用于治疗肿瘤的抗-PD-L1结合物
文献发布时间:2023-06-19 10:29:05
本申请是申请号为201410440824.0、申请日为2014年9月1日、发明名称为“用于治疗肿瘤的抗-PD-L1结合物”的发明专利申请的分案申请。
技术领域
本发明涉及用于靶向免疫疗法的抗-PD-L/PD-1轴抗体结合物以及含有所述结合物的组合物。本发明还涉及本发明的多种化合物在治疗诸如癌症之类的疾病方面的应用。
背景技术
治疗性抗体已用于临床应用二十多年。目前已有十七种抗肿瘤抗体药物用于临床,这些药物包括:美罗华(Rituxan(1997)),赫赛汀(Herceptin(1998)),Mylotarg(2000),Campath(2001),Zevalin(2002),Bexxer(2003),Avastin(2004),Erbitux(2004),Vectibix(2006);Arzerra(2009);Benlysta(2011);Yervoy(2011);Adcetris(2011);Perjeta(2012);Kadcyla(2013);Cyramza(2014),和Sylvant(2014)。这些抗体主要靶定四种分子:EGFR、Her2、CD20和VEGF。
总体而言,治疗性抗体通过三种机制(Scott AM,Wolchok JD,Old LJ.Antibodytherapy of cancer.Nat Rev Cancer.(2012),12:278-87)杀伤肿瘤细胞:(1)抗体直接作用,也就是阻断或激动配体/受体信号转导活性,诱导细胞凋亡并递送药物或细胞毒素剂。抗体受体活化活性可产生直接杀伤肿瘤细胞的作用。例如,一些抗体可与肿瘤细胞表面的受体结合,活化受体,导致细胞凋亡(例如,在线粒体中)。抗体还可通过受体拮抗活性介导肿瘤细胞杀伤。例如,一些抗体可与细胞表面受体结合并阻断二聚化作用、激酶活化作用以及下游信号转导,从而抑制增殖并促进细胞凋亡。抗体与酶的结合可导致中和作用、信号阻断以及细胞死亡。(2)免疫介导的细胞杀伤机制,该机制包括补体依赖性细胞毒性(CDC)、抗体依赖性细胞介导的细胞毒性(ADCC)、T细胞功能调节,等等。免疫介导的肿瘤细胞杀伤可通过如下方式完成:诱导吞噬作用、活化补体、抗体依赖性细胞介导的细胞毒性、通过单链可变片段(scFv)使基因修饰的T细胞靶定肿瘤,通过树突细胞的抗体介导的抗原交叉呈递活化T细胞、抑制T细胞抑制性受体(例如,细胞毒性T淋巴细胞相关抗原4(CTLA4))。其中,抗体的Fc部分的特性对于CDC和ADCC介导的肿瘤细胞杀伤作用尤为重要。(3)抗体对肿瘤脉管系统和基质的特异性效应,通过捕获血管受体拮抗剂或配体诱导血管和基质细胞消融,包括:抑制基质细胞、将毒素递送至基质细胞以及将毒素递送至脉管系统(Scott AM,WolchokJD,Old LJ.Antibody therapy of cancer.Nat Rev Cancer.2012,12(4):278-87)。
治疗性单克隆抗体药物推进了抗癌药物的研究和开发。然而,仍存在一些问题需要进一步研究解决,例如,抗体的免疫原性、长期使用肿瘤靶点的耐受性以及单纯地单一阻断信号转导通路的长期作用。简言之,大多数抗体难以实现对肿瘤细胞长期有效的抑制和杀伤作用。
1964年,“自然”杂志报道了抗体-药物结合(ADC)技术这一新观点,该观点近年来得到突破性发展。ADC使抗体与高毒性药物(毒素)通过化学连接体(连接体)共价连接。抗体识别癌细胞表面抗原分子,内吞作用将ADC带入细胞质内,具体而言,连接体水解之后释放的细胞内环境毒素杀伤细胞。
Seattle Genetics已研发了Brentuximab Vedotin(商品名为Adcetris)这种药物,其已被FDA批准上市。其为单甲基auristatin E(MMAE),一种合成的毒性抗癌药物,其与靶向淋巴瘤细胞特异性CD30分子的抗体连接,具有改善的杀伤肿瘤细胞的效用。
目前,已对几十种这样的ADC药物开展了临床试验。其中,Genentech和Immunogen联合开发了用于治疗乳腺癌的与美登素(maytansine)结合的曲妥珠单抗,一种名为ado-曲妥珠单抗emtansine的药物(Kadcyla),其也被称为T-DM1。2013年2月,FDA已批准T-DM1用于人表皮生长因子受体2(Her2)-阳性转移性乳腺癌。美登素是一种小分子毒素,其可与微管蛋白结合并可通过形成非还原性双-马来酰亚胺-丙二醇复合物防止微管形成。曲妥珠单抗通过靶向人Her2对乳腺癌和胃癌起作用。曲妥珠单抗已被批准用于Her2-阳性癌症。然而,曲妥珠单抗无法促进所有的Her2-阳性细胞的细胞凋亡。T-DM1使选择性靶向Her2受体的曲妥珠单抗与有效的细胞毒性剂美登素结合,从而杀伤肿瘤细胞。T-DM1抗体结合Her2受体,导致从结合物中释放的美登素产生细胞内在化作用,从而杀伤肿瘤细胞。T-DM1具有更好的整体疗效、药代动力学性质以及较低的毒性。
传统的小分子化疗药物具有很强的毒性和药代动力学优势,但是在治疗肿瘤的过程中传统的小分子化疗药物可影响其他生理靶标,产生严重的副作用。抗体-药物结合物使靶向作用和具有特定的药代动力学的小分子药物结合。抗体-药物结合物的结构为具有靶向功能的单克隆抗体与具有特定的药理学性质的化合物的连接。这种技术需要治疗性抗体与靶标特异性结合,与诸如细胞毒素之类的具有治疗作用或其他功能的分子结合。诸如结合的抗体的内吞作用、结合的稳定性以及毒素的释放和杀伤活性之类的许多因素影响这种类型的抗体的作用。
目前正在使用的毒素分子包括微管蛋白抑制剂Auristatin类似物单甲基auristatin E、单甲基auristatin F和美登素。单甲基auristatin E为合成的微管聚合物抑制剂,其可抑制微管聚集,干扰肿瘤细胞有丝分裂并且可诱导细胞凋亡(Naumovski Land Junutula JR.Glembatumumab vedotin,a conjugate of an anti-glycoproteinnon-metastatic melanoma protein B mAb and monomethyl auristatin E fortreatment of melanoma and breast cancer.Curr Opin Mol Ther2003;12(2):248-57.Francisco JA,Cerveny CG等人,cAC10-vcMMAE,an anti-CD30-monomethylauristatin E conjugate with potent and selective antitumor activity.Blood 102(4):1458-65)。单甲基auristatin F为抗有丝分裂Auristatin衍生物,在C末端具有带电荷的苯丙氨酸残基。与不带电荷的MMAE相比,单甲基auristatin F最小化对细胞信号通路的破坏并且最小化细胞毒性。大量CD30细胞测试发现mAb-马来酰亚胺己酰基-缬氨酸-瓜氨酸-p-氨基苄氧基羰基-MMAF(mAb-L1-MMAF)的毒性比单独的MMAF的毒性强2,200倍(Doronina SO等人,Enhanced activity of monomethylauristatin F throughmonoclonal antibody delivery:effects of linker technology on efficacy andtoxicity.Bioconjug Chem,2006;17(1):p114-24)。美登素是一种抗有丝分裂剂,其充当微管蛋白聚合的抑制剂,干扰细胞核内的微管的形成。美登素还抑制DNA、RNA和蛋白质合成,已经发现美登素对于DNA合成的影响最大。
抗体-药物结合物具有直接和间接抗癌作用。抗体阻断或活化配体/受体信号转导,诱导细胞凋亡,并且同时抗体可直接或间接地向肿瘤细胞呈递或递送有效载荷药物(例如,药物、毒素、小干扰RNA或放射性同位素)。治疗性抗体药物结合物使用抗体和结合的药物的双重特性,第一为与靶标分子特异性结合的结合功能,第二为抗体自身的肿瘤细胞杀伤功能,以及第三为结合的药物的特定作用。目前使用的抗体-药物结合药物限于如何直接杀伤肿瘤细胞。然而,由于在抗体、连接体分子、毒素分子、结合方面的严格的技术要求以及能够将毒素带入肿瘤微环境内的分子有限,在实际的临床研究中仍然存在一些难题。
程序性死亡1(PD-1)是CD28家族受体的成员,其包括CD28、CTLA-4、ICOS、PD-1和BTLA。该家族的最初成员CD28和ICOS通过在功能上影响加入单克隆抗体之后T细胞增殖的提高而被发现。PD-1的两个细胞表面糖蛋白配体PD-L1和PD-L2已被识别,并且这两个细胞表面糖蛋白配体已表现出在与PD-1结合之后下调T细胞的活化作用和细胞因子的分泌。PD-L1(B7-H1)和PD-L2(B7-DC)这两者是B7同系物,该B7同系物与PD-1结合。PD-L1还对共刺激分子B7-1具有明显的亲和性。在IFN-γ刺激之后,PD-L1在T细胞、NK细胞、巨噬细胞、髓样DC、B细胞、上皮细胞以及血管内皮细胞上表达。PD-L1基因启动子区域具有针对IRF-1(干扰素调节因子)的响应元件。I型干扰素还可上调鼠类肝细胞、鼠类单核细胞、鼠类DC和鼠类肿瘤细胞上的PD-L1。
已在几种鼠类癌症和包括人肺癌、人卵巢癌和人结肠癌以及各种不同的骨髓瘤在内的人类癌症中发现了PD-L1的表达。PD-L1的上调看起来可使癌症逃脱宿主免疫系统。在对患有肾细胞癌的患者的肿瘤标本的分析中发现,肿瘤中PD-L1的高表达与肿瘤侵袭性增强以及死亡风险提高4.5倍有关。具有较高的PD-L1表达的卵巢癌患者相对于那些具有低PD-L1表达的卵巢癌患者而言具有明显较差的预后。PD-L1表达与上皮内CD8+T-淋巴细胞计数呈负相关,这说明肿瘤细胞上的PD-L1可抑制抗肿瘤CD8+T细胞。
发明内容
一方面,本发明提供一种具有下式(I)的结构的化合物或其药学上可接受的盐或溶剂化物,
TM-L-AM (I),
其中,TM是作为PD-L/PD-1轴拮抗剂(Axis antagonist)的抗体或其功能片段,AM是活化部分,其由下式(II)的结构表示:
其中,虚线表示存在化学键或不存在化学键,
X是S或-NR
W
W
W
W
W
Z是氢,烷基,烯基,炔基,烷氧基,芳基,卤代烷基,杂芳基,杂环基,它们中的每一个可被一个或一个以上选自下列基团的取代基任选地取代:羟基,烷氧基,烷基,烯基,炔基,芳基,杂芳基,杂环基,卤素,氰基,硝基,--N(R
R为氢,烷基,烷氧基,卤代烷基,卤素,芳基,杂芳基,杂环基,它们中的每一个被一个或一个以上选自下列基团的取代基任选地取代:羟基,烷氧基,烷基,烯基,炔基,环烷基,芳基,杂芳基,杂环基,--NH
n为0,1,2,3或4;
Y为–NR
其中,R
任选地,X和Z一同可形成5元至9元环。
在一些实施方式中,AM是式(II)的化合物,其选自表1中的化合物,包括:
2-丙基噻唑并[4,5-c]喹啉-4-胺,
1-(2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-氨基-2-(乙氧基甲基)-a,a-二-甲基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-(4-氨基-2-乙基氨基甲基咪唑并-[4,5-c]喹啉-1-基)-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基-]甲磺酰胺,
4-氨基-2-乙氧基甲基-aa-二甲基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1-乙醇,
4-氨基-aa-二甲基-2-甲氧基乙基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-{2-[3-(苄氧基)丙氧基]乙基}-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-丁基脲,
N1-[2-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)乙基]-2-氨基-4-甲基戊酰胺,
N-(2-{2-[4-氨基-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-苯基脲,
1-(2-氨基-2-甲基丙基)-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
1-{4-[(3,5-二氯苯基)磺酰基]丁基}-2-乙基-1H-咪唑并[4,5-c]喹啉-4-胺,
N-(2-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-环己基脲,
N-{3-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]丙基}-n'-(3-氰基苯基)硫脲,
N-[3-(4-氨基-2-丁基-1H-咪唑并[4,5-c]喹啉-1-基)-2,2-二甲基丙基]苯甲酰胺,
2-丁基-1-[3-(甲基磺酰基)丙基]-1H-咪唑并[4,5-c]喹啉-4-胺,
N-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]-1,1-二甲基乙基}-2-乙氧基乙酰胺,
1-[4-氨基-2-乙氧基甲基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
1-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-{3-[4-氨基-1-(2-羟基-2-甲基丙基)-2-(甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-7-基]苯基}甲磺酰胺,
1-[4-氨基-7-(5-羟基甲基吡啶-3-基)-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
3-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]丙-1,2-二醇,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-丙基脲,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-环戊基脲,
1-[(2,2-二甲基-1,3-二氧戊环-4-基)甲基]-2-(乙氧基甲基)-7-(4-羟基甲基苯基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-[4-氨基-2-乙氧基甲基-1-(2-羟基-2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-7-基]-N-甲氧基-N-甲基苯甲酰胺,
2-乙氧基甲基-N1-异丙基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1,4-二胺,
1-[4-氨基-2-乙基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基]甲磺酰胺,或
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-环己基脲。
在一些实施方式中,L由下式(III)的结构表示:
m为1,2,3,4,5或6,b分别独立地为0或1,并且D由下式(IV)的结构独立地表示:
其中,i分别独立地为0或1;
j分别独立地为0,1,2,3,4,5或6;
A分别独立地为S,O或N-Ra,其中,Ra为氢,烷基,烯基或烷氧基;
B分别独立地为烷基,烯基,--O-烷基--,--烷基-O--,--S-烷基--,--烷基-S--,芳基,杂芳基,杂环基或肽,它们中的每一个被一个或一个以上选自下列基团的取代基任选地取代:羟基,烷氧基,烷基,烯基,炔基,环烷基,--烷基-芳基,--烷基-杂芳基,--烷基-杂环基,--O-R
在一些实施方式中,连接体由下式(V)至(VII)的结构表示:
A、B、i和j为上文中所定义的。
在一些实施方式中,所述连接体选自:S1、S2、S3、S4、S5、S6、S7、–Gly-Phe-Leu-Gly-、-Ala-Leu-Ala-Leu-、-Phe-Arg-、-Phe-Lys-、-Val-Lys-、-Val-Ala-或Val-Cit-,其中,S1至S7由下述结构表示:
其中,m分别独立地为1至20。
在一些实施方式中,TM是抗-PD-L1抗体,其选自:YW243.55.S70,MPDL3280A、MEDI-4736、BMS-936559和MSB0010718C。
在一些实施方式中,TM是抗-PD-1抗体,其选自:MK-3475,AMP-514,AMP-224,BMS-936558和CT-011。
在一些实施方式中,TM是抗-PD-L1抗体、抗-PD-L2抗体或抗-PD-1抗体,L选自:S1、S2、S3、S4、S5、S6、S7、–Gly-Phe-Leu-Gly-、-Ala-Leu-Ala-Leu-、-Phe-Arg-、-Phe-Lys-、-Val-Lys-、-Val-Ala-或Val-Cit-;AM是式(II)的化合物,其选自表1中的化合物,包括:
2-丙基噻唑并[4,5-c]喹啉-4-胺,
1-(2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-氨基-2-(乙氧基甲基)-a,a-二-甲基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-(4-氨基-2-乙基氨基甲基咪唑并-[4,5-c]喹啉-1-基)-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基-]甲磺酰胺,
4-氨基-2-乙氧基甲基-aa-二甲基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1-乙醇,
4-氨基-aa-二甲基-2-甲氧基乙基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-{2-[3-(苄氧基)丙氧基]乙基}-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-丁基脲,
N1-[2-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)乙基]-2-氨基-4-甲基戊酰胺,
N-(2-{2-[4-氨基-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-苯基脲,
1-(2-氨基-2-甲基丙基)-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
1-{4-[(3,5-二氯苯基)磺酰基]丁基}-2-乙基-1H-咪唑并[4,5-c]喹啉-4-胺,
N-(2-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-环己基脲,
N-{3-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]丙基}-n'-(3-氰基苯基)硫脲,
N-[3-(4-氨基-2-丁基-1H-咪唑并[4,5-c]喹啉-1-基)-2,2-二甲基丙基]苯甲酰胺,
2-丁基-1-[3-(甲基磺酰基)丙基]-1H-咪唑并[4,5-c]喹啉-4-胺,
N-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]-1,1-二甲基乙基}-2-乙氧基乙酰胺,
1-[4-氨基-2-乙氧基甲基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
1-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-{3-[4-氨基-1-(2-羟基-2-甲基丙基)-2-(甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-7-基]苯基}甲磺酰胺,
1-[4-氨基-7-(5-羟基甲基吡啶-3-基)-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
3-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]丙-1,2-二醇,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-丙基脲,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-环戊基脲,
1-[(2,2-二甲基-1,3-二氧戊环-4-基)甲基]-2-(乙氧基甲基)-7-(4-羟基甲基苯基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-[4-氨基-2-乙氧基甲基-1-(2-羟基-2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-7-基]-N-甲氧基-N-甲基苯甲酰胺,
2-乙氧基甲基-N1-异丙基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1,4-二胺,
1-[4-氨基-2-乙基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基]甲磺酰胺,或
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-环己基脲,
其中,喹啉环上的胺基团是与连接体连接的连接点。
在一些实施方式中,TM是抗-PD-L1抗体,其选自:YW243.55.S70、MPDL3280A、MEDI-4736、BMS-936559和MSB0010718C。
在一些实施方式中,TM是抗-PD-L1抗体,其选自:YW243.55.S70、MPDL3280A、MEDI-4736、BMS-936559和MSB0010718C,AM是瑞喹莫德或咪喹莫特,其中,喹啉环上的胺基团是与连接体连接的连接点。
在一些实施方式中,TM是抗-PD-1抗体,其选自:MK-3475,AMP-514,AMP-224,BMS-936558和CT-011。
在一些实施方式中,TM是抗-PD-1抗体,其选自:MK-3475,AMP-514,AMP-224,BMS-936558和CT-011;AM是瑞喹莫德或咪喹莫特,其中,喹啉环上的胺基团是与连接体连接的连接点。
另一方面,本发明提供具有下式A至下式C的结构的化合物或其药学上可接受的盐或溶剂化物:
在一些实施方式中,式(II)的化合物是瑞喹莫德或咪喹莫特,其中,喹啉环上的胺基团是与连接体连接的连接点。
在一些实施方式中,PD-L/PD-1轴拮抗剂是抗-PD-L1抗体,其选自:YW243.55.S70、MPDL3280A、MEDI-4736、BMS-936559和MSB0010718C。
在一些实施方式中,PD-L/PD-1轴拮抗剂是抗-PD-L1抗体,其选自:YW243.55.S70、MPDL3280A、MEDI-4736、BMS-936559和MSB0010718C;式(II)的化合物选自表1中的化合物,包括:
2-丙基噻唑并[4,5-c]喹啉-4-胺,
1-(2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-氨基-2-(乙氧基甲基)-a,a-二-甲基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-(4-氨基-2-乙基氨基甲基咪唑并-[4,5-c]喹啉-1-基)-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基-]甲磺酰胺,
4-氨基-2-乙氧基甲基-aa-二甲基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1-乙醇,
4-氨基-aa-二甲基-2-甲氧基乙基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-{2-[3-(苄氧基)丙氧基]乙基}-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-丁基脲,
N1-[2-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)乙基]-2-氨基-4-甲基戊酰胺,
N-(2-{2-[4-氨基-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-苯基脲,
1-(2-氨基-2-甲基丙基)-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
1-{4-[(3,5-二氯苯基)磺酰基]丁基}-2-乙基-1H-咪唑并[4,5-c]喹啉-4-胺,
N-(2-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-环己基脲,
N-{3-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]丙基}-n'-(3-氰基苯基)硫脲,
N-[3-(4-氨基-2-丁基-1H-咪唑并[4,5-c]喹啉-1-基)-2,2-二甲基丙基]苯甲酰胺,
2-丁基-1-[3-(甲基磺酰基)丙基]-1H-咪唑并[4,5-c]喹啉-4-胺,
N-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]-1,1-二甲基乙基}-2-乙氧基乙酰胺,
1-[4-氨基-2-乙氧基甲基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
1-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-{3-[4-氨基-1-(2-羟基-2-甲基丙基)-2-(甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-7-基]苯基}甲磺酰胺,
1-[4-氨基-7-(5-羟基甲基吡啶-3-基)-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
3-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]丙-1,2-二醇,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-丙基脲,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-环戊基脲,
1-[(2,2-二甲基-1,3-二氧戊环-4-基)甲基]-2-(乙氧基甲基)-7-(4-羟基甲基苯基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-[4-氨基-2-乙氧基甲基-1-(2-羟基-2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-7-基]-N-甲氧基-N-甲基苯甲酰胺,
2-乙氧基甲基-N1-异丙基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1,4-二胺,
1-[4-氨基-2-乙基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基]甲磺酰胺,或
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-环己基脲,
其中,喹啉环上的胺基团是与连接体连接的连接点。
在一些实施方式中,PD-L/PD-1轴拮抗剂是抗-PD-1抗体,其选自:MK-3475,AMP-514,AMP-224,BMS-936558和CT-011。
在一些实施方式中,PD-L/PD-1轴拮抗剂是抗-PD-1抗体,其选自:MK-3475,AMP-514,AMP-224,BMS-936558和CT-011;式(II)的化合物是选自表1中的化合物,包括:
2-丙基噻唑并[4,5-c]喹啉-4-胺,
1-(2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-氨基-2-(乙氧基甲基)-a,a-二-甲基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-(4-氨基-2-乙基氨基甲基咪唑并-[4,5-c]喹啉-1-基)-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基-]甲磺酰胺,
4-氨基-2-乙氧基甲基-aa-二甲基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1-乙醇,
4-氨基-aa-二甲基-2-甲氧基乙基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-{2-[3-(苄氧基)丙氧基]乙基}-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-丁基脲,
N1-[2-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)乙基]-2-氨基-4-甲基戊酰胺,
N-(2-{2-[4-氨基-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-苯基脲,
1-(2-氨基-2-甲基丙基)-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
1-{4-[(3,5-二氯苯基)磺酰基]丁基}-2-乙基-1H-咪唑并[4,5-c]喹啉-4-胺,
N-(2-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-环己基脲,
N-{3-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]丙基}-n'-(3-氰基苯基)硫脲,
N-[3-(4-氨基-2-丁基-1H-咪唑并[4,5-c]喹啉-1-基)-2,2-二甲基丙基]苯甲酰胺,
2-丁基-1-[3-(甲基磺酰基)丙基]-1H-咪唑并[4,5-c]喹啉-4-胺,
N-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]-1,1-二甲基乙基}-2-乙氧基乙酰胺,
1-[4-氨基-2-乙氧基甲基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
1-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-{3-[4-氨基-1-(2-羟基-2-甲基丙基)-2-(甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-7-基]苯基}甲磺酰胺,
1-[4-氨基-7-(5-羟基甲基吡啶-3-基)-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
3-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]丙-1,2-二醇,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-丙基脲,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-环戊基脲,
1-[(2,2-二甲基-1,3-二氧戊环-4-基)甲基]-2-(乙氧基甲基)-7-(4-羟基甲基苯基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-[4-氨基-2-乙氧基甲基-1-(2-羟基-2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-7-基]-N-甲氧基-N-甲基苯甲酰胺,
2-乙氧基甲基-N1-异丙基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1,4-二胺,
1-[4-氨基-2-乙基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基]甲磺酰胺,或
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-环己基脲,
其中,喹啉环上的胺基团是与连接体连接的连接点。
又一方面,本发明提供一种药物组合物,其包含有效量的本文提供的化合物(例如,上文中所公开的那些化合物),以及一种或一种以上药学上可接受的载体。
在一些实施方式中,本发明的组合物还包含有效量的其他治疗剂。在一些实施方式中,所述其他治疗剂是抗癌剂,例如,抗代谢药物、拓扑异构酶I和拓扑异构酶II的抑制剂、烷基化剂、微管抑制剂、抗雄性激素剂、GNRh调节剂或者它们的混合物。
再一方面,本发明提供一种抑制肿瘤细胞增殖的方法,所述方法包括将本文提供的化合物施用于所述肿瘤细胞。
又一方面,本发明提供一种治疗受治者体内的肿瘤/癌症的方法,所述方法包括将本文提供的化合物给药于所述受治者。
在一些实施方式中,所述肿瘤/癌症选自:食道癌、胃癌、结肠癌、直肠癌、胰腺癌、包括NSCLC在内的肺癌、乳腺癌、妇科肿瘤(包括子宫颈癌、子宫体癌和卵巢癌)、膀胱癌、包括SCCHN在内的头颈癌、子宫内膜癌、骨肉瘤、前列腺癌、神经母细胞瘤、肾癌、胶质瘤、多形性胶质母细胞瘤以及包括上皮癌在内的皮肤癌。
另一方面,本发明提供本文所述的化合物在制备用于治疗受治者体内的肿瘤/癌症的药物中的应用。
再一方面,本发明提供本文所述的药物组合物在制备用于治疗受治者体内的肿瘤/癌症的药物中的应用。
一方面,本发明提供一种治疗或延迟个体中的癌症的恶化的方法,所述方法包括将有效量的PD-L/PD-1轴拮抗剂以及能够活化人浆细胞样树突细胞、髓样树突细胞、NK细胞、T细胞或肿瘤细胞的免疫治疗剂给药于所述个体。
在一些实施方式中,PD-L/PD-1轴拮抗剂选自:PD-1结合拮抗剂,PD-L1结合拮抗剂和PD-L2结合拮抗剂。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-1结合拮抗剂。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合其配体结合搭档。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L1。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L2。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L1和PD-L2这两者。
在一些实施方式中,PD-1结合拮抗剂是抗体,例如,MDX-1106,Merck3745,CT-011,AMP-224或AMP-514。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-L1结合拮抗剂。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合PD-1。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合B7-1。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合PD-1和B7-1这两者。
在一些实施方式中,PD-L1结合拮抗剂是抗体,例如,选自YW243.55.S70,MPDL3280A、MDX-1105、MEDI-4736和MSB0010718C中的一种抗体。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-L2结合拮抗剂。
在一些实施方式中,PD-L2结合拮抗剂是抗体。
在一些实施方式中,PD-L2结合拮抗剂是免疫粘合素。
在一些实施方式中,所述免疫治疗剂是与不含连接体的活化部分相同的化合物中的一种,例如,不含
在一些实施方式中,在停止治疗之后,所述治疗在个体中产生持续反应。
在一些实施方式中,所述免疫治疗剂连续施用,间歇施用。
在一些实施方式中,所述免疫治疗剂在PD-L/PD-1轴拮抗剂之前施用。
在一些实施方式中,所述免疫治疗剂与PD-L/PD-1轴拮抗剂同时施用。
在一些实施方式中,所述免疫治疗剂在PD-L/PD-1轴拮抗剂之后施用。
在一些实施方式中,所述个体患有结肠直肠癌、黑色素瘤、非小细胞肺癌、卵巢癌、乳腺癌、胰腺癌、恶性血液肿瘤或肾细胞癌。
在一些实施方式中,PD-L/PD-1轴拮抗剂静脉内施用、肌肉内施用、皮下施用、局部施用、口服施用、透皮施用、腹膜内施用、眼窝内施用、通过植入的方式施用、通过吸入的方式施用、鞘内施用、心室内施用或鼻内施用。
另一方面,本发明提供一种组合,其包括:(i)有效量的PD-L/PD-1轴拮抗剂;和(ii)有效量的能够活化人浆细胞样树突细胞、髓样树突细胞、NK细胞、T细胞或肿瘤细胞或者它们的组合的免疫治疗剂。
在一些实施方式中,PD-L/PD-1轴拮抗剂选自:PD-1结合拮抗剂、PD-L1结合拮抗剂和PD-L2结合拮抗剂。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-1结合拮抗剂。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合其配体结合搭档。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L1。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L2。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L1和PD-L2这两者。
在一些实施方式中,PD-1结合拮抗剂是抗体,例如,MDX-1106,Merck3745,CT-011,AMP-224或AMP-514。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-L1结合拮抗剂。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合PD-1。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合B7-1。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合PD-1和B7-1这两者。
在一些实施方式中,PD-L1结合拮抗剂是抗体,例如,选自YW243.55.S70、MPDL3280A、MDX-1105、MEDI-4736和MSB0010718C中的一种抗体。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-L2结合拮抗剂。
在一些实施方式中,PD-L2结合拮抗剂是抗体。
在一些实施方式中,PD-L2结合拮抗剂是免疫粘合素。
在一些实施方式中,所述免疫治疗剂是本文提供的化合物,其是不连接至连接体的活化部分,包括不含
在一些实施方式中,本发明提供一种药物组合物,其包含本文提供的化合物或其药学上可接受的盐,和/或一种或一种以上药学上可接受的载体。
在一些实施方式中,所述药物组合物还包含其他治疗剂。在一些实施方式中,所述其他治疗剂是抗癌剂。在一些实施方式中,所述其他治疗剂是抗代谢药物、拓扑异构酶I和拓扑异构酶II的抑制剂、烷基化剂、微管抑制剂、抗雄性激素剂、GNRh调节剂或者它们的混合物。在一些实施方式中,所述其他治疗剂选自:它莫西芬(tamoxifen),雷洛昔芬(raloxifene),阿那曲唑(anastrozole),依西美坦(exemestane),来曲唑(letrozole),imatanib,紫杉醇,环磷酰胺,洛伐他汀(lovastatin),minosine,吉西他滨(gemcitabine),阿糖胞苷(cytarabine),5-氟尿嘧啶,甲氨蝶呤,多西他赛(docetaxel),戈舍瑞林(goserelin),长春新碱,长春碱,噻氨酯哒唑(nocodazole),替尼泊苷(teniposide),依托泊苷(etoposide),吉西他滨,埃博霉素,长春瑞滨(vinorelbine),喜树碱,道诺霉素(daunorubicin),放线菌素D,米托蒽醌,吖啶,阿霉素,表柔比星,或去甲氧基柔红霉素。
又一方面,本发明提供一种抑制肿瘤细胞增殖的方法,所述方法包括将本发明的化合物施用于所述肿瘤细胞。
在一些实施方式中,本发明提供一种治疗受治者体内的肿瘤/癌症的方法,所述方法包括将本发明的化合物给药于所述受治者。在一些实施方式中,所述肿瘤/癌症选自:食道癌、胃癌、结肠癌、直肠癌、胰腺癌、包括NSCLC在内的肺癌、乳腺癌、妇科肿瘤(包括子宫颈癌、子宫体癌和卵巢癌)、膀胱癌、包括SCCHN在内的头颈癌、子宫内膜癌、骨肉瘤、前列腺癌、神经母细胞瘤、肾癌、胶质瘤、多形性胶质母细胞瘤以及包括上皮癌在内的皮肤癌。
附图说明
本发明的新特点在所附的权利要求书中具体说明。通过结合后面列举的对示例性的实施方式的详细描述可更好地对本发明的特点和优势加以理解,在示例性的实施方式中使用了本发明的原理,并且示例性的实施方式的附图如下:
图1表示B16F10-her2肿瘤及周围细胞。鼠B16F10-her2细胞在体内转染至表达PD-L1的肿瘤细胞,肿瘤周围的细胞表达PD-1。肿瘤及周围细胞与不同量的抗鼠PD-L1或PD-1的抗体一同孵育,使用单独的二抗或不相关鼠IgG作为阴性对照,随后进行藻红蛋白二抗孵育,使用FlowJo软件分析记录的数据。
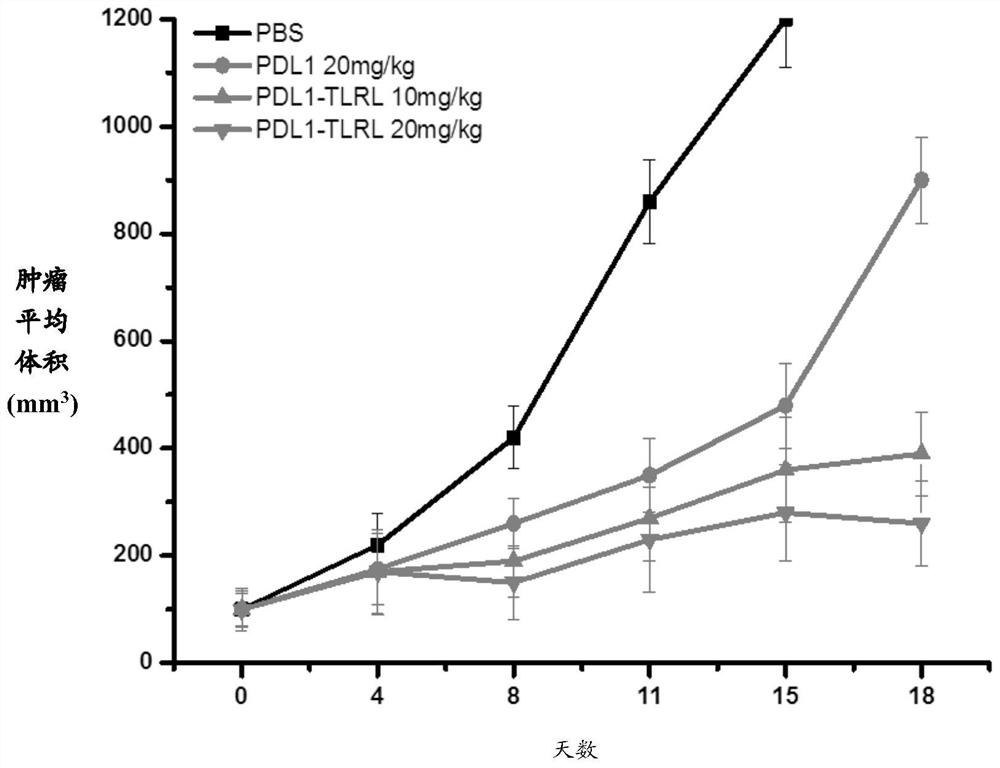
图2显示PD-L1抗体结合物抑制肿瘤生长。比较了待肿瘤生长至尺寸为100mm
图3显示PD-1抗体结合物抑制肿瘤生长。比较了待肿瘤生长至尺寸为100mm
具体实施方式
通过结合用于举例说明的示例性应用,在下文中对本发明的多个方面进行描述。应当理解的是,列举出许多具体的细节、关系以及方法来全面理解本发明。然而,本领域普通技术人员可容易地意识到本发明可不按照具体细节中的一种或一种以上来实施或者可以其他方法来实施。本发明不受举例说明的操作或事件顺序的限制,因为,一些操作或事件可以不同的顺序进行和/或可同时与其他操作或事件一同进行。
进一步地,不是所有举例说明的操作或事件都需要根据本发明的方法来实施。
本文使用的术语是仅仅是为了说明具体实施方式,而无意限定本发明。除非另有明确说明,如本文使用的不指明具体数目的冠词(“a”,“an”和“the”)意在包括复数形式。此外,在具体实施方式部分和/或权利要求中使用的术语“包括(“including”,“include”)、具有(“having”,“has”,“with”)或其变体意在包括与术语“包含(comprising)”类似的方式。
术语“约”或“大约”是指由本领域普通技术人员测定的特定值在可接受的误差范围内,该误差范围部分取决于该值如何测量或确定,即,测量系统的限制。例如,“约”可意味着在本领域中每一操作在1个标准偏差范围内或超过一个标准偏差。可选地,“约”可意味着高达给定值的20%的范围,优选地高达给定值的10%的范围,更优选地高达给定值的5%的范围并且更优选地高达给定值的1%的范围。可选地,尤其对于生物系统或过程而言,该术语可意味着在某个值的某个数量级范围内,优选地在某个值的5倍范围内,更优选地在某个值的2倍范围内。在本申请和权利要求书中描述特定值的条件下,除非另有说明,应当假定术语“约”意味着在特定值的可接受的误差范围内。
除非另有说明,本文使用的所有技术术语和科学术语通常具有与本发明所属技术领域的普通技术人员通常理解的含义相同的含义。一般而言,本文使用的命名系统和细胞培养、分子遗传学、有机化学和核酸化学以及杂交中的实验操作是本领域熟知的且普遍使用的那些命名系统和实验操作。标准技术用于核酸和肽的合成。所述技术和操作通常根据本领域的常规方法和各种不同的常规参考文献进行,本领域的常规方法和各种不同的常规参考文献在本文中提供。本文使用的命名系统和下面描述的分析化学和有机合成中的实验操作是本领域熟知的且普遍使用的命名系统和实验操作。标准技术或其改良用于化学合成和化学分析。
除非另有说明,术语“烷基”其自身或作为另一取代基的一部分是指含有指定数目的碳原子(即,C
术语“亚烷基”其自身或作为另一取代基的一部分是指从烷烃衍生得到的二价自由基,例如,但不限于:-CH
术语“烷氧基”、“烷基氨基”和“烷硫基”(或硫代烷氧基)以其常规含义使用,并且分别是指通过氧原子、氨基基团或硫原子与分子的剩余部分连接的那些烷基基团。
除非另有说明,术语“杂烷基”其自身或与另一术语结合是指由规定数目的碳原子和至少一个选自O、N、Si和S的杂原子构成的、稳定的直链或支链或环状烃自由基或者其组合,并且,其中,氮和硫原子可被任选地氧化并且氮杂原子可被任选地季铵化。杂原子O、N和S以及Si可位于杂烷基的任何内部位置或者位于烷基与分子的剩余部分连接的位置。实例包括但不限于:-CH
一般而言,“酰基取代基”也选自上面列举的基团。如本文使用的术语“酰基取代基”是指连接至本发明的化合物的多环核心的基团且该基团满足直接或间接地连接至本发明的化合物的多环核心的羰基碳的化合价。
除非另有说明,术语“环烷基”和“杂环烷基”其自身或者与其他术语的组合分别表示“烷基”和“杂烷基”的环状形式。此外,对于杂环烷基而言,杂原子可占据杂环与分子的剩余部分连接的位置。环烷基的实例包括但不限于:环戊基、环己基、1-环己烯基、3-环己烯基、环庚基,等等。杂环烷基的实例包括但不限于:1-(1,2,5,6-四氢吡啶基)、1-哌啶基、2-哌啶基、3-哌啶基、4-吗啉基、3-吗啉基、四氢呋喃-2-基、四氢呋喃-3-基、四氢噻吩-2-基、四氢噻吩-3-基、1-哌嗪基、2-哌嗪基,等等。
除非另有说明,术语“卤代”或“卤素”其自身或作为另一取代基的一部分是指氟、氯、溴或碘原子。此外,诸如“卤代烷基”之类的术语意在包括单卤代烷基和多卤代烷基。例如,术语“卤代(C
本文使用的术语“卤代烷基”是指被一个或一个以上本文所定义的卤素基团取代的本文定义的烷基。卤代烷基优选地可为单卤代烷基、二卤代烷基或多卤代烷基(包括全卤代烷基)。单卤代烷基在烷基基团中可具有一个碘、溴、氯或氟。二卤代烷基和多卤代烷基在烷基基团中可具有两个或两个以上相同的卤素原子或不同的卤素基团的组合。优选地,多卤代烷基包含多达12个,10个或8个,或6个,或4个,或3个,或2个卤素基团。卤代烷基的非限定性实例包括氟代甲基、二氟代甲基、三氟代甲基、氯代甲基、二氯代甲基、三氯代甲基、五氟代乙基、七氟代丙基、二氟代氯代甲基、二氯代氟代甲基、二氟代乙基、二氟代丙基、二氯代乙基和二氯代丙基。全卤代烷基是指所有的氢原子均被卤素原子取代的烷基。
本文使用的术语“杂芳基”是指具有1至8个选自N、O、S或Se的杂原子的5元至14元单环或双环或稠合的多环系统。优选地,杂芳基为5元至10元环系统。典型的杂芳基基团包括2-噻吩基或3-噻吩基,2-呋喃基或3-呋喃基,2-吡咯基或3-吡咯基,2-咪唑基、4-咪唑基或5-咪唑基,3-吡唑基、4-吡唑基或5-吡唑基,2-噻唑基、4-噻唑基或5-噻唑基,3-异噻唑基、4-异噻唑基或5-异噻唑基,2-恶唑基、4-恶唑基或5-恶唑基,3-异恶唑基、4-异恶唑基或5-异恶唑基,3-1,2,4-三唑基或5-1,2,4-三唑基,4-1,2,3-三唑基或5-1,2,3-三唑基,四唑基,2-吡啶基、3-吡啶基或4-吡啶基,3-哒嗪基或4-哒嗪基,3-吡嗪基、4-吡嗪基或5-吡嗪基,2-吡嗪基,2-嘧啶基、4-嘧啶基或5-嘧啶基。
术语“杂芳基”还指杂芳香族环与一个或一个以上芳基环、环状脂肪族环或杂环烷基环稠合的基团,其中,自由基或连接点位于杂芳香族环上。非限定性实例包括但不限于:1-中氮茚基(indolizinyl),2-中氮茚基,3-中氮茚基,5-中氮茚基,6-中氮茚基,7-中氮茚基或8-中氮茚基,1-异吲哚基,3-异吲哚基,4-异吲哚基,5-异吲哚基,6-异吲哚基或7-异吲哚基,2-吲哚基,3-吲哚基,4-吲哚基,5-吲哚基,6-吲哚基或7-吲哚基,2-吲唑基,3-吲唑基,4-吲唑基,5-吲唑基,6-吲唑基或7-吲唑基,2-嘌呤基,4-嘌呤基,5-嘌呤基,6-嘌呤基,7-嘌呤基或8-嘌呤基,1-喹嗪基,2-喹嗪基,3-喹嗪基,4-喹嗪基,6-喹嗪基,7-喹嗪基,8-喹嗪基或9-喹嗪基,2-喹啉基,3-喹啉基,4-喹啉基,5-喹啉基,6-喹啉基,7-喹啉基或8-喹啉基,1-异喹啉基,3-异喹啉基,4-异喹啉基,5-异喹啉基,6-异喹啉基,7-异喹啉基或8-异喹啉基,1-2,3-二氮杂萘基,4-2,3-二氮杂萘基,5-2,3-二氮杂萘基,6-2,3-二氮杂萘基,7-2,3-二氮杂萘基或8-2,3-二氮杂萘基,2-1,5-二氮杂萘基,3-1,5-二氮杂萘基,4-1,5-二氮杂萘基,5-1,5-二氮杂萘基或6-1,5-二氮杂萘基,2-喹唑啉基,3-喹唑啉基,5-喹唑啉基,6-喹唑啉基,7-喹唑啉基或8-喹唑啉基,3-噌啉基,4-噌啉基,5-噌啉基,6-噌啉基,7-噌啉基或8-噌啉基,2-蝶啶基,4-蝶啶基,6-蝶啶基或7-蝶啶基,1-4aH咔唑基,2-4aH咔唑基,3-4aH咔唑基,4-4aH咔唑基,5-4aH咔唑基,6-4aH咔唑基,7-4aH咔唑基或8-4aH咔唑基,1-咔唑基,2-咔唑基,3-咔唑基,4-咔唑基,5-咔唑基,6-咔唑基,7-咔唑基或8-咔唑基,1-咔啉基,3-咔啉基,4-咔啉基,5-咔啉基,6-咔啉基,7-咔啉基,8-咔啉基或9-咔啉基,1-菲啶基,2-菲啶基,3-菲啶基,4-菲啶基,6-菲啶基,7-菲啶基,8-菲啶基,9-菲啶基或10-菲啶基,1-吖啶基,2-吖啶基,3-吖啶基,4-吖啶基,5-吖啶基,6-吖啶基,7-吖啶基,8-吖啶基或9-吖啶基,1-萘嵌二氮杂苯基,2-萘嵌二氮杂苯基,4-萘嵌二氮杂苯基,5-萘嵌二氮杂苯基,6-萘嵌二氮杂苯基,7-萘嵌二氮杂苯基,8-萘嵌二氮杂苯基或9-萘嵌二氮杂苯基(perimidinyl),2-菲咯啉基,3-菲咯啉基,4-菲咯啉基,5-菲咯啉基,6-菲咯啉基,8-菲咯啉基,9-菲咯啉基或10-菲咯啉基,1-吩嗪基,2-吩嗪基,3-吩嗪基,4-吩嗪基,6-吩嗪基,7-吩嗪基,8-吩嗪基或9-吩嗪基,1-噻吩嗪基,2-噻吩嗪基,3-噻吩嗪基,4-噻吩嗪基,6-噻吩嗪基,7-噻吩嗪基,8-噻吩嗪基,9-噻吩嗪基或10-噻吩嗪基,1-吩恶嗪基,2-吩恶嗪基,3-吩恶嗪基,4-吩恶嗪基,6-吩恶嗪基,7-吩恶嗪基,8-吩恶嗪基,9-吩恶嗪基或10-吩恶嗪基,l-苯并异喹啉基,3-苯并异喹啉基,4-苯并异喹啉基,5-苯并异喹啉基,6-苯并异喹啉基,7-苯并异喹啉基,8-苯并异喹啉基,9-苯并异喹啉基或10-苯并异喹啉基,2-噻吩并[2,3-b]呋喃基,3-噻吩并[2,3-b]呋喃基,4-噻吩并[2,3-b]呋喃基或5-噻吩并[2,3-b]呋喃基,2-7H-吡嗪并[2,3-c]咔唑基,3-7H-吡嗪并[2,3-c]咔唑基,5-7H-吡嗪并[2,3-c]咔唑基,6-7H-吡嗪并[2,3-c]咔唑基,7-7H-吡嗪并[2,3-c]咔唑基,8-7H-吡嗪并[2,3-c]咔唑基,9-7H-吡嗪并[2,3-c]咔唑基,10-7H-吡嗪并[2,3-c]咔唑基或11-7H-吡嗪并[2,3-c]咔唑基,2-2H-呋喃并[3,2-b]-吡喃基,3-2H-呋喃并[3,2-b]-吡喃基,5-2H-呋喃并[3,2-b]-吡喃基,6-2H-呋喃并[3,2-b]-吡喃基或7-2H-呋喃并[3,2-b]-吡喃基,2-5H-吡啶并[2,3-d]-o-恶嗪基,3-5H-吡啶并[2,3-d]-o-恶嗪基,4-5H-吡啶并[2,3-d]-o-恶嗪基,5-5H-吡啶并[2,3-d]-o-恶嗪基,7-5H-吡啶并[2,3-d]-o-恶嗪基或8-5H-吡啶并[2,3-d]-o-恶嗪基,1-1H-吡唑并[4,3-d]-恶唑基,3-1H-吡唑并[4,3-d]-恶唑基或5-1H-吡唑并[4,3-d]-恶唑基,2-4H-咪唑并[4,5-d]噻唑基,4-4H-咪唑并[4,5-d]噻唑基或5-4H-咪唑并[4,5-d]噻唑基,3-吡嗪并[2,3-d]哒嗪基,5-吡嗪并[2,3-d]哒嗪基或8-吡嗪并[2,3-d]哒嗪基,2-咪唑并[2,1-b]噻唑基,3-咪唑并[2,1-b]噻唑基,5-咪唑并[2,1-b]噻唑基或6-咪唑并[2,1-b]噻唑基,1-呋喃并[3,4-c]噌啉基,3-呋喃并[3,4-c]噌啉基,6-呋喃并[3,4-c]噌啉基,7-呋喃并[3,4-c]噌啉基,8-呋喃并[3,4-c]噌啉基或9-呋喃并[3,4-c]噌啉基,1-4H-吡啶并[2,3-c]咔唑基,2-4H-吡啶并[2,3-c]咔唑基,3-4H-吡啶并[2,3-c]咔唑基,4-4H-吡啶并[2,3-c]咔唑基,5-4H-吡啶并[2,3-c]咔唑基,6-4H-吡啶并[2,3-c]咔唑基,8-4H-吡啶并[2,3-c]咔唑基,9-4H-吡啶并[2,3-c]咔唑基,10-4H-吡啶并[2,3-c]咔唑基或11-4H-吡啶并[2,3-c]咔唑基,2-咪唑并[1,2-b][1,2,4]三嗪基,3-咪唑并[1,2-b][1,2,4]三嗪基,6-咪唑并[1,2-b][1,2,4]三嗪基或7-咪唑并[1,2-b][1,2,4]三嗪基,7-苯并[b]噻吩基,2-苯并恶唑基,4-苯并恶唑基,5-苯并恶唑基,6-苯并恶唑基或7-苯并恶唑基,2-苯并咪唑基,4-苯并咪唑基,5-苯并咪唑基,6-苯并咪唑基或7-苯并咪唑基,2-苯并噻唑基,4-苯并噻唑基,4-苯并噻唑基,5-苯并噻唑基,6-苯并噻唑基或7-苯并噻唑基,1-苯并氧杂革基,2-苯并氧杂革基,4-苯并氧杂革基,5-苯并氧杂革基,6-苯并氧杂革基,7-苯并氧杂革基,8-苯并氧杂革基或9-苯并氧杂革基(benzoxapinyl),2-苯并恶嗪基,4-苯并恶嗪基,5-苯并恶嗪基,6-苯并恶嗪基,7-苯并恶嗪基或8-苯并恶嗪基,1-1H-吡咯并[1,2-b][2]苯并氮杂革基,2-1H-吡咯并[1,2-b][2]苯并氮杂革基,3-1H-吡咯并[1,2-b][2]苯并氮杂革基,5-1H-吡咯并[1,2-b][2]苯并氮杂革基,6-1H-吡咯并[1,2-b][2]苯并氮杂革基,7-1H-吡咯并[1,2-b][2]苯并氮杂革基,8-1H-吡咯并[1,2-b][2]苯并氮杂革基,9-1H-吡咯并[1,2-b][2]苯并氮杂革基,10-1H-吡咯并[1,2-b][2]苯并氮杂革基或11-1H-吡咯并[1,2-b][2]苯并氮杂革基(benzazapinyl)。典型的稠合杂芳基基团包括但不限于:2-喹啉基,3-喹啉基,4-喹啉基,5-喹啉基,6-喹啉基,7-喹啉基或8-喹啉基,1-异喹啉基,3-异喹啉基,4-异喹啉基,5-异喹啉基,6-异喹啉基,7-异喹啉基或8-异喹啉基,2-吲哚基,3-吲哚基,4-吲哚基,5-吲哚基,6-吲哚基或7-吲哚基,2-苯并[b]噻吩基,3-苯并[b]噻吩基,4-苯并[b]噻吩基,5-苯并[b]噻吩基,6-苯并[b]噻吩基或7-苯并[b]噻吩基,2-苯并恶唑基,4-苯并恶唑基,5-苯并恶唑基,6-苯并恶唑基或7-苯并恶唑基,2-苯并咪唑基,4-苯并咪唑基,5-苯并咪唑基,6-苯并咪唑基或7-苯并咪唑基,2-苯并噻唑基,4-苯并噻唑基,5-苯并噻唑基,6-苯并噻唑基或7-苯并噻唑基。
如本文使用的术语“杂环基”或“杂环”是指被任选地取代的完全饱和的或不饱和的、芳香族的或非芳香族的环基团,例如,其为4元至7元单环系统,7元至12元双环系统或者10元至15元三环系统,其在至少一个含有碳原子的环中具有至少一个杂原子。含有杂原子的杂环基团中的每一个环可具有选自氮原子、氧原子和硫原子的1个,2个或3个杂原子,其中,氮原子和硫原子还可被任选地氧化。杂环基团可在杂原子或碳原子处连接。
示例性的单环杂环基团包括:吡咯烷基、吡咯基、吡唑基、氧杂环丁烷基、吡唑啉基、咪唑基、咪唑啉基、咪唑烷基、三唑基、恶唑基、恶唑烷基、异恶唑啉基、异恶唑基、噻唑基、噻二唑基、噻唑烷基、异噻唑基、异噻唑烷基、呋喃基、四氢呋喃基、噻吩基、恶二唑基、哌啶基、哌嗪基、2-氧代哌嗪基、2-氧代哌啶基、2-氧代吡咯烷基、2-氧代氮杂卓基(2-oxoazepinyl)、氮杂卓基(azepinyl)、4-哌啶酮基、吡啶基、吡嗪基、嘧啶基、哒嗪基、四氢吡喃基、吗啉基、噻吗啉基、噻吗啉基亚砜、噻吗啉基砜、1,3-二氧戊环和四氢-1,1-二氧代噻吩基、1,1,4-三氧代-1,2,5-噻二唑烷-2-基,等等。
示例性的双环杂环基团包括:吲哚基、二氢吲哚基、苯并噻唑基、苯并恶嗪基、苯并恶唑基、苯并噻吩基、苯并噻嗪基、奎宁环基、喹啉基、四氢喹啉基、十氢喹啉基、异喹啉基、四氢异喹啉基、十氢异喹啉基、苯并咪唑基、苯并吡喃基、中氮茚基、苯并呋喃基、色酮基(chromonyl)、香豆素基、苯并吡喃基、噌啉基、喹喔啉基、吲唑基、吡咯并吡啶基、呋喃并吡啶基(例如,呋喃并[2,3-c]吡啶基、呋喃并[3,2-b]吡啶基或呋喃并[2,3-b]吡啶基)、二氢异吲哚基、1,3-二氧-1,3-二氢异吲哚-2-基、二氢喹唑啉基(例如,3,4-二氢-4-氧代-喹唑啉基)、2,3-二氮杂萘基,等等。
示例性的三环杂环基团包括:咔唑基、二苯并氮杂卓基、二噻吩并氮杂卓基、苯并吲哚基、菲咯啉基、吖啶基、菲啶基、吩恶嗪基、吩噻嗪基、呫吨基、咔啉基,等等。
术语“杂环基”进一步是指被1个、2个或3个选自下列基团的取代基取代的如上所定义的杂环基团:
(a)烷基;
(b)羟基(或者被保护的羟基);
(c)卤素;
(d)氧,即,=O;
(e)氨基,烷基氨基或二烷基氨基;
(f)烷氧基;
(g)环烷基;
(h)羧基;
(i)杂环氧基,其中,杂环氧基是指通过氧桥键连接的杂环基团;
(j)烷基-O-C(O)--;
(k)巯基;
(l)硝基;
(m)氰基;
(n)氨磺酰基或亚磺酰氨基;
(o)芳基;
(p)烷基-C(O)-O--;
(q)芳基-C(O)-O--;
(r)芳基-S--;
(s)芳氧基;
(t)烷基-S--;
(u)甲酰基,即,HC(O)--;
(v)氨基甲酰基;
(w)芳基-烷基--;以及
(x)被烷基、环烷基、烷氧基、羟基、氨基、烷基-C(O)-NH--、烷基氨基、二烷基氨基或卤素取代的芳基。
本文使用的术语“烯基”是指具有2个至20个碳原子且含有至少一个双键的直链或支链烃基。所述烯基优选地具有约2个至8个碳原子。
除非另有说明,术语“芳基”是指多不饱和芳香族烃取代基,其可为单环或多环(优选地为1个环至3个环),其可为稠合的或共价连接的。术语“杂芳基”是指含有1个至4个选自N、O和S的杂原子的芳基基团(或芳环),其中,氮原子和硫原子可被任选地氧化并且氮原子可被任选地季铵化。杂芳基基团可通过杂原子连接至分子的剩余部分。芳基和杂芳基的非限定性实例包括:苯基、1-萘基、2-萘基、4-联苯基、1-吡咯基、2-吡咯基、3-吡咯基、3-吡唑基、2-咪唑基、4-咪唑基、吡嗪基、2-恶唑基、4-恶唑基、2-苯基-4-恶唑基、5-恶唑基、3-异恶唑基、4-异恶唑基、5-异恶唑基、2-噻唑基、4-噻唑基、5-噻唑基、2-呋喃基、3-呋喃基、2-噻吩基、3-噻吩基、2-吡啶基、3-吡啶基、4-吡啶基、2-嘧啶基、4-嘧啶基、5-苯并噻唑基、嘌呤基、2-苯并咪唑基、5-吲哚基、1-异喹啉基、5-异喹啉基、2-喹喔啉基、5-喹喔啉基、3-喹啉基和6-喹啉基。上述芳基和杂芳基环系统的每一个的取代基选自下述可接受的取代基基团。
为了简洁起见,当与其他术语(例如,芳氧基、芳硫氧基、芳基烷基)组合使用时,术语“芳基”包括上述芳环和杂芳环。因此,术语“芳基烷基”意在包括其中芳基连接至烷基的那些自由基(例如,苄基、苯乙基、吡啶甲基,等等),所述烷基包括碳原子(例如,亚甲基)被例如氧原子取代的那些烷基(例如,苯氧基甲基、2-吡啶基氧甲基、3-(1-萘基氧)丙基,等等)。
上述术语(例如“烷基”、“杂烷基”、“芳基”和“杂芳基”)的每一个包括所指定的自由基的取代形式和未取代形式这两者。每种类型的自由基的优选的取代基在下文提供。
烷基和杂烷基自由基(包括通常称为亚烷基、烯基、杂亚烷基、杂烯基、炔基、环烷基、杂环烷基、环烯基和杂环烯基的那些基团)的取代基通常分别被称为“烷基取代基”和“杂烷基取代基”,并且它们可为选自下列多种基团中的一种或一种以上,但不限于下列基团:-OR’,=O,=NR’,=N-OR’,-NR’R”,-SR’,-卤素,-SiR’R”R”’,-OC(O)R’,-C(O)R’,-CO
类似于对于烷基自由基的取代基的描述,芳基取代基和杂芳基取代基通常被分别称为“芳基取代基”和“杂芳基取代基”,并且为不同的,选自例如,卤素,-OR’,=O,=NR’,=N-OR’,-NR’R”,-SR’,-卤素,-SiR’R”R”’,-OC(O)R’,-C(O)R’,-CO
芳环或杂芳环的相邻原子上的芳基取代基中的两个可任选地被通式–T-C(O)-(CRR’)
本文使用的术语“杂原子”包括氧(O)、氮(N)、硫(S)、磷(P)和硅(Si)。
本文使用的术语“芳氧基”是指-O-芳基和-O-杂芳基这两者,其中,芳基和杂芳基是本文所定义的。
本文使用的术语“药学上可接受的盐”是指保持本发明的化合物的生物功效和性质的盐,其不是生物学上不理想的,也不是其他不理想的。在许多情况下,本发明的化合物能够通过存在的氨基和/或羧基或其类似基团(例如,苯酚或异羟肟酸(hydroxyamicacid))形成酸式盐和/或碱式盐。药学上可接受的酸加成盐可通过无机酸和有机酸形成。可衍生形成盐的无机酸包括,例如,盐酸、氢溴酸、硫酸、硝酸、磷酸,等等。可衍生形成盐的有机酸包括,例如,醋酸、丙酸、乙醇酸、丙酮酸、草酸、马来酸、丙二酸、琥珀酸、富马酸、酒石酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、对甲苯磺酸、水杨酸,等等。药学上可接受的碱加成盐可通过无机碱和有机碱形成。可衍生形成盐的无机碱包括,例如,钠盐、钾盐、锂盐、铵盐、钙盐、镁盐、铁盐、锌盐、铜盐、锰盐、铝盐,等等,特别优选地为铵盐、钾盐、钠盐、钙盐和镁盐。可衍生形成盐的有机碱包括,例如,伯胺、仲胺和叔胺、取代的胺(包括天然生成的取代的胺)、环胺、碱性离子交换树脂,等等,尤其例如,异丙基胺、三甲胺、二乙胺、三乙胺、三丙胺和乙醇胺。本发明的药学上可接受的盐可通过常规化学方法由母体化合物,碱性基团或酸性基团合成。一般而言,这种盐可通过这些化合物的游离酸形式与化学计量量的合适的碱(例如,氢氧化钠,氢氧化钙,氢氧化镁或氢氧化钾,碳酸盐,碳酸氢盐,等等)的反应来制备或者可通过这些化合物的游离碱形式与化学计量量的合适的酸的反应来制备。这些反应通常在水或有机溶剂中进行,或者在水和有机溶剂的混合物中进行。通常,在实际应用中,非水性介质(例如,醚、乙酸乙酯、乙醇、异丙醇或乙腈)为优选的。其他合适的盐的列表可在例如,Remington's Pharmaceutical Sciences,第20版,Mack PublishingCompany,Easton,Pa.,(1985)中找到,该参考文献通过引用并入本文。
本文使用的术语“药学上可接受的载体/赋形剂”包括本领域普通技术人员已知的任何和所有溶剂、分散介质、包衣、表面活性剂、抗氧化剂、防腐剂(例如,抗菌剂、抗真菌剂)、等渗剂、吸收延迟剂、盐、药物、药物稳定剂、结合剂、赋形剂、崩解剂、润滑剂、甜味剂、调味剂、染料,等等,以及它们的组合(参见,例如,Remington's PharmaceuticalSciences,第18版,Mack Printing Company,1990,pp.1289-1329,该参考文献通过引用并入本文)。除了迄今已知的与活性成分不相容的任何常规载体之外,上述药学上可接受的载体/赋形剂可在治疗组合物或药物组合物中使用。
本文使用的术语“受治者”是指动物。优选地,所述动物为哺乳动物。受治者还指例如,灵长类动物(例如,人类),牛、绵羊、山羊、马、狗、猫、兔子、大鼠、小鼠、鱼、鸟,等等。在优选的实施方式中,所述受治者为人类。
一方面,本发明提供具有下式(I)的结构的化合物:
TM-L-AM (I)
其中,TM是靶向部分,例如作为PD-L/PD-1轴拮抗剂的抗体或其功能片段(例如,抗-PD-L1抗体,抗-PD-L2抗体,或抗-PD-1抗体),AM是活化树突细胞、自然杀伤细胞、T细胞或肿瘤细胞或者它们的组合的活化部分,L是连接体。
总体而言,本发明的化合物包含靶向部分。
本文中的“靶向部分(TM)”或“靶向剂”是指特异性地或选择性地与目标分子、细胞、颗粒、组织或聚集体结合的分子、复合物或聚集体,所述目标分子、细胞、颗粒、组织或聚集体通常称为“靶点”或“标志物”并且本文将进一步详细讨论这些目标分子、细胞、颗粒、组织或聚集体。
在一些实施方式中,靶向部分包含免疫球蛋白、蛋白质、肽、小分子、纳米颗粒或核酸。
诸如抗体(例如,嵌合抗体、人源化抗体和人抗体)、受体的配体、植物凝集素和糖类以及一些酶的底物之类的示例性的靶向剂在本领域中被识别并且不受限制地用于实施本发明。其他靶向剂包括一类如下化合物:该化合物不包括特异性分子识别基序,该化合物包括将分子质量加至活化部分的纳米颗粒、诸如聚(乙二醇)之类的大分子、多糖,以及聚氨基酸。额外的分子质量影响活化部分的药代动力学,例如血清半衰期。
在一些实施方式中,靶向部分为抗体,抗体片段,双特异性抗体或其他基于抗体的分子或化合物。然而,靶向部分的其他实例为本领域已知的并且可使用靶向部分的其他实例,例如,适体、avimer、受体结合配体、核酸、生物素-亲和素结合对、结合肽或蛋白质,等等。术语“靶向部分”和“结合部分”在本文中同义使用。
本文中的“靶点”或“标志物”是指任何能够特异性结合特定靶向部分的实体,例如PD-L1,PD-L2或PD-1。
在一些实施方式中,所述靶向部分能够特异性结合PD-L1,PD-L2或PD-1,或者相对于非靶点能够优先结合PD-L1,PD-L2或PD-1。
本文中的“特异性结合”或“优先结合”是指在两个结合搭档之间(例如,在靶向部分及其结合搭档之间)的结合对两个结合搭档具有选择性并且可从不想要的或非特异性相互作用中区分出来。例如,抗原结合部分结合特异性抗原决定簇的能力可通过酶联免疫吸附分析法(ELISA)或其他本领域技术人员熟悉的技术(例如,表面等离子共振技术(在BIAcore仪器上分析)(Liljeblad等人,Glyco J 17,323-329(2000))和传统的结合分析(Heeley,Endocr Res 28,217-229(2002)))测量。术语“抗-[抗原]抗体”和“结合[抗原]的抗体”是指能够通过足够的亲合力结合各自的抗原的抗体,这样,所述抗体用作靶向抗原的诊断剂和/或治疗剂。在一些实施方式中,抗-[抗原]抗体结合不相关的蛋白质的程度小于所测量(例如,通过放射性免疫分析(RIA))的抗体抗原结合程度的约10%。在一些实施方式中,结合[抗原]的抗体的解离常数(KD)小于1μM、小于100nM、小于10nM、小于1nM、小于0.1nM、小于0.01nM或小于0.001nM(例如,10
本文中的“PD-L/PD-1轴拮抗剂”是指如下分子:其抑制PD-L/PD-1轴结合搭档与其结合搭档中的一个或一个以上发生相互作用,从而消除由PD-L/PD-1信号转导轴上的信号转导而产生的T细胞功能障碍,使T细胞功能(例如,增殖、细胞因子的产生,靶细胞杀伤)恢复或提高。本文使用的PD-L/PD-1轴拮抗剂包括PD-1结合拮抗剂,PD-L1结合拮抗剂和PD-L2结合拮抗剂。
本文使用的“PD-1结合拮抗剂”是指如下分子:其降低、阻断、抑制、取消或干扰由PD-1与其结合搭档中的一个或一个以上(例如,PD-L1,PD-L2)的相互作用而产生的信号转导。在一些实施方式中,PD-1结合拮抗剂是抑制PD-1与其结合搭档结合的分子。在特定的方面,PD-1结合拮抗剂抑制PD-1与PD-L1和/或PD-L2的结合。例如,PD-1结合拮抗剂包括抗-PD-1抗体,其抗原结合片段,免疫粘合素,融合蛋白,寡肽以及其他降低、阻断、抑制、取消或干扰由PD-1与PD-L1和/或PD-L2的相互作用而产生的信号转导的分子。在一种实施方式中,PD-1结合拮抗剂减弱阴性共刺激信号,从而使功能障碍性T细胞产生较少的功能障碍(例如,提高对抗原识别的效应物响应),其中,所述阴性共刺激信号由或者通过T淋巴细胞上表达的细胞表面蛋白介导的通过PD-1的信号转导介导。在一些实施方式中,PD-1结合拮抗剂是抗-PD-1抗体。在特定的方面,PD-1结合拮抗剂是本文所述的MDX-1106。在另一特定方面,PD-1结合拮抗剂是本文所述的Merck3745。在另一特定方面,PD-1结合拮抗剂是本文所述的CT-011。
本文使用的“PD-L1结合拮抗剂”是指如下分子:其降低、阻断、抑制、取消或干扰由PD-L1与其结合搭档中的一个或一个以上(例如,PD-1,B7-1)的相互作用而产生的信号转导。在一些实施方式中,PD-L1结合拮抗剂是抑制PD-L1与其结合搭档结合的分子。在特定的方面,PD-L1结合拮抗剂抑制PD-L1与PD-1和/或B7-1的结合。在一些实施方式中,PD-L1结合拮抗剂包括抗-PD-L1抗体,其抗原结合片段,免疫粘合素,融合蛋白,寡肽和其他降低、阻断、抑制、取消或干扰由PD-L1与其结合搭档中的一个或一个以上(例如,PD-1,B7-1)的相互作用而产生的信号转导的分子。在一种实施方式中,PD-L1结合拮抗剂减弱阴性共刺激信号,从而使功能障碍性T细胞产生较少的功能障碍(例如,提高对抗原识别的效应物响应),其中,所述阴性共刺激信号由或通过T淋巴细胞上表达的细胞表面蛋白介导的通过PD-L1的信号转导介导。在一些实施方式中,PD-L1结合拮抗剂是抗-PD-L1抗体。在特定的方面,抗-PD-L1抗体是本文所述的YW243.55.S70。在另一特定的方面,抗-PD-L1抗体是本文所述的MDX-1105。在又一特定的方面,抗-PD-L1抗体是本文所述的MPDL3280A。
本文中的“PD-L2结合拮抗剂”是指如下分子:其降低、阻断、抑制、取消或干扰由PD-L2与其结合搭档中的一个或一个以上(例如,PD-1)的相互作用而产生的信号转导。在一些实施方式中,PD-L2结合拮抗剂是抑制PD-L2与其结合搭档结合的分子。在特定的方面,PD-L2结合拮抗剂抑制PD-L2与PD-1的结合。在一些实施方式中,PD-L2结合拮抗剂包括抗-PD-L2抗体,其抗原结合片段,免疫粘合素,融合蛋白,寡肽和其他降低、阻断、抑制、取消或干扰由PD-L2与其结合搭档中的一个或一个以上(例如,PD-1)的相互作用而产生的信号转导的分子。在一种实施方式中,PD-L2结合拮抗剂减弱阴性共刺激信号,从而使功能障碍性T细胞产生较少的功能障碍(例如,提高对抗原识别的效应物响应),其中,所述阴性共刺激信号由或者通过T淋巴细胞上表达的细胞表面蛋白介导的通过PD-L2的信号转导介导。在一些实施方式中,PD-L2结合拮抗剂是免疫粘合素。
在一些实施方式中,所述靶向部分包含抗体或其功能片段。
本文使用的“免疫球蛋白”或“抗体”是指全长(即,天然生成的或通过正常免疫球蛋白基因片段重组过程形成的)免疫球蛋白分子(例如,IgG抗体)或免疫球蛋白分子的免疫活性(即,特异性结合)部分,例如抗体片段。在本发明要求保护的范围内,抗体或抗体片段可被结合或衍生。这些抗体包括IgGl,lgG2a,IgG3,IgG4(以及IgG4亚型)和IgA同种型。
本文的术语“抗体”以其广义使用并包含各种不同的抗体结构,包括但不限于:单克隆抗体、多克隆抗体、多特异性抗体(例如,双特异性抗体)和抗体片段,只要它们表现出期望的抗原结合活性并且包含免疫球蛋白的Fc区域或等同于该Fc区域的区域。本文中可互换使用的术语“全长抗体”、“完整抗体”和“整个抗体”是指具有与天然抗体结构基本类似的结构的抗体或者具有含本文定义的Fc区域的重链的抗体。
本文的“天然抗体”是指具有不同结构的天然生成的免疫球蛋白分子。例如,天然IgG抗体为约150,000道尔顿的异源四聚体糖蛋白,其由被二硫键连接的两个相同的轻链和两个相同的重链构成。从N端至C端,每一重链具有可变区(VH),也称为可变重链结构域或重链可变结构域,重链之后为三个恒定结构域(CH1、CH2和CH3),也称为重链恒定区。类似地,从N端至C端,每一轻链具有可变区(VL),也称为可变轻链结构域或轻链可变结构域,轻链之后为恒定轻链结构域(CL),也称为轻链恒定区。基于抗体恒定结构域的氨基酸序列,抗体的轻链可指定为两种类型(称为κ和λ)中的一种。
本文中的“抗体片段”是指不同于完整抗体的分子,所述分子包含结合抗原的完整抗体的一部分,所述抗原与完整抗体结合。抗体片段的实例包括但不限于:Fv,Fab,Fab',Fab'-SH,F(ab')2,双体抗体,线性抗体,单链抗体分子(例如,scFv),单结构域抗体和由抗体片段形成的多特异性抗体。关于一些抗体片段的综述请参见,Hudson等人,Nat Med 9,129-134(2003)。关于scFv片段的综述请参见例如,Pliickthun,in The Pharmacology ofMonoclonal Antibodies,vol.113,Rosenburg和Moore编辑,Springer-Verlag,New York,pp.269-315(1994);以及WO 93/16185;和美国专利第5,571,894号和第5,587,458号。关于含有挽救受体结合表位残基且具有提高的体内半衰期的Fab和F(ab’)2片段的讨论,请参见美国专利第5,869,046号。双体抗体为可为二价的或双特异性的具有两个抗原结合位点的抗体片段。请参见例如,EP 404,097;WO1993/01161;Hudson等人,Nat Med 9,129-134(2003);和Hollinger等人,Proc Natl Acad Sci USA 90,6444-6448(1993)。三体抗体和四体抗体也在Hudson等人,Nat Med 9,129-134(2003)中描述。单结构域抗体为含有抗体的所有或一部分重链可变结构域或者含有抗体的所有或一部分轻链可变结构域的抗体片段。在一些实施方式中,单结构域抗体为人单结构域抗体(Domantis,Inc.,Waltham,MA;参见例如,美国专利No.6,248,516Bl)。抗体片段可通过各种不同的技术制备,所述技术包括但不限于:如本文描述的,完整抗体的蛋白水解消化以及通过重组宿主细胞(例如,大肠杆菌或噬菌体)生成。
本文中的“抗原结合结构域”是指包含与抗原的全部或一部分特异性结合且互补的区域的抗体的一部分。抗原结合结构域可通过例如,一个或一个以上抗体可变结构域(也称为抗体可变区)来提供。具体而言,抗原结合结构域包含抗体轻链可变区(VL)和抗体重链可变区(VH)。
本文中的“可变区”或“可变结构域”是指涉及使抗体与抗原结合的抗体重链结构域或轻链结构域。天然抗体的重链和轻链的可变结构域(分别为VH和VL)通常具有类似的结构,其中,每一结构域包含四个保守的框架区(FR)和三个高变区(HVR)。参见例如,Kindt等人,Kuby Immunology,第六版,W.H.Freeman and Co.,第91页(2007)。单个VH或VL结构域可足以带来抗原结合特异性。
本文中的“高变区”或“HVR”是指序列高度可变和/或形成结构限定的环(“高变环”)的抗体可变结构域的各个区域。一般而言,天然四链抗体包含六个HVR,三个在VH(H1、H2、H3)中,三个在VL(L1、L2、L3)中。HVR通常包含来自高变环的氨基酸残基和/或来自互补决定区(CDR)的氨基酸残基,后者具有最高的序列可变性和/或涉及抗原识别。除了VH中的CDR1,CDR通常包含形成高变环的氨基酸残基。高变区(HVR)也被称为“互补决定区(CDR)”并且与形成抗原结合区的可变区部分有关的这些术语在本文中互换使用。该特定区域已由Kabat等人,U.S.Dept.of Health and Human Services,Sequences of Proteins ofImmunological Interest(1983)和Chothia等人,J Mol Biol 196:901-917(1987)描述,其中,当彼此比较时,定义包括氨基酸残基的重叠或子集。然而,关于抗体的CDR或其变体的任何定义的应用意在本文定义的和本文使用的术语的范围内。包含特定CDR的确切的残基数目随CDR的序列和尺寸的不同而不同。在给出抗体的可变区氨基酸序列的条件下,本领域技术人员可常规确定哪些残基包含特定CDR。
本发明的抗体可为嵌合抗体、人源化抗体、人抗体或抗体融合蛋白。
本文中的“嵌合抗体”是指包含抗体重链和轻链这两者的可变结构域的重组蛋白质,所述可变结构域包括来源于一个物种的抗体(优选地为啮齿动物抗体,更优选地为鼠科动物抗体)的互补决定区(CDR),而抗体分子的恒定结构域来源于人抗体的恒定结构域。对于兽医应用而言,嵌合抗体的恒定结构域可来源于其它物种的恒定结构域,所述其它物种例如,类人类灵长类动物、猫或狗。
本文中的“人源化抗体”是指如下重组蛋白:在该重组蛋白中,来自于一个物种的抗体(例如,啮齿动物抗体)的CDR从啮齿动物抗体的可变重链和可变轻链转移至人重链可变结构域和人轻链可变结构域中。抗体分子的恒定结构域来源于人抗体的恒定结构域。在一些实施方式中,人源化抗体的框架区的特定残基,尤其是接触或靠近CDR序列的那些特定残基,可被修饰,例如可被来自于原始啮齿动物、类人类灵长类动物的相应残基或其他抗体代替。
本文中的“人抗体”是指例如从转基因小鼠中获得的抗体,所述转基因小鼠已被“改造”为响应抗原刺激生成特定的人抗体。在该技术中,人重链基因座和人轻链基因座的元件被引入来源于胚胎干细胞系的小鼠品系中,所述胚胎干细胞系包含内源性重链基因座和轻链基因座的靶向断裂。转基因小鼠可合成对人抗原具有特异性的人抗体,并且小鼠可用于生成分泌人抗体的杂交瘤。从转基因小鼠中获得人抗体的方法由Green等人,NatureGenet.7:13(1994),Lonberg等人,Nature 368:856(1994),Taylor等人,Int.Immun.6:579(1994)描述。完全人抗体还可通过基因转染法或染色体转染法以及噬菌体展示技术来构建,所有这些方法为本领域已知的。请参见例如,McCafferty等人,Nature 348:552-553(1990),其中描述了通过来自于未免疫的供体的免疫球蛋白可变结构域基因谱系体外生成人抗体及其片段。在该技术中,抗体可变结构域基因框内克隆至丝状噬菌体的主要或次要外壳蛋白基因中,并且在噬菌体颗粒的表面上展示为功能抗体片段。因为丝状颗粒包含噬菌体基因组的单链DNA拷贝,基于抗体的功能性质的选择还导致对编码表现出那些性质的抗体的基因的选择。通过该方式,噬菌体模仿B细胞的一些性质。噬菌体展示可以多种形式进行,关于噬菌体展示的综述请参见例如,Johnson和Chiswell,Current Opinion inStructural Biology 3:5564-571(1993)。人抗体还可通过体外活化B细胞产生。请参见美国专利第5,567,610号和第5,229,275号,该美国专利的全部内容通过引用并入本文。
本文中的“抗体融合蛋白”是指通过重组产生的抗原结合分子,其中,连接相同或不同的天然抗体、具有相同或不同特异性的单链抗体或抗体片段中的两个或两个以上。融合蛋白包含至少一个特异性结合位点。融合蛋白的化合价表示融合蛋白具有的与抗原或表位结合的结合臂或结合位点的总数,即,单价的、二价的、三价的或多价的。多价的抗体融合蛋白是指该抗体融合蛋白可利用多种与抗原结合的相互作用,因此增加与抗原或不同抗原结合的亲合力。特异性表示抗体融合蛋白能够与多少种不同类型的抗原或表位结合,即,单特异性、双特异性、三特异性、多特异性。使用这些定义,天然抗体(例如,IgG)为二价的,因为其具有两个结合臂,但是其为单特异性的,因为其结合一种类型的抗原或表位。单特异性多价融合蛋白具有一个以上用于相同抗原或表位的结合位点。例如,单特异性双体抗体为具有两个与相同的抗原反应的结合位点的融合蛋白。融合蛋白可包含不同抗体成分的多价或多特异性组合或同一抗体成分的多个拷贝。融合蛋白还可包含治疗剂。
在一些实施方式中,TM是单克隆抗-PD-1抗体。
程序性死亡-1(“PD-1”)是PD-L1(也称为CD274,B7-H1或B7-DC)的受体。PD-1是T细胞调节因子CD28/CTLA4大家族中的大约31kD的I型膜蛋白成员(Ishida,Y.et al.(1992)EMBO J.11:3887-3895;美国专利申请公开第2007/0202100号;第2008/0311117号;第2009/00110667号;美国专利第6,808,710号;第7,101,550号;第7,488,802号;第7,635,757号;第7,722,868号;PCT公开WO 01/14557)。与CTLA4相比,PD-1更加广泛地负向调节免疫反应。
PD-1在活化的T细胞、B细胞和单核细胞上表达(Agata,Y.et al.(1996)Int.Immunol.8(5):765-772;Yamazaki,T.et al.(2002J.Immunol.169:5538-5545)并且在自然杀伤(NK)T细胞中以低水平表达(Nishimura,H.et al.(2000)J.Exp.Med.191:891-898;Martin-Orozco,N.et al.(2007),Semin.Cancer Biol.17(4):288-298)。
PD-1的胞外区域由单独的免疫球蛋白(Ig)V结构域构成,该结构域与CTLA4中的对应的结构域具有23%的一致性(Martin-Orozco,N.et al.(2007)Semin.Cancer Biol.17(4):288-298)。胞外IgV结构域之后为跨膜区域和胞内尾。胞内尾包含位于基于免疫受体酪氨酸的抑制性基序和基于免疫受体酪氨酸的转换基序中的两个磷酸化位点,这表明PD-1负向调节TCR信号(Ishida,Y.et al.(1992EMBO J.11:3887-3895;Blank,C.et al.(Epub2006Dec.29)Immunol.Immunother.56(5):739-745)。
能够免疫特异性结合鼠PD-1的抗体已被报道(参见,例如,Agata,T.et al.(1996)Int.Immunol.8(5):765-772)。
抗-PD-1抗体结合PD-1并且提高T细胞功能,从而上调细胞介导的免疫反应并用于治疗T细胞功能失调性疾病,例如,肿瘤免疫。
在一些实施方式中,抗-PD-1抗体是MK-3475(之前称为lambrolizumab,Merck),AMP-514,AMP-224(MedImmune/AstraZeneca),BMS-936558(MDX-1106,Bristol-MyersSquibb)或CT-011(Curetech)。
Pembrolizumab(MK-3475)是人源化的单克隆抗-PD-1抗体,其被设计为重新活化抗肿瘤免疫性。Pembrolizumab通过抑制T细胞上PD-1与其配体PD-L1和PD-L2的相互作用而发挥双配体阻断PD-1通路的作用。
在一些实施方式中,抗-PD-1抗体是US8,354,509和US8,168,757中公开的抗体中的一种,该美国专利文献的全部内容通过引用并入本文。
Nivolumab(也称为BMS-936558或MDX1106)是完全人IgG4单克隆抗体,由Bristol-Myers Squibb研发,用于治疗癌症。
在一些实施方式中,抗-PD-1抗体是WO2004/056875、US7,488,802和US8,008,449中公开的抗体中的一种,这些专利文献的全部内容通过引用并入本文。
AMP-514和AMP-224是抗-程序性细胞死亡1(PD-1)单克隆抗体(mAb),其由Amplimmune研发,被MedImmune获得。
在一些实施方式中,抗-PD-1抗体是美国申请公开第20140044738号中公开的抗体中的一种,该美国申请公开的全部内容通过引用并入本文。
在一些实施方式中,六个CDR是:(A)抗-PD-1抗体1E3的三个轻链CDR和三个重链CDR;(B)抗-PD-1抗体1E8的三个轻链CDR和三个重链CDR;或(C)抗PD-1抗体1H3的三个轻链CDR和三个重链CDR。
Pidilizumab(CT-011)是由以色列的Curetech Ltd公司研发的抗-PD-1单克隆抗体。
在一些实施方式中,抗-PD-1抗体是美国专利申请公开第20080025980号和第20130022595号中公开的抗体中的一种,该美国专利申请公开的全部内容通过引用并入本文。
在一些实施方式中,TM是单克隆抗-PD-L1抗体。
程序性细胞死亡1配体1(PD-L1,也称为CD274和B7-H1)是PD-1的配体,其在活化的T细胞、B细胞、骨髓细胞和巨噬细胞上被发现。虽然PD-1有两个内源性配体PD-L1和PD-L2,但是抗肿瘤疗法关注抗-PD-L1抗体。PD-1和PD-L1的复合物抑制CD8+T细胞的增殖并降低免疫反应(Topalian et al.,2012,N Engl J Med 366:2443-54;Brahmer et al.,2012,NEng J Med 366:2455-65)。抗-PD-L1抗体已用于治疗非小细胞肺癌、黑色素瘤、结肠直肠癌、肾细胞癌、胰腺癌、胃癌、卵巢癌、乳腺癌和恶性血液病(Brahmer et al.,N Eng J Med366:2455-65;Ott et al.,2013,Clin Cancer Res 19:5300-9;Radvanyi et al.,2013,Clin Cancer Res 19:5541;Menzies&Long,2013,Ther Adv Med Oncol 5:278-85;Bergeret al.,2008,Clin Cancer Res 14:13044-51)。PD-L1是B7家族成员,该B7家族成员在包括APC和活化的T细胞在内的许多类型的细胞上表达(Yamazaki et al.(2002)J.Immunol.169:5538)。PD-L1与PD-1和B7-1这两者结合。通过PD-L1与T细胞表达的B7-1的结合以及通过B7-1与T细胞表达的PD-L1的结合均导致T细胞受到抑制(Butte et al.(2007)Immunity 27:111)。本领域已有证据表明,类似于其他B7家族成员,PD-L1还可向T细胞提供共刺激信号(Subudhi et al.(2004)J.Clin.Invest.113:694;Tamura et al.(2001)Blood 97:1809)。
除非另有明确说明,本文中的“PD-L1”是指包括具有全长多肽的至少一种生物活性的由细胞自然表达的任何变体或同种型和/或它们的片段。此外,术语“PD-L1”包括PD-L1(Freeman et al.(2000)J.Exp.Med.192:1027)和具有全长多肽的至少一种生物活性的由细胞自然表达的任何变体或同种型和/或它们的片段。例如,本领域已知来自包括人类在内的不同物种的PD-L1序列(参见,例如,Chen et al.,美国专利第6,803,192号,其公开了人和小鼠PD-L1序列,Wood et al.,美国专利第7,105,328号,其公开了人PD-L1序列,这两篇参考文献的全部内容通过引用并入本文)。
抗-PD-L1抗体与PD-L1结合并增强T-细胞功能,从而上调细胞介导的免疫反应,用于治疗T细胞功能失调性疾病,例如,肿瘤免疫。
在一些实施方式中,抗-PD-L1抗体是MPDL3280A和YW243.55.S70(Genentech/Roche),MEDI-4736(MedImmune/AstraZeneca),BMS-936559(MDX-1105,Bristol-MyersSquibb)和MSB0010718C(EMD Serono/Merck KGaA)。
MPDL3280A(Genentech)是一种设计为靶向在肿瘤细胞和肿瘤浸润的免疫细胞上表达的PD-L1的基因工程抗-PD-L1抗体。MPDL3280A被设计为阻止PD-L1与PD-1和B7.1结合。对PD-L1的这种阻断能够活化T细胞,恢复T细胞检测和攻击肿瘤细胞的能力。MPDL3280A包含设计为通过最小化抗体依赖性细胞毒性(ADCC)优化疗效和安全性的基因工程片段可结晶(Fc)结构域。
在一些实施方式中,抗-PD-L1抗体是US7,943,743中公开的抗体中的一种,该美国专利的全部内容通过引用并入本文。
BMS-936599(MDX-1105,Bristol-Myers Squibb)是抑制PD-L1配体与PD-1和CD80这两者结合的完全人IgG4抗-PD-L1 mAb。
在一些实施方式中,抗-PD-L1抗体是US7,943,743中公开的抗体中的一种,该美国专利的全部内容通过引用并入本文。
MSB0010718C(EMD Serono of Merck KGaA)是与PD-L1结合的完全人IgG1单克隆抗体。
在一些实施方式中,抗-PD-L1抗体是WO2013079174A1中公开的抗体中的一种,该专利文献的全部内容通过引用并入本文。
MEDI4736(MedImmune/AstraZeneca)是特异性结合PD-L1,阻止PD-L1与PD-1和CD80结合的人IgG1抗体。
在一些实施方式中,抗-PD-L1抗体是WO2011066389A1和US8,779,108中公开的抗体中的一种,该专利文献的全部内容通过引用并入本文。
在一些实施方式中,抗-PD-L1抗体是US8,552,154中公开的抗体中的一种,该美国专利的全部内容通过引用并入本文。
在一些实施方式中,靶向部分包含Fab,Fab’,F(ab’)2,单结构域抗体,T和Abs二聚物,Fv,scFv,dsFv,ds-scFv,Fd,线性抗体,微小抗体、双体抗体、双特异性抗体片段、bibody、tribody、sc-双体抗体、κ(λ)body,BiTE,DVD-Ig,SIP,SMIP,DART,或者含有一个或一个以上CDR的抗体类似物。
在一些实施方式中,靶向部分包含颗粒(靶向颗粒),优选地为纳米颗粒,任选地为连接至靶向分子的靶向纳米颗粒,所述靶向分子可特异性结合或优先结合靶点。在一些实施方式中,靶向颗粒其自身引导本发明的化合物(例如,通过在肿瘤细胞或肿瘤组织中的富集),而无需与其连接额外的靶向分子。
本文中的“纳米颗粒”是指直径小于1000nm的任何颗粒。在一些实施方式中,治疗剂和/或靶向分子可与聚合物基体结合。在一些实施方式中,靶向分子可与聚合物基体的表面共价结合。在一些实施方式中,共价结合由连接体介导。在一些实施方式中,治疗剂可与聚合物基体表面结合,封装在聚合物基体内,被聚合物基体包围,和/或分散于整个聚合物基体。美国专利第8,246,968号,该美国专利的全部内容在此通过引用并入本文。
总体而言,本发明的纳米颗粒包含任何类型的颗粒。根据本发明可使用任何颗粒。在一些实施方式中,颗粒是可生物降解的且生物相容的。总体而言,生物相容性物质对细胞无毒。在一些实施方式中,如果将某种物质加至细胞中产生小于细胞死亡的某一阈值的结果,那么认为该物质是生物相容的。在一些实施方式中,如果将某种物质加至细胞中不诱导副作用,那么认为该物质为生物相容的。总体而言,可生物降解的物质是经过治疗相关时间段(例如,数周、数月或者数年)在生理学条件下发生分解的物质。在一些实施方式中,可生物降解的物质为可通过细胞机制进行分解的物质。在一些实施方式中,可生物降解的物质是可通过化学过程分解的物质。在一些实施方式中,颗粒是生物相容且可生物降解的物质。在一些实施方式中,颗粒是生物相容性物质,但不是可生物降解的物质。在一些实施方式中,颗粒是可生物降解的物质,但不是生物相容性物质。
在一些实施方式中,颗粒的粒度大于肾排泄极限(例如,直径大于6nm的颗粒)。在一些实施方式中,颗粒尺寸为足以避免通过肝脏从血流中清除的大小(例如,直径小于1000nm的颗粒)。总体而言,颗粒的生理化学特性应当允许靶向颗粒通过降低肾排泄和肝脏清除在血浆中长期循环。
通常,理想的是使用尺寸、形状和/或组成相对均匀的颗粒群,这样,每一颗粒具有类似的性质。例如,至少80%的颗粒,至少90%的颗粒或至少95%的颗粒的直径或最大尺寸为平均直径或最大尺寸加减5%,10%或20%。在一些实施方式中,颗粒群的尺寸、形状和/或组成可为不均一的。
ζ(Zeta)电位为颗粒表面电势的测量值。在一些实施方式中,颗粒的ζ电位为-50mV至+50mV。在一些实施方式中,颗粒的ζ电位为-25mV至+25mV。在一些实施方式中,颗粒的ζ电位为-10mV至+10mV。在一些实施方式中,颗粒的ζ电位为-5mV至+5mV。在一些实施方式中,颗粒的ζ电位为0mV至+50mV。在一些实施方式中,颗粒的ζ电位为0mV至+25mV。在一些实施方式中,颗粒的ζ电位为0mV至+10mV。在一些实施方式中,颗粒的ζ电位为0mV至+5mV。在一些实施方式中,颗粒的ζ电位为-50mV至0mV。在一些实施方式中,颗粒的ζ电位为-25mV至0mV。在一些实施方式中,颗粒的ζ电位为-10mV至0mV。在一些实施方式中,颗粒的ζ电位为-5mV至0mV。在一些实施方式中,颗粒的ζ电位基本为中性(即,约0mV)。
根据本发明可使用多种不同的颗粒。在一些实施方式中,颗粒为球形或类球形。在一些实施方式中,颗粒为球形或类球形。在一些实施方式中,颗粒为扁平的或板状的。在一些实施方式中,颗粒为立方体或类立方体。在一些实施方式中,颗粒为卵形或椭圆形。在一些实施方式中,颗粒为圆柱形、圆锥形或金字塔形。
在一些实施方式中,颗粒为微颗粒(例如,微球)。总体而言,“微颗粒”是指直径小于1000μm的任何颗粒。在一些实施方式中,颗粒为微微型颗粒(picoparticle)(例如,微微球体)。总体而言,“微微型颗粒”是指直径小于1nm的任何颗粒。在一些实施方式中,颗粒为脂质体。在一些实施方式中,颗粒为胶束。
颗粒可为实心的或中空的并且可包含一个或一个以上层(例如,纳米壳,纳米环)。在一些实施方式中,每层相对于其他各层具有独特的组成和独特的性质。例如,颗粒可具有核/壳结构,其中,核为一层,壳为另一层。颗粒可包含多个不同的层。在一些实施方式中,一层可为充分交联的,另一层不充分交联,等等。在一些实施方式中,不同层中的一层,几层或所有层可包含一种或一种以上待递送的治疗剂或诊断剂。在一些实施方式中,一层包含待递送的药剂,另一层不含待递送的药剂,等等。在一些实施方式中,每个单独的层包含不同的待递送的药剂或药剂的集合。
在一些实施方式中,颗粒为多孔的,其是指颗粒包含孔或通道,所述孔或通道通常比颗粒的尺寸小。例如,颗粒可为多孔二氧化硅颗粒,例如,介孔二氧化硅纳米颗粒,或者颗粒可具有介孔二氧化硅涂层(Lin等人,2005,J.Am.Chem.Soc.,17:4570)。颗粒可具有直径为约1nm至约50nm的孔,例如,直径为约1nm至20nm的孔。颗粒体积的约10%至95%可由孔或通道内的空隙构成。
颗粒可具有涂层。例如,如果颗粒包含对细胞具有毒性的物质,那么生物相容性涂层的使用可为有优势的。合适的涂层物质包括但不限于:诸如牛血清白蛋白(BSA)之类的天然蛋白质、诸如聚乙二醇(PEG)或PEG衍生物之类的生物相容性亲水聚合物、磷脂-(PEG)、二氧化硅、脂质、聚合物、诸如葡萄聚糖之类的碳水化合物、可与本发明的纳米颗粒结合的其他纳米颗粒,等等。涂层可通过诸如浸蘸、使用层-层技术、自组装、共轭作用等的多种方式涂敷或组装。自组装是指自发地组装成高级结构的过程,该过程依赖于高级结构的成分(例如,分子)彼此之间的自然吸引作用。该过程通常基于尺寸、形状、组成或化学性质通过分子的随机运动和键的形成而发生。
聚合物的实例包括聚亚烷(例如,聚乙烯),聚碳酸酯(例如,聚(1,3-二氧杂环己烷-2酮)),聚酐(例如,聚(癸二酸酐)),聚羟基酸(例如,聚(β-羟基链烷酸酯)),聚延胡索酸酯,聚己酸内酯,聚酰胺(例如,聚己内酰胺),聚缩醛树脂,聚醚,聚酯(例如,聚乳酸,聚乙醇酸交酯),聚(原酸酯),聚乙烯醇,聚氨酯,聚磷腈,聚丙烯酸酯,聚甲基丙烯酸酯,聚氰基丙烯酸酯,聚脲,聚苯乙烯和聚胺。在一些实施方式中,根据本发明的聚合物包括已由美国食品药品管理局(FDA)根据21C.F.R.§177.2600批准用于人体的聚合物,包括但不限于:聚酯(例如,聚乳酸,聚乙醇酸,聚(乳酸-co-乙醇酸),聚己酸内酯,聚戊内酯,聚(1,3-二氧杂环己烷-2酮)),聚酐(例如,聚(癸二酸酐)),聚醚(例如,聚乙二醇),聚氨酯,聚甲基丙烯酸酯,聚丙烯酸酯和聚氰基丙烯酸酯。
在一些实施方式中,颗粒可为非聚合颗粒(例如,金属颗粒,量子点,陶瓷颗粒,含有无机材料的聚合物,骨衍生的材料,骨代用品,病毒颗粒,等等)。在一些实施方式中,待递送的治疗剂或诊断剂可与这样的非聚合颗粒的表面结合。在一些实施方式中,非聚合颗粒为非聚合成分的聚集体,例如,金属原子(例如,金原子)的聚集体。在一些实施方式中,待递送的治疗剂或诊断剂可与非聚合成分的聚集体的表面结合和/或封装在非聚合成分的聚集体内、被非聚合成分的聚集体围绕和/或分散于整个非聚合成分的聚集体。
颗粒(例如,纳米颗粒,微颗粒)可使用本领域已知的任何方法制备。例如,颗粒剂型可通过下列方法以及本领域普通技术人员熟知的其他方法形成:例如纳米沉淀,流动聚焦流体通道,喷雾干燥,单乳液和双乳液溶剂蒸发,溶剂萃取,相分离,研磨,微乳液操作,微制造,纳米制造,牺牲层,简单和复合凝聚。可选地或额外地,已经描述了用于单分散半导体纳米颗粒,导电性纳米颗粒,磁性纳米颗粒,有机纳米颗粒和其他纳米颗粒的水性和有机溶剂合成方法(Pellegrino等人,2005,Small,1:48;Murray等人,2000,Ann.Rev.Mat.Sci.,30:545;以及Trindade等人,2001,Chem.Mat.,13:3843)。
制备用于递送封装的药剂的微颗粒的方法在文献中描述(参见,例如,Doubrow,编辑,“Microcapsules and Nanoparticles in Medicine and Pharmacy,”CRC Press,BocaRaton,1992;Mathiowitz等人,1987,J.Control.Release,5:13;Mathiowitz等人,1987,Reactive Polymers,δ:275;以及Mathiowitz等人,1988,J.Appl.Polymer Sci.,35:755)。
在一些实施方式中,靶向部分包含核酸靶向部分。
总体而言,核酸靶向部分是结合与器官、组织、细胞、细胞外基质成分和/或细胞内腔室有关的成分(靶点)的任何多核苷酸。
在一些实施方式中,核酸靶向部分为适体。
适体通常为与特定目标结构结合的多核苷酸,所述特定目标结构与特定器官、组织、细胞、细胞外基质成分和/或细胞内腔室有关。总体而言,适体的靶向功能基于适体的三维结构。在一些实施方式中,适体与靶点的结合通常由适体和靶点这两者的二维和/或三维结构之间的相互作用介导。在一些实施方式中,适体与靶点的结合不仅仅基于适体的基本序列,还取决于适体和/或靶点的三维结构。在一些实施方式中,适体通过Watson-Crick互补碱基配对与其靶点结合,所述Watson-Crick碱基配对被破坏碱基配对的结构(例如,发夹环)阻碍。
在一些实施方式中,核酸靶向部分为spiegelmer(PCT公布WO 98/08856,WO 02/100442和WO 06/117217)。总体而言,spiegelmer为合成的镜像核酸,其可特异性结合靶点(即,镜像适体)。spiegelmer通过如下结构特征表征:所述结构特征使得它们不易受外切-核酸酶和内切-核酸酶的影响。
本领域普通技术人员会意识到的是,根据本发明可使用任何能够特异性结合靶点的核酸靶向部分(例如,适体或spiegelmer)。在一些实施方式中,根据本发明待使用的核酸靶向部分可靶定与疾病、失调和/或病症有关的标志物。在一些实施方式中,根据本发明待使用的核酸靶向部分可靶向癌相关靶点。在一些实施方式中,根据本发明待使用的核酸靶向部分可靶向肿瘤标志物。使用根据本发明的核酸靶向部分可靶定任何类型的癌症标志物和/或任何肿瘤标志物。举例而言,核酸靶向部分可靶定与前列腺癌、肺癌、乳腺癌、直肠结肠癌、膀胱癌、胰腺癌、子宫内膜癌、卵巢癌、骨癌、食管癌、肝癌、胃癌、脑肿瘤、皮肤黑色素瘤和/或白血病有关的标志物。
本发明的核酸(包括核酸靶向部分和/或待递送的功能性RNA,例如,RNAi-诱导实体,核酶,tRNA,等等,下面进一步详细描述)可根据任何可获得的技术制备,包括但不限于:化学合成、酶合成、较长的前体的酶裂解或化学裂解,等等。合成RNA的方法为本领域已知的(参见例如,Gait,M.J.(编辑)Oligonucleotide synthesis:a practical approach,Oxford[Oxfordshire],Washington,D.C.:IRL Press,1984;和Herdewijn,P.(编辑)Oligonucleotidesynthesis:methods and applications,Methods in molecularbiology,v.288(Clifton,N.J.)Totowa,N.J.:Humana Press,2005)。
形成核酸靶向部分的核酸可包含天然生成的核苷,修饰的核苷,具有在一个或一个以上核苷之间插入的烃连接体(例如,亚烷基)或聚醚连接体(例如,PEG连接体)的天然生成的核苷,具有在一个或一个以上核苷之间插入的烃连接体或PEG连接体的修饰的核苷,或它们的组合。在一些实施方式中,核酸靶向部分的核苷酸或修饰的核苷酸可被烃连接体或聚醚连接体取代,只要核酸靶向部分的结合亲合力和选择性基本不会由于取代而降低(例如,核酸靶向部分对靶点的解离常数不应大于约1×10
本领域普通技术人员已知,根据本发明的核酸可包含天然生成的核酸中发现的全部类型的核苷酸或者可包括一种或一种以上核苷酸类似物或具有与天然生成的核酸的结构不同的结构。美国专利第6,403,779号、第6,399,754号、第6,225,460号、第6,127,533号、第6,031,086号、第6,005,087号、第5,977,089号,这些美国专利中的参考文献公开了多种不同的具体核苷酸类似物和可使用的修饰。参见Crooke,S.(编辑)Antisense DrugTechnology:Principles,Strategies,and Applications(第一版),Marcel Dekker;ISBN:0824705661;第一版(2001)以及其中的参考文献。例如,2’-修饰包括卤代、烷氧基和烯丙氧基。在一些实施方式中,2’-OH基团被选自下列的基团取代:H,OR,R,卤素,SH,SR,NH2,NHR,NR2或CN,其中,R为C1-C6烷基,烯基,或炔基,并且卤素为F,Cl,Br或I。修饰的连接键的实例包括硫代磷酸酯和5’-N-亚磷酰胺连接键。
根据本发明,可使用包含多种不同的核苷酸类似物、修饰的骨架或非天然生成的核苷间连接键的核酸。本发明的核酸可包括天然核苷(即,腺苷、胸苷、鸟苷、胞苷、尿苷、脱氧腺苷、脱氧胸苷、脱氧鸟苷、脱氧胞苷)或修饰的核苷。修饰的核苷酸的实例包括碱基修饰的核苷(例如,阿糖胞苷(aracytidine)、肌核苷、异鸟苷、水粉荤素(nebularine)、假尿苷、2,6-二氨基嘌呤、2-氨基嘌呤、2-硫代胸苷、3-脱氮-5-氮杂胞苷、2’-脱氧尿苷、3-硝基吡咯、4-甲基吲哚、4-硫代尿苷、4-硫代胸苷、2-氨基腺苷、2-硫代胸苷、2-硫代尿苷、5-溴代胞苷、5-碘代尿苷、肌核苷、6-氮尿苷、6-氯代嘌呤、7-脱氮腺苷、7-脱氮鸟苷、8-氮杂腺苷、8-叠氮腺苷、苯并咪唑、M1-甲基腺苷、吡咯并嘧啶、2-氨基-6-氯代嘌呤、3-甲基腺苷、5-丙炔基胞苷、5-丙炔基尿苷、5-溴代尿苷、5-氟代尿苷、5-甲基胞苷、7-脱氮腺苷、7-脱氮鸟苷、8-氧腺苷、8-氧鸟苷、O(6)-甲基鸟嘌呤和2-硫代胞苷),化学或生物修饰的碱基(例如,甲基化的碱基),修饰的糖类(例如,2’-氟代核糖、2’-氨基核糖、2’-叠氮核糖、2’-O-甲基核糖、L-对映异构体核苷阿糖和己糖),修饰的磷酸酯基团(例如,硫代磷酸酯和5’-N-亚磷酰胺连接键)以及它们的组合。用于核酸的化学合成的天然核苷酸单体和修饰的核苷酸单体易于获得。在一些情况下,含有这些修饰的核酸相对于仅由天然生成的核苷酸构成的核酸表现出改善的性质。在一些实施方式中,本文所述的核酸修饰被用于降低和/或防止核酸酶(例如,核酸外切酶,核酸内切酶,等等)消化。例如,核酸的结构可通过在一条链或两条链的3’端包括核苷酸类似物以降低消化来稳定。
修饰的核酸不需要沿着分子的全长进行统一修饰。不同的核苷酸修饰和/或骨架结构可存在于核酸的各个不同位置。本领域普通技术人员可理解的是,核苷酸类似物或其他修饰可位于使核酸的功能基本不受影响的核酸的任何位置。举例而言,修饰可位于使核酸靶向部分特异性结合靶点的能力基本不受影响的核酸靶向部分的任何位置。修饰的区域可位于一条链或两条链的5’端和/或3’端。例如,已使用如下修饰的核酸靶向部分:位于该修饰的核酸靶向部分中的两条链中的任一条链的5’端和/或3’端处的大约1至5个残基为核苷酸类似物和/或具有骨架修饰。所述修饰可为5’或3’末端修饰。一条或两条核酸链可包含至少50%未修饰的核苷酸,至少80%未修饰的核苷酸,至少90%未修饰的核苷酸或100%未修饰的核苷酸。
例如,根据本发明的核酸可包含对糖类、核苷或核苷间连接键的修饰,例如,美国专利申请公开第2003/0175950号,第2004/0192626号,第2004/0092470号,第2005/0020525号以及第2005/0032733号中描述的那些。本发明包括具有本文所述的修饰中的任何一种或一种以上的任何核酸的应用。例如,已报道了多种末端结合物(例如,脂质(例如,胆固醇)、石胆酸、月桂酸(aluric acid)、长支链烷基)改善细胞摄取。例如,可使用本领域已知的任何合适的测试方法检测类似物和修饰,从而选择使治疗剂或诊断剂的递送得以改善、使核酸的靶向部分与靶点的特异性结合得以改善等等的那些类似物和修饰。在一些实施方式中,根据本发明的核酸可包括一个或一个以上非天然核苷连接键。在一些实施方式中,一个或一个以上位于核酸靶向部分的3’端、5’端或3’端和5’端这两端的内在核苷酸被倒转生成诸如3’-3’连接键或5’-5’连接键之类的连接键。
在一些实施方式中,根据本发明的核酸不是合成的,其为已从其天然环境中分离出来的天然生成的实体。
可使用任何方法来设计新的核酸靶向部分(请参见例如下列美国专利:6,716,583;6,465,189;6,482,594;6,458,543;6,458,539;6,376,190;6,344,318;6,242,246;6,184,364;6,001,577;5,958,691;5,874,218;5,853,984;5,843,732;5,843,653;5,817,785;5,789,163;5,763,177;5,696,249;5,660,985;5,595,877;5,567,588和5,270,163以及下列美国专利申请公开:2005/0069910,2004/0072234,2004/0043923,2003/0087301,2003/0054360和2002/0064780)。本发明提供一种用于设计新的核酸靶向部分的方法。本发明还提供一种用于从候选核酸靶向部分的混合物中分离或识别新的核酸靶向部分的方法。
可设计和/或识别与蛋白质、碳水化合物、脂质和/或核酸结合的核酸靶向部分。在一些实施方式中,核酸靶向部分可被设计和/或识别为在与蛋白质和/或其特征部分结合的本发明的复合物中使用,所述蛋白质和/或其特征部分例如,肿瘤标志物、整合素、细胞表面受体、跨膜蛋白、细胞间蛋白质、离子通道、膜转运蛋白、酶、抗体、嵌合蛋白,等等。在一些实施方式中,核酸靶向部分可被设计和/或识别为在与碳水化合物和/或其特征部分结合的本发明的复合物中使用,所述碳水化合物和/或其特征部分例如,糖蛋白、糖类(例如,单糖、二糖和多糖)、多糖包被(即,大多数真核细胞的外表面上的碳水化合物富集的外周区域),等等。在一些实施方式中,核酸靶向部分可被设计和/或识别为在与脂质和/或其特征部分结合的本发明的复合物中使用,所述脂质和/或其特征部分例如,油、饱和脂肪酸、不饱和脂肪酸、甘油酯、激素、类固醇(例如,胆固醇、胆汁酸)、维生素(例如,维生素E)、磷脂、神经鞘脂、脂蛋白,等等。在一些实施方式中,核酸靶向部分可被设计和/或识别为在与核酸和/或其特征部分结合的本发明的复合物中使用,所述核酸和/或其特征部分例如,DNA核酸、RNA核酸、修饰的DNA核酸、修饰的RNA核酸和包括DNA、RNA、修饰的DNA和修饰的RNA的任何组合的核酸,等等。
可使用任何可获得的方法设计和/识别核酸靶向部分(例如,适体或spiegelmer)。在一些实施方式中,核酸靶向部分通过从候选的核酸混合物中识别核酸靶向部分来设计和/或识别。指数富集配体系统进化(SELEX)或其改良方法为从候选的核酸混合物中识别与靶点结合的核酸靶向部分的常用方法。
选择性结合任何靶点的核酸靶向部分可通过SELEX方法或其改良方法分离,条件是所述靶点可用作SELEX方法中的靶点。
总体而言,本发明的化合物包含活化部分。
本文中的“活化部分”是指能够刺激或增强人体的免疫系统或肿瘤细胞的分子或试剂。总体而言,所述活化部分直接或间接地作用于toll样受体、核苷酸-寡聚结构域样受体、RIG-I-样受体、c型凝集素受体或胞质DNA感受器,或者它们的组合。
在一些实施方式中,所述活化部分活化人免疫细胞或肿瘤细胞,或者它们的组合,所述人免疫细胞包括但不限于:树突细胞、巨噬细胞、单核细胞、髓样抑制细胞、NK细胞、B细胞、T细胞。
树突细胞为最强的抗原呈递细胞。树突细胞在启动先天性免疫反应和获得性免疫反应中发挥主要作用。树突细胞还在诱导和维持免疫耐受方面发挥关键作用。
本文中的“树突细胞(DC)”是指异质细胞群,其包括两个主要的亚型,即髓样DC(mDC)和浆细胞样DC(pDC)(Steinman等人,1979,J.Exp.Med.,149,1-16)。这两种血液DC亚组最初通过它们的CD11c(整合素补体受体)和CD123(IL-3Rα)的表达来区分。pDC和mDC群中的每一种构成人体内PBMC群的约0.2%至约0.6%。
本文中的“pDC”是指浆细胞样树突细胞,并且它们代表了在血液和外周淋巴器官中发现的树突细胞的亚型。这些细胞表达表面标志物CD123、BDCA-2(CD303)和BDCA-4(CD304)和HLA-DR,但是不表达CD11c,CD14,CD3,CD20或CD56,这使pDC与一般树突细胞、单核细胞、T细胞、B细胞和NK细胞得以区分。作为先天性免疫系统的成分,这些细胞表达细胞内Toll样受体7和9,这使病毒和细菌核酸能够得到检测,所述病毒和细菌核酸例如,ssRNA或CpG DNA基序。在刺激和随后的活化之后,这些细胞产生大量I型干扰素(主要为IFN-α和IFN-β)和III型干扰素(例如,IFN-λ),这两种干扰素是介导多种作用的重要的多效性抗病毒化合物。通过产生大量I型干扰素、细胞因子和趋化因子,浆细胞样树突细胞广泛参与人体先天性免疫反应和获得性免疫反应。它们可调节NK细胞、T细胞、B细胞和其他涉及免疫反应强度、持续期和反应模式的细胞,因此,它们在肿瘤、感染和自体免疫疾病中发挥非常重要的作用(Liu YJ.IPC:professional type 1interferon-producing cells andplasmacytoid dendritic cell precursors.Annu Rev Immunol.2005;23:275-306.Gilliet M,Cao W,Liu YJ.Plasmacytoid dendritic cells:sensing nucleic acidsin viral infection and autoimmune diseases.Nat Rev Immunol.2008Aug;8(8):594-606)。
本文中的“mDC”是指髓样树突细胞,并且它们表示血液和外周淋巴器官中发现的循环树突细胞的亚型。这些细胞表达表面标志物CD11c,CD1a,HLA-DR以及BDCA-1(CD1c)和BDCA-3(CD141)中的任一种。它们不表达BDCA-2或CD123,这使mDC与pDC得以区分。mDC也不表达CD3,CD20或CD56。作为先天性免疫系统的成分,mDC表达Toll样受体(TLR),该受体包括TLR2、TLR3、TLR4、TLR5、TLR6和TLR8,其使细菌和病毒成分能够得到检测。在刺激和随后的活化之后,这些细胞为最有效的抗原呈递细胞,从而活化抗原特异性CD4和CD8 T细胞。此外,mDC具有产生大量IL-12和IL23的能力,这种能力对于诱导Th1介导的或Th17细胞介导的免疫非常重要。
研究发现许多实体瘤(例如,乳腺癌和头颈癌,卵巢癌)中具有pDC的浸润(Treilleux I,Blay JY,Bendriss-Vermare N等人,Dendritic cell infiltration andprognosis of early stage breast cancer.Clin Cancer Res 2004;10:7466-7474,Hartmann E,Wollenberg B,Rothenfusser S等人,Identification and functionalanalysis of tumor-infiltrating plasmacytoid dendritic cells in head and neckcancer.Cancer Res 2003;63:6478-6487.Zou WP,Machelon V,Coulomb-L'Hermin A,等人,Stromal-derived factor-1in human tumors recruits and alters the functionof plasmacytoid precursor dendritic cells.Nat Med 2001;7:1339-1346),并且研究还发现由肿瘤细胞分泌的因子抑制DC成熟(Gabrilovich DI,Corak J,Ciernik IF等人,Decreased antigen presentation by dendritic cells in patients with breastcancer.Clin Cancer Res 1997;3:483-490.Bell D,Chomarat P,Broyles D等人,Inbreast carcinoma tissue,immature dendritic cells reside within the tumor,whereas mature dendritic cells are located in peritumoral areas.J Exp Med1999;190:1417-1425.Menetrier-Caux C,Montmain G,Dieu MC等人,Inhibition of thedifferentiation of dendritic cells from CD34(+)progenitors by tumor cells:role of interleukin-6and macrophage colony-stimulating factor.Blood 1998;92:4778-4791)。这些未成熟的DC细胞无法在提高抗肿瘤免疫方面发挥作用。相比之下,肿瘤微环境中的DC通过抑制抗肿瘤免疫和促进血管生成而促进肿瘤生长。有证据表明Toll样受体7激动剂咪喹莫特和Toll样受体9激动剂CpG药物可刺激肿瘤微环境中的pDC,从而抑制肿瘤发展(Dummer R,Urosevic M,Kempf W等人,Imiquimod in basal cell carcinoma:howdoes it work?Br J Dermatol 2003;149:57-58.Miller RL,Gerster JF,Owens ML等人,Imiquimod applied topically:a novel immune response modifier and new class ofdrug.Int J Immunopharmacol 1999;21:1-14.Hofmann MA,Kors C,Audring H等人,Phase1evaluation of intralesionally injected TLR9-agonist PF-3512676in patientswith basal cell carcinoma or metastatic melanoma.J Immunother 2008;31:520-527)。
在一些实施方式中,所述人树突细胞是浆细胞样树突细胞。
在一些实施方式中,所述人树突细胞是髓样树突细胞。
在一些实施方式中,所述活化部分能够特异性结合人TLR7或TLR8,由此能够活化pDC或mDC。
在一些实施方式中,所述活化部分能够特异性结合人TLR7和TLR8,由此能够活化pDC和mDC。pDC选择性表达内涵体Toll样受体(TLR)7和TLR9,其中,mDC选择性表达TLR8。BaoM,Liu YJ.Regulation of TLR7/9signaling in plasmacytoid dendritic cells,Protein Cel.2013Jan;4(1):40-52。Hémont C,Neel A,Heslan M,Braudeau C,JosienR.Human blood mDC subsets exhibit distinct TLR repertoire andresponsiveness.J Leukoc Biol.2013Apr;93(4):599-609。
在一些实施方式中,所述活化部分能够活化自然杀伤细胞(NK细胞),优选人NK细胞。
自然杀伤(NK)细胞为一类细胞毒性淋巴细胞,其构成免疫系统的主要成分。NK细胞为由CD56或CD16的表达和T细胞受体(CD3)的缺乏界定的外周血液淋巴细胞的一个亚型。所述自然杀伤细胞以MHC非限制性方式识别并杀伤转化的细胞系而无需引发。NK细胞在抑制肿瘤和防止细胞受到病毒感染方面发挥重要作用。NK细胞识别靶细胞并递送足够的信号以触发靶点溶解的过程由细胞表面上的大量抑制性受体和活化受体确定。将NK自身与改变的NK自身区分开涉及抑制性受体对MHC-1分子和诸如CD48和Clr-1b之类的非MHC配体的识别。感染的或损伤的细胞(改变的自身)的NK识别通过由各种活化受体(包括,NKG2D,Ly49H和NKp46/Ncr1)识别的应激诱导的配体(例如,MICA,MICB,Rael,H60,Mult1)或病毒编码的配体(例如,m157,血凝素)调节。
NK细胞代表异体或自体干细胞移植之后数月外周血液中的主要淋巴样细胞,并且它们在这个时间段对病原体免疫发挥主要作用(Reittie等人(1989)Blood 73:1351-1358;Lowdell等人(1998)Bone Marrow Transplant 21:679-686)。NK细胞在移植、移植物抗宿主疾病、抗白血病活性和移植后感染方面的作用在如下文献中回顾:Lowdell(2003)Transfusion Medicine 13:399-404。
人NK细胞通过天然细胞毒性和抗体依赖性细胞毒性(ADCC)介导肿瘤细胞的溶解和病毒感染的细胞的溶解。
人NK细胞由阳性和阴性细胞溶解信号控制。阴性(抑制性)信号通过包含受体CD94/NKG2A的C-植物凝集素结构域和一些杀伤免疫球蛋白样受体(KIR)转导。通过抑制性信号对NK溶解的调节被称为“自我缺失”假说,其中,特异性HLA(在靶细胞表面表达的I类等位基因)与NK细胞上的抑制性受体结合。肿瘤细胞和一些病毒感染的细胞(例如,CMV)上的HLA分子的下调使这种抑制降低至目标阈值以下并且如果靶细胞还携带NK引发和活化分子,那么靶细胞可变得易受NK细胞介导的溶解影响。TLR7、TLR8或TLR9激动剂可活化mDC和pDC这两者,从而生成I型IFN并表达诸如GITR配体之类的共刺激分子,随后活化NK细胞,从而生成IFN-g并有效促进NK细胞杀伤功能。
抑制性受体归为两组,一组为称为杀伤免疫球蛋白样受体(KIR)的Ig-超家族,另一组为植物凝集素家族(NKG2,在细胞表面与CD94形成二聚体)。KIR具有2个结构域的细胞外结构或3个结构域的细胞外结构并且与HLA-A、HLA-B或HLA-C结合。NKG2/CD94复合物结合HLA-E。
抑制性KIR具有多达4个细胞内结构域,所述结构域包含ITIM并且被表征得最好的抑制性KIR为已知与HLA-C分子结合的KIR2DL1、KIR2DL2和KIR2DL3。KIR2DL2和KIR2DL3结合第一组HLA-C等位基因,而KIR2DL1结合第二组等位基因。一些白血病/淋巴瘤细胞表达第一组HLA-C等位基因和第二组HLA-C等位基因这两者并且已知这些白血病/淋巴瘤细胞为抗NK介导的细胞溶解的。
关于阳性活化信号,ADCC被认为是通过CD16介导的,并且已识别了大量导致天然细胞毒性的触发受体,包括CD2,CD38,CD69,NKRP-I,CD40,B7-2,NK-TR,NKp46,NKp30和NKp44。此外,带有短胞浆内尾部的几种KIR分子也为刺激性的。已知这些KIR(KIR2DS1,KIR2DS2和KIR2DS4)与HLA-C结合,它们的细胞外结构域与它们的相关抑制性KIR相同。活化KIR缺乏ITIM,反而与导致NK细胞活化的DAP12结合。控制抑制性KIR和活化KIR的表达的机制仍然还不清楚。
在一些实施方式中,所述活化部分能够活化肿瘤细胞。一些报道已描述了TLR在小鼠或人癌症或癌症细胞系中的表达。例如,TLR1至TLR6通过结肠、肺、前列腺和黑色素瘤小鼠肿瘤细胞系表达(Huang B等人,Toll-like receptors on tumor cells facilitateevasion of immune surveillance.Cancer Res.2005;65(12):5009–5014),TLR3在人乳腺癌细胞中表达(Salaun B,Coste I,Rissoan MC,Lebecque SJ,Renno T.TLR3 candirectly trigger apoptosis in human cancer cells.J Immunol.2006;176(8):4894–4901),肝癌和胃癌细胞表达TLR2和TLR4(Huang B等人,Listeria monocytogenespromotes tumor growth via tumor cell toll-like receptor 2signaling.CancerRes.2007;67(9):4346–4352),并且TLR9(Droemann D等人,Human lung cancer cellsexpress functionally active Toll-like receptor 9.Respir Res.2005;6:1)和TLR4(He W,Liu Q,Wang L,Chen W,Li N,Cao X.TLR4 signaling promotes immune escape ofhuman lung cancer cells by inducing immunosuppressive cytokines and apoptosisresistance.Mol Immunol.2007;44(11):2850–2859)通过人肺癌细胞表达。在人肺癌肿瘤细胞中发现TLR7和TLR8(Cherfils-Vicini J,Platonova S,Gillard M,Laurans L,Validire P,Caliandro R,Magdeleinat P,Mami-Chouaib F,Dieu-Nosjean MC,FridmanWH,Damotte D,Sautès-Fridman C,Cremer I.J.Clin Invest.2010;120(4):1285–1297)。
对肿瘤细胞的作用可以是各种各样的,例如,在人乳腺癌和黑色素瘤细胞中,通过Poly I:C刺激TLR3直接触发肿瘤细胞凋亡(Salaun B,Coste I,Rissoan MC,Lebecque SJ,Renno T.TLR3 can directly trigger apoptosis in human cancer cells.JImmunol.2006;176(8):4894–4901.,Salaun B,Lebecque S,Matikainen S,Rimoldi D,Romero P.Toll-like receptor 3expressed by melanoma cells as a target fortherapy?Clin Cancer Res.2007;13(15Pt 1):4565–4574)。另一方面,人肺癌细胞表达具有功能活性的Toll-样受体9并且通过CpG-ODN对TLR-9表达细胞系A549和HeLa的刺激表现出抗细胞凋亡作用。尽管如此,累积的数据说明TLR很好地与癌细胞结合并且可直接或间接地影响癌细胞。
总体而言,本发明的活化部分包含蛋白质、抗体、核酸、小分子或配体。
在一些实施方式中,AM能够特异性结合人TLR7和/或人TLR8。
在一些实施方式中,活化部分包含:(a)单链RNA(ssRNA),优选地为ORN02,ORN06,ssPoly(U),ssRNA40,ssRNA41,ssRNA-DR或Poly(dT),或者(b)配体类似物,优选地为CL075,CL097,CL264,CL307,Gardiquimod、洛索立宾(Loxoribine)、咪喹莫特或瑞喹莫德。
在一些实施方式中,活化部分包含TLR配体(TLRL)、Nod-样受体配体、RIG-样受体配体、CLR配体、CDS配体或炎性体诱导物。
在一些实施方式中,活化部分包含一种或一种以上选自以下的TLR的配体:TLR2,TLR3,TLR4,TLR5,TLR7,TLR8,TLR7/TLR8,TLR9和TLR10。
本领域的一些报道已描述了TLR在小鼠或人癌症或癌细胞系中的表达。例如,TLR1至TLR6在结肠小鼠肿瘤细胞系、肺小鼠肿瘤细胞系、前列腺小鼠肿瘤细胞系和黑色素瘤小鼠肿瘤细胞系中表达(Huang B,et al.Toll-like receptors on tumor cellsfacilitate evasion of immune surveillance.Cancer Res.2005;65(12):5009–5014),TLR3在人乳腺癌细胞(Salaun B,Coste I,Rissoan MC,Lebecque SJ,Renno T.TLR3 candirectly trigger apoptosis in human cancer cells.J Immunol.2006;176(8):4894–4901)中表达,肝癌和胃癌细胞表达TLR2和TLR4(Huang B,et al.Listeria monocytogenespromotes tumor growth via tumor cell toll-like receptor 2signaling.CancerRes.2007;67(9):4346–4352),并且TLR9(Droemann D,et al.Human lung cancer cellsexpress functionally active Toll-like receptor 9.Respir Res.2005;6:1)和TLR4(He W,Liu Q,Wang L,Chen W,Li N,Cao X.TLR4 signaling promotes immune escape ofhuman lung cancer cells by inducing immunosuppressive cytokines and apoptosisresistance.Mol Immunol.2007;44(11):2850–2859)在人肺癌细胞中表达。TLR7和TLR8在人肺癌肿瘤细胞中发现(Cherfils-Vicini J,Platonova S,Gillard M,Laurans L,Validire P,Caliandro R,Magdeleinat P,Mami-Chouaib F,Dieu-Nosjean MC,FridmanWH,Damotte D,Sautès-Fridman C,Cremer I.J.Clin Invest.2010;120(4):1285–1297)。
TLR为感应微生物产物和/或启动获得性免疫反应的蛋白质家族。TLR活化树突细胞(DC)。TLR为含有富含亮氨酸的重复单元胞外结构域、跨膜结构域和细胞内TIR(Toll/白介素受体)结构域的保守跨膜分子。TLR识别微生物中的不同结构,通常称为“PAMP”(病原体相关分子模式)。与TLR结合的配体引起细胞内信号通路的级联反应,该级联反应诱导参与炎症和免疫的因子的生成。
可用于这些受体的示例性的激动剂包括但不限于:脂蛋白、脂多肽、肽聚糖、酵母聚糖、脂多糖、奈瑟氏球菌孔蛋白、鞭毛蛋白、profiilin、galactoceramide、胞壁酰二肽、giucopyranosyl脂质A(GLA)和瑞喹莫德(R848)。肽聚糖、脂蛋白和脂磷壁酸为革兰氏阳性细胞壁成分。脂多糖由大多数细菌表达。鞭毛蛋白为细菌鞭毛的结构成分,所述细菌鞭毛的结构成分由病原细菌和共生细菌分泌。半乳糖苷神经酰胺(Galactosylceramide(α-GalCer))为天然杀伤T(NKT)细胞的活化剂。胞壁酰二肽为所有细菌共有的生物活性肽聚糖基序。这些激动剂通过Toll-样受体介导先天性免疫活化。激动剂与其同源受体的特异性结合通常根据亲和性表达。本发明的配体可以约10
已发现了人体中至少八种TLR。在细胞表面表达的TLR包括TLR2,TLR3,TLR4,TLR5,TLR7,TLR8或TLR7/TLR8,TLR9和TLR10,并且TLR通过ER腔室表达。人树突细胞亚类可基于不同的TLR表达模式来识别。例如,髓样DC或“常规”DC亚类(mDC)当被刺激时表达TLR2至TLR8,并且产生一连串活化标志物(例如,CD80,CD86,I类和II类MHC,CCR7),促炎细胞因子和趋化因子。这种刺激和所产生的表达的结果为抗原特异性CD4+T细胞和抗原特异性CD8+T细胞引发。这些DC获得提高的摄取抗原的能力并且以合适的形式将抗原呈递于T细胞。相反,DC的浆细胞样亚类(pDC)在活化后仅仅表达TLR7和TLR9,由此活化NK细胞和T细胞。不受任何特定理论的限制,死亡的肿瘤细胞可对DC功能产生不良影响,假设通过TLR激动剂活化DC在治疗癌症的免疫疗法中对于引发抗肿瘤免疫有益。使用放疗和化疗成功治疗乳腺癌也说明了需要TLR4活化。
本领域已知的且在本发明中有用的TLR激动剂包括但不限于:Pam3Cys(TLR-1/2激动剂),细胞壁成分产物(LAM,LM,LPS,LTA,LTA)(TLR-2激动剂),MALP2(TLR-2激动剂),Pam2Cys(TLR-2激动剂),FSL(TLR-2激动剂),酵母聚糖(TLR-2激动剂),PGN(TLR-2激动剂),polyribosinic,polyribocytidic acid(Poly I:C)(TLR-3激动剂),聚腺苷-聚尿苷酸(polyAU,TLR-3激动剂),通过聚-L-赖氨酸和羧甲基纤维素稳定的聚肌苷-聚胞苷酸
在一些实施方式中,活化部分包含:
(i)TLR2的配体,其选自:(a)热杀死的细菌产物,优选地为HKAL,HKEB,HKHP,HKLM,HKLP,HKLR,HKMF,HKPA,HKPG,或HKSA,HKSP,和(b)细胞壁成分产物,优选地为LAM,LM,LPS,LTA,LTA,PGN,FSL,Pam2CSK4,Pam3CSK4,或酵母聚糖;
(ii)TLR3的配体,其选自:Poly(I:C)和Poly(A:U);
(iii)TLR4的配体,其选自:LPS和MPLA;
(iv)TLR5的配体,其选自:FLA和鞭毛蛋白;
(v)TLR7,TLR 8,或TLR 7和TLR的配体,其选自:ORN02(TLR8),ORN06(TLR8),ssPoly(U)(TLR8),ssRNA40(TLR8),ssRNA41(TLR8),ssRNA-DR(TLR8),Poly(dT)(TLR7/8),CL075(TLR7/8),CL097(TLR7/8),CL264(TLR7),CL307(TLR7),Gardiquimod(TLR7),洛索立宾(TLR7),咪喹莫特(TLR7/8),和瑞喹莫德(TLR7/8);
(vi)TLR9的配体,其选自:ODN1585,ODN1668,ODN1826,ODN2006,ODN2007,ODN2216,ODN2336,ODN2395,和ODN M362;
(vii)TLR10的配体;
(viii)核苷酸-寡聚结构域(NOD)样配体,其选自:NOD1激动剂(C12-iE-DAP,iE-DAP,Tri-DAP)、NOD2激动剂(L18-MDP,MDP,M-TriLYS,M-TriLYS-D-ASN,莫拉丁酯(Murabutide),N-羟乙酸基-MDP)和NOD1/NOD2激动剂(M-TriDAP,PGN);
(ix)一种或一种以上RIG-I样受体(RLR)的配体,其选自:5’ppp-dsRNA,Poly(dA:dT),Poly(dG:dC),和Poly(I:C);
(x)一种或一种以上C型植物凝集素受体(CLR)的配体,其选自:凝胶多糖AL,HKCA,HKSC,WGP,酵母聚糖和海藻糖-6,6-二山嵛酸盐;
(xi)一种或一种以上细胞溶质DNA传感器(CDS)的配体,其选自:c-GMP,c-G-AMP,c-G-GMP,c-A-AMP,c-di-AMP,c-di-IMP,c-di-GMP,c-di-UMP,HSV-60,ISD,pCpG,Poly(dA:dT),Poly(dG:dC),Poly(dA),和VACV-70;以及
(xii)炎性体诱导物,其选自:(a)NLRP3炎性体蛋白质复合物,优选地为明矾晶体,ATP,CPPD晶体,Hermozoin,MSU晶体,纳米SiO2,尼日利亚菌素(Nigericin),以及(b)AIM2炎性体蛋白质复合物,优选地为Poly(dA:dT)。
在一些实施方式中,活化部分包括IMO2134,IMO205,MGN-1703,MGN-1704,阿托莫得(Agatolimod),SD-101,QAX-935,DIMS0150,Pollinex Quattro,OM-174,Eritoran,TAK-242,NI-0101,I型干扰素,II型干扰素,III型干扰素,IL-12,IL-23,IL-18,IL-7或IL-15。
在一些实施方式中,活化部分是TLR7和/或TLR8激动剂。
在一些实施方式中,活化部分能够特异性结合人TLR7和/或人TLR8,优选地特异性结合TLR7和TLR8这两者(例如,瑞喹莫德(R848)),由此活化部分能够活化pDC和mDC这两者。TLR7和TLR8是与系统发育和结构相关的。TLR7由人pDC和B细胞选择性表达。TLR8主要由mDC、单核细胞、巨噬细胞和骨髓抑制细胞表达。TLR7特异性激动剂活化浆细胞样DC(pDC),从而生成大量1型IFN并且表达高水平的共刺激分子,所述共刺激分子促进T细胞、NK细胞、B细胞和mDC的活化。TLR8特异性激动剂活化髓样DC、单核细胞、巨噬细胞或骨髓衍生的抑制细胞,从而生成大量1型IFN、IL-12和IL-23,并且表达高水平的I类MHC、II类MHC和共刺激分子,所述I类MHC、II类MHC和共刺激分子促进抗原特异性CD4和CD8+T细胞的活化。
通过TLR活化人免疫细胞的其他部分或分子可由TLR报告体细胞系(例如,来自InvivoGen的293/TLR报告体细胞)识别。
可活化TLR7和/或TLR8的其他部分或分子可通过使用来自InvivoGen的293/TLR报告体细胞而被识别,并且可活化TLR7和/或TLR8的其他部分或分子可通过这些其他部分刺激pDC生成1型IFN的能力、刺激mDC生成1型IFN和IL-12的能力以及上调CD80和CD86的表达的能力而被识别。
能够通过1型IFN和共刺激分子(例如,由活化的pDC和/或mDC表达的GITR-配体)活化NK细胞的活化部分可通过总外周血单核细胞检测中NK细胞增殖和活化标志物CD69的表达来识别。
能够活化肿瘤细胞以使其经历细胞死亡并生成诸如1型IFN、IL-1a/b、TNF或IL-6之类的细胞因子的活化部分可使用本领域已知的方法来识别。
能够活化DC细胞、单核细胞、巨噬细胞、骨髓抑制细胞、B细胞、NK细胞、T细胞或肿瘤细胞的其他活化部分或分子可使用本领域已知的方法识别。
在一些实施方式中,当靶向部分包括抗PD-L1或PD-1抗体(或其片段)或适体时,活化部分包括CpG寡核苷酸。
在一些实施方式中,尤其是当靶向部分包括抗体或其片段或者适体时,活化部分不包括治疗剂,所述治疗剂例如,化疗剂或毒素(例如,抗体-药物结合物中通常使用的毒素)。因此,一般而言,本发明的化合物不包括抗体-药物结合物(ADC)。
在一些实施方式中,所述活化部分是由下式(II)的结构表示的TLR7和/或TLR8激动剂或其药学上可接受的盐或溶剂化物:
其中,虚线表示存在化学键或不存在化学键,
X是S或-NR
W
W
W
W
W
Z是氢,烷基,烯基,炔基,烷氧基,芳基,卤代烷基,杂芳基,杂环基,它们中的每一个可被一个或一个以上选自下列基团的取代基任选地取代:羟基,烷氧基,烷基,烯基,炔基,芳基,杂芳基,杂环基,卤素,氰基,硝基,--N(R
R为氢,烷基,烷氧基,卤代烷基,卤素,芳基,杂芳基,杂环基,它们中的每一个被一个或一个以上选自下列基团的取代基任选地取代:羟基,烷氧基,烷基,烯基,炔基,环烷基,芳基,杂芳基,杂环基,--NH
n为0,1,2,3或4;
Y为–NR
其中,R
任选地,X和Z一同可形成5至9元环。
在一些实施方式中,式(II)中的X是S。
在一些实施方式中,式(II)中的X是–NR
在一些实施方式中,式(II)中的Z为氢、烷基、烷氧基、芳基、杂芳基、卤代烷基,它们中的每一个被一个至三个选自下列基团的取代基任选地取代:羟基、烷基、芳基、杂芳基、杂环基、氰基、--烷氧基-烷基、硝基和—N(R
在一些实施方式中,式(II)中的Y为—NH
在一些实施方式中,式(II)中的n为1或2。
在一些实施方式中,式(II)中的R为芳基或杂芳基,它们中的每一个被一个至三个选自下列基团的取代基任选地取代:羟基,烷氧基,--烷基-羟基,--O-R
在一些实施方式中,活化部分是选自表1的TLR7和/或TLR8激动剂。表1中的化合物在下列参考文献中详细描述并表征:US4,689,338,US5,389,640,US5,226,575,US6,110,929,US6,194,425,US5,352,784,US6,331,539,US5,482,936,US6,451810,WO2002/46192,WO2002/46193,WO2002/46194,US2004/0014779和US2004/0162309。
表1:代表性的TLR7和/或TLR8激动剂
优选地,AM是瑞喹莫德或咪喹莫特。
在一些实施方式中,活化部分是下式(VIII)的结构所代表的TLR7和/或TLR8激动剂或其药学上可接受的盐或溶剂化物:
其中,
其中,V是–NR
R
在一些实施方式中,活化部分是下式(IX)的结构所代表的TLR7和/或TLR8激动剂:
其中,
并且,R
在一些实施方式中,式(IX)的化合物中的R
此外,在式(IX)的化合物中,R
在一些可选的实施方式中,N
在一些实施方式中,式(IX)的化合物中的R
在一些实施方式中,活化部分是下式(XI)的结构所代表的TLR7和/或TLR8激动剂:
其中,R
在一些可选的实施方式中,式(IX)或(XI)中的R
并且余下的R
在一些可选的实施方式中,TLR是选自以下的化合物:
可选地,所述化合物选自:
在一些可选的实施方式中,TLR激动剂化合物是:
在一些可选的实施方式中,TLR激动剂是选自以下的化合物:
在一些可选的实施方式中,TLR激动剂是:
在一些可选的实施方式中,TLR激动剂是选自以下的化合物:
在一些实施方式中,活化部分是下式(XII)的结构所代表的TLR7和/或TLR8激动剂:
其中,Y是CF
R
或,R
R
R
R
或,R
在式(XII)的化合物的一些实施方式中,R
在式(XII)的化合物的一些实施方式中,R
在式(XII)的化合物的一些实施方式中,Y是CF
在一些实施方式中,活化部分是下式(XIII)的结构所代表的TLR7和/或TLR8激动剂:
其中:Z是H、烷基、烯基、炔基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、OR
R
或,R
或,R
R
R
或,R
在一些实施方式中,活化部分是下式(XIV)的结构所代表的TLR7和/或TLR8激动剂:
其中:Z是H、烷基、烯基、炔基、杂烷基、环烷基、杂环烷基、芳基、杂芳基、OR
R
或,R
或,R
R
R
或,R
n是0、1、2、3或4。连接体连接至所述激动剂的可能的连接位点中的一个,例如-NH
在一些实施方式中,Z是OR
在一些实施方式中,Z是NR
在一些实施方式中,R
在一些实施方式中,活化部分是下式(XV)的结构所表示的TLR7和/或TLR8激动剂:
其中,环A代表6元至10元芳香碳环或5元至10元杂芳香环。
R代表卤素原子、烷基、羟基烷基、卤代烷基、烷氧基、羟基烷氧基、卤代烷氧基、氨基、烷基氨基、二烷基氨基或含有1至2个环杂原子的4元至7元环,所述1至2个环杂原子选自:1个至2个氮原子以及任选地0至1个氧原子或0至1个硫原子;
n代表0至2的整数,当n是2时,R可以相同或可以不同。
Z
X
Y
X
R
R
在一些实施方式中,R
Y
X
Z
X
Y
Y
n是整数0、1或2;
R代表卤素或C
R
R
R
R
(i)3元至8元杂环,其包含1个或2个选自环基团NR
(ii)C
(iii)C
R
R
R
R
R
R
R
R
R
R
m、p、q和r分别独立地代表整数0、1或2;并且
A代表C
在一些实施方式中,活化部分是具有下述结构的TLR7和/或TLR8激动剂:
其中,R是Me或H。
在一些实施方式中,活化部分是具有下述结构的TLR7和/或TLR8激动剂:
在一些实施方式中,活化部分是具有下式(XVI)的结构的TLR7和/或TLR8激动剂:
其中,R
R
在一些实施方式中,R
在一些实施方式中,R
在一些实施方式中,R
在一些实施方式中,TLR7和/或TLR8激动剂选自下列化合物:
以及
在一些实施方式中,活化部分是具有下述结构的TLR7和/或TLR8激动剂:
其中:
R
R
R
X是O或S;
Y是H,卤素,OH,OR
Z是H,卤素,OH,OR
在一些实施方式中,活化部分是具有下式(XVIII)的结构的TLR7和/或TLR8激动剂:
其中:
Y—Z是—CR
L
R
X
D是碳环,取代的碳环,杂环或取代的杂环,其中,所述碳环、所述取代的碳环、所述杂环或所述取代的杂环是被一个或两个-L
D是杂环,取代的杂环,杂芳基或取代的杂芳基,其中,所述杂环,所述取代的杂环,所述杂芳基或所述取代的杂芳基包含一个至四个氮原子;
L
R
n是0,1,2,3,4或5;
R
R
当R
位于相邻碳原子上的两个R
R
R
R
R
R
R
其中,每个取代的烷基,取代的烯基,取代的炔基,取代的杂烷基,取代的碳环,取代的碳环烷基,取代的杂环,取代的杂环烷基,取代的芳基烷基,取代的杂芳基烷基,取代的碳环杂烷基,取代的杂环杂烷基,取代的芳基杂烷基,取代的杂芳基杂烷基,取代的亚烷基,取代的杂亚烷基,取代的亚烯基,取代的亚炔基,取代的碳环烯基或取代的杂环烯基被一个至四个取代基独立地取代,所述取代基选自:卤素,—R,—O
在一些实施方式中,活化部分为具有下述结构的TLR7和/或TLR8激动剂:
其中:
L
R
R
X
D是苯基,联苯基,吡啶基,其中,所述苯基,所述联苯基或所述吡啶基被-L
D是吡啶基,哌啶基,吡嗪基或1,2,3,4-四氢异喹啉基;
n是0或1;
R
L
R
R
在一些实施方式中,活化部分是具有下述结构的TLR7和/或TLR8激动剂:
在一些实施方式中,活化部分不包括WO2014012479A1中公开的TLR拮抗剂,尤其是其中公开的瑞喹莫德(R848)及其类似物。
总体而言,本发明的化合物包含连接体,所述连接体连接靶向部分和活化部分。尽管,一些化合物不含连接体,活化部分和靶向部分直接连接。
本文中的“连接体”是指使第一分子与第二分子通过化学键连接的部分。在本发明的连接体中,可割断连接,从而释放第一和/或第二分子的生物活性形式。连接体的优选实例为包含在中性pH条件下稳定但在较低pH条件下易于发生裂解的化学键的部分。具体而言,优选的连接体的实例为包含在pH为7至8的条件下稳定但在pH为4至6的条件下易于裂解的化学键的部分。连接体的另一实例为包含在存在酶的条件下易于裂解的化学键的部分。这些对酶敏感的连接体的优选实例为肽,所述肽包含内涵体肽酶的识别序列。连接体的另一实例为对氧化还原电位敏感的连接体,该连接体在低还原电位(例如,低浓度的硫醇或谷胱甘肽)条件下稳定但在高还原电位(例如,高浓度的硫醇或谷胱甘肽)条件下裂解。这些对氧化还原电位敏感的连接体的优选实例包括二硫化物和次磺酰胺。具体而言,优选的实例包括取代的芳基-烷基二硫化物,其中,芳基被有空间需求的且吸电子或给电子的取代基取代,从而控制趋于与硫醇反应的二硫键的敏感性。连接体的另一实例为包含在暴露于辐射之后易于裂解的化学键的部分。这些对辐射敏感的连接体的优选实例为2-硝基苄基醚,其在暴露于光之后裂解。具体而言,这些连接体的优选实例为如下部分:在连接键被割断之前该部分掩盖两个连接的分子中的一个的生物活性。
在一些实施方式中,本发明的化合物包含选自如下基团的连接体:肼基团、多肽、二硫化物基团和硫醚基团。
本文中的“肼基团”或“肼连接体”或“自环化肼连接体”是指在改变条件(例如,pH改变)之后可发生环化反应并形成一个或一个以上环的连接体部分。当连接时,肼基团被转化为腙。这种连接可通过例如在L4部分与酮基团反应而发生。因此,术语肼连接体也可用于描述本发明的连接体,因为这种向腙的转化发生在连接之后。
本文中的“五元肼连接体”是指含有肼的分子部分,该部分在条件发生改变(例如,pH发生改变)之后会进行环化反应并形成一个或一个以上五元环。可选地,该五元连接体可被类似地描述为五元肼连接体。
本文中的“六元肼连接体”是指含有肼的分子部分,该部分在条件发生改变(例如,pH发生改变)之后会进行环化反应并形成一个或一个以上六元环。该六元连接体可被类似地描述为六元肼连接体。
本文中的“环化反应”是指肽、肼或二硫化物连接体的环化,该“环化反应”表示连接体环化形成环并且启动药物配体复合物的分离。环化速率可异地测量并且当至少90%、95%或100%的产物形成时完成环化。
在一些实施方式中,本发明的化合物包含位于靶向部分和活化部分之间的连接体区域,并且所述连接体可被存在于细胞内环境(例如,溶酶体或内涵体或小凹内)中的裂解剂裂解。连接体可为例如,肽基连接体,该肽基连接体由细胞内肽酶或蛋白酶(包括但不限于:溶酶体蛋白酶或内涵体蛋白酶)裂解。通常,肽基连接体的长度为至少两个氨基酸的长度或至少三个氨基酸的长度。裂解剂可包括组织蛋白酶B、组织蛋白酶D和纤溶酶,已知组织蛋白酶B、组织蛋白酶D和纤溶酶均可水解二肽药物衍生物,导致靶细胞内部的活性药物释放(参见例如,Dubowchik and Walker,1999,Pharm.Therapeutics83:67-123)。最典型的连接体为肽基连接体,其可由存在于靶细胞或组织中的酶裂解。例如,可使用可由硫醇依赖性蛋白酶组织蛋白酶-B裂解的肽基连接体(例如,Phe-Leu或(Gly-Phe-Leu-Gly)连接体),所述硫醇依赖性蛋白酶组织蛋白酶-B在癌组织中高表达。其他这类连接体例如在美国专利第6,214,345号中描述。在一些实施方式中,可由细胞内蛋白酶裂解的肽基连接体为Val-Cit连接体或Phe-Lys连接体(参见例如,美国专利第6,214,345号,该美国专利描述了带有val-cit连接体的阿霉素的合成)。使用细胞内蛋白水解释放治疗剂的一个优势为所述治疗剂通常在偶联时被弱化并且结合物的血清稳定性通常较高。
在一些实施方式中,可裂解的连接体为pH敏感的,即对某一pH值条件敏感而发生水解。通常,pH敏感的连接体在酸性条件下可水解。例如,可使用在溶酶体中可水解的酸不稳定连接体(例如,腙、缩氨基脲、硫代缩氨基脲、顺式乌头酰胺、原酸酯、缩醛、缩酮,等等),参见例如,美国专利第5,122,368号,第5,824,805号,第5,622,929号,Dubowchik andWalker,1999,Pharm.Therapeutics 83:67-123;Neville等人,1989,Biol.Chem.264:14653-14661。这些连接体在中性pH条件下相对稳定,例如,在血液中的那些连接体,但是在低于5.5或5.0的pH条件(大约为溶酶体的pH)下不稳定。在一些实施方式中,可水解的连接体为硫醚连接体(例如,通过酰基腙化学键与治疗剂连接的硫醚,参见例如,美国专利第5,622,929号)。
在其他实施方式中,连接体为在还原性条件下可裂解的(例如,二硫化物连接体)。本领域已知多种二硫化物连接体,包括例如,可使用SATA(N-琥珀酰亚胺基-5-乙酰基硫代乙酸酯),SPDP(N-琥珀酰亚胺基-3-(2-吡啶基二硫代)丙酸酯),SPDB(N-琥珀酰亚胺基-3-(2-吡啶基二硫代)丁酸酯)和SMPT(N-琥珀酰亚胺基-氧羰基-α-甲基-α-(2-吡啶基二硫代)甲苯),SPDB和SMPT(参见例如,Thorpe等人,1987,Cancer Res.47:5924-5931;Wawrzynczak等人,In Immunoconjugates:Antibody Conjugates in Radioimagery and Therapy ofCancer(C.W.Vogel编辑),Oxford U.出版,1987.还参见美国专利第4,880,935号)形成的那些连接体。
在其他具体实施方式中,连接体为丙二酸酯连接体(Johnson等人,1995,Anticancer Res.15:1387-93),马来酰亚胺苯甲酰基连接体(Lau等人,1995,Bioorg-Med-Chem.3(10):1299-1304)或3’-N-酰胺类似物(Lau等人,1995,Bioorg-Med-Chem.3(10):1305-12)。
通常,连接体对细胞外环境基本不敏感。本文在连接体的部分使用的“对细胞外环境基本不敏感”是指在本发明的化合物样品中,当本发明的化合物存在于细胞外环境(例如,血浆中)时,不超过约20%的连接体裂解,典型地不超过约15%的连接体裂解,更加典型地不超过约10%连接体裂解,甚至更加典型地不超过约5%的连接体裂解,不超过约3%的连接体裂解,或者不超过约1%的连接体裂解。例如,可通过将(a)本发明的化合物(“化合物样品”)和(b)等摩尔量的未偶联的抗体或治疗剂(“对照样品”)独立地与血浆一同孵育一段预定的时间段(例如,2小时、4小时、8小时、16小时或24小时)并随后比较存在于化合物样品中的未偶联的抗体或治疗剂的量和存在于对照样品中的未偶联的抗体或治疗剂的量(例如通过高效液相色谱测量)来测定连接体是否对细胞外环境基本不敏感。
在其他不相互排斥的实施方式中,连接体促进细胞内在化。在一些实施方式中,当连接体偶联于活化部分时,连接体促进细胞内在化。在其他实施方式中,当连接体偶联于靶向部分和活化部分这两者时,连接体促进细胞内在化。
可在本发明的组合物和方法中使用的多种连接体在WO2004010957(名称为“DrugConjugates and Their Use for Treating Cancer,An Autoimmune Disease or anInfectious Disease”)以及US20120141509A1和US20120288512A1(其公开的内容通过引用并入本文)中描述。
在一些实施方式中,连接体单元具有如下通式:
-Ta-Ww-Yy-
其中,-T-为支架单元,a为0或1,-W-分别独立地为氨基酸单元,w独立地为2至12的整数,-Y-为间隔单元,y为0、1或2。
当存在支架单元(-T-)时,该支架单元将靶向部分连接至氨基酸单元(-W-)。可天然地或通过化学操作存在于靶向部分(例如,抗体)上的有用的官能团包括但不限于:巯基、氨基、羟基、碳水化合物的异头羟基和羧基。合适的官能团为巯基和氨基。巯基可通过还原抗体的分子内二硫键生成。可选地,巯基可通过抗体的赖氨酸基团的氨基与2-亚氨基硫杂环戊烷(Traut试剂)或其他巯基生成试剂的反应生成。在一些实施方式中,抗体为重组抗体并且被设计为带有一个或一个以上赖氨酸。在其他实施方式中,重组抗体被设计为带有额外的巯基基团,例如,额外的半胱氨酸。
在一些实施方式中,支架单元与抗体的硫原子形成化学键。所述硫原子可来源于还原的抗体(A)的巯基基团(-SH)。这些实施方式的代表性的支架单元在式(XXa)和(XXb)的方括号中描述,其中,A-、-W-、-Y-、-D、w和y为如上所定义的,并且R
示例性的支架单元为R
另一示例性的支架单元为R
另一示例性的支架单元为R
在一些其他具体实施方式中,支架单元通过抗体单元的硫原子和支架单元的硫原子之间的二硫键连接至抗体单元(A)。本实施方式的代表性的支架单元在式(XXI)的方括号中描述,其中,R
在其他具体实施方式中,支架的反应性基团包含可与抗体的氨基基团反应的反应位点。氨基基团可为精氨酸或赖氨酸。合适的胺反应位点包括但不限于:活化的酯(例如,琥珀酰亚胺酯、4-硝基苯基酯、五氟代苯基酯),酸酐、酰氯、磺酰氯、异氰酸酯和异硫氰酸酯。这些实施方式的代表性的支架单元在式(XXIIa)和(XXIIb)的方括号中描述,其中,R
另一方面,支架的反应性官能团包含与可存在于抗体上的修饰的碳水化合物基团反应的反应位点。在一些实施方式中,抗体通过酶的方式被糖基化,从而生成碳水化合物基团。碳水化合物可被诸如高碘酸钠之类的试剂温和地氧化,得到的氧化的碳水化合物的羰基单元可与含有诸如酰肼、肟、反应性胺、肼、硫代氨基脲、肼羧酸酯和芳基酰肼(例如Kaneko等人,1991,Bioconjugate Chem 2:133-41中所描述的那些)之类的官能团的支架缩合。本实施方式的代表性的支架单元在式(XXIIIa)至(XXIIIc)的方括号中描述,其中,R
如果间隔单元存在的话,氨基酸单元(-W-)将支架单元(-T-)连接至间隔单元(-Y-),如果间隔单元不存在的话,氨基酸单元将支架单元连接至细胞毒性剂或细胞抑制剂(活化部分,D)。-Ww-为二肽、三肽、四肽、五肽、六肽、七肽、八肽、九肽、十肽、十一肽或十二肽单元。每个-W-单元独立地具有如下方括号中所述的通式,并且w为2至12的整数。
其中,R
连接体单元的氨基酸单元可通过酶(包括但不限于:肿瘤相关蛋白酶)的方式被酶裂解,从而释放出活化部分(-D),释放后该活化部分在体内被质子化,从而产生活化分子(D)。
示例性的W
其中,R
其中,R
其中,R
合适的氨基酸单元包括,但不限于:式(XXIVa)单元,其中,R
一些实施方式中,-Ww-为二肽、三肽或四肽单元。
在R
在一些实施方式中,氨基酸单元为苯基丙氨酸-赖氨酸二肽(Phe-Lys或FK连接体)。在一些实施方式中,氨基酸单元为缬氨酸-瓜氨酸二肽(Val-Cit或VC连接体)。在一些实施方式中,氨基酸单元为5-氨基戊酸、均苯基丙氨酸赖氨酸、四异喹啉羧酸酯赖氨酸、环己基丙氨酸赖氨酸、异哌啶酸(isonepecotic acid)赖氨酸、β-丙氨酸赖氨酸、甘氨酸丝氨酸缬氨酸谷酰胺或异哌啶酸。
氨基酸单元可包含天然氨基酸。在其他实施方式中,氨基酸单元可包含非天然氨基酸。
当存在间隔单元(-Y-)时,该间隔单元将氨基酸单元连接至药物单元。间隔单元为两个大类:自切除的(self-immolative)和非自切除的。非自切除的间隔单元为在TM-连接体-AM结合物或药物连接体化合物中的氨基酸单元酶裂解之后间隔单元的一部分或全部仍然连接于活化部分单元的间隔单元。非自切除间隔单元的实例包括但不限于:(甘氨酸-甘氨酸)间隔单元和甘氨酸间隔单元。当含有甘氨酸-甘氨酸间隔单元或甘氨酸间隔单元的TM-连接体-AM结合物通过肿瘤细胞相关蛋白酶、癌细胞相关蛋白酶或淋巴细胞相关蛋白酶发生酶裂解时,甘氨酸-甘氨酸-药物部分或甘氨酸-药物部分从A-T-Ww-中裂解出来。为了释放出AM,应当在靶细胞中进行独立的水解反应,从而使甘氨酸-药物单元化学键裂解。
在典型的实施方式中,-Yy-为可被Qm取代的对氨基苄基醚,其中,Q为-C
在一些实施方式中,非自切除间隔单元(-Y-)为-Gly-Gly-。
在一些实施方式中,非自切除间隔单元(-Y-)为-Gly-。
在一种实施方式中,AM-连接体化合物或TM-连接体-AM结合物缺乏间隔单元(y=0)。
可选地,含有自切除间隔单元的TM-连接体-AM结合物可释放AM(D),无需单独的水解步骤。在这些实施方式中,-Y-为对氨基苄基醇(PAB)单元,该对氨基苄基醇(PAB)单元通过PAB基团的氮原子与-Ww-连接并且通过碳酸酯基团、氨基甲酸酯基团或醚基团直接连接于-D。
自切除间隔单元的其他实例包括,但不限于:与PAB基团电子等价的芳香族化合物,例如,2-氨基咪唑基-5-甲醇衍生物(参见例如,Hay等人,1999,Bioorg.Med.Chem.Lett.9:2237)和邻位或对位-氨基苄基缩醛。可使用酰胺键水解后易于发生环化的间隔单元,例如,取代和未取代的4-氨基丁酸酰胺(Rodrigues等人,1995,Chemistry Biology 2:223),适当取代的双环[2.2.1]和双环[2.2.2]环系统(Storm等人,1972,J.Amer.Chem.Soc.94:5815)以及2-氨基苯基丙酸酰胺(Amsberry等人,1990,J.Org.Chem.55:5867)。甘氨酸的α-位被取代的含胺药物的消除(Kingsbury等人,1984,J.Med.Chem.27:1447)也是自切除间隔单元的策略的实例,该策略可用于TM-连接体-AM结合物。
在可选的实施方式中,间隔单元为支链双(羟甲基)苯乙烯(BHMS)单元,其可用于合并多个部分。
典型的间隔单元(-Yy-)由下式(XXVa)-(XXVc)表示:
其中,Q为C
在一些实施方式中,连接体是酶可裂解的。在一些实施方式中,连接体不是酶可裂解的。
在一些实施方式中,所述连接体由下式(III)的结构表示:
m为1,2,3,4,5或6,b分别独立地为0或1,并且D由下式(IV)的结构独立地表示:
其中,i分别独立地为0或1;
j分别独立地为0,1,2,3,4,5或6;
A分别独立地为S,O或N-Ra,其中,Ra为氢,烷基,烯基或烷氧基;
B分别独立地为烷基,烯基,--O-烷基--,--烷基-O--,--S-烷基--,--烷基-S--,芳基,杂芳基,杂环基或肽,它们中的每一个被一个或一个以上选自下列基团的取代基任选地取代:羟基,烷氧基,烷基,烯基,炔基,环烷基,--烷基-芳基,--烷基-杂芳基,--烷基-杂环基,--O-R
在一些实施方式中,所述连接体由下述式(V)至式(VII)的结构表示:
A,B,i和j是上文所定义的。
在一些实施方式中,所述连接体选自:S1、S2、S3、S4、S5、S6、S7、–Gly-Phe-Leu-Gly-、-Ala-Leu-Ala-Leu-、-Phe-Arg-、-Phe-Lys-、-Val-Lys-、-Val-Ala-或Val-Cit-,其中,S1至S7由下述结构表示:
其中,m分别独立地为1至20。优选地,m为1至3,1至5,1至10或2至5。
因此,本发明提供一种式(I)的化合物,其中,TM是抗-PD-L1抗体,L选自:S1、S2、S3、S4、S5、S6、S7、–Gly-Phe-Leu-Gly-、-Ala-Leu-Ala-Leu-、-Phe-Arg-、-Phe-Lys-、-Val-Lys-、-Val-Ala-或Val-Cit-;AM是式(II)的化合物。在一种实施方式中,TM是MPDL3280A,MEDI-4736,BMS-936559或MSB0010718C,AM是选自表1的化合物,其中,喹啉环上的胺基团是与连接体连接的连接点。在一种实施方式中,TM是MPDL3280A,MEDI-4736,BMS-936559或MSB0010718C,L是S1,S2或S3;AM是选自表1的化合物,其中,喹啉环上的胺基团是与连接体连接的连接点。在一种实施方式中,TM是西妥昔单抗,L是S1,S2或S3;AM是瑞喹莫德或咪喹莫特,其中,喹啉环上的胺基团是与连接体连接的连接点。
另一方面,本发明提供一种式(I)的化合物,其中,TM是抗-PD-1抗体;L选自:S1、S2、S3、S4、S5、S6、S7、–Gly-Phe-Leu-Gly-、-Ala-Leu-Ala-Leu-、-Phe-Arg-、-Phe-Lys-、-Val-Lys-、-Val-Ala-或Val-Cit-;AM是式(II)的化合物。在一种实施方式中,TM是MK-3475,AMP-514,AMP-224,BMS-936558或CT-011,AM是选自表1的化合物,其中,喹啉环上的胺基团是与连接体连接的连接点。在一种实施方式中,TM是MK-3475,AMP-514,AMP-224,BMS-936558或CT-011,L是S1,S2或S3;AM是选自表1的化合物,其中,喹啉环上的胺基团是与连接体连接的连接点。在一种实施方式中,TM是西妥昔单抗,L是S1,S2或S3;AM是瑞喹莫德或咪喹莫特,其中,喹啉环上的胺基团是与连接体连接的连接点。
另一方面,本发明提供具有下式A至下式C的结构的化合物或其药学上可接受的盐或溶剂化物:
在一些实施方式中,式(II)的化合物是瑞喹莫德或咪喹莫特,其中,喹啉环上的胺基团是与连接体连接的连接点。
在一些实施方式中,PD-L/PD-1轴拮抗剂是抗-PD-L1抗体,其选自:YW243.55.S70,MPDL3280A,MEDI-4736,BMS-936559和MSB0010718C。
在一些实施方式中,PD-L/PD-1轴拮抗剂是抗PD-L1抗体,其选自:YW243.55.S70,MPDL3280A,MEDI-4736,BMS-936559和MSB0010718C;式(II)的化合物选自表1中的化合物,其包括:
2-丙基噻唑并[4,5-c]喹啉-4-胺,
1-(2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-氨基-2-(乙氧基甲基)-a,a-二-甲基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-(4-氨基-2-乙基氨基甲基咪唑并-[4,5-c]喹啉-1-基)-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基-]甲磺酰胺,
4-氨基-2-乙氧基甲基-aa-二甲基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1-乙醇,
4-氨基-aa-二甲基-2-甲氧基乙基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-{2-[3-(苄氧基)丙氧基]乙基}-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-丁基脲,
N1-[2-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)乙基]-2-氨基-4-甲基戊酰胺,
N-(2-{2-[4-氨基-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-苯基脲,
1-(2-氨基-2-甲基丙基)-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
1-{4-[(3,5-二氯苯基)磺酰基]丁基}-2-乙基-1H-咪唑并[4,5-c]喹啉-4-胺,
N-(2-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-环己基脲,
N-{3-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]丙基}-n'-(3-氰基苯基)硫脲,
N-[3-(4-氨基-2-丁基-1H-咪唑并[4,5-c]喹啉-1-基)-2,2-二甲基丙基]苯甲酰胺,
2-丁基-1-[3-(甲基磺酰基)丙基]-1H-咪唑并[4,5-c]喹啉-4-胺,
N-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]-1,1-二甲基乙基}-2-乙氧基乙酰胺,
1-[4-氨基-2-乙氧基甲基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
1-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-{3-[4-氨基-1-(2-羟基-2-甲基丙基)-2-(甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-7-基]苯基}甲磺酰胺,
1-[4-氨基-7-(5-羟基甲基吡啶-3-基)-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
3-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]丙-1,2-二醇,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-丙基脲,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-环戊基脲,
1-[(2,2-二甲基-1,3-二氧戊环-4-基)甲基]-2-(乙氧基甲基)-7-(4-羟基甲基苯基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-[4-氨基-2-乙氧基甲基-1-(2-羟基-2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-7-基]-N-甲氧基-N-甲基苯甲酰胺,
2-乙氧基甲基-N1-异丙基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1,4-二胺,
1-[4-氨基-2-乙基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基]甲磺酰胺,或
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-环己基脲,
其中,喹啉环上的胺基团是与连接体连接的连接点。
在一些实施方式中,PD-L/PD-1轴拮抗剂是抗-PD-1抗体,其选自:MK-3475,AMP-514,AMP-224,BMS-936558和CT-011。
在一些实施方式中,PD-L/PD-1轴拮抗剂是抗-PD-1抗体,其选自:MK-3475,AMP-514,AMP-224,BMS-936558和CT-011;式(II)的化合物选自表1中的化合物,其包括:
2-丙基噻唑并[4,5-c]喹啉-4-胺,
1-(2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-氨基-2-(乙氧基甲基)-a,a-二-甲基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-(4-氨基-2-乙基氨基甲基咪唑并-[4,5-c]喹啉-1-基)-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基-]甲磺酰胺,
4-氨基-2-乙氧基甲基-aa-二甲基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1-乙醇,
4-氨基-aa-二甲基-2-甲氧基乙基-1H-咪唑并[4,5-c]喹啉-1-乙醇,
1-{2-[3-(苄氧基)丙氧基]乙基}-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-丁基脲,
N1-[2-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)乙基]-2-氨基-4-甲基戊酰胺,
N-(2-{2-[4-氨基-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-苯基脲,
1-(2-氨基-2-甲基丙基)-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-4-胺,
1-{4-[(3,5-二氯苯基)磺酰基]丁基}-2-乙基-1H-咪唑并[4,5-c]喹啉-4-胺,
N-(2-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]乙氧基}乙基)-n'-环己基脲,
N-{3-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]丙基}-n'-(3-氰基苯基)硫脲,
N-[3-(4-氨基-2-丁基-1H-咪唑并[4,5-c]喹啉-1-基)-2,2-二甲基丙基]苯甲酰胺,
2-丁基-1-[3-(甲基磺酰基)丙基]-1H-咪唑并[4,5-c]喹啉-4-胺,
N-{2-[4-氨基-2-(乙氧基甲基)-1H-咪唑并[4,5-c]喹啉-1-基]-1,1-二甲基乙基}-2-乙氧基乙酰胺,
1-[4-氨基-2-乙氧基甲基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
1-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-{3-[4-氨基-1-(2-羟基-2-甲基丙基)-2-(甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-7-基]苯基}甲磺酰胺,
1-[4-氨基-7-(5-羟基甲基吡啶-3-基)-2-(2-甲氧基乙基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
3-[4-氨基-2-(乙氧基甲基)-7-(吡啶-3-基)-1H-咪唑并[4,5-c]喹啉-1-基]丙-1,2-二醇,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-丙基脲,
1-[2-(4-氨基-2-乙氧基甲基-1H-咪唑并[4,5-c]喹啉-1-基)-1,1-二甲基乙基]-3-环戊基脲,
1-[(2,2-二甲基-1,3-二氧戊环-4-基)甲基]-2-(乙氧基甲基)-7-(4-羟基甲基苯基)-1H-咪唑并[4,5-c]喹啉-4-胺,
4-[4-氨基-2-乙氧基甲基-1-(2-羟基-2-甲基丙基)-1H-咪唑并[4,5-c]喹啉-7-基]-N-甲氧基-N-甲基苯甲酰胺,
2-乙氧基甲基-N1-异丙基-6,7,8,9-四氢-1H-咪唑并[4,5-c]喹啉-1,4-二胺,
1-[4-氨基-2-乙基-7-(吡啶-4-基)-1H-咪唑并[4,5-c]喹啉-1-基]-2-甲基丙-2-醇,
N-[4-(4-氨基-2-乙基-1H-咪唑并[4,5-c]喹啉-1-基)丁基]甲磺酰胺,或
N-[4-(4-氨基-2-丁基-1H-咪唑并[4,5-c][1,5]萘啶-1-基)丁基]-n'-环己基脲,
其中,喹啉环上的胺基团是与连接体连接的连接点。
总体而言,式(II)的结构表示的活化部分可使用下列合成步骤制备。在步骤(1)中,式A的4-氯代-3-硝基喹啉与通式R
可选地,式(II)的化合物可根据US6,331,539B1,US6,451,810B1,US7,157,452和US7,301027B2中所述的合成方法制备。
另一方面,式(I)的化合物可通过使用连接体以连接靶向部分和活化部分这两者来制备。所述连接体使用其反应位点与靶向部分和活化部分结合。在一些实施方式中,所述结合通过在连接体与靶向部分和活化部分之间形成共价键进行。在一些实施方式中,所述反应位点为亲核基团。在一些实施方式中,所述反应位点为亲电基团。连接体中有用的亲核基团包括但不限于:酰肼基团、肟基团、氨基基团、肼基团、缩氨基硫脲基团、肼羧酸酯基团和芳基酰肼基团。有用的亲电基团包括但不限于:马来酰亚胺基团、碳酸酯基团和卤代乙酰胺基团。
一方面,本发明提供一种治疗或延迟个体中的癌症恶化的方法,所述方法包括将有效量的PD-L/PD-1轴拮抗剂和能够活化人浆细胞样树突细胞,髓样树突细胞,NK细胞、T细胞或肿瘤细胞的免疫治疗剂给药于所述个体。总体而言,在该联合疗法中,PD-L/PD-1轴拮抗剂和免疫治疗剂没有连接在一起,例如没有通过连接体共价连接在一起。
本文中的“免疫治疗剂”是指能够激活或增强体内的免疫系统或肿瘤细胞的化合物、分子或药剂。免疫治疗剂用于通过诱导、增强或抑制免疫反应来治疗疾病。本发明的免疫治疗剂通常被设计为诱发或增强免疫反应,而非抑制免疫反应。
总体而言,本发明的免疫治疗剂直接或间接地作用于toll样受体、核苷酸-寡聚结构域样受体、RIG-I-样受体、c型凝集素受体或胞质DNA感受器,或者它们的组合。具体而言,本发明的免疫治疗剂能够活化人浆细胞样树突细胞,髓样树突细胞,NK细胞、T细胞或肿瘤细胞,或者它们的组合。
所述免疫治疗剂是不与连接体或靶向部分连接的与本发明公开的活化部分相同的化合物中的一种,例如,不含
在一些实施方式中,PD-L/PD-1轴拮抗剂选自:PD-1结合拮抗剂,PD-L1结合拮抗剂和PD-L2结合拮抗剂。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-1结合拮抗剂。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合其配体结合搭档。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L1。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L2。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L1和PD-L2这两者。
在一些实施方式中,PD-1结合拮抗剂是抗体,例如,MDX-1106,Merck 3745,CT-011,AMP-224或AMP-514。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-L1结合拮抗剂。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合PD-1。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合B7-1。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合PD-1和B7-1这两者。
在一些实施方式中,PD-L1结合拮抗剂是抗体,例如,选自:YW243.55.S70,MPDL3280A,MDX-1105,MEDI-4736和MSB0010718C中的一种抗体。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-L2结合拮抗剂。
在一些实施方式中,PD-L2结合拮抗剂是抗体。
在一些实施方式中,PD-L2结合拮抗剂是免疫粘合素。
在一些实施方式中,在停止治疗之后,所述治疗在个体体内产生持续反应。
在一些实施方式中,所述免疫治疗剂连续施用,间歇施用。
在一些实施方式中,所述免疫治疗剂在PD-L/PD-1轴拮抗剂之前施用。
在一些实施方式中,所述免疫治疗剂与PD-L/PD-1轴拮抗剂同时施用。
在一些实施方式中,所述免疫治疗剂在PD-L/PD-1轴拮抗剂之后施用。
在一些实施方式中,所述个体患有结肠直肠癌、黑色素瘤、非小细胞肺癌、卵巢癌、乳腺癌、胰腺癌、恶性血液肿瘤或肾细胞癌。
在一些实施方式中,PD-L/PD-1轴拮抗剂静脉内施用、肌肉内施用、皮下施用、局部施用、口服施用、透皮施用、腹膜内施用、眼窝内施用、通过植入的方式施用、通过吸入的方式施用、鞘内施用、心室内施用或鼻内施用。
另一方面,本发明提供一种组合,其包括:(i)有效量的PD-L/PD-1轴拮抗剂;和(ii)有效量的能够活化人浆细胞样树突细胞、髓样树突细胞、NK细胞、T细胞或肿瘤细胞或者它们的组合的免疫治疗剂。
在一些实施方式中,PD-L/PD-1轴拮抗剂选自:PD-1结合拮抗剂、PD-L1结合拮抗剂和PD-L2结合拮抗剂。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-1结合拮抗剂。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合其配体结合搭档。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L1。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L2。
在一些实施方式中,PD-1结合拮抗剂抑制PD-1结合PD-L1和PD-L2这两者。
在一些实施方式中,PD-1结合拮抗剂是抗体,例如,MDX-1106,Merck3745,CT-011,AMP-224或AMP-514。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-L1结合拮抗剂。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合PD-1。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合B7-1。
在一些实施方式中,PD-L1结合拮抗剂抑制PD-L1结合PD-1和B7-1这两者。
在一些实施方式中,PD-L1结合拮抗剂是抗体,例如,选自YW243.55.S70,MPDL3280A,MDX-1105,MEDI-4736和MSB0010718C中的一种抗体。
在一些实施方式中,PD-L/PD-1轴拮抗剂是PD-L2结合拮抗剂。
在一些实施方式中,PD-L2结合拮抗剂是抗体。
在一些实施方式中,PD-L2结合拮抗剂是免疫粘合素。
本发明还涉及包含本发明的化合物或其药学上可接受的盐,以及一种或一种以上药学上可接受的载体的药物制剂。
本文所述的化合物(例如,其加成盐或水合物)以及药学上可接受的载体可使用多种不同的给药模式或途径递送至患者。合适的给药途径包括但不限于:吸入给药、透皮给药、口服给药、直肠给药、透过粘膜给药、肠道给药和肠胃外给药(包括肌肉内、皮下和静脉注射)。优选地,包含作为靶向部分的抗体或抗体片段的本发明的化合物以肠胃外的方式给药,更优选地,以静脉注射方式给药。
本文使用的术语“给药”意在包括直接和间接地将化合物递送至期望的作用位点的所有方式。
本文所述的化合物或其药学上可接受的盐和/或其水合物可单独给药,与本发明的其他化合物联合给药和/或与其他治疗剂联合以混合剂的形式给药。当然,对可与本发明的化合物联合给药的治疗剂的选择部分取决于治疗中的病症。
例如,当向患有由依赖于自诱导物的生物体引起的疾病病症的患者给药时,本发明的化合物可以混合剂的形式给药,所述混合剂含有用于治疗疼痛、感染和其他症状以及通常与疾病有关的副作用的药剂。这些药剂包括例如,镇痛剂、抗生素,等等。
当向正在进行癌症治疗的患者给药时,化合物可以混合剂的形式给药,所述混合剂包含抗癌剂和/或补充增效剂。所述化合物还可以含有治疗放疗的副作用的药剂的混合剂的形式给药,所述治疗放疗的副作用的药剂例如,止吐药,防辐射剂,等等。
可与本发明的化合物联合给药的补充增效剂包括例如,三环类抗抑郁药(例如,米帕明(imipramine)、地昔帕明(desipramine)、阿米替林(amitriptyline)、氯米帕明(clomipramine)、曲米帕明(trimipramine)、多塞平(doxepin)、去甲替林(nortriptyline)、普罗替林(protriptyline)、阿莫沙平(amoxapine)和马普替林(maprotiline)),非三环类抗抑郁药(例如,舍曲林(sertraline)、曲唑酮(trazodone)和西酞普兰(citalopram)),Ca2+拮抗剂(例如,维拉帕米(verapamil)、硝苯地平(nifedipine)、尼群地平(nitrendipine)和卡罗维林(caroverine)),两性霉素,三苯乙醇类似物(例如,它莫昔芬(tamoxifen)),抗心律失常药物(例如,奎尼丁(quinidine)),抗高血压药物(例如,利血平(reserpine)),硫醇消耗物(例如,丁硫氨酸和亚砜亚胺)以及甲酰四氢叶酸钙。
本发明的活性化合物以其自身的形式给药或者以药物组合物的形式给药,在所述药物组合物中,所述活性化合物与一种或一种以上药学上可接受的载体、赋形剂或稀释剂混合。根据本发明使用的药物组合物通常使用一种或一种以上生理学可接受的载体以常规方式配制,所述生理学可接受的载体包含赋形剂和佐剂,所述生理学可接受的载体有利于将活性化合物加工成可在药学上使用的制剂。合适的剂型取决于所选择的给药途径。
对于透过粘膜给药而言,在剂型中使用适于待穿透的屏障的渗透剂。这些渗透剂通常为本领域已知的。
对于口服给药而言,化合物易于通过将活性化合物与本领域已知的药学上可接受的载体结合配制。这些载体能够使本发明的化合物配制成由待治疗的患者口服的片剂、药丸、糖衣药丸、胶囊、液体、凝胶、糖浆、浆状物和悬浮液。口服的药物制剂可通过如下方式获得:将固体赋形剂与活性剂化合物混合,任选地研磨得到的混合物,并对颗粒混合物进行加工,如果想要得到片剂或糖衣片芯的话,需要在该过程之前加入合适的佐剂。具体而言,合适的赋形剂为填充剂(例如,包括乳糖、蔗糖、甘露醇或山梨醇的糖),纤维素制剂(例如,玉米淀粉、小麦淀粉、大米淀粉、土豆淀粉、明胶、黄耆胶、甲基纤维素、羟丙基甲基纤维素、羧乙基纤维素钠)和/或聚乙烯吡咯烷酮(PVP)。如果需要的话,可加入崩解剂,例如,交联的聚乙烯吡咯烷酮、琼脂或海藻酸或其盐(例如,海藻酸钠)。
向糖衣片芯提供合适的包衣。为了实现这个目的,可使用浓缩的糖溶液,该溶液可任选地含有阿拉伯树胶、滑石粉、聚乙烯吡咯烷酮、卡波姆凝胶、聚乙二醇和/或二氧化钛、漆用溶液和合适的有机溶剂或溶剂混合物。可将染料或颜料加至片剂或糖衣药丸包衣中,用于识别或表征不同的活性化合物剂量组合。
可口服使用的药物制剂包括由明胶制成的推合式胶囊和由明胶和增塑剂(例如,丙三醇或山梨糖醇)制成的软的密封的胶囊。推合式胶囊可含有与诸如乳糖之类的填充剂、诸如淀粉之类的粘合剂和/或诸如滑石粉或硬脂酸镁之类的润滑剂和任选地稳定剂混合的活性成分。在软胶囊中,活性化合物可溶解于或悬浮于合适的液体中,例如,脂肪油、液体石蜡或液体聚乙二醇。此外,可加入稳定剂。用于口服给药的所有剂型的剂量应当适于这种给药方式。
对于口腔给药而言,组合物可以通过常规方式配制的片剂或含片的形式给药。
对于吸入给药而言,根据本发明使用的化合物便于通过使用合适的推进剂(例如,二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷、二氧化碳或其他合适的气体)以由加压包或喷雾器提供的喷雾的形式递送。在使用加压喷雾的情况下,剂量单位可通过提供递送计量的量的阀来确定。在吸入器或吹入器中使用的胶囊和药筒(例如,明胶胶囊和药筒)可被配制为含有化合物和合适粉末基体(例如,乳糖或淀粉)的粉末混合物的剂型。
化合物可被配制为通过注射(例如,快速注射或连续输注)方式肠胃外给药的剂型。注射为本发明的组合物的优选的给药方法。用于注射的剂型可以带有添加的防腐剂的单位剂量形式提供,例如,以安瓶或多剂量容器的形式提供。组合物可采用诸如油性载体或水性载体中的悬浮液、溶液或乳液之类的形式并且可含有诸如悬浮剂、稳定剂和/或分散剂之类的调配剂(formulatory),并且可加入例如交联的聚乙烯吡咯烷酮、琼脂或海藻酸或其盐(例如,海藻酸钠)。
用于肠胃外给药的药物剂型包括水溶性形式的活性化合物的水溶液。此外,活性化合物的悬浮液可制备成合适的油性注射悬浮液。合适的亲脂性溶剂或载体包括脂肪油(例如,芝麻油)或合成的脂肪酸酯(例如,油酸乙酯或甘油三酯)或脂质体。水性注射悬浮液可包含如下物质:该物质增加悬浮液的粘度,例如,羧甲基纤维素钠、山梨糖醇或葡聚糖。任选地,悬浮液还可含有合适的稳定剂或药剂,该稳定剂或药剂增加化合物的溶解度,从而制备高度浓缩的溶液。对于注射而言,本发明的药剂可被配制成水性溶液,优选地配制成生理相容性缓冲液(例如,Hanks溶液、林格氏溶液或生理盐水缓冲液)。
可选地,活性成分可为使用前溶解于合适的载体中的粉末形式,所述合适的载体例如,无菌无热原水。
化合物还可被配制成直肠用组合物,例如,栓剂或保留灌肠剂型,例如含有常规栓剂基体(例如可可油或其他甘油酯)。
除了前述剂型之外,化合物还可被配制成长效制剂。这种长期起效的剂型可通过植入或经皮递送(例如,皮下递送或肌肉内递送)、肌肉内注射或透皮贴剂给药。因此,例如,化合物可通过合适的聚合材料或疏水性材料(例如,可接受的油中的乳液)或离子交换树脂配制或可将化合物配制成微溶衍生物,例如,配制成微溶的盐。
药物组合物还可包含合适的固体或凝胶相载体或赋形剂。这些载体或赋形剂的实例包括碳酸钙、磷酸钙、各种糖类、淀粉、纤维素衍生物、明胶和诸如聚乙二醇之类的聚合物。
优选的药物组合物为配制成注射剂型的组合物,例如,静脉注射剂型,并且该优选的药物组合物包含基于总的100%重量的药物组合物的约0.01%重量至约100%重量的本发明的化合物。药物配体结合物可为抗体-细胞毒素结合物,其中,选择靶向特定癌症的抗体。
在一些实施方式中,本发明的药物组合物还包含其他治疗剂。
在一些实施方式中,所述其他治疗剂为抗癌剂。
在一些实施方式中,其他抗癌剂选自:抗代谢药物、拓扑异构酶I和拓扑异构酶II的抑制剂、烷基化剂、微管抑制剂、抗雄性激素剂、GNRh调节剂或者它们的混合物。
在一些实施方式中,所述其他治疗剂为化疗剂。
本文中的“化疗剂”是指在治疗癌症中有用的化学化合物。实例包括但不限于:吉西他滨(Gemcitabine),伊立替康(Irinotecan),阿霉素(Doxorubicin),5-氟尿嘧啶(5-Fluorouracil),阿糖胞苷(Cytosine arabinoside,“Ara-C”),环磷酰胺(Cyclophosphamide),塞替派(Thiotepa),白消安(Busulfan),细胞毒素,紫杉酚,甲氨蝶呤(Methotrexate),顺铂(Cisplatin),美法仑(Melphalan),长春碱(Vinblastine)和卡铂(Carboplatin)。
在一些实施方式中,第二化疗剂选自:他莫昔芬(tamoxifen),雷洛昔芬(raloxifene),阿那曲唑(anastrozole),依西美坦(exemestane),来曲唑(letrozole),伊马替尼(imatanib),紫杉醇(paclitaxel),环磷酰胺(cyclophosphamide),洛伐他汀(lovastatin),含羞草素(minosine),吉西他滨(gemcitabine),阿糖胞苷(cytarabine),5-氟尿嘧啶,甲氨蝶呤,多西他赛(docetaxel),戈舍瑞林(goserelin),长春新碱(vincristine),长春碱(vinblastine),诺考达唑(nocodazole),替尼泊苷(teniposide),依托泊苷(etoposide),吉西他滨,埃博霉素(epothilone),长春瑞滨(vinorelbine),喜树碱(camptothecin),柔红霉素(daunorubicin),放线菌素D(actinomycin D),米托蒽醌(mitoxantrone),吖啶(acridine),阿霉素(doxorubicin),表柔比星(epirubicin)或去甲氧基柔红霉素(idarubicin)。
另一方面,本发明提供一种试剂盒,所述试剂盒包含本发明的化合物或组合物中的一种或一种以上以及使用所述化合物或组合物的说明书。在示例性的实施方式中,本发明提供用于将本发明的连接体臂与另一分子偶联的试剂盒。所述试剂盒包括连接体和用于使连接体与特定的官能团连接的说明书。所述试剂盒还包括细胞毒性药物、靶向剂、可检测的标记、药学上可接受的盐或缓冲剂中的一种或一种以上。所述试剂盒还可包括容器和任选地一种或一种以上小瓶、测试管、烧瓶、瓶子或注射器。其他形式的试剂盒对于本领域技术人员而言是显而易见的并且在本发明的范围内。
本发明的化合物包含抗PD-L1抗体,连接体和作为TLR7/8激动剂的式(II)表示的活化部分。如现有技术中所述,在抗体药物结合物中只发现了几种主要的化学类型的细胞毒性剂。它们分为以下两种类型:DNA损伤剂和微管破坏剂。本发明提供一类新的在抗体药物结合物中使用的化学细胞毒性剂——调节免疫反应并随后活化树突细胞和自然杀伤细胞且促进抗肿瘤活性的TLR7/8激动剂。本发明意想不到地发现,本发明的化合物表现出优异的抗肿瘤活性。在用本发明的化合物进行常规治疗的过程中,抗体部分起到类似导弹的作用,其将细胞毒性剂递送至表达PD-L1的肿瘤细胞/癌细胞,在该表达PD-L1的肿瘤细胞/癌细胞中,所述细胞毒性剂对肿瘤细胞/癌细胞直接或间接地发挥作用。这种治疗的有利之处在于:毒性副作用降低且药代动力学曲线得到改善。而且,从载体抗体中缓慢释放细胞毒性剂(药物)产生持续较高的肿瘤内活性细胞毒性剂浓度以及较低的活性细胞毒性剂血药浓度。
因此,另一方面,本发明提供一种抑制表达PD-L1的肿瘤细胞/癌细胞的增殖的方法,所述方法包括将本发明的化合物施用于所述肿瘤细胞/癌细胞。在一些实施方式中,所述肿瘤可以是转移的或非转移的。
本文中的“癌症”或“肿瘤”是指人体内的病理状态,其特征为不受控制的细胞增殖。实例包括但不限于:癌症、淋巴瘤、母细胞瘤和白血病。癌症的更加具体的实例包括但不限于:肺(小细胞和非小细胞)癌、乳腺癌、前列腺癌、类癌性癌症、膀胱癌、胃癌、胰腺癌、肝癌(肝细胞癌)、肝母细胞瘤、结肠直肠癌、头颈癌、鳞状细胞癌、食管癌、卵巢癌、子宫癌、子宫内膜癌、间皮瘤、黑色素瘤、肉瘤、骨肉瘤、脂肪肉瘤、甲状腺癌、硬纤维瘤、急性髓细胞白血病(AML)和慢性髓细胞白血病(CML)。
本文中的“抑制”或“治疗”是指降低、治疗性和预防性治疗,其中,目的是降低或预防指定的病理性失调或病症。在一个实施例中,给药本发明的化合物之后,癌症患者可经历肿瘤尺寸减小。“治疗”包括(1)抑制患有或表现出疾病的病理或症状的受治者体内的疾病;(2)缓解患有或表现出疾病的病理或症状的受治者体内的疾病;和/或(3)影响患有或表现出疾病的病理或症状的受治者或患者体内的疾病的任何可测量的降低。本发明的化合物在一定程度上可防止癌细胞生长和/或杀死癌细胞,在该程度上的本发明的化合物可为抑制细胞生长的和/或细胞毒性的。
本文中的“治疗有效量”是指有效“治疗”受治者或哺乳动物体内的失调的本文提供的化合物的量。在治疗癌症的情况下,治疗有效量的药物可降低癌细胞的数目,使肿瘤尺寸减小,抑制癌细胞浸润进入外周器官,抑制肿瘤转移,抑制肿瘤生长至某一程度,和/或一定程度地减轻与癌症相关的症状中的一种或一种以上。
另一方面,本发明提供一种治疗受治者体内表达PD-L1的肿瘤/癌症的方法,所述方法包括将治疗有效量的本发明的化合物给药于所述受治者。在一些实施方式中,肿瘤或癌症可以是处于任何阶段的肿瘤或癌症,例如,早期或晚期肿瘤或癌症,例如,I期、II期、III期、IV期或V期肿瘤或癌症。在一些实施方式中,肿瘤或癌症可为转移的或非转移的。在转移性肿瘤或癌症的情况下,本发明的方法可减缓或抑制原发性肿瘤或癌症向其他位点转移,或者减缓或抑制在远离原发性肿瘤或癌症治疗的其他位点形成或产生转移性肿瘤或癌症。因此,本发明的方法还包括:1)减缓或抑制可能发生转移或已经发生转移的肿瘤细胞或癌细胞(例如,播散性肿瘤细胞,DTC)的生长、增殖、迁移或入侵;2)减缓或抑制原发性肿瘤或癌症向一个或一个以上不同于原发性肿瘤或癌症的其他位点、位置或区域形成转移或建立转移;3)在转移已形成或已建立之后,减缓或抑制一个或一个以上不同于原发性肿瘤或癌症的其他位点、位置或区域的转移的生长或增殖;以及4)在转移已形成或已建立之后,减缓或抑制额外的转移的形成或建立。
在一些实施方式中,肿瘤或癌症为实体或液体细胞团块。“实体”瘤是指通常聚集在一起并形成团块的癌症、瘤或转移。具体的非限定性实例包括:乳腺肿瘤/乳腺癌、卵巢肿瘤/卵巢癌、子宫肿瘤/子宫癌、子宫颈肿瘤/子宫颈癌、胃肿瘤/胃癌、肺肿瘤/肺癌、胃肿瘤/胃癌、结肠肿瘤/结肠癌、膀胱肿瘤/膀胱癌、胶质肿瘤/胶质癌和子宫内膜肿瘤/子宫内膜癌等。“液体肿瘤”是指自然分散或扩散的瘤,其通常不会形成实体团块。具体的实例包括:网状内皮系统瘤或造血系统瘤,例如,淋巴瘤、骨髓瘤和白血病。白血病的非限定性实例包括:急性和慢性淋巴母细胞性白血病、成髓细胞白血病和多发性骨髓瘤。通常,这些疾病由低分化急性白血病(例如,成红细胞白血病和急性巨核细胞白血病)引起。具体的骨髓性疾病包括,但不限于:急性早幼粒细胞白血病(APML)、急性粒细胞白血病(AML)和慢性粒细胞白血病(CML)。淋巴系统恶性肿瘤包括,但不限于:急性淋巴细胞性白血病(ALL),其包括B细胞系ALL(B-ALL)和T细胞系ALL(T-ALL),慢性淋巴细胞性白血病(CLL),幼淋巴细胞白血病(PLL),多毛细胞白血病(HLL)和Waldenstroem巨球蛋白血症(WM)。具体的恶性淋巴瘤包括:非霍奇金淋巴瘤及其变体,外周T细胞淋巴瘤,成人T细胞白血病/淋巴瘤(ATL),皮肤T细胞淋巴瘤(CTCL),大颗粒淋巴细胞白血病(LGF),霍奇金疾病和Reed-Sternberg疾病。
在一些实施方式中,本发明的方法可与其他治疗方法或疗法(例如,手术切除术、放疗、离子或化学辐射疗法、化疗、免疫疗法、局部或区域热疗(过高热疗法)或接种疫苗)一同实施。这些其他治疗方法或疗法可在本发明的化合物的给药之前、基本同时(分开或以混合物的形式)或之后给予。
在一些实施方式中,本发明的方法包括将治疗有效量的本发明的化合物与其他治疗剂联合给药。在一些实施方式中,所述其他治疗剂是抗癌剂/抗肿瘤剂。在一些实施方式中,所述其他治疗剂是抗代谢物、拓扑异构酶I和拓扑异构酶II的抑制剂、烷基化剂、微管抑制剂、抗雄性激素剂、GNRh调节剂或者它们的混合物。在一些实施方式中,所述其他治疗剂选自:它莫西芬(tamoxifen)、雷洛昔芬(raloxifene)、阿那曲唑(anastrozole)、依西美坦(exemestane)、来曲唑(letrozole)、imatanib、紫杉醇、环磷酰胺、洛伐他汀(lovastatin)、minosine、吉西他滨(gemcitabine)、阿糖胞苷(cytarabine)、5-氟尿嘧啶、甲氨蝶呤、多西他赛(docetaxel)、戈舍瑞林(goserelin)、长春新碱、长春碱、噻氨酯哒唑(nocodazole)、替尼泊苷(teniposide)、依托泊苷(etoposide)、吉西他滨、埃博霉素、长春瑞滨(vinorelbine)、喜树碱、道诺霉素(daunorubicin)、放线菌素D、米托蒽醌、吖啶、阿霉素、表柔比星或去甲氧基柔红霉素。
与一种或一种以上其他治疗剂“联合”给药包括同时给药和以任何顺序连续给药。本文使用的术语“药物组合”是指通过混合活性成分或组合活性成分得到的产品,并且包括活性成分的固定组合和非固定组合这两者。术语“固定组合”是指活性成分(例如式(I)的化合物)和联合药剂以单个实体或剂量同时给药于患者。术语“非固定组合”是指活性成分(例如式(I)的化合物)和联合药剂作为分开的实体同时或按顺序(没有特定的时间限制)给药于患者,这种给药方式向患者体内提供治疗有效量的活性成分。后者还用于混合剂治疗,例如,给药三种或三种以上活性成分。
适用于本发明的药物组合物包括包含治疗有效量的活性成分的组合物,所述治疗有效量即为有效实现期望目的的量。对特定应用有效的实际量将(尤其)取决于治疗中的病症。有效量的确定恰好在本领域技术人员的能力范围内(尤其是根据本文具体公开的内容)。
对于本文所述的任何化合物而言,治疗有效量可最先通过细胞培养检测法确定。目标血药浓度为能够抑制细胞生长或分裂的活性化合物的浓度。在优选的实施方式中,细胞活性的至少25%被抑制。能够诱导至少约30%、50%、75%或甚至90%或者更高的细胞活性抑制的活性化合物的目标血药浓度为目前优选的。可监测患者体内的细胞活性抑制的百分数,从而评估所达到的血药浓度的适当性,并且可上调或下调剂量以实现期望的抑制百分数。
如本领域已知的,用于人体的治疗有效量还可通过动物模型来确定。例如,用于人体的剂量可被配制成实现已在动物体内发现的有效循环浓度。如上所述,人体内的剂量可通过监测细胞抑制和上调或下调剂量来调节。
治疗有效剂量还可通过已知的表现出类似药理学活性的化合物的人体数据来确定。所使用的剂量可基于与已知化合物比较的所给药的化合物的相对生物利用度和效力来调节。
基于上述方法和本领域熟知的其他方法为实现人体内的最大效力对剂量进行调节恰好在本领域普通技术人员的能力范围内。
在局部给药的情况下,所给药的化合物的全身循环浓度不是特别重要。在这种情况下,给药化合物以达到在局部区域有效实现期望的结果的浓度。
对于预防和/或治疗与异常细胞增殖有关的疾病方面的应用而言,优选的是,所给药的化合物的循环浓度为约0.001μM至20μM,优选地约0.01μM至5μM。
本文所述的化合物的口服给药的患者剂量典型地为约1mg/天至约10,000mg/天,更加典型地为约10mg/天至约1,000mg/天,最典型地为约50mg/天至约500mg/天。按照患者体重而言,典型的剂量为约0.01mg/kg/天至约150mg/kg/天,更加典型地为约0.1mg/kg/天至约15mg/kg/天,最典型地为约1mg/kg/天至约10mg/kg/天,例如,5mg/kg/天或3mg/kg/天。
在至少一些实施方式中,延迟或抑制肿瘤生长的患者剂量可为1μmol/kg/天或更少。例如,患者剂量可为0.9μmol/kg/天、0.6μmol/kg/天、0.5μmol/kg/天、0.45μmol/kg/天、0.3μmol/kg/天、0.2μmol/kg/天、0.15μmol/kg/天或0.1μmol/kg/天或更少(参考药物的摩尔数)。优选地,当以每日剂量给药持续至少五天的时间时,抗体药物结合物延迟肿瘤生长。
对于其他给药模式而言,剂量和间隔可分开调节,从而提供对于治疗中的特定临床适应症有效的给药的化合物的血药浓度。例如,在一种实施方式中,根据本发明的化合物可以相对高的浓度每天多次给药。可选地,更加理想的是,以最小有效浓度给药本发明的化合物并且使用较低频率的给药方案。这会提供与个体的疾病的严重程度相称的治疗方案。
使用本文的教导可安排有效的治疗方案,而不导致较大的毒性,并且还完全有效地治疗特定患者表现出的临床症状。这种安排应当包括通过考虑多种因素仔细选择活性化合物,所述多种因素例如,化合物效力、相对生物利用度、患者体重、存在的不良副作用及其严重性、优选的给药模式和所选择的药剂的毒性特征。
虽然本文描述并显示了本发明的优选的实施方式,但是对于本领域技术人员而言显而易见的是,这些实施方式仅仅是举例说明。本领域技术人员可对这些实施方式作出多种改变、修改和替换,而不背离本发明。应当理解的是,本文所述的本发明的可选实施方式可用于实践本发明。通过所附的权利要求来限定本发明的范围,并且这些权利要求范围内的方法和结构及其等同物也被所附的权利要求涵盖。
本发明进一步通过下述实施例举例说明,但本发明不限于此,下述实施例举例说明本发明的化合物的制备方法。
试剂:来自ATCC(Manassas,VA;Cat No.CRL2539和Cat No.CRL6475)的4T1和B16F10细胞,鼠neu/pEZ-Lv105 cDNA,葡萄糖DMEM,L-谷酰胺,Lipofectamine 2000(Invitrogen;Carlsbad,CA)。
为了制备用于筛选偶联的抗鼠neu和抗鼠PD-L1的细胞系,生成表达标记的Her2的4T1或B16F10细胞。鼠neu/pEZ-Lv105 cDNA构建体通过标准Lipofectamine 2000操作规程转染至4T1或B16F10细胞(该细胞在高浓度葡萄糖DMEM+10%FBS+2mM L-谷酰胺中生长)。
为了确定偶联的抗鼠PD-L1的结合能力,对表达鼠her2的4T1或B16F10细胞在体内成肿瘤后进行FACS分析。简言之,将2×10
试剂:抗PD-L1或PD-1抗体(Bio X cell,West Lebanon,NH)、与瑞喹莫德连接的MC-val-cit-PAB(MC-vc-PAB-TLRL)(在Contract Research Organization,China合成)、硼酸钠、氯化钠、二硫苏糖醇(DTT)、Sephadex G25、DTPA、DTNB和马来酰亚胺基己酰基-单甲基(Sigma-Aldrich,Milwaukee,Wisconsin)。
用于生成抗体TLR配体偶联物的药物包括PD-L1抗体和瑞喹莫德(TLRL)。用于生成TLAC的连接体为可裂解的连接体MC-vc-PAB或非可裂解的连接体MC。
PD-L1或PD-1抗体通过20mg/mL浓度下的缓冲液交换从中纯化出来,在pH为8.0的条件下溶解于500mM硼酸钠和500mM氯化钠的抗体用过量的100mM二硫苏糖醇(DTT)处理。在37℃孵育约30分钟之后,通过在Sephadex G25树脂上的洗脱交换缓冲液并用含有1mM DTPA的PBS进行洗脱。硫醇/Ab值通过测定还原的抗体的浓度和硫醇浓度来检测,所述还原的抗体的浓度通过溶液在280nm的吸光度来检测,所述硫醇浓度通过与DTNB(Sigma-Aldrich,Milwaukee,Wisconsin)反应以及在412nm处的吸光度来确定。溶解于PBS中的还原的抗体在冰上冷冻。在已知浓度下在乙腈和水中稀释溶解于DMSO的药物连接体试剂(与瑞喹莫德连接的Mc-),并且将其加至PBS中的冷冻的还原抗体中。约1小时后,加入过量的马来酰亚胺淬灭反应并遮盖任何未反应的抗体硫醇基团。在一些情况下,通过标准步骤与免疫球蛋白的赖氨酸进行偶联。反应混合物通过离心超滤浓缩,偶联的抗体通过PBS中的G25树脂的洗脱进行纯化和脱盐,在无菌条件下通过0.2μm的过滤器过滤,并冷冻储存。
抗体通过半胱氨酸由马来酰亚胺基己酰基-缬氨酸-瓜氨酸(vc)-p-氨基苄氧基羰基(MC-vc-PAB)连接至TLRL。MC-vc-PAB连接体可通过诸如组织蛋白酶B之类的细胞间蛋白酶裂解,当裂解时,释放游离药物(Doronina等人,Nat.Biotechnol.,21:778-784(2003)),而MC连接体为抗细胞间蛋白酶裂解的。PD-L1或PD-1抗体在pH为8.0的条件下溶解于500mM硼酸钠和500mM氯化钠中,并且进一步用过量的100mM二硫苏糖醇(DTT)处理。在37℃下孵育约30分钟后,通过Sephadex G25树脂上的洗脱交换缓冲液并且用含有1mM DTPA的PBS洗脱。硫醇/Ab值通过测定还原的抗体的浓度和硫醇浓度来检测,所述还原的抗体的浓度通过溶液在280nm处的吸光度来检测,所述硫醇浓度通过与DTNB(Sigma-Aldrich,Milwaukee,Wisconsin)反应以及在412nm处的吸光度(消光系数=13600cm
通常,抗体与MC-vc-TLRL的偶联反应生成非均一混合物,该混合物包含与不同数量的TLRL药物连接的、偶联的抗体,即,药物分配为1至约8的药物装载。因此,抗体MC-vc-TLRL包括分离的纯化的物种分子和平均药物装载为1至8的混合物。通过控制加工过程,药物装载为3至5。通过偶联反应得到的化合物制剂中,每个抗体中TLRL药物部分的平均数目可由常规方式表征,例如质谱法、ELISA分析法、电泳分离法和HPLC。也可确定根据药物的抗体MC-vc-TLRL的量分配。通过ELISA,可确定抗体与TLRL偶联的特定制剂中的药物有效装载数目的平均值(Hamblett等人(2004)Clinical Cancer Res.10:7063-7070;Sanderson等人(2005)Clinical Cancer Res.11:843-852)。然而,药物的分配值不能通过抗体-抗原结合和ELISA检测限辨别。而且,用于检测抗体药物偶联物的ELISA分析不能确定药物部分与抗体在哪个位置连接,例如,重链或轻链片段或特定的氨基酸残基。在一些情况下,均一的抗鼠PD-L1 MC-vc-TLRL的分离、纯化和表征(其中,具有其他药物装载的抗鼠PD-L1 MC-vc-TLRL中的药物为特定值)可通过诸如反相HPLC或电泳分离之类的方式实现。
对于源自肿瘤细胞株4T1-her2或B16F10-her2在免疫健全小鼠肿瘤模型的研究而言,将6至8周龄的雌性Balb/c裸鼠(获自SLAC,Beijing,China)用于肿瘤细胞移植。根据动物护理和使用委员会(Institutional Animal Care and Use Committee)的实验室动物护理和使用指南和规程,用正常裸鼠饮食饲养动物并且将动物养在SPF动物设施中。将1x10
在肿瘤生长达到预定值时第一天和第五天,通过静脉注射途径给药含有10mg/kg至20mg/kg抗体的药物或参比药物。通过卡尺每周测量一次肿瘤以确定其皮下生长。用卡尺每周测量两次肿瘤的二维尺寸。使用如下方程式计算肿瘤体积:肿瘤体积=(长度×宽度
所有比较的显著性使用Student双尾t检测计算,假设模拟组和样品组之间的方差不相等,当p<0.05时认为结果是显著的。参数之间的相关性使用斯皮尔曼秩相关测试评价,P值<0.05被认为是统计学上显著的。
- 用于治疗肿瘤的抗-PD-L1结合物
- 用于治疗肿瘤的抗-PD-L1结合物