双端氨基硅醚化合物的制备方法及双端氨基硅醚化合物
文献发布时间:2023-06-19 12:08:44
技术领域
本申请涉及硅醚制备技术领域,尤其涉及一种双端氨基硅醚化合物的制备方法,以及经由该制备方法制得的双端氨基硅醚化合物。
背景技术
在化工行业中,双端氨基硅醚化合物能够作为二胺单体以用于生产嵌段共聚物,例如双端氨基硅醚化合物与二酐反应可以将硅醚引入至聚酰亚胺中,由此获得的含硅聚酰亚胺兼具聚酰亚胺优良的耐热性和化学稳定性,以及硅醚的易加工性、低吸湿性以及良好的粘接性。此外,双端氨基硅醚化合物具有良好的分子柔性和反应性,其可以在头发护理和纺织品配制物中用作柔软剂。
目前,双端氨基硅醚化合物的工业应用范围受到其生产成本的制约。现已报道的双端氨基硅醚化合物的合成方法具有生产成本较高、步骤繁琐、大位阻双端氨基硅氧烷产物收率低的问题。例如,在申请公布号为CN 102276639A的中国专利申请中,其提供了一种双端氨基(聚)硅醚的制备方法,该制备方法先采用邻二甲酸酐保护烯脂肪烃基伯氨的氨基以获得N-烯脂肪烃基酰亚胺,然后在铂催化剂的作用下,将N-烯脂肪烃基酰亚胺与含氢硅醚发生加成反应以构建C-Si键以获得双酞酰亚胺硅醚,接着在酸催化作用下与硅单体发生聚合反应以获得双酞酰亚胺聚硅醚,最后将双酞酰亚胺聚硅醚置于水合肼的乙醇溶液中以回流去保护,获得双端氨基聚硅醚。该方法存在的缺点在于:第一方面,构建C-Si键的过程中需使用昂贵的铂催化剂,具有生产成本高的缺点;第二方面,铂催化剂催化加成构建C-Si键具有明显的局限性,当硅原子上连接有大位阻的基团(如芳基)时产物收率较低,不适合大规模工业生产;第三方面,对于在酸催化作用下,双酞酰亚胺硅醚和硅单体发生聚合反应获得双酞酰亚胺聚硅醚的步骤,强酸会损伤硅氧键,从而存在破坏产物结构完整性的风险。
发明内容
针对现有技术中存在的问题,本申请提供了一种双端氨基硅醚化合物的制备方法,以及经由该制备方法制得的双端氨基硅醚化合物,其中,双端氨基硅醚化合物包括采用氨基封端的硅醚以及采用氨基封端的硅醚聚合物。
第一方面,本申请提供了一种双端氨基硅醚化合物的制备方法,所述制备方法包括如下步骤:
(A)将式(1)所示的化合物a与式(2)所示的化合物b进行亲核取代反应,以获得式(3)所示的化合物c;
(B)在碱性条件下,将所述化合物c、有机硼化合物以及过氧化氢混合进行硼氢化氧化反应,以获得式(4)所示的化合物d;
(C)将所述化合物d与式(5)所示的化合物e进行反应,以获得式(6)所示的化合物f;以及
(D)将所述化合物f进行还原反应,以获得式(7)所示的最终产物;
其中,式(1)至式(5)如下:
在式(1)至式(7)中,n≥0;
R
R
R
R
本申请的一些实施例中,在式(1)至式(7)中,1≤n≤50。
在本申请的一些实施方式中,在式(1)至式(7)中,1≤n≤10。
在本申请的一些实施方式中,所述R
在本申请的一些实施方式中,当n大于0时,所述R
在本申请的一些实施方式中,所述R
在本申请的一些实施方式中,所述R
在本申请的一些实施方式中,所述R
在本申请的一些实施方式中,步骤(A)包括步骤:将所述化合物a溶于有机溶剂以获得含有化合物a的有机溶液,然后在0℃以下的无水无氧条件下将所述化合物b与所述含有化合物a的有机溶液相混合,以进行亲核取代反应。
在本申请的一些实施方式中,所述化合物a与所述化合物b的摩尔当量为1:(2~8)。
在本申请的一些实施方式中,步骤(B)包括以下步骤:
(B1)将所述化合物c溶于有机溶剂以获得含有化合物c的有机溶液,然后在无水无氧条件下,将所述含有化合物c的有机溶液与所述有机硼化合物相混合以进行反应;以及
(B2)当步骤(B1)中所述化合物c消耗完毕后,控温35℃以下,然后加入碱性试剂和过氧化氢以进行反应,以获得式(4)所示的化合物d。
在本申请的一些实施方式中,所述有机硼化合物如下面的式(8)所示:
在式(8)中,R
在本申请的一些实施方式中,所述有机硼化合物选自以下结构式所示化合物中的至少一种:
在本申请的一些实施方式中,所述有机硼化合物选自以下结构式所示化合物中的至少一种:
在本申请的一些实施方式中,在步骤(B)中,所述碱性试剂、所述有机硼化合物与所述化合物c的摩尔当量为(1~20):(1~6):1。
在本申请的一些实施方式中,在步骤(B)中,所述碱性试剂与过氧化氢的体积比为(1~3):(1~3)。
在本申请的一些实施方式中,在步骤(C)中,在试剂g的作用下,所述化合物d与所述化合物e进行反应,其中,所述试剂g包括化合物g
其中,在式(9)中,R
在式(10)中,R
在本申请的一些实施方式中,在式(9)中,R
在本申请的一些实施方式中,在式(10)中,R
在本申请的一些实施方式中,所述步骤(C)包括以下步骤:
(C1)将所述化合物d、所述化合物e以及所述化合物g
(C2)在10℃~100℃的保护气体氛围下,向步骤(C1)的所述有机混合溶液中加入所述化合物g
在本申请的一些实施方式中,在步骤(C1)中,所述化合物d、所述化合物e以及所述化合物g
在本申请的一些实施方式中,在步骤(C2)中,所述化合物g1与所述有机混合溶液化合物d的摩尔比为(1~8):1。
在本申请的一些实施方式中,步骤(D)中,所述将化合物f进行氢化还原去保护基还原反应是采用水合肼处理化合物f。
式第二方面,本申请提供了一种如第一方面中所述的制备方法制得的双端氨基硅醚化合物,该双端氨基硅醚化合物包括采用氨基封端的硅醚、硅醚低聚物以及硅醚高聚物。
本申请提供了一种双端氨基硅醚化合物的制备方法,以及经由该制备方法制得的双端氨基硅醚化合物,该制备方法可以用于制备各种分子量大小的硅醚化合物,例如双端氨基硅醚,又如双端氨基聚硅醚。该制备方法通过可获得的氯硅醚化合物或硅醚化合物作为起始原料,将起始原料与化合物b反应来构建C-Si键,无需贵金属催化剂催化,具有生产成本低、反应条件温和可控、安全环保、高效以及适合大规模工业化生产的优点。此外,该制备方法尤其适用于制备含有大位阻芳基、有吸电子或推电子效应的杂环取代基的双端氨基硅醚化合物,能够高收率获得目标产物。
附图说明
下面结合附图,通过对本申请的具体实施方式详细描述,将使本申请的技术方案及其它有益效果显而易见。
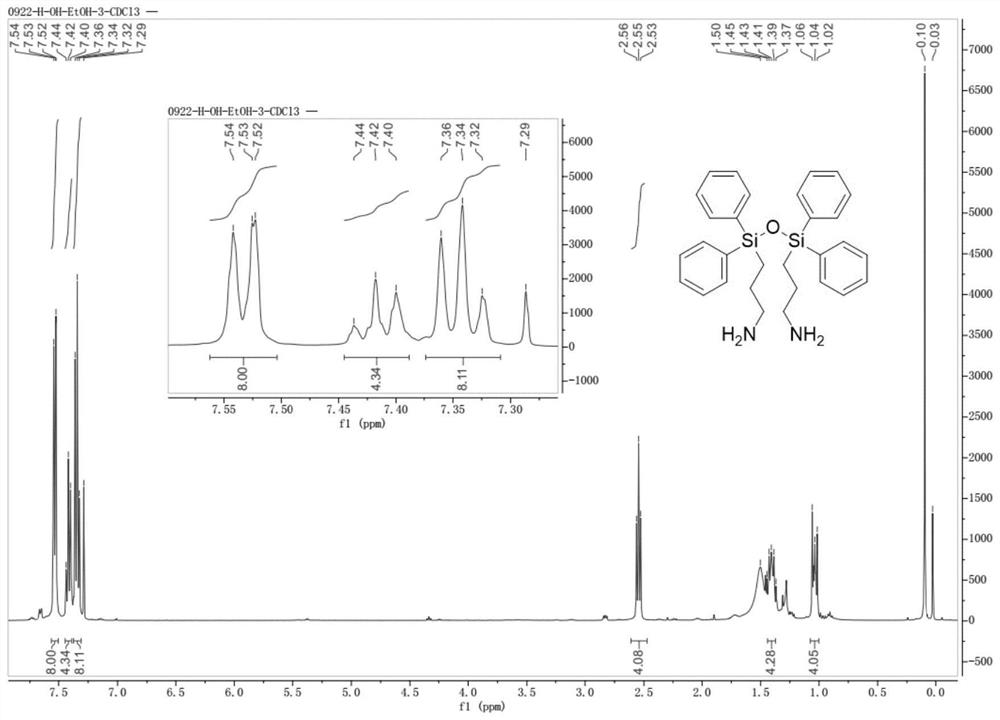
图1为本申请实施例1中制得的最终产物的核磁检测图谱。
具体实施方式
本申请旨在提供一种双端氨基硅醚化合物的制备方法及双端氨基硅醚化合物,其能够用于制备各种分子量大小的硅醚化合物,该制备方法尤其适用于制备含有大位阻芳基、有吸电子或推电子效应的杂环取代基的双端氨基硅醚化合物,能够高收率获得目标产物。
在本申请的描述中,以一范围型式的描述仅仅是因为方便及简洁,不应理解为对本发明范围的硬性限制;因此,应当认为范围型式的描述已经具体公开所有可能的子范围以及该范围内的单一数值,例如对于聚合度n的范围为1≤n≤10,应当认为从1到10的范围描述已经具体公开子范围,例如1≤n≤3、1≤n≤8、2≤n≤4、5≤n≤10等,以及所属范围内的单一数字,例如1、2、3、4、5、6、7、8、9以及10,此不管任何范围为何皆适用。另外,本申请中涉及的数值范围是指包括所指范围内的任何引用的数字(分数或整数)。
术语“聚合度”是指量聚合物分子大小的指标,以重复单元数为基准,聚合度用于表征聚合物大分子链上所含有的重复单元数目的数值,以n表示。可以理解的是,当制备的产物为双端氨基聚硅醚时,尤其是当双端氨基聚硅醚为分子量大于1500的高聚物时,该产物可以是由一组不同聚合度(处于一特定范围内)和不同结构形态的同系物的混合物所组成,该产物的聚合度n是指其平均聚合度。
本申请“双端氨基硅醚化合物”是指具有式(7)所示结构的化合物,其包括双端氨基硅醚和双端氨基聚硅醚,其中,“双端氨基硅醚”是指式(7)中聚合度n为1的化合物,例如1,3-双(3’-胺丙基)-1,1,3,3-四苯基二硅醚,又如1,3-双(3’-胺丙基)-1,3-二甲基-1,3-二苯基二硅醚;“双端氨基聚硅醚”是指式(7)中聚合度n为大于1的数值的化合物,例如1,5-双(3’-胺丙基)-聚二苯基硅醚,又如双(3-胺丙基)-多聚二苯基硅醚。对于式(7),其聚合度n例如满足条件1≤n≤10,或是满足条件1≤n≤100,或是满足条件100≤n≤1000,或是满足条件1000≤n≤10000。
式术语“脂肪烃基”是指只含碳、氢两种原子的官能团,为对应的烃失去一个氢原子后剩下的自由基,脂肪烃基为除芳烃基以外的所有烃基的总称,包括饱和脂肪烃基和不饱和脂肪烃基。“脂肪烃基”包含但不限于甲基、乙基、丙基、异丙基等。
术语“烷基”是指饱和脂肪烃基,其是烷烃分子中失去一个氢原子而形成的脂肪烃基。
术语“取代烷基”是指烷基上的一个氢原子或多个氢原子任选地被其他基团取代,其他基团例如可以是卤素原子,允许存在多重取代度。
术语“烷氧基”是指是指基团R
术语“烃氧基”是指R
术语“芳基”是指芳烃分子的芳核碳上去掉一个氢原子后,剩下一价基团的总称,通常用Ar-表示。“芳基”包括但不限于苯基。
术语“取代芳基”是指芳基上的一个氢原子或多个氢原子任选地被其他基团取代,其他基团例如可以是卤素原子,允许存在多重取代度。
术语“芳氧基”是指基团Ar-O-,其中Ar-为芳基。“芳氧基”包括但不限于苯氧基。
术语“取代芳氧基”是指芳氧基上的一个氢原子或多个氢原子任选地被其他基团取代,其他基团例如可以是卤素原子,允许存在多重取代度。
术语“杂环基”是指由碳原子及非碳原子构成的环状结构化合物失去一个氢原子而形成的基团,非碳原子例如可以是氮原子、氧原子、硫原子等。
术语“取代杂环基”是指杂环基上的一个氢原子或多个氢原子任选地被其他基团取代,其他基团例如可以是卤素原子,允许存在多重取代度。
术语“有机硼化合物”是指一类含有碳硼键或硼原子的有机化合物,例如能够参与硼氢化反应且含有硼原子的有机化合物,在本申请实施例中,有机硼化合物具有式(8)所示的结构。
除非另行定义,文中所使用的所有专业与科学用语与本领域技术人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明中。文中所述的较佳实施方法与材料仅作示范之用,但不能限制本申请的内容。
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过本领域已知方法制备。
本申请涉及一种双端氨基硅醚化合物的制备方法,所述制备方法包括如下步骤:
(A)将式(1)所示的化合物a与式(2)所示的化合物b进行亲核取代反应,以获得式(3)所示的化合物c;
(B)在碱性条件下,将化合物c、有机硼化合物以及过氧化氢混合进行加成反应,以获得式(4)所示的化合物d;
(C)将化合物d与式(5)所示的化合物e进行反应,以获得式(6)所示的化合物f;以及
(D)将化合物f进行去保护基还原反应,以获得式(7)所示的最终产物;
其中,式(1)至式(5)如下:
对式(1)至式(7)作出如下说明:
其中,n≥0,例如1≤n≤50,又如1≤n≤10。
R
R
R
R
R
在本申请的一些实施方式中,步骤(A)包括以下步骤:
(A1)将化合物a溶于有机溶剂以获得含有化合物a的有机溶液,然后在0℃以下的无水无氧条件下,将化合物b与含有化合物a的有机溶液相混合以进行亲核取代反应。
具体的,有机溶剂例如可以是甲醇、乙醇、异丙醇、乙酸乙酯、二氯甲烷、甲苯、四氢呋喃以及正己烷中的至少一种。
(A2)步骤(A1)中的亲核取代反应结束后,对反应物进行后处理,以获得所述化合物c。
具体的,步骤(A2)的后处理包括淬灭、洗涤、干燥、浓缩以及纯化操作。纯化例如可以是减压蒸馏、硅胶柱层析以及结晶纯化中的至少一种。
示例性的,步骤(A)具体为:(A1)将化合物a溶于四氢呋喃以获得含有1.0摩尔当量化合物b的四氢呋喃溶液;在无水无氧条件下,将2~8摩尔当量的化合物b冷却至0℃以下,缓慢滴加含有1.0摩尔当量化合物a的四氢呋喃溶液,采用高效液相色谱(HPLC)监控反应过程;(A2)步骤(A1)中的亲核取代反应结束后,向反应物中加入饱和氯化铵溶液或0.5mol/L的盐酸溶液进行淬灭,然后用饱和氯化钠溶液洗涤,再用无水硫酸镁干燥,并将有机相浓缩至无溶剂,最后在小于100Pa、10~120℃的条件下蒸馏纯化获得化合物c。
作为替代性的实施方案,步骤(A)具体为:(A1)将化合物a溶于四氢呋喃以获得含有1.0摩尔当量化合物b的四氢呋喃溶液;在无水无氧条件下,将2~8摩尔当量的化合物b冷却至0℃以下,缓慢滴加含有1.0摩尔当量化合物b的四氢呋喃溶液,采用高效液相色谱(HPLC)监控反应过程;(A2)步骤(A1)中的亲核取代反应结束后,向反应物中加入饱和氯化铵溶液或0.5mol/L的盐酸溶液进行淬灭,然后用饱和氯化钠溶液洗涤,再用无水硫酸镁干燥,并将有机相浓缩至无溶剂以获得浓缩物,将浓缩物过8~10倍的化合物a重量的硅胶后再次浓缩至无溶剂,然后加入甲醇、乙醇、异丙醇、乙酸乙酯、二氯甲烷、甲苯、四氢呋喃以及正己烷中的至少一种有机溶剂以结晶纯化获得化合物c。
在本申请的一些实施方式中,步骤(B)包括以下步骤:
(B1)将化合物c溶于有机溶剂以获得含有化合物c的有机溶液,然后在无水无氧条件下,将含有化合物c的有机溶液与有机硼化合物相混合以进行反应。
具体的,有机溶剂例如可以是甲醇、乙醇、异丙醇、乙酸乙酯、二氯甲烷、甲苯、四氢呋喃以及正己烷中的至少一种。在一些实施方式中,有机硼化合物与化合物c的摩尔比为(1~6):1。
在本申请的一些实施方式中,有机硼化合物如下面的式(8)所示:
在式(8)中,R
在一些实施方式中,有机硼化合物选自以下结构式所示化合物中的至少一种:
在一些实施方式中,有机硼化合物选自以下结构式所示化合物中的至少一种:
在一些实施方式中,步骤(B)包括以下步骤:
(B1)将所述化合物c溶于有机溶剂以获得含有化合物c的有机溶液,然后在无水无氧条件下,将所述含有化合物c的有机溶液与所述有机硼化合物相混合以进行反应;以及
(B2)当步骤(B1)中化合物c消耗完毕后,控温35℃以下,然后加入碱性试剂和过氧化氢以进行混合反应。
具体的,碱性试剂例如可以是碳酸氢钠、碳酸钠、碳酸钾、氢氧化钠以及氢氧化钾中的至少一种,优选为1mol/L~6mol/L的碳酸氢钠水溶液、碳酸钠水溶液、碳酸钾水溶液、氢氧化钠水溶液以及氢氧化钾水溶液中的至少一种。在一些实施方式中,碱性试剂与化合物c的摩尔比为(1~20):1。在一些实施方式中,碱性试剂与过氧化氢的体积比为1:1。
(B3)当步骤(B2)中的混合反应结束后,对反应物进行后处理,以获得化合物d。
具体的,步骤(A2)的后处理包括洗涤、干燥、浓缩以及纯化操作。纯化例如可以是减压蒸馏、硅胶柱层析以及结晶纯化中的至少一种。
示例性的,步骤(B)具体为:(B1)将1.0摩尔当量的化合物c溶于5~30倍体积的干燥的四氢呋喃中以获得含有化合物c的有机溶液,然后在无水无氧条件下,向含有化合物c的有机溶液中加入1~6摩尔当量的有机硼化合物,在回流下搅拌反应,采用HPLC监控整个反应过程;(B2)当步骤(B1)中化合物c消耗完毕后,控温35℃以下,向反应体系中加入1~20摩尔当量的碱性试剂,然后加入与碱性试剂等体积的30%(质量百分数)过氧化氢溶液,搅拌反应1~8h;(B3)当步骤(B2)的反应结束后,向反应物中加入饱和氯化钠溶液进行洗涤,再用无水硫酸镁干燥,并将有机相浓缩至无溶剂,最后在小于100Pa、10~100℃的条件下蒸馏纯化获得化合物d。其中,步骤(B3)中的蒸馏纯化操作可用硅胶柱层析和结晶纯化替代,具体参照上述步骤(A)的替代性实施方案进行。
在本申请的一些实施方式中,在步骤(C)中,在试剂g的作用下,化合物d与化合物e进行反应,其中,试剂g包括化合物g
其中,在式(9)中,R
在式(10)中,R
(C1)将所述化合物d、所述化合物e以及所述化合物g2相混合,然后溶于有机溶剂中,以获得有机混合溶液。
具体的,有机溶剂例如可以是甲醇、乙醇、异丙醇、乙酸乙酯、二氯甲烷、甲苯、四氢呋喃或正己烷中的至少一种。优选化合物e、化合物g
(C2)在10℃~100℃的保护气体氛围下,向步骤(C1)的有机混合溶液中加入化合物g
具体的,保护气体优选为氮气,化合物g
(C3)对步骤(C2)获得的反应物进行后处理,以获得化合物f。
具体的,步骤(C3)的后处理包括过滤除固体、洗涤、干燥、浓缩以及纯化操作。纯化例如可以是结晶纯化。
示例性的,步骤(C)具体为:(C1)将化合物d、化合物e以及化合物g
在本申请的一些实施方式中,步骤(D)中包括以下步骤:
(D1)将化合物f溶于有机溶剂中以获得含有化合物f的有机溶液,然后加入水合肼,在保护气体氛围下进行回流反应直至化合物f消耗完毕,以获得反应物。
具体的,有机溶剂例如可以是甲醇、乙醇、异丙醇、乙酸乙酯、二氯甲烷、甲苯、四氢呋喃以及正己烷中的至少一种。保护气体例如可以是氮气。例如化合物f与水合肼的摩尔比为1:(2~10)。
(D2)对步骤(D1)获得的反应物进行后处理,以获得式(7)所示的最终产物。
具体的,步骤(D2)的后处理包括过滤、浓缩以及纯化操作,其中,纯化例如可以是减压蒸馏、盐析纯化等。
示例性的,步骤(D)具体为:(D1)将化合物f溶于四氢呋喃中以获得含有化合物f的有机溶液,然后加入85%水合肼,其中,化合物f与85%水合肼的摩尔比为1:5,在50~120℃的氮气保护氛围下,回流反应3~24h,HPLC监控整个反应过程,直至化合物f消耗完毕结束反应,获得反应物;(D2)将步骤(D1)的反应物过滤以除去固体,然后将滤液浓缩至无有机溶剂,最后在小于100Pa、10~120℃的条件下蒸馏纯化获得最终产物。
作为步骤(D2)的替代性实施方案,步骤(D2)还可以为:将步骤(D1)的反应物过滤以除去固体,然后将滤液浓缩至无有机溶剂,接着加入10%(质量浓度)硫酸以成盐,过滤以收集滤饼,将滤饼溶于甲苯中并用碱性溶液调整pH为10~14,然后采用无水硫酸镁干燥有机相,再次过滤,并将滤液浓缩至无溶剂以获得最终产物。
以下结合各个具体的实施例对本申请的双端氨基硅醚化合物的制备方法进行详细说明。
实施例1:制备1,3-双(3’-胺丙基)-1,1,3,3-四苯基二硅醚
本实施例提供了一种1,3-双(3’-胺丙基)-1,1,3,3-四苯基二硅醚的制备方法,具体包括如下步骤:
S1、取一装有温度计、恒压滴液漏斗的三口瓶,在氮气的保护氛围下,向该三口瓶中加入600mL的1mol/L的烯丙基溴化镁(CAS号为1730-25-2,对应为化合物b),冷却至0℃;将100g(221.49mmol)的1,3-二氯-1,1,3,3-四苯基二硅醚(CAS号为7756-87-8,对应为化合物a)溶于300mL的无水四氢呋喃中以获得四氢呋喃溶液,将四氢呋喃溶液加入至滴液漏斗中,在搅拌的条件下缓慢滴加入烯丙基溴化镁以发生格氏反应,在整个格氏反应过程中,三口瓶内的温度不超过10℃,并且不断搅拌直至停止反应;反应结束后,向三口瓶内的反应物中缓慢滴加300mL的饱和氯化铵溶液淬灭,然后搅拌分液并取有机相;采用饱和氯化钠溶液洗涤有机相两次,每次消耗300mL的饱和氯化钠溶液,然后用无水硫酸镁干燥有机相,将干燥后的有机相浓缩至无溶剂以制得淡黄色的油状液体,最后将油状液体在80Pa的条件下减压蒸馏获得95.3g的无色油状液体(对应为化合物c),无色油状液体久置可得白色固体,收率为92.9%。
其中,步骤S1的反应式如下:
S2、取一装有温度计,恒压滴液漏斗和回流管的四口瓶,在氮气的保护氛围下,向该四口瓶中加入95g(205.30mmol,1摩尔当量)步骤S1制得的化合物c,并用950mL的无水四氢呋喃溶解以获得四氢呋喃溶液;向所述四氢呋喃溶液中分批次加入2.5摩尔当量的1mol/L硼烷四氢呋喃溶液(商购,CAS号为14044-65-6),然后加热整个混合体系以进行回流反应;当HPLC检测到回流反应体系中化合物c消耗完毕时,将整个回流反应体系降温至30℃,然后向其中缓慢滴加520mL的3mol/L碳酸钠水溶液,整个滴加过程中确保四口瓶内的温度不超过35℃;碳酸钠(Na
其中,步骤S2的反应式如下:
S3、另取一装有温度计、恒压滴液漏斗的三口瓶,在氮气的保护氛围下,向该三口瓶中加入89g(178.44mmol,1摩尔当量)步骤S2制得的化合物d,然后加入2.5摩尔当量的三环己基膦(CAS号为2622-14-2)和2.5摩尔当量的邻苯二甲酰亚胺(CAS号为85-41-6),再加入890mL的无水甲苯,使得化合物d、三环己基膦和邻苯二甲酰亚胺三者充分溶解于无水甲苯中以制得有机混合溶液;将有机混合溶液冷却至10℃,然后缓慢滴加2.5摩尔当量的偶氮二甲酸二异丙酯(CAS号为2446-83-5),滴加结束后将反应体系自然升温至室温,并在搅拌条件下反应直至化合物d消耗完毕,以获得反应物;将反应物过滤以除去固体,然后采用饱和氯化钠溶液洗涤有机相两次,每次消耗300mL的饱和氯化钠溶液,接着用无水硫酸镁干燥有机相,再将干燥后的有机相浓缩至无溶剂,最后用有机溶剂结晶纯化获得111.7g的化合物f,化合物f为白色固体,收率为82.6%,其中有机溶剂为四氢呋喃和石油醚按照1:3的体积比配制而成的混合剂。
其中,步骤S3的反应式如下:
S4、另取一装有温度计的三口瓶,向其中加入111g(146.63mmol,1摩尔当量)步骤S3制得的化合物f,然后加入1.1L的甲醇以使化合物f充分溶于甲醇中,获得含有化合物f的甲醇溶液;向含有化合物f的甲醇溶液中加入5当量的85%水合肼,然后在氮气的保护氛围下加热以进行回流反应,回流反应在搅拌条件下进行,直至所述化合物f消耗完毕而停止反应,获得反应物;将反应物自然降温至室温,然后过滤以除去固体,将滤液浓缩至无溶剂,最后在65Pa的条件下减压蒸馏获得64.6g的最终产物,收率为88.6%,最终产物的核磁检测结果为如图1所示,
其中,步骤S4的反应式如下:
实施例2:制备1,3-双(3’-胺丙基)-1,3-二甲基-1,3-二苯基二硅醚
本实施例提供了一种1,3-双(3’-胺丙基)-1,3-二甲基-1,3-二苯基二硅醚的制备方法,具体包括如下步骤:
S1、取一装有温度计、恒压滴液漏斗的三口瓶,在氮气的保护氛围下,向该三口瓶中加入400mL的1mol/L的烯丙基溴化镁(CAS号为1730-25-2,对应为化合物b),冷却至0℃;将50g(152.74mmol)的1,3-二氯-1,3-二甲基-1,3-二苯基二硅醚(CAS号为3582-72-7,对应为化合物a)溶于300mL的无水四氢呋喃中以获得四氢呋喃溶液,将四氢呋喃溶液加入至滴液漏斗中,在搅拌的条件下缓慢滴加入烯丙基溴化镁以发生格氏反应,在整个格氏反应过程中,三口瓶内的温度不超过10℃,并且不断搅拌直至停止反应;反应结束后,向三口瓶内的反应物中缓慢滴加150mL的饱和氯化铵溶液淬灭,然后搅拌分液并取有机相;采用饱和氯化钠溶液洗涤有机相两次,每次消耗200mL的饱和氯化钠溶液,然后用无水硫酸镁干燥有机相,将干燥后的有机相浓缩至无溶剂以制得淡黄色的油状液体,最后将油状液体在100Pa的条件下减压蒸馏获得46.7g的无色油状液体(对应为化合物c),收率为90.3%。
其中,步骤S1的反应式如下:
S2、取一装有温度计,恒压滴液漏斗和回流管的四口瓶,在氮气的保护氛围下,向该四口瓶中加入46g(135.85mmol,1摩尔当量)步骤S1制得的化合物c,并用460mL的无水四氢呋喃溶解以获得四氢呋喃溶液;向所述四氢呋喃溶液中分批次加入2.5摩尔当量的1mol/L硼烷四氢呋喃溶液(商购,CAS号为14044-65-6),然后加热整个混合体系以进行回流反应;当HPLC检测到回流反应体系中化合物c消耗完毕时,将整个回流反应体系降温至30℃,然后向其中缓慢滴加340mL的3mol/L碳酸钠水溶液,整个滴加过程中确保四口瓶内的温度不超过35℃;碳酸钠(Na
其中,步骤S2的反应式如下:
S3、另取一装有温度计、恒压滴液漏斗的三口瓶,在氮气的保护氛围下,向该三口瓶中加入43g(114.78mmol,1摩尔当量)步骤S2制得的化合物d,然后加入2.5摩尔当量的三环己基膦(CAS号为2622-14-2)和2.5摩尔当量的邻苯二甲酰亚胺(CAS号为85-41-6),再加入430mL的无水甲苯,使得化合物d、三环己基膦和邻苯二甲酰亚胺三者充分溶解于无水甲苯中以制得有机混合溶液;将有机混合溶液冷却至10℃,然后缓慢滴加2.5摩尔当量的偶氮二甲酸二异丙酯(CAS号为2446-83-5),滴加结束后将反应体系自然升温至室温,并在搅拌条件下反应直至化合物d消耗完毕,以获得反应物;将反应物过滤以除去固体,然后采用饱和氯化钠溶液洗涤有机相两次,每次消耗130mL的饱和氯化钠溶液,接着用无水硫酸镁干燥有机相,再将干燥后的有机相浓缩至无溶剂,最后用有机溶剂结晶纯化获得61.3g的化合物f,化合物f为白色固体,收率为84.4%,其中有机溶剂为四氢呋喃和石油醚按照1:5的体积比配制而成的混合剂。
其中,步骤S3的反应式如下:
S4、另取一装有温度计的三口瓶,向其中加入61g(96.39mmol,1摩尔当量)步骤S3制得的化合物f,然后加入610mL的甲醇以使化合物f充分溶于甲醇中,获得含有化合物f的甲醇溶液;向含有化合物f的甲醇溶液中加入5摩尔当量的85%水合肼,然后在氮气的保护氛围下加热以进行回流反应,回流反应在搅拌条件下进行,直至所述化合物f消耗完毕而停止反应,获得反应物;将反应物自然降温至室温,然后过滤以除去固体,将滤液浓缩至无溶剂,最后在90Pa的条件下减压蒸馏获得30.8g的最终产物,收率为85.7%,最终产物的核磁检测结果为:
其中,步骤S4的反应式如下:
实施例3:制备1,5-双(3’-胺丙基)-聚二苯基硅醚
本实施例提供了一种1,5-双(3’-胺丙基)-聚二苯基硅醚的制备方法,具体包括如下步骤:
S1、取一装有温度计、恒压滴液漏斗的三口瓶,在氮气的保护氛围下,向该三口瓶中加入380mL的1mol/L的烯丙基溴化镁(CAS号为1730-25-2,对应为化合物b),冷却至0℃;将90g(138.51mmol)的1,5-二氯-聚二苯基硅醚(CAS号为7756-88-9,对应为化合物a)溶于300mL的无水四氢呋喃中以获得四氢呋喃溶液,将四氢呋喃溶液加入至滴液漏斗中,在搅拌的条件下缓慢滴加入烯丙基溴化镁以发生格氏反应,在整个格氏反应过程中,三口瓶内的温度不超过10℃,并且不断搅拌直至停止反应;反应结束后,向三口瓶内的反应物中缓慢滴加270mL的饱和氯化铵溶液淬灭,然后搅拌分液并取有机相;采用饱和氯化钠溶液洗涤有机相两次,每次消耗270mL的饱和氯化钠溶液,然后用无水硫酸镁干燥有机相,将干燥后的有机相浓缩至无溶剂以制得淡黄色的油状液体,再将油状液体溶于900mL的有机溶剂中,其中有机溶剂为石油醚和二氯甲烷按照3:1的体积比配制而成的混合剂,然后过720g的硅胶柱纯化,最后将纯化后的有机相浓缩至无溶剂,获得80.7g的化合物c,化合物c为白色固体,收率为88.1%。
其中,步骤S1的反应式如下:
S2、取一装有温度计,恒压滴液漏斗和回流管的四口瓶,在氮气的保护氛围下,向该四口瓶中加入80g(121.02mmol,1摩尔当量)步骤S1制得的化合物c,并用800mL的无水四氢呋喃溶解以获得四氢呋喃溶液;向所述四氢呋喃溶液中分批次加入2.5摩尔当量的1mol/L硼烷四氢呋喃溶液(商购,CAS号为14044-65-6),然后加热整个混合体系以进行回流反应;当HPLC检测到回流反应体系中化合物c消耗完毕时,将整个回流反应体系降温至30℃,然后向其中缓慢滴加300mL的3mol/L碳酸钠水溶液,整个滴加过程中确保四口瓶内的温度不超过35℃;碳酸钠(Na
其中,步骤S2的反应式如下:
S3、另取一装有温度计、恒压滴液漏斗的三口瓶,在氮气的保护氛围下,向该三口瓶中加入70g(100.42mmol,1摩尔当量)步骤S2制得的化合物d,然后加入2.5摩尔当量的三环己基膦(CAS号为2622-14-2)和2.5摩尔当量的邻苯二甲酰亚胺(CAS号为85-41-6),再加入700mL的无水甲苯,使得化合物d、三环己基膦和邻苯二甲酰亚胺三者充分溶解于无水甲苯中以制得有机混合溶液;将有机混合溶液冷却至10℃,然后缓慢滴加2.5摩尔当量的偶氮二甲酸二异丙酯(CAS号为2446-83-5),滴加结束后将反应体系自然升温至室温,并在搅拌条件下反应直至化合物d消耗完毕,以获得反应物;将反应物过滤以除去固体,然后采用饱和氯化钠溶液洗涤有机相两次,每次消耗210mL的饱和氯化钠溶液,接着用无水硫酸镁干燥有机相,再将干燥后的有机相浓缩至无溶剂,最后用有机溶剂结晶纯化获得83.5g的化合物f,化合物f为白色固体,收率为87.1%,其中有机溶剂为乙酸乙酯和石油醚按照1:4的体积比配制而成的混合剂。
其中,步骤S3的反应式如下:
S4、另取一装有温度计的三口瓶,向其中加入83g(86.88mmol,1当量)步骤S3制得的化合物f,然后加入830mL的甲醇以使化合物f充分溶于甲醇中,获得含有化合物f的甲醇溶液;向含有化合物f的甲醇溶液中加入5当量的85%水合肼,然后在氮气的保护氛围下加热以进行回流反应,回流反应在搅拌条件下进行,直至所述化合物f消耗完毕而停止反应,获得反应物;将反应物自然降温至室温,然后过滤以除去固体,将滤液浓缩至无溶剂以获得淡黄粘稠油状液体,向该油状液体中加入200mL的10%(质量分数)硫酸以成盐,然后过滤收集滤饼,再将滤饼溶于无水甲苯中,并用饱和氢氧化钾水溶液调整pH至10,取有机相用无水硫酸镁干燥,然后再次过滤以收集滤液,再将滤液浓缩至无溶剂以获得白色块状固体的最终产物,收率为80.6%,最终产物的核磁检测结果为:
其中,步骤S4的反应式如下:
实施例4:制备双(3’-胺丙基)-多聚二苯基硅醚
本实施例提供了一种双(3’-胺丙基)-多聚二苯基硅醚的制备方法,其中,双(3’-胺丙基)-多聚二苯基硅醚的聚合度n为5~10,具体包括如下步骤:
S1、取一装有温度计、恒压滴液漏斗的三口瓶,在氮气的保护氛围下,向该三口瓶中加入250mL的1mol/L的烯丙基溴化镁(CAS号为1730-25-2,对应为化合物b),冷却至0℃;将100g(152.74mmol)的二氯-多聚二苯基硅醚(对应为化合物a,商购获得)溶于1000mL的无水四氢呋喃中以获得四氢呋喃溶液,将四氢呋喃溶液加入至滴液漏斗中,在搅拌的条件下缓慢滴加入烯丙基溴化镁以发生格氏反应,在整个格氏反应过程中,三口瓶内的温度不超过10℃,并且不断搅拌直至停止反应;反应结束后,向三口瓶内的反应物中缓慢滴加200mL的饱和氯化铵溶液淬灭,然后搅拌分液并取有机相;采用饱和氯化钠溶液洗涤有机相两次,每次消耗500mL的饱和氯化钠溶液,然后用无水硫酸镁干燥有机相,将干燥后的有机相浓缩至无溶剂以制得淡黄色固体,将该淡黄色固体溶于2000mL的有机溶剂以获得有机溶液,其中有机溶剂为四氢呋喃和石油醚按照1:1的体积比配制而成的混合剂,然后将该有机溶液过800g的硅胶柱以纯化,最后将纯化收集的有机相浓缩至无溶剂以获得90.6g的化合物c(CAS号为97947-62-1),化合物c为白色固体。
其中,步骤S1的反应式如下:
S2、取一装有温度计,恒压滴液漏斗和回流管的四口瓶,在氮气的保护氛围下,向该四口瓶中加入90g的步骤S1制得的化合物c,并用900mL的无水四氢呋喃溶解以获得四氢呋喃溶液;向所述四氢呋喃溶液中分批次加入2.5摩尔当量的1mol/L硼烷四氢呋喃溶液(商购,CAS号为14044-65-6),然后加热整个混合体系以进行回流反应;当HPLC检测到回流反应体系中化合物c消耗完毕时,将整个回流反应体系降温至30℃,然后向其中缓慢滴加200mL的3mol/L碳酸钠水溶液,整个滴加过程中确保四口瓶内的温度不超过35℃;碳酸钠(Na
其中,步骤S2的反应式如下:
S3、另取一装有温度计、恒压滴液漏斗的三口瓶,在氮气的保护氛围下,向该三口瓶中加入87g的步骤S2制得的化合物d,然后加入55g的三环己基膦(CAS号为2622-14-2)和30g的邻苯二甲酰亚胺(CAS号为85-41-6),再加入870mL的无水甲苯,使得化合物d、三环己基膦和邻苯二甲酰亚胺三者充分溶解于无水甲苯中以制得有机混合溶液;将有机混合溶液冷却至10℃,然后缓慢滴加35g的偶氮二甲酸二异丙酯(CAS号为2446-83-5),滴加结束后将反应体系自然升温至室温,并在搅拌条件下反应直至化合物d消耗完毕,以获得反应物;将反应物过滤以除去固体,然后采用饱和氯化钠溶液洗涤有机相两次,每次消耗300mL的饱和氯化钠溶液,接着用无水硫酸镁干燥有机相,再将干燥后的有机相浓缩至无溶剂,获得100.3g的化合物f,化合物f为米白色固体。
其中,步骤S3的反应式如下:
S4、另取一装有温度计的三口瓶,向其中加入100g的步骤S3制得的化合物f,然后加入500mL的异丙醇以使化合物f充分溶于异丙醇中,获得含有化合物f的异丙醇溶液;向含有化合物f的异丙醇溶液中加入20mL的85%水合肼,然后在氮气的保护氛围下加热以进行回流反应,回流反应在搅拌条件下进行,直至所述化合物f消耗完毕而停止反应,获得反应物;将反应物自然降温至室温,然后过滤以除去固体,将滤液浓缩至无溶剂以获得淡黄油状液体,向该油状液体中加入300mL的10%(质量分数)硫酸并剧烈搅拌以成盐,然后过滤收集滤饼,再将滤饼溶于无水甲苯中,并用饱和氢氧化钾水溶液调整pH至10,取有机相用无水硫酸镁干燥,然后再次过滤以收集滤液,再将滤液浓缩至无溶剂以获得54.9g米白色块状固体的最终产物,最终产物的核磁检测结果为:
其中,步骤S4的反应式如下:
以上对本申请实施例所提供的一种双端氨基硅醚化合物的制备方法及双端氨基硅醚化合物,进行了详细介绍。本文中使用了具体个例对本申请的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本申请的技术方案及其核心思想;本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的脱离本申请各实施例的技术方案的范围。
- 双端氨基硅醚化合物的制备方法及双端氨基硅醚化合物
- 用基于双(氨基-3-丙基)醚或(氨基-2-乙基)-(氨基-3-丙基)醚的吸收剂溶液从气态流出物除去酸化合物的方法