一种达可替尼杂质及其制备方法
文献发布时间:2023-06-19 12:18:04
技术领域
本发明属于药品质量控制领域,具体涉及一种达可替尼杂质及其制备方法和检测方法。
背景技术
Dacomitinib(中文名:达可替尼,又叫达克替尼)是美国辉瑞公司(Pfizer)研制的第二代、不可逆的EGFR酪氨酸激酶抑制剂(TKI),该药作用机制类似阿法替尼,能不可逆抑制三种不同ERBB家族分子成员,包括EGFR(HER1),HER2和HER4。Dacomitinib于2018年9月27日获美国FDA批准上市,商品名为
Dacomitinib结构式如下:
公开号为CN112707845A的中国专利申请(申请人为本发明的申请人)报道了达克替尼的关键中间体(式P05-3A所示)的制备方法:以2-氨基-4-氟苯甲酸(式P05-11B)为原料,先进行乙酰化反应得到P05-11B-1A,再进行硝化反应得到P05-11B-2A,接着进行成醚反应同时脱去乙酰基保护得到P05-10A-3A,然后进行酰胺化反应得到P05-10A-4A,再经三氯氧磷脱水得到P05-4A,最后与N,N-二甲基甲酰胺二甲基缩醛反应到达克替尼的关键中间体(式P05-3A)。反应流程如下:
利用上述专利申请中报道的方法能够成功制得达克替尼的关键中间体(式P05-3A),该方法反应条件温和,反应时间短,产物收率较高。但是,申请人在进一步的研究中发现,在达克替尼的关键中间体(式P05-3A)的制备过程中会产生一种工艺杂质(命名为杂质b),影响达克替尼的关键中间体(式P05-3A)的纯度和质量。该杂质b的产生是因为步骤5残留的P05-10A-4A在进行下一步反应(步骤6)时先生成杂质a,由于酰胺基的存在,生成的杂质a易环化而转化成杂质b。路线如下:
本领域技术人员公知的,原料药或成品药物中的杂质含量对人体用药安全有显著影响。在原料药或成品药物的研究、生产、贮存和临床应用中,也必须保持药物的纯度,降低药物的杂质含量,这样才能保证药物的有效性和安全性。药物中含有的杂质是影响药物纯度的主要因素,如果药物中含有超过限量的杂质,就有可能使药物的理化常数变动,外观性状产生变异,并影响药物的稳定性;杂质增多也必然使药物的含量偏低或活性降低,毒副作用增加。因此,药物的杂质检查是控制药物纯度,提高药品质量的一个非常重要的环节。
为了保证商业化产品质量,完善达克替尼及其关键中间体的质量控制方法,提高达克替尼及其关键中间体的纯度和质量,亟需研究出制备杂质b的方法,将制得的杂质b作为标准品对达克替尼及其关键中间体进行质量控制。
发明内容
本发明的一个目的在于提供一种达可替尼新杂质,以及该杂质的制备方法和检测方法。本发明的另一个目的在于提供一种提高达可替尼及其关键中间体纯度的方法。
本发明具体提供了以下化合物或其药学上可接受的盐:
本发明还提供了上述化合物的制备方法,所述方法包括以下步骤:以化合物P05-10A-4A和N,N-二甲基甲酰胺二甲基缩醛为原料进行反应,制得杂质b:
进一步地,化合物P05-10A-4A和N,N-二甲基甲酰胺二甲基缩醛的摩尔比为1:(1~5);
所述反应的溶剂为有机溶剂;
化合物P05-10A-4A与有机溶剂的质量体积比为1:(5~20)g/mL;
所述反应的温度为80~120℃,时间为1~5h。
进一步地,化合物P05-10A-4A和N,N-二甲基甲酰胺二甲基缩醛的摩尔比为1:1.5;
所述有机溶剂为N,N-二甲基甲酰胺;
化合物P05-10A-4A与有机溶剂的质量体积比为1:10g/mL;
所述反应的温度为100℃,时间为3h。
进一步地,所述方法还包括以下提纯步骤:反应结束后,向反应液中加入水,析出固体,过滤得粗品,然后加入正庚烷,加热回流,然后降至室温搅拌,过滤,取固体干燥。
本发明还提供了一种检测达可替尼或其中间体中杂质b含量的方法,所述方法包括以下步骤:
(1)制备供试品溶液:称取达可替尼或其中间体,用溶剂溶解,得到供试品溶液;
(2)制备对照品溶液:称取杂质b,用溶剂溶解,得到对照品溶液;杂质b的结构为
(3)进样检测:分别吸取供试品溶液、对照品溶液,利用高效液相色谱法进行检测,色谱条件如下:
色谱柱:以十八烷基硅烷键合硅胶为填充剂;
流动相:由流动相A和流动相B组成,其中,流动相A为磷酸二氢钾溶液,流动相B为乙腈。
进一步地,所述达可替尼中间体的结构为
进一步地,步骤(1)和步骤(2)中,所述溶剂为乙腈和水的混合液体;
和/或,步骤(3)所述磷酸二氢钾溶液的浓度为0.005~0.02M;
和/或,步骤(3)所述检测的波长为220~240nm,柱温为30~45℃,流速为0.5~2.0mL/min。
进一步地,所述乙腈和水的混合液体中,乙腈和水的体积比为1∶9;
和/或,所述磷酸二氢钾溶液的浓度为0.01M,pH值为8.0;
和/或,所述检测的波长为230nm,柱温为40℃,流速为1mL/min。
进一步地,步骤(3)中,所述流动相的梯度洗脱条件为:
其中,流动相A和流动相B的百分数为体积百分数。
本发明还提供了一种提高达可替尼或其中间体纯度的方法,所述方法包括以下步骤:
(a)制备达可替尼或其中间体溶液:称取达可替尼或其中间体,用溶剂溶解,得到达可替尼或其中间体溶液;所述达可替尼中间体的结构为
(b)制备对照品溶液:称取杂质b,用溶剂溶解,得到对照品溶液;杂质b的结构为
(c)分别吸取达可替尼或其中间体溶液、对照品溶液,注入高效液相色谱柱进行分离,将达可替尼或其中间体溶液中与对照品溶液保留时间相同的组分除去,收集剩余组分;
其中,高效液相色谱柱的色谱条件如上所述,溶剂如上所述。
本发明首次发现达克替尼关键中间体(式P05-3A)的制备过程中会产生新杂质:杂质b,并提供了该新杂质的制备方法。以本发明制得的杂质b为标准品,能够检测达克替尼关键中间体中杂质b的含量,本发明为达克替尼关键中间体的质量控制提供了一种新的方法,为达克替尼的安全用药提供了保证。
另外,以本发明制得的杂质b为对照品,还能够除去达克替尼关键中间体中的杂质b,进一步提高达克替尼关键中间体的纯度,提高达克替尼及其关键中间体的产品质量。
此外,根据本发明制得的杂质b的结构和理化性质,能够通过优化达克替尼关键中间体的制备工艺来减少杂质b的产生,从源头上减少达克替尼关键中间体中杂质b的含量。
本发明杂质b的制备方法原料易得,合成路线短,操作简单,可以为达克替尼及其关键中间体的质量研究提供杂质对照品。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
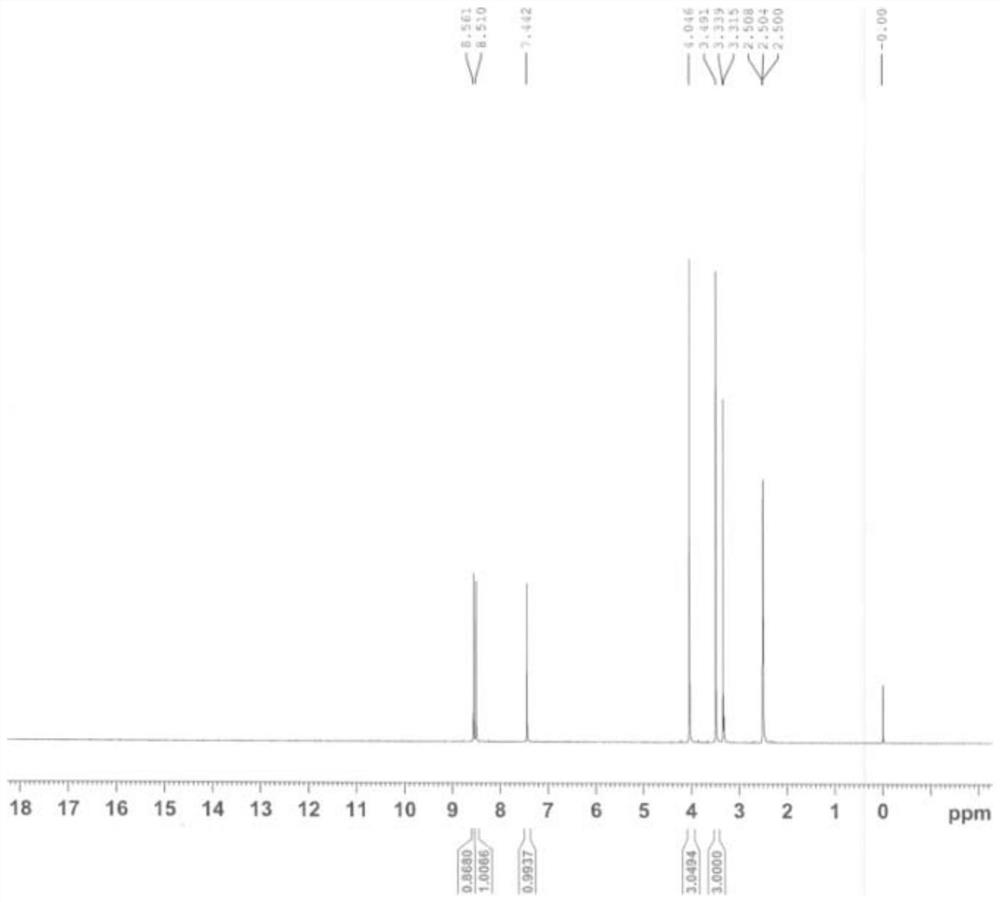
图1:本发明实施例1制得的杂质b的
图2:本发明实施例1制得的杂质b的LCMS谱图。
图3:达克替尼关键中间体中杂质b含量的检测结果。
具体实施方式
如无特别说明,本发明所用原料与设备均为已知产品,通过购买市售产品所得。
式化合物P05-10A-4A是按照公开号为CN112707845A的中国专利申请实施例1记载的方法制得的。
实施例1:杂质b的制备方法
按照下述合成路线,制备杂质b:
将P05-10A-4A(5.0g,0.0237mol)、N,N-二甲基甲酰胺二甲基缩醛(4.23g,0.0355mol)和N,N-二甲基甲酰胺(简称DMF,50mL)加入到反应瓶中,加热至100℃反应3h,TLC监控反应完全。降温至室温,向反应液中加入100ml水,析出固体,过滤得粗品。加入正庚烷,加热回流30min,降至室温搅拌1h,过滤,烘干,得到杂质b(4.74g,收率85%),HPLC纯度99.79%。
杂质b的结构表征如图1和图2所示:
LCMS:m/z 236.2[M+H]
实施例2:检测达克替尼关键中间体中杂质b含量的方法
1、检测方法:
(1)制备供试品溶液:按照公开号为CN112707845A的中国专利申请实施例1记载的方法制得达克替尼关键中间体(式P05-3A):
取上述制得的式P05-3A所示化合物,精密称定,加稀释剂[水:乙腈=90:10(v/v)的混合溶液]超声溶解并稀释制成每1mL中含1.0mg式P05-3A所示化合物的溶液,作为供试品溶液。
(2)制备对照品溶液:取本发明实施例1制得的杂质b,精密称定,加稀释剂[水:乙腈=90:10(v/v)的混合溶液]超声溶解并稀释制成每1mL中含1.0mg杂质b的溶液,作为对照品溶液。
(3)进样检测:分别吸取供试品溶液、对照品溶液各10μL注入液相色谱仪,利用高效液相色谱法(中国药典2015年版四部通则0512)进行检测。色谱条件如下:
色谱柱:用十八烷基硅烷键合硅胶为填充剂,ODS-3(4.6mm×250mm,5μm);
流动相:A相:0.01M磷酸氢二钾(pH=8.0),B相:乙腈;
检测波长:230nm;
流速:1mL/min;
柱温:40℃;
进样量:10μL;
进样浓度:1.0mg/mL;
梯度洗脱条件如表1所示。
表1梯度洗脱条件
2、检测结果
结果如图3所示,杂质b的保留时间为23.927min,达克替尼关键中间体(式P05-3A)的保留时间为28.468min。计算得出,受检达克替尼关键中间体中,杂质b的含量为2.28wt.%。
上述实验表明,以本发明制得的杂质b为标准品,利用本发明的检测方法,能够检测达克替尼关键中间体(式P05-3A)中杂质b的含量。
实施例3:提高达克替尼关键中间体纯度的方法
(1)按照本发明实施例2步骤(1)的方法制备供试品溶液,作为待提纯的达可替尼关键中间体溶液;
(2)按照本发明实施例2步骤(1)的方法制备供试品溶液;
(3)分别吸取供试品溶液、对照品溶液,注入高效液相色谱柱,利用高效液相色谱法进行分离。色谱条件同实施例2。
将待提纯的达可替尼关键中间体溶液中保留时间为23.927min的组分去除后,收集保留时间为28.468min的组分,浓缩干燥,即得到高纯度的达克替尼关键中间体(式P05-3A)。
上述实验表明,以本发明制得的杂质b为对照品,利用本发明的色谱条件,能够除去达克替尼关键中间体(式P05-3A)中的杂质b,进一步提高达克替尼关键中间体(式P05-3A)的纯度。
综上,本发明首次发现达克替尼关键中间体(式P05-3A)的制备过程中会产生新杂质:杂质b,并提供了该新杂质的制备方法。以本发明制得的杂质b为标准品,能够检测达克替尼关键中间体中杂质b的含量,本发明为达克替尼关键中间体的质量控制提供了一种新的方法。以本发明制得的杂质b为对照品,还能够除去达克替尼关键中间体中的杂质b,进一步提高达克替尼关键中间体的纯度,为制备高纯度的达克替尼及其关键中间体提供了一种新的方法。
- 一种达可替尼降解杂质化合物、制备方法及应用
- 一种达可替尼杂质及其制备方法