~2h。
由于本发明涉及加热,因此,本发明步骤(1)中选用沸点相对高的石油醚、苯、甲苯、二甲苯、吡啶中的一种作为溶剂,同时,不建议使用二氯甲烷、甲醇、乙醇等溶剂沸点的溶剂。乙醇、二氯甲烷、二氯乙烷、石油醚与乙醇胺溶解性较好,容易脱除乙醇胺,降低乙醇胺的含量,因此,步骤(2)中选择乙醇、二氯甲烷、二氯乙烷、石油醚中的一种为溶剂。步骤(3)中选择乙酸乙酯、二氯甲烷、二氯乙烷、正己烷中的一种为溶剂,这几种溶剂在低温下对原料的溶解性较好,但对产品溶解性较差,便于后续的重结晶。
本发明对各步骤的反应时间及温度进行探究,发现:步骤(1)中,当反应温度大于120℃时,反应副产品含量增加,当温度小于100℃时,原料未完全反应;当反应时间小于4h时,存在原料残留,反应时间大于6h时,副反应增加;步骤(2)中,当反应温度小于30℃时,原料无法完全反应,当反应温度大于50℃时,反应会发生副反应,导致副产品含量增加;反应时间小于10h时,原料残留量较大,反应时间大于12h时,副产品含量增加;步骤(3)中,当反应温度小于10℃时,反应速度较慢,原料未完全反应,当反应温度大于10℃时,发生多种副反应,副产物增加;当反应时间小于1h时,原料还未完全反应,当反应时间大于2h时,反应产生的副产物增多,且副产物含量增加。因此,优选步骤(1)反应温度为100~120℃,反应时间为4-6h;步骤(2)反应温度为30~50℃,反应时间为10-12h;步骤(3)反应温度为10℃,反应时间为1~2h,此时所得神经酰胺E的总产率和纯度最高,杂质最少。
本发明进一步对各步骤所用量进行探究,发现:步骤(1)中,当十六醇与氯丙二醇的摩尔比大于1:3时,副产品种类及含量增多,当其摩尔比小于1:1时,原料未完全反应;步骤(2)中,乙醇胺与SN1中间体摩尔比小于1:1时,未完全反应,其摩尔比大于2:1时,会生成其他杂质,影响后续反应;步骤(3)中,当棕榈酰氯与SN2仲胺衍生物摩尔比大于2:1时,酰氯残留会导致产品发黄且呈黏稠状,反应过程中会生成副产品,影响反应结果,其摩尔比小于1:1时,产品收率低。因此,优选步骤(1)十六醇与氯丙二醇的摩尔比为1:1~1:3;步骤(2)乙醇胺与SN1中间体摩尔比为1:1~2:1;步骤(3)棕榈酰氯与SN2仲胺衍生物摩尔比为1:1~2:1,此时所得神经酰胺E的总产率和纯度最高,杂质最少。
本发明的有益效果在于:
(1)与现有神经酰胺E(鲸蜡基-PG羟乙基棕榈酰胺)合成工艺相比,使用本发明的合成方法制备的神经酰胺E(鲸蜡基-PG羟乙基棕榈酰胺)收率和收率均得到了大幅提高,产物总收率在75%以上,纯度达到了99.5%。
(2)本发明的合成工艺路线简单,生产周期短,且原料易得,成本低,经济效益高,各步反应副产物含量较低,更适于工业化生产。
(3)本发明的整个合成工艺中不存在较高压力和较高温度,过程中催化剂、溶剂为常规试剂,安全性强,可操作性高,反应在常规反应釜内即可进行。
附图说明
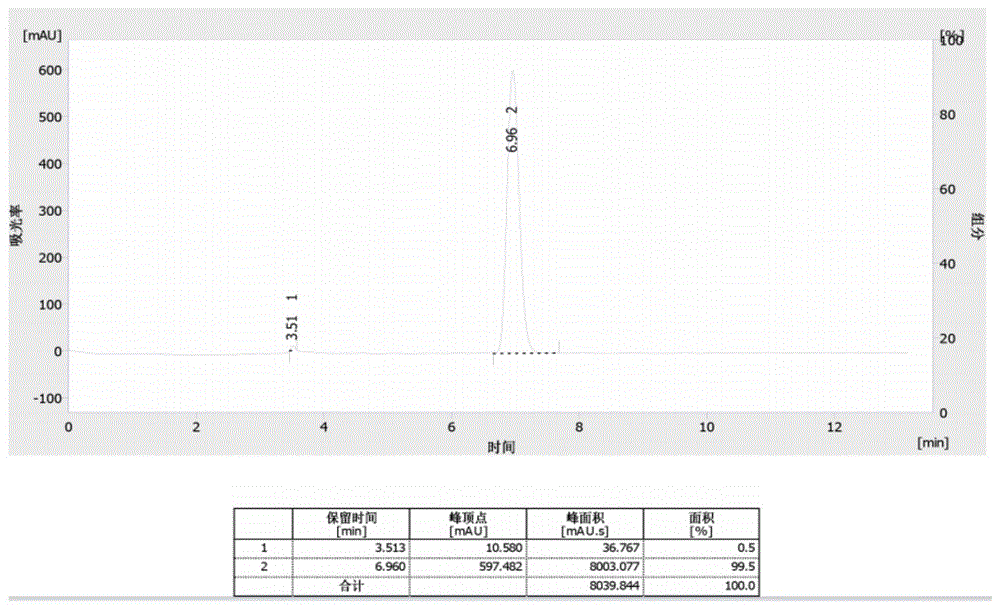
图1为实施例1提纯后目标产品的液相色谱图;
图2为对比例4提纯后目标产品的液相色谱图;
图3为对比例5提纯后目标产品的液相色谱图;
图4为对比例11按照传统工艺生产的目标产品的液相色谱图。
具体实施方式
为了能使本领域技术人员更好的理解本发明,现结合具体实施方式对本发明进行更进一步的阐述。
实施例1
一种鲸蜡基-PG羟乙基棕榈酰胺的合成方法,包括以下步骤:
(1)向500mL三口烧瓶中添加十六醇0.1mol(24.24g)、石油醚50mL、浓硫酸0.001mol(0.13g),搅拌升温至100℃,待物料全部溶解,溶液呈无色透明时,缓慢滴加氯丙二醇0.2mol(22.00g),反应5.5h后,TLC检测,无十六醇显像,停止反应将石油醚旋蒸脱除,得到SN1中间体(1-氯-3-(十五烷氧基)-2-丙醇),中间体含量约为95%以上。
(2)将SN1中间体0.095mol(25.12g),加入至500mL三口烧瓶中,加入50mL二氯乙烷,搅拌升温至45℃,开始滴加乙醇胺0.2mol(12.21g),滴加完毕后,反应11h,减压蒸干,得到白色固体得到SN2仲胺衍生物,含量在90%以上,乙醇胺含量在3%以下。
(3)将0.09mol(32.31g)SN2仲胺衍生物加入500mL三口烧瓶中,加入100ml二氯乙烷、三乙胺0.09mol(9.09g)搅拌降温至10℃,样品溶解后,滴加棕榈酰氯0.09mol(24.74g),溶液中出现白色固体,反应2h抽滤,固体采用甲醇进行重结晶,得到神经酰胺E,总产率在77%,纯度在99.5%。
实施例2
与实施例1的不同之处在于:步骤(1)中,使用二甲苯作为有机溶剂;步骤(2)中,使用二氯甲烷作为反应溶剂;
步骤(3)为:将0.09mol(32.21g)SN2仲胺衍生物加入500mL三口烧瓶中,加入50ml二氯甲烷,搅拌降温至10℃,样品溶解后,滴加棕榈酰氯0.09mol(24.74g)反应1h后滴加三乙胺0.09mol(9.09g),溶液中出现白色固体,反应1h抽滤,固体采用甲醇进行重结晶,得到神经酰胺E,总产率在75%,纯度在99.2%。
实施例3
与实施例1的不同之处在于:步骤(1)中,质子酸为浓硝酸,反应时间为6h;步骤(2)中,反应时间为10h;
步骤(3)为:将0.09mol(32.21g)SN2仲胺衍生物加入500mL三口烧瓶中,加入50ml二氯甲烷,搅拌降温至10℃,样品溶解后,滴加棕榈酰氯0.09mol(24.74g)反应1h后滴加碳酸氢钠0.09mol(7.56g),溶液中出现白色固体,反应1h抽滤,固体采用甲醇进行重结晶,得到神经酰胺E,总产率在57%,纯度在98.5%。
实施例4
与实施例1的不同之处在于:步骤(1)中,反应温度为110℃。
神经酰胺E总产率在60%,纯度在99.1%。
实施例5
与实施例1的不同之处在于:步骤(1)中,反应温度为120℃。
神经酰胺E总产率在59%,纯度在99.1%。
实施例6
与实施例1的不同之处在于:步骤(1)中,反应时间为4h。
神经酰胺E总产率在65%,纯度在99.0%。
实施例7
与实施例1的不同之处在于:步骤(1)中,十六醇与氯丙二醇的摩尔比为:1:1。
神经酰胺E总产率在61%,纯度在98.5%。
实施例8
与实施例1的不同之处在于:步骤(2)中,乙醇胺与SN1中间体摩尔比为1:1。
神经酰胺E总产率在59%,纯度在99.3%。
实施例9
与实施例1的不同之处在于:步骤(2)中,反应时间为10h。
神经酰胺E总产率在65%,纯度在99.3%。
实施例10
与实施例1的不同之处在于:步骤(3)中,棕榈酰氯与SN2仲胺衍生物摩尔比为:1.5:1。
神经酰胺E总产率在67%,纯度在98.5%。
对比例1
合成方法同实施例1,不同之处在于,步骤(1)的反应时间为2h。神经酰胺E的总产率为55%,纯度为99.5%。
对比例2
合成方法同实施例1,不同之处在于,步骤(1)的反应时间为7h。神经酰胺E的总产率为53%,纯度为99.5%。
对比例3
合成方法同实施例1,不同之处在于,步骤(1)中十六醇与氯丙二醇的摩尔比为1:2.5。神经酰胺E的总产率为51%,纯度为99.2%。
对比例4
合成方法同实施例1,不同之处在于,步骤(1)中反应温度为130℃。神经酰胺E的总产率为45%,纯度为98.9%。
对比例5
合成方法同实施例1,不同之处在于,步骤(2)的反应时间为14h。神经酰胺E的总产率为46%,纯度为98.6%。
对比例6
合成方法同实施例1,不同之处在于,步骤(2)中乙醇胺与SN1中间体摩尔比为3:1。神经酰胺E的总产率为50%,纯度为99.0%。
对比例7
合成方法同实施例1,不同之处在于,步骤(2)中反应温度为55℃。神经酰胺E的总产率为52%,纯度为99.0%。
对比例8
合成方法同实施例1,不同之处在于,步骤(3)中,棕榈酰氯与SN2仲胺衍生物摩尔比为1:2。神经酰胺E的总产率为47%,纯度为98.5%。
对比例9
合成方法同实施例1,不同之处在于,步骤(3)中,棕榈酰氯与SN2仲胺衍生物摩尔比为3:1。神经酰胺E的总产率为40%,纯度为99.1%。
对比例10
合成方法同实施例1,不同之处在于,步骤(3)中,反应时间为3h。神经酰胺E的总产率为47%,纯度为98.5%。
对比例11
按照传统方法,先制备缩水甘油醚,再进一步制备鲸蜡基-PG羟乙基棕榈酰胺,具体合成方法如下:
步骤一:向三口瓶中加入十六醇0.1mo1(24.2g)及石油醚120mL,升温35℃,搅拌溶解;再加入四丁基溴化铵0.0005mol(0.16g),NaOH0.2mol(8.000g);搅拌片刻,滴加环氧氯丙烷0.15mol(13.8g)。滴毕,继续反应约8小时,反应结束。过滤,滤饼少量石油醚洗涤,合并滤液,减压蒸干即得淡黄色油状粗品,该缩水甘油醚不需纯化直接备用下一步。
步骤二:取上述全部缩水甘油醚粗品,加入乙醇300mL,乙醇胺0.2mol(18.32g)室温搅拌,约14小时反应完全。减压蒸干,得到白色固体。石油醚重结晶得到仲胺衍生物粗品,不需要进一步纯化直接备用下一步。步骤三:取上述所有仲胺衍生物粗品,将KOH 0.01mo1(0.561g)加入到圆底烧瓶中,再加入100mL乙醇,升温80℃搅拌溶解;随后,减压蒸除乙醇。再次升温至80℃,减压下缓慢滴加0.12mol(32.400g)棕榈酸甲酯,约3小时滴毕;继续反应4小时,停止反应。正已烷/乙醇重结晶,得到神经酰胺E,总产率40%,纯度98%。
实施例1-10与对比例1-11制备得到的神经酰胺E的总产率与纯度如表1所示。
表1不同条件下神经酰胺E的总产率与纯度
由表1可知,实施例1所制备的神经酰胺E的总收率和纯度最高,相较于使用传统方法制备神经酰胺E的总收率,其收率将近是传统方法收率的2倍,纯度相较于传统方法所得神经酰胺E的纯度也有提高;所用的溶剂、原料之间的摩尔比以及反应时间的温度和时间均会对最终的总收率产生较大的影响,当反应温度增加至本发明所限最高温度以上时,反应副产品含量增加,其总收率减小,而当反应温度减小至本发明所限最低温度以下时,会导致原料无法完全反应进而总收率也相应减小;当反应时间增加至本发明所限最高时间以上时,副产品含量会相应增加,当反应时间缩短至本发明所限最低时间以下时,原料残留量较大进而总收率相应减小;当十六醇与氯丙二醇的摩尔比大于1:3时,副产品种类及含量增多,当其摩尔比小于1:1时,原料未完全反应;乙醇胺与SN1中间体摩尔比小于1:1时,未完全反应,其摩尔比大于2:1时,会生成其他杂质,影响后续反应;当棕榈酰氯与SN2仲胺衍生物摩尔比大于2:1时,酰氯残留会导致产品发黄且呈黏稠状,反应过程中会生成副产品,影响反应结果,其摩尔比小于1:1时,产品收率低。即,在本发明的温度、时间、摩尔比的限定范围内可以得到神经酰胺E的最高总收率和纯度。
试验例
使用液相色谱对实施例1、对比例4、对比例5、对比例11的目标产品进行测定,其目标产品的谱图分别如图1-图4所示,图1即实施例1仅有两个出峰,第一个出峰为副产物,其含量为0.5%,第二个出峰为目标产物即神经酰胺E,其含量为99.5%,可知副产物很少;图2即对比例4有三个出峰,第一个和第二个出峰为副产物,其含量分别为0.5%、0.8%,第三个出峰为目标产物即神经酰胺E,其含量为98.6%,可知,反应温度的升高会导致副产物的增多,使神经酰胺E的总产率降低;图3即对比例5有三个出峰,第一个和第二个出峰为副产物,其含量分别为0.5%、0.6%,第三个出峰为目标产物即神经酰胺E,其含量为98.9%,可知,反应时间的延长同样会导致副产物的增多,使神经酰胺E的总产率降低;图4即对比例11有四个出峰,第一个、第二个和第三个出峰为副产物,其含量分别为0.2%、1.5%、0.3%,第三个出峰为目标产物即神经酰胺E,其含量为98%,可见,使用传统方法制备神经酰胺E时的副产物较多,影响整体收率和纯度,其总收率较低。
- 一种用于建筑铺设的电力电缆转弯半径测量工器具
- 一种电力电缆转弯半径测量工器具及其测量方法